

Abstract
Our prior studies demonstrated that exogenous supplements of pure hyaluronan (HA) tetramers (HA4) dramatically upregulate elastin matrix synthesis by adult vascular smooth muscle cells (SMCs). Some studies suggest that exogenous HA likely only transiently contacts and signals cells, and may elicit different cell responses when presented on a substrate (e.g., scaffold surface). To clarify such differences, we used a carbodiimide-based chemistry to tether HA4 onto glass, and compared elastin matrix synthesis by SMCs cultured on these substrates, with those cultured with equivalent amounts of exogenous HA4. Tethered HA4-layers were first characterized for homogeneity, topography, and hydrolytic stability using SEM, XPS, AFM, and FACE. In general, mode of HA4 presentation did not influence its impact on SMC proliferation, or cell synthesis of tropoelastin and matrix elastin, relative to non-HA controls; however, surface-tethered HA4 stimulated SMCs to generate significantly greater amounts of elastin-stabilizing desmosine crosslinks, which partially accounts for the greater resistance to enzymatic breakdown of elastin derived from these cultures. Elastin derived from both sets of cultures contained peptide masses that correspond to the predominant peptides present in rat aortic elastin. SEM and TEM showed that HA4 stimulated fibrillin-mediated elastin matrix deposition, and organization into fibrils. Surface-immobilized HA4 was particularly conducive to organization of elastin into aggregating fibrils, and their networking to form closely-woven sheets of elastin fibers, as seen in cardiovascular tissues. The results suggest that incorporation of elastogenic HA4 mers onto cell culture substrates or scaffolds is a better approach than exogenous supplementation for in vitro or in vivo regeneration of architecturally and compositionally faithful-, and more stable mimics of native vascular elastin matrices.
1. Introduction
Elastin is a crucial structural protein distributed in the extracellular matrix of blood vessels. Vascular smooth muscle cells (SMCs) typically synthesize elastin as soluble tropoelastin, which is then post-translationally crosslinked by lysyl oxidase into a structural matrix. Vascular elastin allows blood vessels to recoil to their original dimensions during diastole to propel blood forward, and also vitally regulates cell-signaling pathways involved in morphogenesis, injury response, inflammation, and tissue calcification [1-5]. The preservation or restoration of vascular elastin, when it is degraded by disease, or when congenitally absent or malformed, is thus crucial to reinstating vascular homeostasis.
Current elastin preservative strategies aim to (a) protect existing elastin against degradation by matrix metalloproteinases (MMP) [6] and elastases [7], (b) replace lost elastin structures (e.g., with synthetic elastomers [8, 9], peptide-derived elastomers [10, 11], allogeneic elastomers [12]), or (c) regenerate elastin matrices by providing appropriate elastogenic cues (e.g., scaffolds, growth factors). However these approaches have thus far met with only limited success due to non-identification of suitable biochemical cues that can upregulate inherently poor tropoelastin synthesis by adult vascular cells. Our approach thus focuses on the use of scaffolds composed of a sub-set of ECM molecules, namely, glycosaminoglycans (GAGs), that are purported to facilitate elastogenesis in vivo, and which may evoke integrin-ECM interactions to preserve the native cell phenotype.
Specifically, hyaluronan (HA), a non-sulfated GAG, has been implicated in the synthesis and organization of microfibrils (fibrillin), a precursor for elastic fiber deposition [13] and has also been indirectly linked to elastin synthesis through its intimate binding of versican [14]. Via these interactions, HA may associate with microfibrillar proteins (fibulin-1, 2) and elastin-associated proteins to form higher-order macromolecular structures important for elastic fiber assembly [15]. In previous publications, we reported successful synthesis and recruitment of elastin by SMCs seeded atop bioactivated HA hydrogels containing a surface-mixture of bio-inert high molecular weight (HMW) HA (>1 MDa) and shorter, more bioactive HA fragments (including oligomers) generated by UV exposure [16]. When cultured with exogenous HA of defined molecular weights [17, 18], we showed the elastogenic effect of HA on SMCs to be size- and dose- dependant [17]. Exogenous supplements of HA tetramers (MW ∼ 756 Da) up-regulated elastin synthesis by SMCs and enhanced the formation of an elastin fiber-rich matrix [18]. The quality of the elastin matrix however, differed from that generated by SMCs atop the bioactivated hydrogels, where dense elastic fiber networks were formed. These qualitative differences in elastin matrix structure raise questions as to whether the mode of presentation of HA to SMCs (i.e. exogenous supplementation vs. as a substrate) differentially impacts tropoelastin synthesis, the incorporation of elastin into the cell layer, and the ultrastructural organization and stability of this elastin matrix.
Studies have suggested that exogenous HA only transiently signals cells and may inadequately simulate the response of cells grown on HA substrates [19]; thus, the true effects of HA scaffolds on cell response might be better predicted by long-term cell contact with surface-immobilized HA. Therefore, investigation of the differential effects of exogenous vs. surface-tethered HA on SMC elastin synthesis is important to establish the validity of an exogenous supplementation model. A 2-D model of surface- immobileized, uncrosslinked HA will also eliminate other influencing parameters associated with a 3-D scaffolding architecture (e.g., crosslinking, derivatization, inter-polymer networks) [20-22] enabling us to directly compare its elastogenic effects with that of exogenous HA. In this study, we used a previously developed carbodiimide chemistry to tether HA tetramers onto a glass substrate [23], and compared the elastin matrix generated by SMCs cultured on these substrates, with those cultured with exogenous HA tetramers (HA4).
2. Materials and Methods
2.1 Preparation of HA tetramers (HA4)
HA tetramers (756 Da; 75% w/w), which we found previously to elastogenically stimulate SMCs, were prepared by an enzymatic digestion protocol wherein HMW HA was broken down [23]. Briefly, HA (MW ∼ 1.5 × 106 Da; 20 mg; Sigma-Aldrich, St. Louis, MO) was enzymatically digested (3.6 mg testicular hyaluronidase, 451 U/ mg) in 4 ml of digest buffer (150 mM NaCl, 100 mM CH3COONa, 1mM Na2-EDTA, pH 5.0) at 37 °C for 18 hours. Enzyme activity was terminated by boiling the mixture in a water bath for 5 minutes following digestion. The mixture was dialyzed in water (12 hours), freeze dried, and stored at −20 °C until use. The mixtures were analyzed with Poly-Acrylamide Gel Electophoresis (PAGE) and Matrix Assisted Laser Desorption/ Ionization Time-Of-Flight (MALDI-TOF) spectroscopy to confirm HA oligomer size distribution and relative abundance within the digests. The enzymatic digests primarily containing HA tetramers will be henceforth referred to as HA4.
2.2 Preparation and characterization of HA4- tethered surfaces
HA4 was immobilized onto 4-well glass chamber slides (Nalgene Nunc International, Napersville, IL) and characterized using methods we described in a prior article [23]. Briefly, glass chamber slides were incubated (1 h) in 1 M NaOH, rinsed with DI water and 95% v/v ethanol (Sigma, St Louis, MO), and then reacted with 1 ml of a 3% v/v solution of 3-aminopropyl-trimethoxysilane (APTMS; Fluka Chemical Corp., Milwaukee, WI) in 95% v/v ethanol (30 min, 20 °C) while agitated on an orbital shaker. The slides were then briefly rinsed in ethanol and dried under a steady stream of argon gas, then heated in an oven (1 hr, 115 °C), again rinsed three times with 95% v/v ethanol, and dried under argon.
The exposed amine groups of APTMS were covalently reacted with the carboxyl groups present on HA, using a carbodiimide reaction. Briefly, HA4 (3 mg/ ml) was prepared with 200 mM 1-Ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride (EDC; Pierce Biotechnology Inc., Rockford, IL) and 100 mM N-hydroxysuccinimide (NHS; Pierce Biotechnology Inc.). A 1 ml solution aliquot was applied to each well and allowed to react for 16 hours with constant shaking. The slides were then soaked in a bath of DI water for 2 hours and finally air-dried under a steady stream of argon gas.
The immobilization of amine groups was confirmed using a spectrophotometric s-STDB assay (λ = 498 nm). Successful immobilization of HA4 was demonstrated using X-ray photoelectron spectroscopy (XPS), with high resolution of the C1 peak; likewise, the immobilization of homogenous layers of HA4 was confirmed using scanning electron microscopy (SEM) and atomic force microscopy (AFM). Finally, the hydrolytic stability of coated HA4 layers was confirmed by quantifying the amount of HA4 retained on the glass surfaces after 1,14, and 21 days of incubation with culture medium, using Flurorphore Assisted Carbohydrate Electrophoresis (FACE) analysis.
2.3 Cell Culture
Low Passage SMCs (P4−8) were obtained by passaging primary cells isolated from adult rat aortal explants. Stock cells were trypsinized (0.25% trypsin/ 0.1% v/v EDTA; Invitrogen), pelletted by centrifugation (500g, 7 min), re-suspended in DMEM: F12 containing 10% v/v FBS and 1% v/v penicillin-streptomycin and seeded onto single-well HA4 coated glass slides (Nalge NUNC International, Naperville, IL; Culture area = 8 cm2) at a density of 104 cells/ cm2. Other untreated wells were identically seeded with SMCs, which were then cultured with exogenous of HA4 at a concentration (4 μg/ well) equivalent to the amount immobilized. The negative controls were SMCs seeded on uncoated glass surfaces and cultured with HA-free medium. In all cases, cells were harvested at 21 days of culture (n = 9 wells/case). Spent medium was replaced twice weekly during the duration of culture, and pooled at the end of the 21-day culture period, The cell layers were harvested at the same time.
2.4 DNA assay for cell proliferation
The DNA content of SMCs cultured with exogenous or surface-tethered HA4 or in their absence (control), were compared to determine impact of HA4, and of their mode of presentation, on cell proliferation. The measured amounts of synthesized matrix were also normalized to the DNA content to provide a reliable basis of comparison between samples. Broadly, the cell layers were harvested at 1 and 21 days of culture, sonicated, and DNA contents quantified using the fluorometric method described by Labarca and Paigen [24]. Actual cell count were then calculated on the basis of 6 pg DNA/cell.
2.5 Fastin assay for elastin
A Fastin assay (Accurate Scientific and Chemical Corporation, Westbury, NY) was used to quantify the total amount of elastin within cell layers (matrix elastin), and that released into the culture medium as a soluble precursor (tropoelastin, E1). For each condition, tropoelastin in the spent medium was collected and pooled over 21 days of culture and frozen at −20 °C. To isolate matrix elastin, cell layers were thoroughly scrapped into NaCl/ Pi buffer and centrifuged (2500 RPM, 10 min). The supernatant was removed and the remaining cell pellet was digested with 0.1 N NaOH (98 °C, 1 h), and centrifuged (10,000g, 10 min) to yield an insufficiently crosslinked, alkali soluble fraction (E2), and a mature, mostly highly crosslinked, insoluble pellet (E3). Since the Fastin assay quantifies only soluble α-elastin, the insoluble elastin was first converted to a soluble form prior to assay. To do this, the elastin pellet (E3) was dried to a constant weight, solubilized with three cycles of treatment with 0.25 N oxalic acid (1 h/cycle, 95 °C), and the pooled digests then filtered in microcentrifuge tubes fitted with low molecular weight (10 kDa) cut-off membranes. All three elastin components (E1 - tropoelastin, E2 – loosely crosslinked elastin, E3 – densely crosslinked elastin) were quantified using the Fastin assay. The weight of insoluble elastin using the Fastin assay was compared to that determined by weighing the dry elastin pellet (E3), to affirm low/ no loss of elastin material during matrix processing. In each case, the ratio of matrix elastin (E2 +E3 fractions) to total elastin (tropo+ matrix; i.e., E1+ E2+ E3 fractions) was compared to gauge efficiency of crosslinking into a structural elastin matrix.
2.6 Desmosine assay for detection of crosslinks within cultured elastin matrices
Desmosine content was assayed by an ELISA method [25] to confirm the extent of crosslinking within the matrix elastin. Briefly, cell layers were scraped off, re-suspended in 1 ml of 5% v/v trichloroacetic acid, and centrifuged (3000g, 10 min, 4 °C). The pellet was digested with collagenase type VII (Sigma-Aldrich; 12 h, 37 °C) and recentrifuged (3000g, 10 min, 4 °C) to obtain a supernatant (D1) and a pellet. The pellet was digested with pancreatic porcine elastase type III (Sigma-Aldrich; 12 h, 37 °C) to obtain soluble peptide fractions (D2). Fractions D1 and D2 were pooled together, hydrolyzed with 6 N HCl at 110 °C and dried to powder under inert nitrogen over 18−24 hours. The dried samples were reconstituted in double deionized water and diluted for assay. The wells in micro-titer plates to be used for assay were pre-blocked using desmosine-albumin conjugate (EPC, Owensville, Missouri) in 0.05 M sodium carbonate buffer (pH 9.6, 4 °C), then washed with 0.05% v/v Tween-20 and phosphate-buffered saline (PBS) solution (1 h, 25 °C). Desmosine standards/ samples were incubated (12 h, 25 °C) with rabbit antiserum to desmosine-hemocyanin conjugate (Elastin Products Company, Owensville, Missouri). After removal of primary antibody solution, the wells were successively incubated with peroxidase-conjugated anti-rabbit IgG 0.05% v/v Tween 20-PBS solution (2 h, 25 °C). Finally, 0.08 mg of the colorimetric compound 2,2—Œ-Azino-bis (3-ethylbenzothiazoline-6-sulfonic acid)) (Sigma), dissolved in 0.1 M citrate phosphate buffer containing 0.003% v/v hydrogen peroxide, (pH 4), was added to the wells and incubated (1 h, 25 °C). Absorbances were read in a UV spectrophotometer at λ = 405 nm.
2.7 Matrix ultrastructure
Transmission electron microscopy (TEM) was used to selectively compare distribution and ultra-structure of matrix elastin between cell layers cultured with exogenous or surface-tethered HA4, or in their absence (control). Adhered cell layers (n = 3/ case) were fixed in 2.5% v/v glutaraldehyde, post-fixed in 1% v/v OsO4, dehydrated with ethanol, embedded in resin, cut into 70 nm thick sections, stained with uranyl acetate and lead citrate, and visualized on a TEM (JEOL USA., Peabody, MA).
2.8 Fibrillin-mediated organization of matrix elastin
Immunogold labeling was used to detect the presence of fibrillin, a scaffolding protein that normally precedes the deposition of elastin in the ECM [26]. Cell layers were incubated with a rabbit vs. rat primary fibrillin-I antibody (20 % v/v; 14 hours, dark; EPC), then rinsed and treated with a goat anti-rabbit IgG conjugated with gold nanoparticles (10 nm; Structure Probe, Inc., West Chester, PA) diluted 1:3 v/v in 20 mM TRIS-HCl, pH 8.2 in PBS with 0.1% v/v BSA. Labeled specimens were processed as described above, stained with 2% v/v uranyl acetate, and finally resin embedded and sectioned. Cell layers untreated with the primary antibody served as negative controls.
2.9 SEM analysis of elastin matrix ultrastructure
SEM analysis was performed to compare the elastin matrix secreted by HA4 treated cell layers with that of native rat aortal elastin, and to identify the presence of fenestrated sheets or lamellae of elastin. HA4 was exogenously supplemented to cell cultures at a dose of 4 μg/ 5 ml or was surface-tethered and cultured with SMCs over 21 days (n = 3/ case/ time point). Spent media was removed from atop cell layers after 21 days of culture, washed once with d.d. water and allowed to stand for 30 mins with 1% w/v sodium dodecyl sulfate detergent (Sigma). The first wash was discarded, and the dishes then were treated for a further 2−6 hours with the detergent. The dishes were then washed with a stream of distilled water, rinsed four times with 70% v/v ethanol, and allowed to dry.
Adult rat aortal sections that served as controls for this study were pre-processed before ethanol dehydration (n = 3/ case/ time point; 3 repeats). The Institutional Animal Care and Use Committee (IACUC) at the Medical University of South Carolina approved all animal protocols. Briefly, the aortal sections were de-fatted, then treated with collagenase type II (Sigma; 0.5 units/ mg of wet tissue) dissolved in 50 mM Tris buffer, 10 mM CaCl2, pH 8.0 for 2 hours at 37 °C. Collagenase-treated sections were then digested in 100 mM NaOH (30 min, 37 °C), rinsed clean with d.d.water and dehydrated in a graded series of 50%, 70%, 95% and 100% ethanol and processed for SEM. The specimens were then dried in hexamethyldisilazane under a fume hood. The samples were finally mounted on aluminum stubs, sputter coated and examined using SEM.
2.10 Stability of elastin matrix against enzymatic degradation
Enzymatic degradation studies were performed to compare the stabilities of elastin matrices synthesized by SMCs exposed to exogenous and surface-tethered HA4. The cell-matrix layer at the end of 21 days of culture, was scraped off the culture dish surface, and centrifuged (2000 g, 15 min) to form a pellet, which was stored at –20 °C in PBS until further use (n = 3/ case/ time point × 3 repeats). Control adult rat aortal sections were rinsed in cold saline to remove fat and adherent tissues and sectioned into smaller pieces and stored at –20 °C in PBS until further use (n = 3/ case/ time point × 3 repeats). The test and control specimens were suspended in 100 mM NaOH and incubated at 37 °C for 4 hours on a shaker at 180 rpm to extract all cellular material, non-collagenous components, and some of the collagen, leaving the insoluble elastin matrix intact. These samples were then rinsed in d.d.water and incubated with collagenase II (0.5 units/ mg of wet tissue; Worthington Biochemical Corporation, Lakewood, NJ) in 50 mM TRIS buffer, 10 mM CaCl2, pH 8.0 on a shaker at 180 rpm (37 °C, 2h) to remove residual collagen, leaving behind pure elastin. These samples were lyophilized, their dry weights recorded, and incubated with 1ml of 20 units/ ml of elastase (Worthington) solutions prepared in 100 mM Tris buffer, 1 mM CaCl2, 0.02% NaN3, pH 7.8 on a shaker at 600 rpm (37 °C, up to 8h). Residual pellets (n = 9/case) obtained by centrifugation (2000g, 15 min, 4 °C) were rinsed in DI water, lyophilized, weighed and the % loss of elastin calculated from dry weights of pellets before (0 h) and after (8 h) exposure to elastase, using the equation 2.1.
| Equation (1) |
Digestate solutions were also analyzed via SDS-PAGE for soluble elastin products (n = 9/ case/ time point). Briefly, all solutions were incubated at 95 °C for 10 min and loaded onto 10% w/v Tris-Glycine gels (Invitrogen) under reduced conditions. Elastin degradation products entrapped within the gel were visualized using silver staining with 0.1 % w/v AgNO3 solution (Sigma). Purified elastin (EPC) was used as a staining control. The band intensities of the elastin degradation products were quantified using Chemi-Imager IS 4400 system (Alpha Innotech).
2.11 Peptide analysis of insoluble matrix elastin
Peptide analysis was performed using mass spectrometry to compare peptides within purified elastin derived from cell layers exposed to exogenous and surface-tethered HA4, or no additives (control) and to compare the quality of cultured elastin and that of native matrix elastin extracted from adult rat aorta (n = 9/ case for all experimental samples and control samples). For elastin peptide analysis, the cell pellets, and defatted aortal sections (obtained as in section 2.10) were homogenized, treated with 100 mM NaOH (37 °C, 5 hours) on a shaker at 180 rpm, and the alkali-insoluble elastin (E3) converted into a soluble form with elastase (50:1 v/v tissue to enzyme, prepared in 1 mM Tris buffer, 37 °C, 24 h). These soluble peptides were then precipitated with acetone to remove unwanted salts (PBS solution) and detergents (SDS), which could interfere with analysis by decreasing the signal/ noise ratio. Finally, the peptides were loaded onto a c18 trap column (Thermo Electron Corporation, Waltham, MA), de-salted and concentrated, loaded onto 75 μm analytical C18 RP columns (Thermo Electron Corporation), and eluted off by a gradient from 2%- 70% v/v acetonitrile in the presence of 0.6% v/v heptafluorobutyric acid (HFBA). Eluted peptides were analyzed on a Thermo Finnigan LTQ mass spectrometer (Thermo Electron Corporation) using a data-dependant MSMS acquisition. Resulting data was searched against a database applying filters to identify and sequence as many peptides as possible from any given sample.
2.12 Statistical methods
Statistical significance between and within groups was determined using MS Excel's statistical function for one-way ANOVA. Differences were deemed statistically significant for p < 0.05. Quantitative results are reported as mean ± standard deviation.
3. Results
3.1 Preparation and analysis of HA oligosaccharide mixtures
As reported previously [18], testicular hyaluronidase digests of long-chain HA (1.5 MDa) were analyzed using PAGE and MALDI-TOF to confirm the resulting content of HA oligomers. These analyses indicated that 20 mg of HA digested with 81 U of enzyme/ mg HA for 18 h yields a mixture containing a narrow size range of HA oligomers (4−8 mers) with 4 mers as the major component (HA4, 75 ± 0.4 % w/w).
3.2 Characterization of tethered layers of HA4
The s-STDB assay showed APTMS treatment of glass to incorporate amine groups at a density of 9 ± 3 amines/ nm2. SEM showed mostly homogenous sheets of tethered HA4 (Figure 1, panels A-C). Atomic force micrographs also confirmed homogenous coverage of the functionalized glass with HA4 (Figure 1, panels D-F). The presence of HA4 was also confirmed by altered elemental profiles detected through XPS; specifically, a lower % content of N and Si atoms and higher % content of C-N bonding ratios were detected on HA-treated surfaces relative to APTMS-treated and -untreated glass (Table 1). Quantification of surface-bound HA4, following 21 days of incubation in culture medium, in the absence of any seeded cells, showed that these layers were hydrolytically stable over the first 2 weeks (6.4 ± 3.0% loss), but exhibited greater loss (59 ± 34%) between weeks 2 and 3 of incubation.
Figure 1.

Scanning electron micrographs of untreated glass, aminosilane-treated glass, and HA4mer-tethered glass. Glass surfaces appear mostly smooth with occasional spots that are owed to commercial plasma coating (A). Glass surfaces treated with the aminosilane show the presence of APTMS molecules (B). Panel (C) shows aminated surfaces further reacted with enzymatic digests of long-chain HA, containing primarily HA4 (75% w/w). Panels D-F show atomic force micrographs and corresponding peak heights of uncoated glass (D), APTMS (E), and HA4-tethered (F) surfaces. As seen, the HA4mers reacted uniformly across the surface. Magnification: 1500× (A and B) and 900× (C).
Table 1.
Elemental profiles analysis of glass, aminosilane-treated, and HA oligomer-bound surfaces determined by XPS analysis. Glass is composed primarily of oxygen and silicon. An increase in carbon and decrease in silicon is observed with the introduction of APTMS, a carbon based structure. Oligomers (predominantly HA4 mers) contained in enzymatic digests of long-chain HA masked the detection of N atoms from amine groups presented by the APTMS, but allowed marginal detection of elemental silicon from exposed glass in-between the short, tethered HA4mers. Results represent mean ± SD of readings obtained from n = 2 regions/ sample with a total of n = 4 samples/ substrate.
| XPS Atomic Composition (%) | ||||
|---|---|---|---|---|
| Surface | Si | O | C | N |
| Glass | 26.3 ± 0.2 | 66.5 ± 0.4 | 5.9 ± 0.4 | — |
| APTMS | 11.0 ± 0.2 | 23.8 ± 1.4 | 53.0 ± 1.0 | 12.1 ± 0.4 |
| HA digests | 2.9 ± 0.3 | 23.0 ± 1.2 | 65.4 ± 0.6 | 8.3 ± 0.9 |
3.3 Cell proliferation
The number of adherent cells on immobilized HA4 surfaces was only 59 ± 0.0 % of the theoretical seeding number (105 cells/ well). This actual seeding density was hence used for the control and exogenous experiments. Control cell layers proliferated 1.9 ± 0.04 times the original seeded number, at 21 days of culture. Cell layers exposed to HA4, both exogenous and surface-tethered, showed proliferation levels similar to controls (1.95 ± 0.01 and 1.95 ± 0.02, respectively; p = 0.50).
3.4 Tropoelastin synthesis
In general, cell layers cultured on surface-immobilized HA4 substrates, and with exogenous HA4 produced 1.8 ± 0.03 and 1.7 ± 0.04 times the tropoelastin output relative to their respective control cultures (156,509 ± 151 ng/ ng, and 155,590 ± 116 ng/ ng respectively) and were statistically deemed to be similar (p = 0.67).
3.5 Matrix elastin synthesis
In general, the total DNA-normalized output of matrix elastin for cell layers cultured with surface-tethered HA4 was 2.5 ± 0.04 times that produced by controls (931 ± 26 ng/ ng; p = 1.37E-5 for surface-tethered vs. control). Similarly the DNA-normalized matrix elastin output by cells cultured with exogenous HA4 was 2.7 ± 0.02 times that produced in controls. Differences in the amounts of total matrix elastin synthesized by the two sets of test cultures were deemed statistically significant (p = 0.018). However, relative to non-HA controls, a much greater fold-increase was observed in the amount of alkali-soluble fraction of matrix elastin produced by cells cultured with HA4; also, these amounts were greater within cell layers cultured on surface-tethered HA4 than within cell layers supplemented exogenously with HA4 (11 ± 0.05 vs. 9.5 ± 0.06 times control; 71 ± 2ng/ng for control; p = 0.003 for surface-tethered vs. exogenous).
3.6 Desmosine crosslinks
Cell layers cultured with surface-tethered HA4 contained a greater amount of desmosine than cell layers cultured with exogenous HA digests with respect to non-HA controls (5.6 ± 0.05 vs. 5.16 ± 0.01 times control; p = 0.0015). Cell layers containing surface-tethered and exogenous HA4 contained 2.17 ± 0.02 and 1.92 ± 0.00 ng of desmosine/ng of matrix elastin, respectively (p = 0.002).
3.7 Elastin pellet stability against enzymatic degradation
Over an 8-hour period of digestion with elastase, aortal (control) cell pellets showed much more rapid and almost complete loss of elastin relative to that from HA4-supplemented cultures (Figure 2A). Elastin derived from exogenous HA4r-supplemented cultures were more rapidly digested by elastase than that derived from cell layers cultured on surface-tethered HA4 substrates (Figure 2A). Digests run through an SDS-PAGE gel showed higher band intensities for aortal samples than from cultures containing HA4 (Figure 2B), reflecting a higher content of soluble elastin peptides.
Figure 2.

Comparison of enzymatic degradation profiles of insoluble rat aortal elastin, and that isolated from HA4-treated and untreated cell layers. In each case, elastin pellets were digested with 1 ml of elastase (20 U/ml) over 8 hours at 37 °C (n = 9/ case/ time point). In panel A, values shown represent mean ± SD of fractions of the respective pre-digestion elastin pellet amounts retained after the designated period of enzymatic breakdown. Note that the error bars are too small to be visible on this plot.
Silver staining of soluble elastin peptides within the digestate solutions in each case show greater intensity, signifying greater amounts of elastin degradation products generated in the case of elastin from aortae and HA-free cultures than upon elastase treatment of elastin pellets from HA4-exposed cell layers.
3.8 Peptide analysis
Eluting samples of cultured and aortal (control) elastin analyzed via mass spectrometry generated all the peaks corresponding to the predominant peptides present in rat elastin, as listed in a commercial database (Bioworks 3.2 rat database). The data also showed that both elastin produced by cells cultured in presence of HA4, and aortal elastin, contained peptides of sizes within similar ranges of mass within samples eluted out of a C18 Pepmap trap column (Figure 3A-C). On the basis of the identical mass/charge ratios and the absolute masses corresponding to each of the eluted peaks obtained for all samples, the elastin produced in the presence of HA4 can be broadly assumed to contain identical peptide masses and hence AA sequences to aortal elastin.
Figure 3.
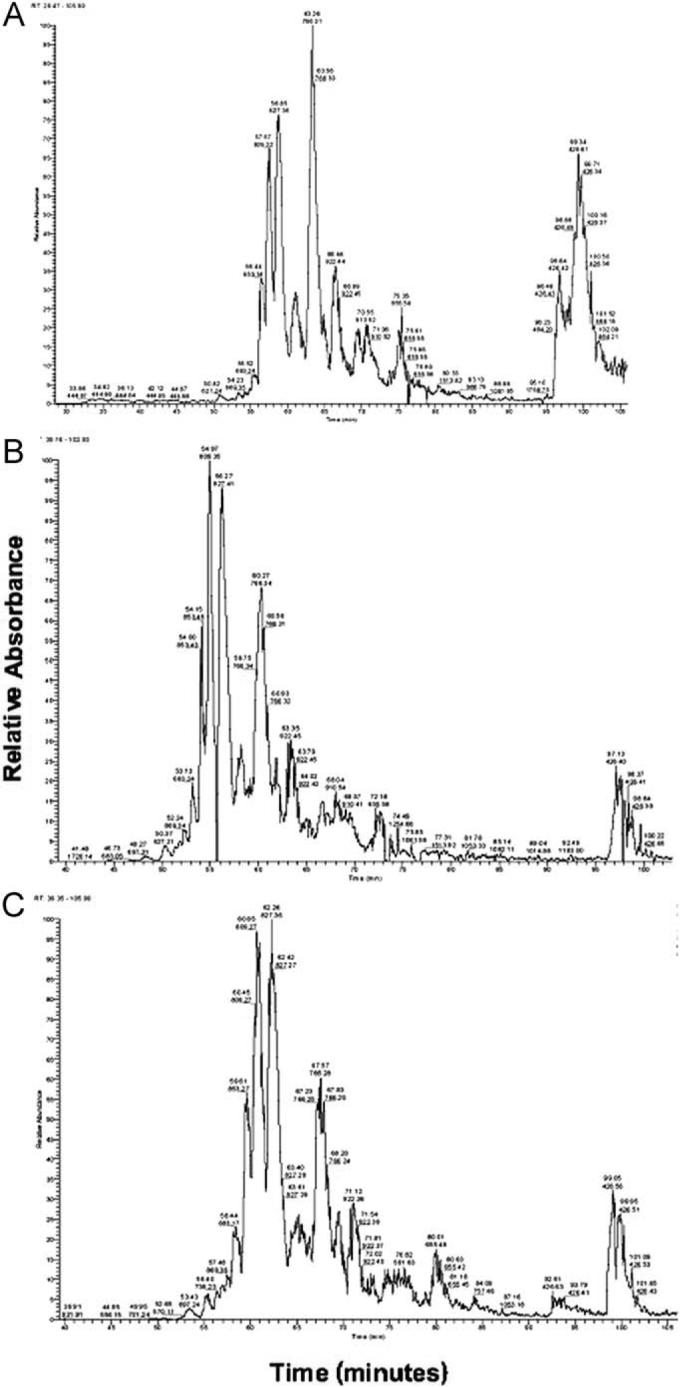
Representative mass spectroscopy spectra for peptides derived by elastase digestion of elastin matrices isolated from native rat aortae (A), and smooth muscle cell layers exposed to exogenous HA4 supplements (B) and cultured on HA4-tethered substrates. Irrespective of source, the spectra contain almost identical peaks, primarily contained within a mass range of 100−922 Da, that reflect similar relative abundance of content of peptides of designated mass or mass/charge ratios contained in a rat protein database accessed through Bioworks 3.2 software for protein identification. A total of n = 9 samples/case were analyzed and good repeatability of outcomes was observed.
3.9 Ultrastructural analysis of elastin matrix
SEM micrographs captured from aortal controls showed tightly continuous and fenestrated elastin sheets (Figure 4A). Elastin matrices within cell layers cultured in absence of HA4 were mostly clumpy and featureless and contained few elastin fibers and no fiber networks (Figure 4B). However, elastin within cell layers cultured on HA4-tethered glass were mostly organized into fibers that were tightly interwoven to create continuous and fenestrated sheets, with fibrils clearly connecting with adjacent sheet-like structures and visible at the periphery of the sheets (Figure 4C, D). Although cell layers supplemented exogenously with HA4 contained continuous sheets of elastin, no clear elastin fiber networks/ woven sheets were seen, although individual elastic fibers were commonly seen. Transmission electron micrographs supported the broad observations made through SEM: as opposed to within control cell layers (Panel 5A), elastin within cell layers cultured with exogenously supplemented (Panel 5B) or surface-tethered HA4 (Panel 5C) contained numerous elastin fibrils. These elastin fibrils were seen to aggregate into bundles, and contained darkly-stained, peripherally-distributed fibrillin microtubules (Panel 5D). In general, cell layers exogenously supplemented with HA4 contained visibly fewer fibrils than those cultured on HA4-tethered surfaces..
Figure 4.

Scanning electron micrographs comparing ultrastructures of alkali-insoluble matrix elastin isolated from rat aortae, and cell layers cultured for 3 weeks. Aortal elastin was mostly organized into continuous (A) and fenestrated sheets (B) with a few visible areas exhibiting elastin fibers closely networked into sheets. Elastin isolated from HA-free control cell layers were however mostly clumpy and formless with no fibers or distinct organization visible (C). Cell layers that received exogenous HA4 supplements or were cultured atop HA4-tethered glass appeared mostly similar, organized into continuous and fenestrated sheets (D). However, in addition, the latter cultures contained sheets of tightly intertwined/ woven elastin fibers (E), with individual fibers visible at the sheet periphery (F); only individual elastin fibers, not woven sheets were seen cell layers that received exogenous HA4.
4. Discussion
In light of the results of our prior studies and that of others, which suggested that exogenous HA fragments/oligomers elicit different cellular and matrix synthesis responses than do the same fragments when presented on a substratum (e.g., an HA scaffold or gel surface), we performed the current study to compare responses of SMCs to exogenous and surface-tethered HA4 in the context of elastin matrix synthesis. Specifically, we sought to compare the quality and stabilities of elastin, generated in presence of both exogenous and tethered HA 4mers, and its resistance to breakdown and degradation following crosslinking into an elastin matrix.
The most straight-forward method of presenting HA4 on a substratum, is to create a hydrogel composed of HA4 oligomers. Since 4mer solutions are poorly viscous, such gels would require chemical crosslinking, which could potentially alter basal cellular responses (e.g., elastin matrix synthesis) elicited by the uncrosslinked HA oligomers. To circumvent this problem, we currently use a silane-coupling agent (aminotrialkoxysilane) that presents an exposed -NH2 group, to bind HA4 as a thin layer onto glass via their carboxyl (-COOH) moieties. Such tethered layers may be deemed to represent uncrosslinked HA oligomer hydrogels of infinitesimal thickness. The proliferation and quantitative matrix synthesis outcomes of SMCs cultured atop HA4 surfaces clearly show that silane coupling of HA oligomers preserves the innate cellular interaction of these fragments. Such a model of presenting HA fragments to cells thus enables us to merely evaluate the standalone impact of 2D HA substrates on cell phenotype and matrix synthesis, avoiding the scaffold chemistry-dependent effects.
In the current study, we selected the dose of exogenous HA4 (4 μg) to be equivalent to the amount of surface-tethered HA calculated as an average amount (0.5 μg/ cm2; 8 cm2 total area) tethered onto the glass slide during 21 days of culture. We observed that overall hydrolytic stability of the tethered HA4 layers were compromised after 2 weeks of culture. However, it is to be noted that this was measured in the absence of a seeded cell layer that could shield and hence protect the underlying tethered HA4 layers from hydrolytic degradation. Based on the 59% loss of HA4 we observed over 3 weeks, theoretically, only 3.3 μg/well (not 8 μg/well) should remain tethered at the end of this period. This loss is not expected to alter our observed cell responses, since in our prior study, we clearly demonstrated that such elastogenic and other responses of cells are not dose-specific over such small dose ranges of HA4 [18].
Biochemical analyses of test and control cell layers showed that total cellular elastin output, as represented by the sum of tropoelastin (E1), and crosslinked matrix Elastin (E2 + E3), is almost doubled in the presence of HA4; however, mode of presentation of HA4, i.e., as a substratum or as an exogenous bolus, does not appear to influence this total cellular elastin output. The amounts of crosslinked matrix elastin in test cultures were however dramatically up-regulated relative to non-HA control cell layers. We can infer from this finding that HA4 identically enhances elastin crosslinking, irrespective of the method of presentation to SMCs. The total amount of matrix elastin does not appear to be influenced by the mode of HA4 delivery; however, surface-tethered HA4 appears to significantly promote deposition of crosslinked matrix elastin beyond that of exogenous HA4 (11 ± 0.05 vs. 9.5 ± 0.06-fold increase over controls).
The drastic increase in crosslinked matrix elastin may be due to two possibilities. The first is that the pre-deposition of a scaffold of fibrillin microfibrils is promoted by close-range, long-term cell interaction with substratum-bound HA4; previous studies have shown soluble tropoelastin precursors to preferentially deposit on such scaffolds, even in the absence of lysyl oxidase-mediated crosslinking, to form a robust structural matrix [27]. A second possibility is that an anionic substratum, as represented by surface-tethered HA4, coacervates tropoelastin molecules and facilitates more efficient lysyl oxidase mediated stabilization via desmosine crosslinking, to form a densely crosslinked elastin matrix. The results of our desmosine assays support the second possibility. Cell layers cultured on HA4-tethered substrates contain greater amounts of desmosine (5.6 times controls) relative to those receiving exogenous boluses of the HA4 (5.1 times controls), suggesting benefits of HA4 to elastin stabilization. On the basis of this small, yet statistically significant difference in desmosine content, we would expect only marginal increase in stability of elastin matrices isolated from cell layers cultured on HA4-tethered substrates. The results of our elastase degradation studies, however, show that elastin pellets isolated from cell layers cultured on HA4-tethered surfaces were much more resistant to enzymatic degradation (23 ± 3 % loss) relative to elastin isolated from cultures that exogenously received HA4 (92 ± 3% loss). We previously reported that exogenous HA4 (2 μg/ml) induced a nearly 50% loss in activity of the Elastin-Laminin Receptor (ELR) relative to HA-free SMC cultures [18] resulting in improved stability of the synthesized elastin matrix by preventing its breakdown. Here, it is certainly possible that prolonged interaction of SMCs with surface-tethered HA4 suppresses ELR activity beyond what occurs through likely transient signaling by exogenous HA4, and hence further stabilizes elastin matrix to levels exceeding that achieved through enhanced desmosine crosslinking alone.
Studies on the primary structure of elastin are very useful to help understand the biochemical basis of pathological conditions by unmasking any environmentally-induced genetic alterations in the primary structure, i.e., the peptide sequence. To do this, the identities of elastin-derived peptides extracted from the above test samples (surface-tethered, exogenous HA4) and controls (rat aorta), respectively were compared by MS analysis. In this study, we used elastase, instead of the more commonly used trypsin, to digest elastin, due to the extremely hydrophobic and trypsin-resistant nature of this protein. The data obtained from these results revealed that cells cultured with HA4 synthesized elastin that did not differ significantly from native aortal elastin in the masses of their contained peptides. Almost identical peaks, with a mass range between 100 and 922 Da were identified in each spectrum, irrespective of the sample run for MS analysis. However, this method has limitations in that it does not permit identification of amino acid sequences; only the presence and relative abundance of peptides can be identified. In future studies, we plan to compare the amino acid sequence profile of elastin cultured under the conditions we have described here, and that of native rat aortic elastin.
Scanning electron micrographs of elastin isolated from both cell layers cultured with exogenous and surface-tethered HA4 showed only minor differences between them in elastin morphology or density. In cell layers cultured on HA-tethered surfaces, numerous elastin fibers, woven tightly into sheets were seen, while in cell layers that exogenously received HA4, only isolated elastin fibers, not woven sheets were seen. Rat aortic elastin contained continuous and fenestrated sheets of elastin but only sporadical individual elastin fibers likely because they were too densely meshed together for easy visualization; other studies [28] show cardiovascular elastin to contain closely meshed elastin fiber networks. No fibrous networks could be located in elastin sourced from control condition, which only contained formless clumps of amorphous elastin. Thus elastin matrices isolated from HA4 treated SMC produced all the structural forms generally associated with elastin in cardiovascular tissues [28], such as continuous and fenestrated elastin sheets, woven sheets of elastin fibers, and loose elastin fibers; surface-tethered HA4 appears to encourage elastin fiber formation, and organization into fiber networks to levels beyond that enhanced by HA4 exogenously supplemented to the culture medium.
5. Conclusions
Results of this study show that irrespective of their mode of presentation to cells (exogenous or surface-tethered), HA4 greatly up-regulate synthesis of soluble tropoelastin relative to non-HA control cell layers, and enhance recruitment and crosslinking of these precursors to form structural matrix elastin. The results also show that irrespective of their mode of delivery, HA4 encourages elastic fiber formation relative to non-HA controls. However, the study also clearly shows that presentation of HA4 on a 2D surface as opposed to exogenous supplementation, is more conducive to deposition of matrix elastin in the highly crosslinked form, likely by encouraging more efficient elastin crosslinking via desmosine. Finally, our studies also suggest that non-transient, intimate interaction between tethered HA4 and SMCs, enhances elastin stability against enzymatic degradation possibly due to improved desmosine crosslinking and/ or suppression of ELR activity. The current outcomes encourage incorporation of HA oligosaccharides as elastogenic agents, into cellular scaffolds, which may be used to restore elastin matrix homeostasis in blood vessels.
Figure 5.

Representative transmission electron micrographs showing elastin within 21 day-old cell layers cultured HA-free (Panel A) in the presence of exogenous HA4 (4 μg/ well; panel B), and on HA4-tethered substrates (2 μg/cm2, 8 cm2 culture area; panel C-E).
In each case, representative images were selected from nearly 20 captured micrographs. A qualitative comparison of elastin matrix within the control and test cell layers reveals differences in the amount and nature of elastin deposited. HA free cultures contained only small clumps of amorphous elastin and no fibers. Cultures that received HA4 exogenously however contained larger clumps of amorphous elastin and more elastin fibers than controls, while still greater amounts of elastin were seen within cell layers cultured on HA4-tethered surfaces, which were predominantly assembled into aggregating fiber bundles. Immunogold labeling confirmed deposition of fibrillin microtubules (deep staining spots), scaffolds that precede elastin deposition and fibril organization (see arrows in panel E).
Acknowledgements
This study was funded by the American Heart Association (SDG 0335085N), the National Science Foundation (0132573) and National Institutes of Health (CO6 RR018823 and 1R21EB006078-01A1). The authors would also like to acknowledge the help obtained from Jennifer Bethard, at the MUSC proteomics core facility, for peptide-sequencing of elastin protein.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ross R, Bornstein P. Elastic fibers in the body. Sci Am. 1971;224(6):44–52. doi: 10.1038/scientificamerican0671-44. [DOI] [PubMed] [Google Scholar]
- 2.Li DY, Brooke B, Davis EC, Mecham RP, Sorensen LK, Boak BB, Eichwald E, Keating MT. Elastin is an essential determinant of arterial morphogenesis. Nature. 1998;393(6682):276–80. doi: 10.1038/30522. [DOI] [PubMed] [Google Scholar]
- 3.Karnik SK, Brooke BS, Bayes-Genis A, Sorensen L, Wythe JD, Schwartz RS, Keating MT, Li DY. A critical role for elastin signaling in vascular morphogenesis and disease. Development. 2003;130(2):411–23. doi: 10.1242/dev.00223. [DOI] [PubMed] [Google Scholar]
- 4.Kimura Y, Okuda H. Inhibitory effects of soluble elastin on intraplatelet free calcium concentration. Thromb Res. 1988;52(1):61–4. doi: 10.1016/0049-3848(88)90041-2. [DOI] [PubMed] [Google Scholar]
- 5.Robert L, Jacob MP, Fulop T. Elastin in blood vessels. Ciba Found Symp. 1995;192:286–99. doi: 10.1002/9780470514771.ch15. [DOI] [PubMed] [Google Scholar]
- 6.Stock UA, Sakamoto T, Hatsuoka S, Martin DP, Nagashima M, Moran AM, Moses MA, Khalil PN, Schoen FJ, Vacanti JP, Mayer JE., Jr Patch augmentation of the pulmonary artery with bioabsorbable polymers and autologous cell seeding. J Thorac Cardiovasc Surg. 2000;120(6):1158–67. doi: 10.1067/mtc.2000.109539. [DOI] [PubMed] [Google Scholar]
- 7.Courtney JM, Plant BJ, Morgan K, Rendall J, Gallagher C, Ennis M, Kalsheker N, Elborn S, O'connor CM. Association of improved pulmonary phenotype in irish cystic fibrosis patients with a 3' enhancer polymorphism in alpha-1-antitrypsin. Pediatr Pulmonol. 2006;41(6):584–91. doi: 10.1002/ppul.20416. [DOI] [PubMed] [Google Scholar]
- 8.Pego AP, Poot AA, Grijpma DW, Feijen J. Biodegradable elastomeric scaffolds for soft tissue engineering. J Control Release. 2003;87(1−3):69–79. doi: 10.1016/s0168-3659(02)00351-6. [DOI] [PubMed] [Google Scholar]
- 9.Wang Y, Ameer GA, Sheppard BJ, Langer R. A tough biodegradable elastomer. Nat Biotechnol. 2002;20(6):602–6. doi: 10.1038/nbt0602-602. [DOI] [PubMed] [Google Scholar]
- 10.Bellingham CM, Lillie MA, Gosline JM, Wright GM, Starcher BC, Bailey AJ, Woodhouse KA, Keeley FW. Recombinant human elastin polypeptides self-assemble into biomaterials with elastin-like properties. Biopolymers. 2003;70(4):445–55. doi: 10.1002/bip.10512. [DOI] [PubMed] [Google Scholar]
- 11.Mithieux SM, Rasko JE, Weiss AS. Synthetic elastin hydrogels derived from massive elastic assemblies of self-organized human protein monomers. Biomaterials. 2004;25(20):4921–7. doi: 10.1016/j.biomaterials.2004.01.055. [DOI] [PubMed] [Google Scholar]
- 12.Langerak SE, Groenink M, van der Wall EE, Wassenaar C, Vanbavel E, van Baal MC, Spaan JA. Impact of current cryopreservation procedures on mechanical and functional properties of human aortic homografts. Transpl Int. 2001;14(4):248–55. doi: 10.1007/s001470100309. [DOI] [PubMed] [Google Scholar]
- 13.Reinboth BJ, Finnis ML, Gibson MA, Sandberg LB, Cleary EG. Developmental expression of dermatan sulfate proteoglycans in the elastic bovine nuchal ligament. Matrix Biol. 2000;19(2):149–62. doi: 10.1016/s0945-053x(00)00060-3. [DOI] [PubMed] [Google Scholar]
- 14.Wight TN. Versican: a versatile extracellular matrix proteoglycan in cell biology. Curr Opin Cell Biol. 2002;14(5):617–23. doi: 10.1016/s0955-0674(02)00375-7. [DOI] [PubMed] [Google Scholar]
- 15.Isogai Z, Aspberg A, Keene DR, Ono RN, Reinhardt DP, Sakai LY. Versican interacts with fibrillin-1 and links extracellular microfibrils to other connective tissue networks. J Biol Chem. 2002;277(6):4565–72. doi: 10.1074/jbc.M110583200. [DOI] [PubMed] [Google Scholar]
- 16.Ramamurthi A, Vesely I. Evaluation of the matrix-synthesis potential of crosslinked hyaluronan gels for tissue engineering of aortic heart valves. Biomaterials. 2005;26(9):999–1010. doi: 10.1016/j.biomaterials.2004.04.016. [DOI] [PubMed] [Google Scholar]
- 17.Joddar B, Ramamurthi A. Dose- and fragment size specific effects of exogenous hyaluronan on elastin matrix synthesis. Biomaterials. 2006;27(15):2994–3004. doi: 10.1016/j.biomaterials.2006.01.020. [DOI] [PubMed] [Google Scholar]
- 18.Joddar B, Ramamurthi A. Elastogenic effects of exogenous hyaluronan oligosaccharides on vascular smooth muscle cells. Biomaterials. 2006;27(33):5698–707. doi: 10.1016/j.biomaterials.2006.07.020. [DOI] [PubMed] [Google Scholar]
- 19.Deed R, Rooney P, Kumar P, Norton JD, Smith J, Freemont AJ, Kumar S. Early-response gene signalling is induced by angiogenic oligosaccharides of hyaluronan in endothelial cells. Inhibition by non-angiogenic, high-molecular-weight hyaluronan. Int J Cancer. 1997;71(2):251–6. doi: 10.1002/(sici)1097-0215(19970410)71:2<251::aid-ijc21>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 20.Saltzman WM, Parkhurst MR, Parsons-Wingerter P, Zhu WH. Three-dimensional cell cultures mimic tissues. Ann N Y Acad Sci. 1992;665:259–73. doi: 10.1111/j.1749-6632.1992.tb42590.x. [DOI] [PubMed] [Google Scholar]
- 21.Kanda K, Matsuda T, Oka T. Mechanical stress induced cellular orientation and phenotypic modulation of 3-D cultured smooth muscle cells. ASAIO J. 1993;39(3):M686–90. [PubMed] [Google Scholar]
- 22.O'Callaghan CJ, Williams B. Mechanical strain-induced extracellular matrix production by human vascular smooth muscle cells: role of TGF-beta (1) Hypertension. 2000;36(3):319–24. doi: 10.1161/01.hyp.36.3.319. [DOI] [PubMed] [Google Scholar]
- 23.Ibrahim S, Joddar B, Craps M, Ramamurthi A. A surface-tethered model to assess size-specific effects of hyaluronan (HA) on endothelial cells. Biomaterials. 2007;28(5):825–35. doi: 10.1016/j.biomaterials.2006.09.030. [DOI] [PubMed] [Google Scholar]
- 24.Labarca C, Paigen K. A simple, rapid, and sensitive DNA assay. Anal Biochem. 1980;102(2):344–52. doi: 10.1016/0003-2697(80)90165-7. [DOI] [PubMed] [Google Scholar]
- 25.Viglio S, Iadarola P, Lupi A, Trisolini R, Tinelli C, Balbi B, Grassi V, Worlitzsch D, Doring G, Meloni F, Meyer KC, Dowson L, Hill SL, Stockley RA, Luisetti M. MEKC of desmosine and isodesmosine in urine of chronic destructive lung disease patients. Eur Respir J. 2000;15(6):1039–45. doi: 10.1034/j.1399-3003.2000.01511.x. [DOI] [PubMed] [Google Scholar]
- 26.Rock MJ, Cain SA, Freeman LJ, Morgan A, Mellody K, Marson A, Shuttleworth CA, Weiss AS, Kielty CM. Molecular basis of elastic fiber formation. Critical interactions and a tropoelastin-fibrillin-1 cross-link. J Biol Chem. 2004;279(22):23748–58. doi: 10.1074/jbc.M400212200. [DOI] [PubMed] [Google Scholar]
- 27.Kozel BA, Ciliberto CH, Mecham RP. Deposition of tropoelastin into the extracellular matrix requires a competent elastic fiber scaffold but not live cells. Matrix Biol. 2004;23(1):23–34. doi: 10.1016/j.matbio.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 28.Scott MJ, Vesely I. Morphology of porcine aortic valve cusp elastin. J Heart Valve Dis. 1996;5(5):464–71. [PubMed] [Google Scholar]