

Abstract
The present study was undertaken to investigate the role of CD40 ligation in the expression of inducible nitric-oxide synthase (iNOS) in mouse BV-2 microglial cells and primary microglia. Ligation of CD40 alone by either cross-linking antibodies against CD40 or a recombinant CD40 ligand (CD154) was unable to induce the production of NO in BV-2 microglial cells. The absence of induction of NO production by CD40 ligation alone even in CD40-overexpressed BV-2 microglial cells suggests that a signal transduced by the ligation of CD40 alone is not sufficient to induce NO production. However, CD40 ligation markedly stimulated interferon-γ (IFN-γ)-mediated NO production. Ligation of CD40 in CD40-overexpressed cells further stimulated IFN-γ-induced production of NO. This stimulation of NO production was accompanied by stimulation of the iNOS protein and mRNA. In addition to BV-2 glial cells, CD40 ligation also stimulated IFN-γ-mediated NO production in mouse primary microglia and peritoneal macrophages. To understand the mechanism of induction/stimulation of iNOS, we investigated the roles of nuclear factor κB (NF-κB) and CCAAT/enhancer-binding protein β (C/EBPβ), transcription factors responsible for the induction of iNOS. IFN-γ alone was able to induce the activation of NF-κB as well as C/EBPβ. However, CD40 ligation alone induced the activation of only NF-κB but not of C/EBPβ, suggesting that the activation of NF-κB alone by CD40 ligation is not sufficient to induce the expression of iNOS and that the activation of C/EBPβ is also necessary for the expression of iNOS. Consistently, dominant-negative mutants of p65 (Δp65) and C/EBPβ (ΔC/EBPβ) inhibited the expression of iNOS in BV-2 microglial cells that were stimulated with the combination of IFN-γ and CD40 ligand. Stimulation of IFN-γ-mediated activation of NF-κB but not of C/EBPβ by CD40 ligation suggests that CD40 ligation stimulates the expression of iNOS in IFN-γ-treated BV-2 microglial cells through the stimulation of NF-κB activation. This study illustrates a novel role for CD40 ligation in stimulating the expression of iNOS in microglial cells, which may participate in the pathogenesis of neuroinflammatory diseases.
Nitric oxide (NO), a diffusible gas, plays an important role in many physiological and diverse pathophysiological conditions (1, 2). At low concentrations NO has been shown to play a unique role in neurotransmission and vasodilation, whereas at higher concentrations it is neurotoxic (1, 2). Consistently, NO, which is derived in excessive amounts from the activation of inducible nitric-oxide synthase (iNOS)1 in glial cells (microglia and astrocytes), is assumed to contribute to oligodendrocyte degeneration in demyelinating diseases and neuronal death during neurodegenerative diseases (3-7). Evidence from several laboratories emphasizes the involvement of NO in the pathophysiology of multiple sclerosis (MS) and experimental allergic encephalomyelitis (EAE), the animal model of MS (4, 8, 9). Analysis of cerebrospinal fluid from MS patients has shown increased levels of nitrite and nitrate compared with normal controls (11). The reaction of NO with forms peroxynitrite (ONOO−), a strong nitrosating agent capable of nitrosating tyrosine residues of a protein to nitrotyrosine. Increased levels of nitrotyrosine have been found in demyelinating lesions of MS brains as well as in spinal cords of mice with EAE (12, 13). Subsequently, a semiquantitative reverse transcription-PCR for iNOS mRNA in MS brains also shows a markedly higher expression of iNOS mRNA in MS brains than in normal brains (6, 14).
CD40, a 45–50-kDa receptor, is a member of the tumor necrosis factor (TNF) receptor superfamily and is expressed in a wide range of both immune and non-immune cell types (16-18). Recently, several investigators have shown that enhanced CD40-CD40 ligation can be dangerous to the host as it is involved in the pathogenesis of a number of autoimmune inflammatory diseases (e.g. MS, arthritis, insulin-dependent diabetes) (19-21). High levels of CD40 ligand (CD40L)-expressing cells co-localize with CD40-positive cells in both MS and EAE; most of these CD40-positive cells are of the monocytic lineage (macrophages or microglia) (19, 20). Furthermore, the importance of CD40-CD40L interaction in the disease process of EAE/MS has been highlighted by the fact that administration of anti-CD40L monoclonal antibody attenuates the development of EAE (20, 21) and that EAE cannot be induced in CD40L(−/−) mice (19). However, the molecular events surrounding the marked increase of CD40 ligation in neural tissues of MS patients and EAE animals are not completely understood.
We herein report that CD40 ligation markedly stimulates the expression of iNOS by augmenting the activation of NF-κB in IFN-γ-treated microglial cells.
MATERIALS AND METHODS
Reagents
Fetal bovine serum, Hank’s balanced salt solution, and Dulbecco’s modified Eagle’s medium/F-12 were from Life Technologies, Inc. l-NG-Monomethylarginine (l-NMA) and d-NG-monomethylarginine (d-NMA) were purchased from Biomol. Arginase was purchased from Sigma. Antibodies against mouse macrophage iNOS were obtained from Calbiochem. Cross-linking antibodies against CD40 were obtained from BD PharMingen. Mouse recombinant IFN-γ was obtained from R&D Systems. Human recombinant CD40L was obtained from Alexis Biochemicals. 125I-labeled protein A and [α-32P]dCTP were obtained from PerkinElmer Life Sciences. The dominant-negative mutant of CCAAT/enhancer-binding protein β (C/EBPβ), ΔC/EBPβ, was kindly provided by Dr. Steve Smale of the University of California at Los Angeles.
Isolation of Mouse Primary Microglia
Microglial cells were isolated from mixed glial cultures according to the procedure of Guilian and Baker (22). Briefly, on day 7–9 the mixed glial cultures were washed three times with Dulbecco’s modified Eagle’s medium/F-12 and subjected to shaking at 240 rpm for 2 h at 37 °C in a rotary shaker. The floating cells were washed and seeded into plastic tissue culture flasks and incubated at 37 °C for 2 h. The attached cells were removed by trypsinization and seeded onto new plates for further studies. Ninety to ninety-five percent of this preparation was found to be positive for Mac-1 surface antigen. For the induction of NO production, cells were stimulated with cytokines or CD40L in serum-free Dulbecco’s modified Eagle’s medium/F-12. Mouse BV-2 microglial cells (a kind gift from Virginia Bocchini of University of Perugia, Italy) were also maintained and induced with different stimuli as indicated above.
Isolation of Mouse Peritoneal Macrophages
Macrophages were obtained from mice by peritoneal lavage with sterile RPMI 1640 medium containing 1% fetal bovine serum (23, 24). Macrophages at a concentration of 4 × 105/ml in RPMI 1640 medium containing l-glutamine and gentamicin were added in volumes of 1 ml to each well of six-well plates. After 1 h, nonadherent cells were removed by washing, and 1 ml of serum-free RPMI 1640 medium with various stimuli was added to the adherent cells.
Construction of Mouse CD40 cDNA Expression Construct
Murine CD40 cDNA was produced by reverse transcription-PCR from RNA isolated from murine RAW264.7 cells that were stimulated with 10 ng/ml IFN-γ and 1 μg/ml lipopolysaccharide for 24 h. The PCR product was cloned into the pTARGET (Promega) mammalian expression vector utilizing 5′- and 3′-T overhangs according to the recommendations provided by the manufacturer. PCR amplification of CD40 cDNA was done by adding 2 μl of the cDNA with sense primer 5′-CTGCATGGTGTCTTTGCCTCGGCTG-3′ and antisense primer 5′-TTCAGACCAGGGGCCTCAAGGCTAT-3′ (Life Technologies, Inc.). PCR was carried out in 50-μl reaction volumes with 20 mm Tris-HCl (pH 8.0); 2 mm MgCl2; 10 mm KCl; 6 mm (NH4)2SO4; 0.1% Triton X-100; 10 μg/ml nuclease-free bovine serum albumin; 0.2 mm each of dATP, dGTP, dCTP, and dTTP; and 1.25 units of Taq polymerase (Life Technologies, Inc.). PCR amplifications were performed for 30 cycles at 94 °C for 1 min, 60 °C for 30 s, and 75 °C for 2 min. The PCR reaction products were size-fractionated on 1.5% agarose, 0.5× Tris acetate/EDTA (TAE) and visualized by ethidium bromide fluorescence. A band of ~1000 base pairs was isolated and cloned into precut pTARGET derived from the parent vector pCIneo (Promega). The CD40-pTARGET construct was used to transform competent Escherichia coli JM109. Several transformed clones were isolated, and plasmids were prepared from each clone. The sequence of the inserted DNA in several plasmid constructs, which were prepared at the core facilities of the Beadle Center for Biotechnology, University of Nebraska, confirmed the presence of CD40 cDNA in the pTARGET vector.
Expression of Mouse CD40 cDNA in BV-2 Glial Cells
Cells at 50–60% confluence were transfected with different concentrations of CD40 cDNA by LipofectAMINE Plus (Life Technologies, Inc.) following the manufacturer’s protocol (24-26). Twenty-four hours after transfection, cells were stimulated with cross-linking antibodies against CD40 and/or IFN-γ under serum-free conditions. After 24 h of incubation, culture supernatants were transferred to measure NO production.
Assay for NO Synthesis
Synthesis of NO was determined by an assay of culture supernatants for nitrite, a stable reaction product of NO with molecular oxygen. Briefly, 400 μl of culture supernatant was allowed to react with 200 μl of Griess reagent (27-30) and incubated at room temperature for 15 min. The optical density of the assay samples was measured spectrophotometrically at 570 nm. Fresh culture medium served as the blank in all experiments. Nitrite concentrations were calculated from a standard curve derived from the reaction of NaNO2 in the assay. Protein was measured by the procedure of Bradford (31).
Immunoblot Analysis for iNOS
Immunoblot analysis for iNOS was carried out as described earlier (28-30). Briefly, cells were detached by scraping, washed with Hank’s buffer, and homogenized in 50 mM Tris-HCl (pH 7.4) containing protease inhibitors (1 mM phenylmethylsulfonyl fluoride, 5 μg/ml aprotinin, 5 μg/ml pepstatin A, and 5 μg/ml leupeptin). After electrophoresis, the proteins were transferred onto a nitrocellulose membrane, and the iNOS band was visualized by immunoblotting with antibodies against mouse macrophage iNOS and 125I-labeled protein A (27-29).
RNA Isolation and Northern Blot Analysis
Cells were taken out of the culture dishes directly by adding Ultraspec-II RNA reagent (Biotecx Laboratories, Inc.), and total RNA was isolated according to the manufacturer’s protocol. For Northern blot analyses, 20 μg of total RNA was electrophoresed on 1.2% denaturing formaldehyde-agarose gels, electrotransferred to Hybond nylon membrane (Amersham Pharmacia Biotech), and hybridized at 68 °C with a 32P-labeled cDNA probe using ExpressHyb hybridization solution (CLONTECH) as described by the manufacturer. The cDNA probe was made by PCR amplification using two primers (forward primer, 5′-CTCCTTCAAAGAGGCAAAAATA-3′; reverse primer, 5′-CACTTCCTCCAGGATGTTGT-3′) (27-29). After hybridization, the filters were washed two or three times in solution I (2× SSC, 0.05% SDS) for 1 h at room temperature followed by solution II (0.1× SSC, 0.1% SDS) at 50 °C for another hour. The membranes were then dried and exposed to x-ray films (Eastman Kodak Co.). The same amount of RNA was hybridized with probe for glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
Generation of C/EBPβ-dependent Reporter Construct (pC/EBPβ-Luc)
The C/EBPβ-sensitive promoter was generated as follows to include four C/EBPβ binding sites. A single primer set was generated with the following sense sequence: 5′-ATAGAGCTCATACGTCACATTGCACAATCATAGAGCTCATTACGTCACATTGCACAATCATACAATACGTCACATTGCACAATCATTCATAACGTCACATTGCACAATCTAATCTCGAGTAA-3′. The antisense primer was as follows: 5′-ATACTCGAGGATTGTGCAATGTGACGTTTACTCGAGATTAGATTGTGCAATGTGACGTTATGAATGATTGTGCAATGTGACGTATTGTATGATTGTGCAATGTGACGTAATGAGCTCTAT-3′. The underlined areas correspond to the C/EBPβ binding site of the mouse iNOS gene promoter. At the distal end of the primers, a SacI restriction site was included and a XhoI site was included at the proximal end of the primers. After annealing and SacI/XhoI digestion, the fragment was ligated into a pGL-3 basic vector (Promega) according to the manufacturer’s protocol, and the resulting reporter construct was confirmed by sequencing.
Assay of Transcriptional Activities of NF-κB and C/EBPβ
To assay the transcriptional activities of NF-κB and C/EBPβ, cells at 50–60% confluence were transfected with either pBIIX-Luc, an NF-κB-dependent reporter construct (32), or pC/EBPβ-Luc by using the LipofectAMINE Plus method (Life Technologies, Inc.) (24-26). All transfections included 50 ng/μg total DNA of pRL-TK (a plasmid encoding Renilla luciferase used as transfection efficiency control; Promega). After 24 h of transfection, cells were treated with different stimuli for 6 h. Firefly and Renilla luciferase activities were obtained by analyzing total cell extract according to standard instructions provided by the Dual-Luciferase kit (Promega) in a TD-20/20 luminometer (Turner Designs). Relative luciferase activity of cell extracts was typically represented as the ratio of firefly luciferase value/Renilla luciferase value × 10−3.
RESULTS
Cross-linking of CD40 Stimulates the Production of NO and the Expression of iNOS in BV-2 Microglial Cells
Ligation of CD40 was achieved by using cross-linking antibodies against CD40 (anti-CD40). Recently, it has been shown that CD40 is constitutively expressed at low levels on cultured glial cells (33, 34). The results depicted in Fig. 1 show that IFN-γ alone was capable of inducing the production of NO in mouse BV-2 microglial cells at different hours of stimulation. On the other hand, ligation of CD40 by anti-CD40 alone was unable to induce the production of NO. However, anti-CD40 markedly stimulated IFN-γ-induced production of NO at different hours of stimulation. The stimulation of NO production by anti-CD40 was also dose-dependent, resulting in maximal stimulation of IFN-γ-induced production of NO at a concentration of 2 μg/ml (Fig. 2A). The inhibition of NO production by arginase (an enzyme that degrades the substrate, l-arginine, of NOS) and l-NMA (a competitive inhibitor of NOS) but not by d-NMA (a negative control of l-NMA) suggests that IFN-γ/anti-CD40-induced NO production in BV-2 microglial cells is dependent on NOS-mediated arginine metabolism (data not shown). To understand the mechanism of stimulation of NO production, we examined the effect of anti-CD40 on protein and mRNA levels of iNOS in the presence or absence of IFN-γ Western blot analysis with antibodies against murine macrophage iNOS and Northern blot analysis for iNOS mRNA clearly showed that IFN-γ but not CD40 ligation alone induced the expression of iNOS protein (Fig. 2B) and iNOS mRNA (Fig. 2C). However, consistent with the stimulation of NO production, ligation of CD40 markedly stimulated IFN-γ-induced expression of iNOS protein and iNOS mRNA.
Fig. 1. Stimulation of IFN-γ-mediated production of NO by CD40 ligation in BV-2 microglial cells.
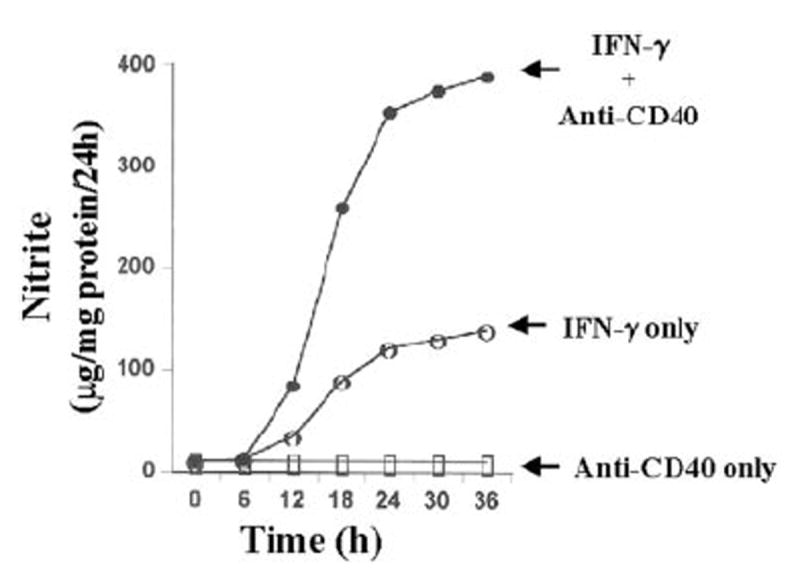
Cells were treated with 2 μg/ml cross-linking antibodies against CD40 in the presence or absence of IFN-γ (25 units/ml) under serum-free conditions. At different hours of incubation supernatants were used for the nitrite assay using Griess reagent as described under “Materials and Methods.” Data are expressed as the mean of two separate experiments.
Fig. 2. CD40 ligation stimulates the expression of iNOS in IFN-γ-treated BV-2 microglial cells.
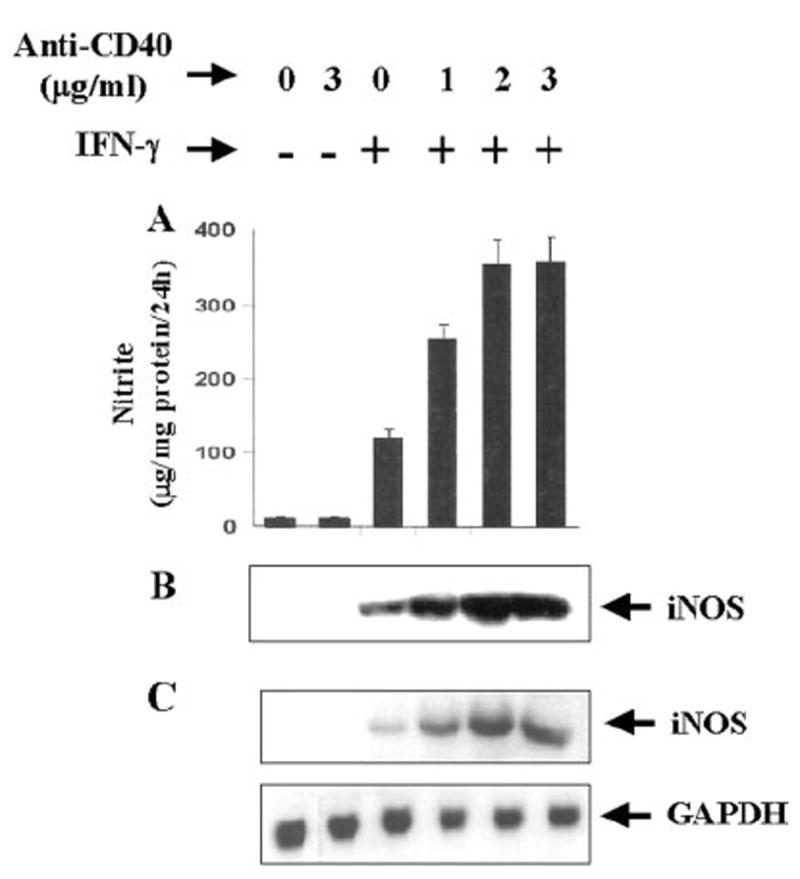
Cells were treated with different concentrations of cross-linking antibodies against CD40 in the presence or absence of IFN-γ (25 units/ml) under serum-free conditions. A, after 24 h, supernatants were used for the nitrite assay as mentioned under “Materials and Methods.” Data are mean ± S.D. of three different experiments. B, cell homogenates were electrophoresed, transferred on nitrocellulose membrane, and immunoblotted with antibodies against mouse macrophage iNOS as described under “Materials and Methods.” C, after 6 h of incubation, cells were taken out directly by adding Ultraspec-II RNA reagent (Biotecx Laboratories Inc.) to the plates for isolation of total RNA, and Northern blot analysis for iNOS mRNA was carried out as described under “Materials and Methods.”
Human Soluble Recombinant CD40L (CD154) Stimulates the Expression of iNOS in BV-2 Microglial Cells
To further prove from a different perspective that CD40-CD40L interaction augments the induction of iNOS, we examined the effect of human soluble recombinant CD40L (CD154) on the induction of iNOS in BV-2 microglial cells. Consistent with the effect of anti-CD40, recombinant CD40L alone was unable to induce the production of NO (Fig. 3A) and the expression of iNOS mRNA (Fig. 3B). However, CD40L markedly stimulated IFN-γ-induced production of NO and the expression of iNOS mRNA, confirming that CD40-CD40L interaction has a stimulatory effect on the expression of iNOS.
Fig. 3. Dose-dependent stimulation of iNOS expression by human recombinant CD40L in IFN-γ-treated BV-2 microglial cells.
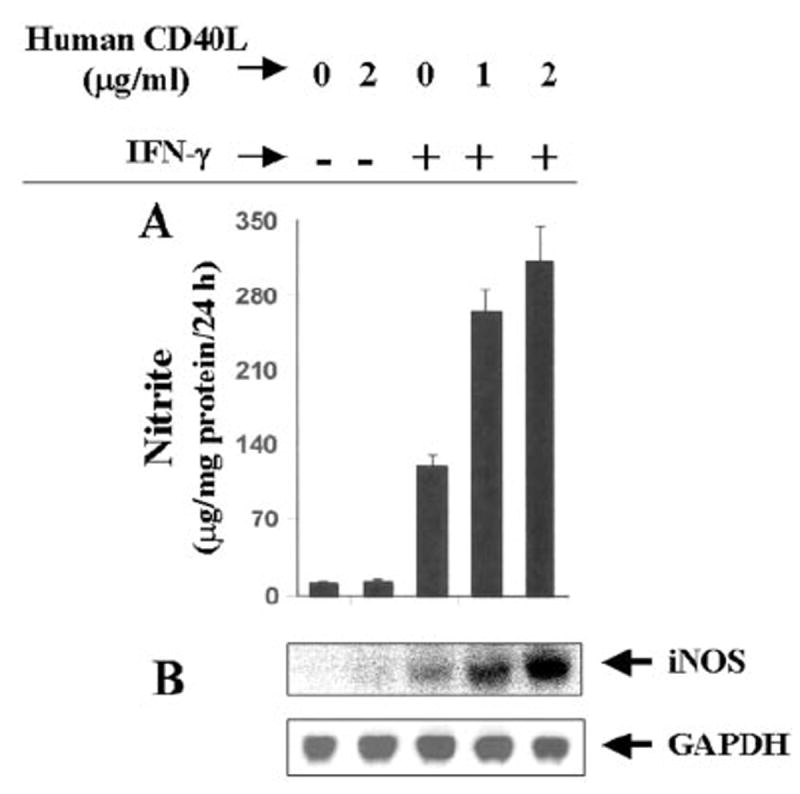
Cells were treated with different concentrations of human recombinant CD40L (CD154) in the presence or absence of IFN-γ (25 units/ml) under serum-free conditions. A, after 24 h, supernatants were used for the nitrite assay. Data are mean ± S.D. of three different experiments. B, after 6 h of incubation, total RNA was isolated from cells, and Northern blot analysis for iNOS mRNA was carried out.
Effect of Overexpression of CD40 on the Expression of iNOS in BV-2 Microglial Cells
The inability of recombinant CD40L and anti-CD40 to induce the expression of iNOS in IFN-γ-untreated cells suggests that either signals transduced by CD40 ligation alone are not sufficient for the induction of iNOS, or the expression of CD40 is too low to be ligated enough by anti-CD40 or CD40L to induce the expression of iNOS. To examine the validity of the latter possibility, we studied the effect of CD40 ligation on the induction of iNOS in CD40-overexpressed BV-2 microglial cells. Cells were transfected with different concentrations of mouse CD40 cDNA expression construct followed by the ligation of CD40 in the presence or absence of IFN-γ. Ligation of CD40 in the presence of IFN-γ induced the production of NO by ~70-fold in CD40 cDNA-transfected BV-2 glial cells as compared with a ~16-fold induction in empty vector-transfected cells (Fig. 4). On the other hand, CD40 ligation by anti-CD40 was unable to induce the production of NO even in CD40 cDNA-transfected BV-2 microglial cells (Fig. 4). The increase of IFN-γ/anti-CD40-induced NO production by CD40 cDNA was dose-dependent, and the maximal increase of NO production was observed at 0.2 μg. However, IFN-γ/anti-CD40-induced NO production decreased in cells transfected with higher doses of CD40 cDNA (Fig. 4). This decrease in NO production was due to the increase in cell death in the presence of IFN-γ and anti-CD40 at higher concentrations of CD40 cDNA (data not shown), suggesting that too much CD40 ligation is cytotoxic to the cells. Northern blot analysis also showed that CD40 ligation alone was unable to induce the expression of iNOS mRNA in CD40 cDNA-transfected cells (Fig. 5). However, a marked stimulation of IFN-γ-induced expression of iNOS mRNA by CD40 ligation was observed in CD40 cDNA-transfected cells (Fig. 5). Stimulation of IFN-γ-induced expression of iNOS mRNA by overexpression of CD40 even in the absence of CD40 ligation (Fig. 5) suggests that overexpression of CD40 probably mimics the clustering associated with CD40 ligation. Taken together, these studies suggest that the inability of CD40 ligation to induce the expression of iNOS in IFN-γ-untreated cells is not due to the weak expression of CD40. Therefore, it is likely that signals transduced by CD40 ligation alone are not sufficient for the induction of iNOS.
Fig. 4. Effect of overexpression of CD40 on the induction of NO production in BV-2 microglial cells.
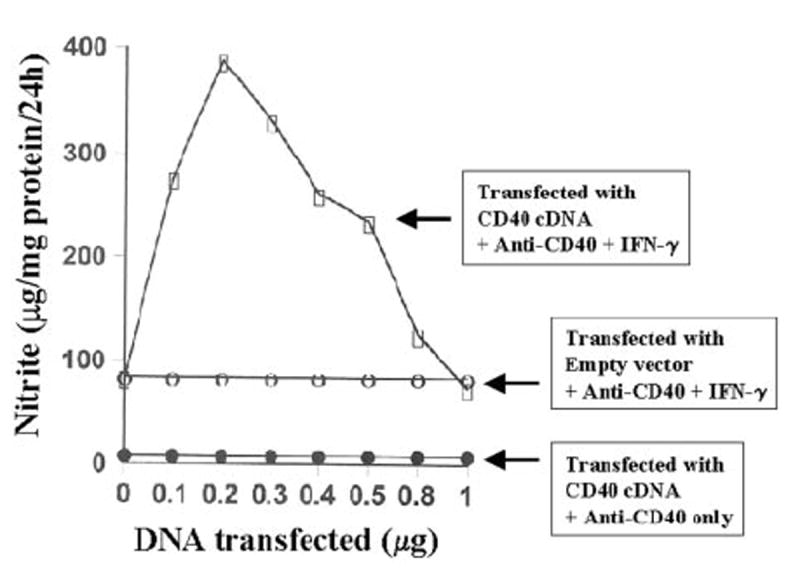
Cells plated at 50–60% confluence in six-well plates were transfected with different amounts of either CD40 cDNA or an empty vector using LipofectAMINE Plus as described under “Materials and Methods.” After 24 h of transfection, cells were stimulated with anti-CD40 (1 μg/ml) in the presence or absence of IFN-γ (10 units/ml) under serum-free conditions. After 24 h of stimulation, supernatants were used for the nitrite assay. Data are expressed as the mean of two separate experiments.
Fig. 5. Effect of overexpression of CD40 on the expression of iNOS mRNA in BV-2 microglial cells.
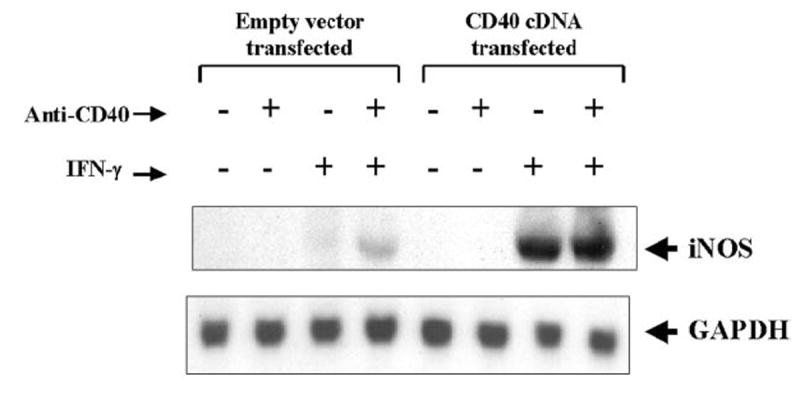
Cells plated at 50–60% confluence in a 100-mm dish were transfected with 0.8 μg of either CD40 cDNA or an empty vector. After 24 h of transfection, cells were stimulated with anti-CD40 (1 μg/ml) in the presence or absence of IFN-γ (10 units/ml) under serum-free conditions. After 6 h of incubation, total RNA was isolated from cells, and Northern blot analysis for iNOS mRNA was carried out.
Role of NF-κB and C/EBPβ in the Induction of iNOS in BV-2 Microglial Cells
The presence of NF-κB DNA-binding sites in the promoter of iNOS (35, 36) and the inhibition of expression of iNOS by the inhibitors of NF-κB suggest that NF-κB plays an important role in the expression of iNOS (23-26, 28-30, 35-38). Recent studies have also shown that the activation of C/EBPβ is also important for the induction of iNOS (39-42). Therefore, we analyzed the role of NF-κB and C/EBPβ in the induction of iNOS in IFN-γ- and anti-CD40-stimulated BV-2 microglial cells. Activation of these transcription factors was monitored by transcriptional activities using the expression of luciferase from reporter constructs like pBIIX-Luc (for NF-κB) and pC/EBPβ-Luc (for C/EBPβ) as an assay. IFN-γ alone induced the activation of both NF-κB (Fig. 6A) and C/EBPβ (Fig. 6B) in a dose-dependent fashion, and the maximal activation of these transcription factors occurred at 25 units/ml IFN-γ (Fig. 6). On the other hand, the ligation of CD40 alone by anti-CD40 induced the activation of only NF-κB but not of C/EBPβ (Fig. 7A). The induction of NF-κB activation by anti-CD40 was also dose-dependent, and the maximal induction was observed at 2 μg/ml of anti-CD40 (Fig. 7A). It is possible that the level of CD40 in normal BV-2 glial cells is too low to be ligated enough by anti-CD40 to induce the activation of C/EBPβ. To address this possibility, we examined whether the ligation of CD40 alone is capable of inducing C/EBPβ in CD40-overexpressed cells. The ligation of CD40 alone markedly induced the activation of NF-κB (>75-fold) in BV-2 glial cells transfected with CD40 cDNA (Fig. 7B) as compared with a ~3.5-fold induction found in empty vector-transfected cells (data not shown). The stimulation of anti-CD40-induced NF-κB activation by CD40 cDNA was dose-dependent, and the maximal stimulation of NF-κB activation was observed at 0.2 μg. However, under the same conditions CD40 ligation was unable to induce the activation of C/EBPβ (Fig. 7B). These studies suggest that signals transduced by CD40-CD40 ligation alone are sufficient for the activation of NF-κB but not C/EBPβ. Next we examined whether the activation of both NF-κB and C/EBPβ is important for the expression of iNOS. NF-κB was inhibited by a dominant-negative mutant of p65 (Δp65) (32). The expression of Δp65 but not the empty vector inhibited the production of NO (Fig. 8A), and the expression of iNOS mRNA (Fig. 8B) in cells stimulated with the combination of IFN-γ and anti-CD40. A naturally occurring alternate C/EBPβ translation product, known as liver-enriched inhibitory protein (LIP), lacks an activation domain yet retains the ability to inhibit the function of C/EBPβ (43). LIP therefore acts as a dominant negative of C/EBPβ (43). Consistent with an important role of C/EBPβ, the expression of ΔC/EBPβ also inhibited the production of NO (Fig. 8A) and the expression of iNOS mRNA (Fig. 8B) in cells stimulated with the combination of IFN-γ and anti-CD40. These studies suggest that activation of both NF-κB and C/EBPβ is important for the expression of iNOS and that CD40 ligation alone is unable to induce the expression of iNOS because of its inability to induce the activation of C/EBPβ. Next we examined the effect of CD40 ligation on IFN-γ-mediated activation of NF-κB and C/EBPβ. Ligation of CD40 stimulated IFN-γ-mediated activation of NF-κB (Fig. 9A) but not C/EBPβ (Fig. 9B), suggesting that the ligation of CD40 stimulates IFN-γ-induced expression of iNOS by stimulating the activation of NF-κB.
Fig. 6. IFN-γ induces the activation of both NF-κB (A) and C/EBPβ (B) in BV-2 microglial cells.
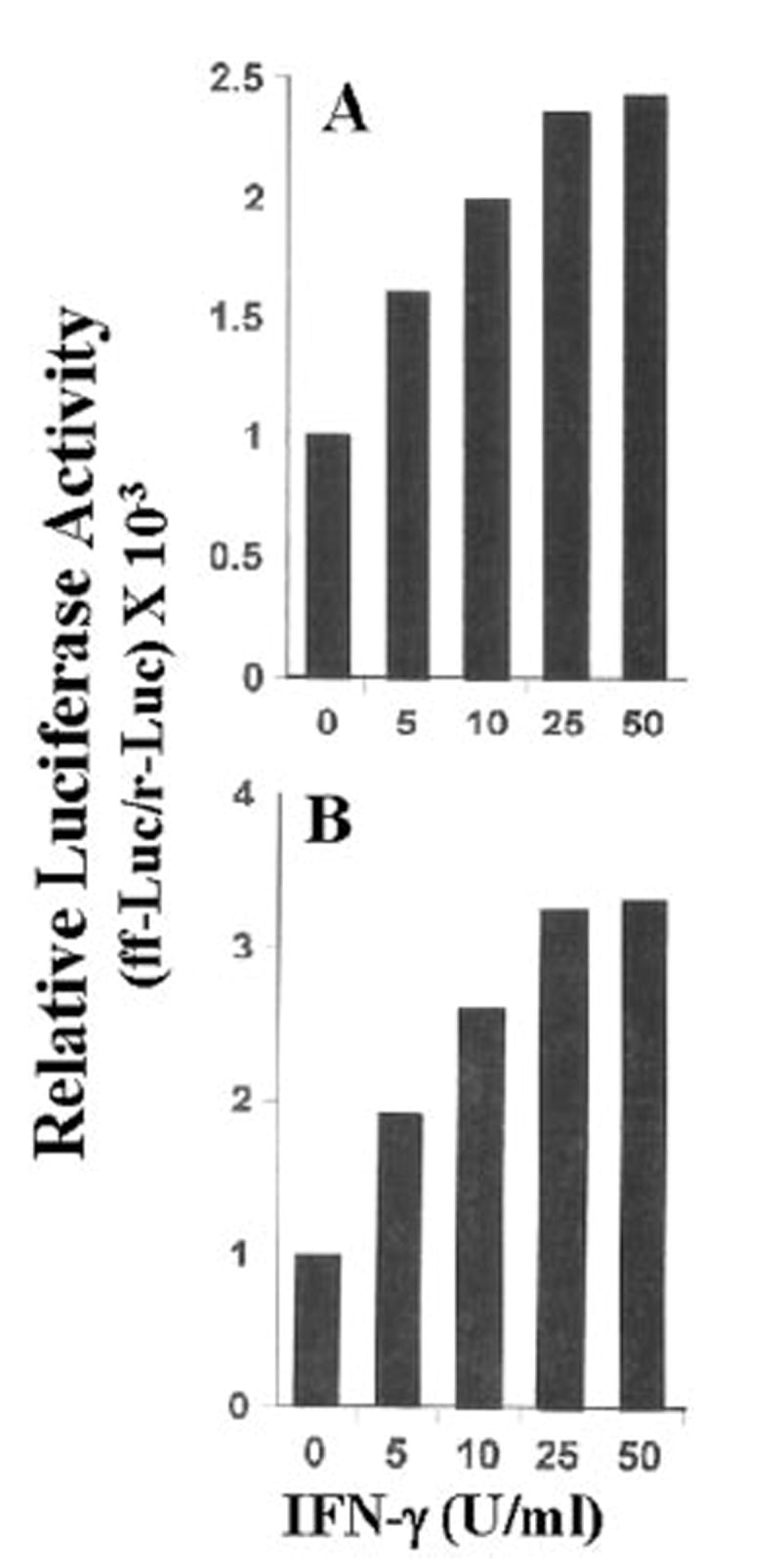
Cells plated at 50–60% confluence in six-well plates were cotransfected with 1 μg of either pBIIX-Luc(anNF-κB-dependentreporterconstruct)orpC/EBPβ-Luc(aC/EBPβ-dependent reporter construct) and 50 ng of pRL-TK (a plasmid encoding Renilla luciferase, used as a transfection efficiency control). After 24 h of transfection, cells were stimulated with different concentrations of IFN-γ for 6 h under serum-free conditions. Firefly (ff-Luc) and Renilla luciferase (r-Luc) activities were obtained by analyzing the total cell extract as described under “Materials and Methods.” Data are expressed as the mean of two separate experiments. U, units.
Fig. 7. Ligation of CD40 induces the activation of NF-κB but not of C/EBPβ in BV-2 microglial cells.
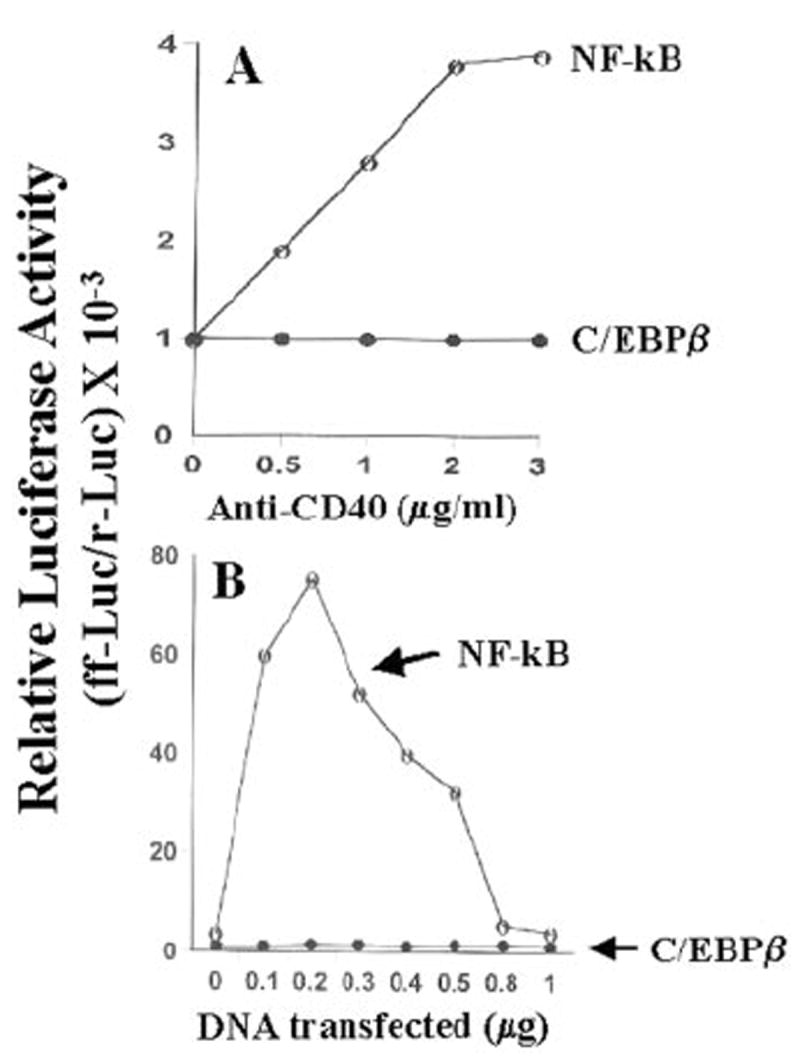
A, cells plated at 50–60% confluence in six-well plates were cotransfected with 1 μg of either pBIIX-Luc or pC/EBPβ-Luc and 50 ng of pRL-TK. After 24 h of transfection, cells were stimulated with different concentrations of anti-CD40 for 6 h under serum-free conditions. Firefly (ff-Luc) and Renilla luciferase (r-Luc) activities were assayed as described above. Data are expressed as the mean of two separate experiments. B, cells plated at 50–60% confluence in six-well plates were cotransfected with different concentrations of CD40 cDNA and either 1 μg of either pBIIX-Luc or pC/EBPβ-Luc. All transfections also included 50 ng of pRL-TK. After 24 h of transfection, cells were stimulated with 1 μg/ml anti-CD40 for 6 h under serum-free conditions. Firefly and Renilla luciferase activities were obtained by analyzing the total cell extract as described above. Data are expressed as the mean of two separate experiments.
Fig. 8. Dominant-negative mutants of p65 (Δp65) and C/EBPβ (ΔC/EBPβ) inhibit the expression of iNOS in CD40-stimulated BV-2 microglial cells.
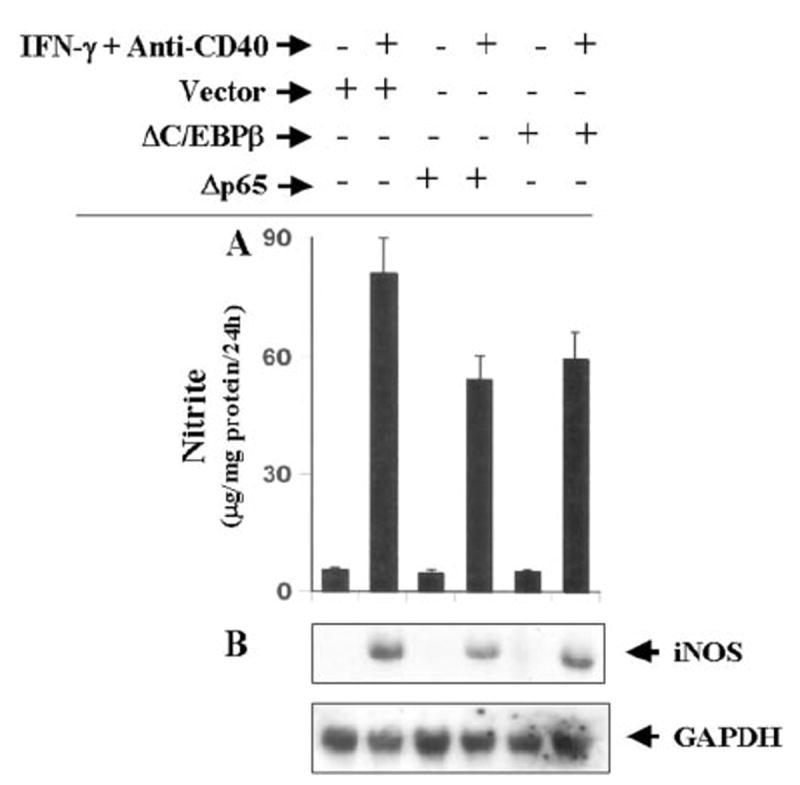
A, cells plated at 50–60% confluence in six-well plates were transfected with 1 μg of either Δp65 or ΔC/EBPβ. After 24 h of transfection, cells were stimulated with the combination of anti-CD40 and IFN-γ for 24 h under serum-free conditions, and supernatants were used for the nitrite assay. Data are mean ± S.D. of three different experiments. B, similarly after 24 h of transfection, cells were stimulated with the combination of anti-CD40 and IFN-γ under serum-free conditions. After 6 h of incubation, total RNA was isolated, and Northern blot analysis for iNOS mRNA was carried out.
Fig. 9. Ligation of CD40 stimulates the activation of NF-κB but not of C/EBPβ in IFN-γ-stimulated BV-2 microglial cells.
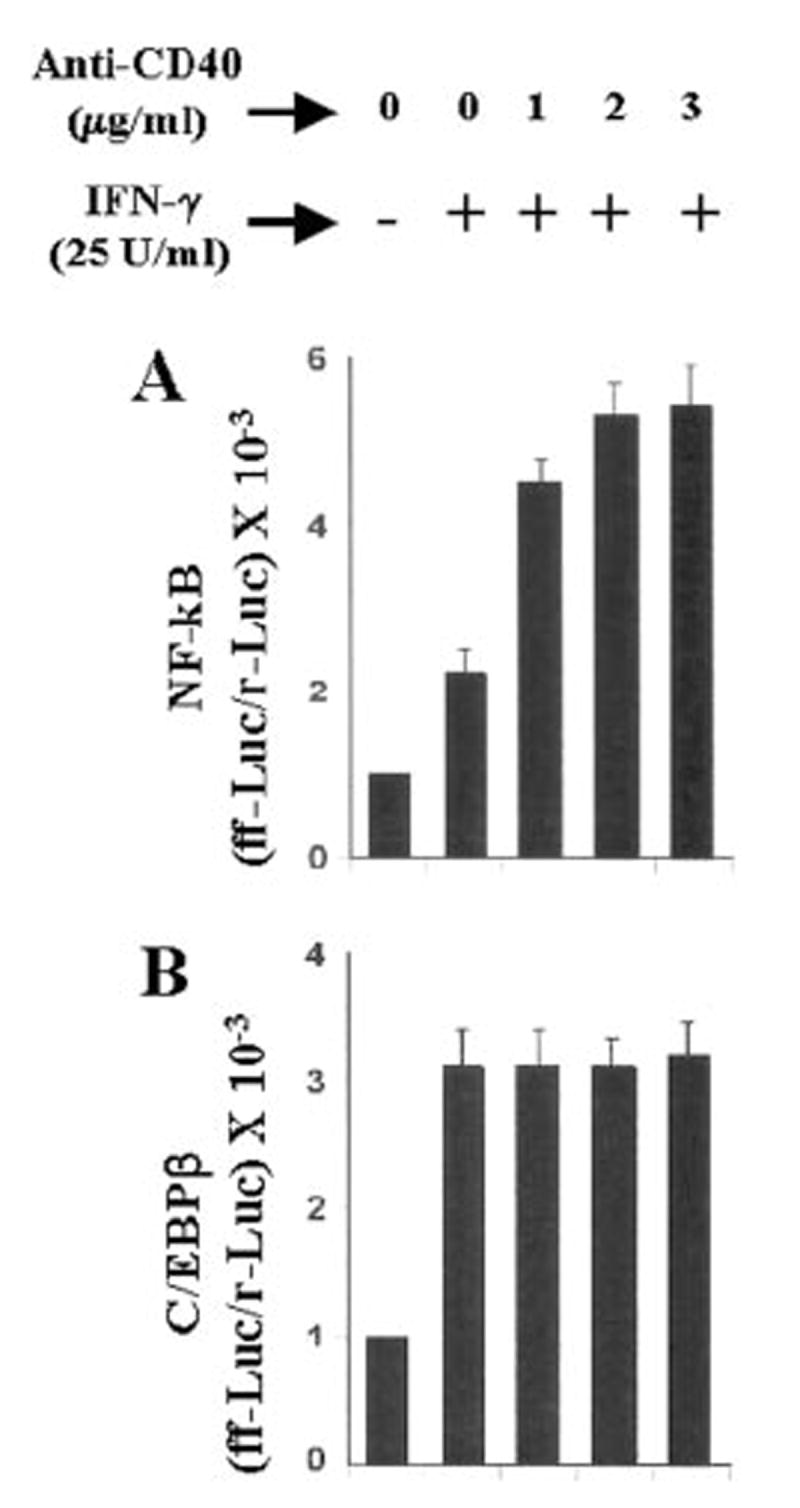
Cells plated at 50–60% confluence in six-well plates were cotransfected with 1μg of either pBIIX-Luc (A) or pC/EBPβ-Luc (B) and 50 ng of pRL-TK. After 24 h of transfection, cells were stimulated with 25 units/ml IFN-γ and different concentrations of anti-CD40 for 6 h under serum-free conditions. Firefly (ff-Luc) and Renilla luciferase (r-Luc) activities were obtained by analyzing the total cell extract. Data are mean ± S.D. of three different experiments.
CD40 Ligation Stimulates the Production of NO in IFN-γ-treated Mouse Primary Microglia and Peritoneal Macrophages
To understand the effect of CD40 ligation on the induction of NO production in primary cells, we examined the effect of anti-CD40 and human recombinant CD40L on the production of NO in mouse primary microglia and peritoneal macrophages in the presence or absence of IFN-γ (Table I). Consistent with the induction of NO production in BV-2 microglial cells, the ligation of CD40 alone was unable to induce the production of NO in both mouse primary microglia and peritoneal macrophages. However, the ligation of CD40 markedly stimulated the production of NO in IFN-γ-treated mouse primary microglia and macrophages.
TABLE I. Ligation of CD40 by cross-linking antibodies against either CD40 or human recombinant CD40L stimulates the production of NO in IFN-γ-treated mouse primary microglia and macrophages.
Mouse primary microglia and peritoneal macrophages were cultured for 24 h with IFN-γ (25 units/ml) and different concentrations of either cross-linking antibodies against CD40 or human recombinant CD40L under serum-free conditions and nitrite concentration in the supernatants was measured as described under “Materials and Methods.” Data are mean ± S.D. of three different experiments. ND, not determined.
| Treatments | Nitrite | |
|---|---|---|
| Microglia | Macrophages | |
| μg/mg protein/24 h | ||
| Control | 11.7 ± 1.5 | 12.5 ± 1.6 |
| IFN-γ alone | 146.8 ± 16 | 176.5 ± 21.5 |
| Anti-CD40 (2 μg/ml) alone | 12.9 ± 1.1 | 14.6 ± 1.2 |
| IFN-γ + anti-CD40 (1 μg/ml) | 321 ± 36 | 348 ± 29 |
| IFN-γ + anti-CD40 (2 μg/ml) | 412 ± 31 | 482 ± 55 |
| CD40L (2 μg/ml) alone | ND | 15.1 ± 1.9 |
| IFN-γ + CD40L (1 μg/ml) | ND | 312 ± 23 |
| IFN-γ + CD40L (2 μg/ml) | ND | 407 ± 34 |
DISCUSSION
CD40 participates in inflammatory processes after binding to its cognate ligand (CD40L). Several lines of evidence presented in this manuscript clearly support the conclusion that the ligation of CD40 markedly stimulates the expression of iNOS in activated microglial cells. This conclusion is based on the following observations. First, anti-CD40 (cross-linking antibodies) or human recombinant CD40L alone was unable to induce the production of NO and the expression of iNOS. However, anti-CD40 or recombinant CD40L markedly stimulated IFN-γ-induced production of NO and the expression of iNOS in mouse BV-2 microglial cells, primary microglia, and peritoneal macrophages. Second, the stimulation of IFN-γ-induced expression of iNOS by CD40 ligation was greater in cells transfected with the CD40 cDNA expression construct as compared with that in cells transfected with the empty vector. However, CD40 ligation alone was unable to induce iNOS even in CD40-overexpressed BV-2 microglial cells. These observations suggest that a signal transduced by CD40 ligation alone is not sufficient to induce the expression of iNOS but is sufficient to stimulate the expression of iNOS in microglial cells. Because NO produced from the activation of iNOS in the central nervous system participates in the pathophysiology of MS (3-14), the enhanced ligation of CD40 in the central nervous system of MS patients (19-21) and the marked stimulation of iNOS expression by the ligation of CD40 suggest that CD40-CD40L interaction may participate in the pathophysiology of MS through the enhancement of iNOS expression.
The signaling events in the induction of iNOS have not yet been completely established. Proinflammatory cytokines (TNF-α, IL-1β, or IFN-γ) bind to their respective receptors and induce the expression of iNOS via NF-κB activation (23-26, 28-30, 35-38). The presence of a consensus sequence in the promoter region of iNOS for the binding of NF-κB (35, 36) and the inhibition of iNOS expression with the inhibition of NF-κB activation establishes an essential role for NF-κB activation in the induction of iNOS. Activation of NF-κB by various cellular stimuli involves the proteolytic degradation of IκB, the inhibitory subunit of the NF-κB complex, and the concomitant nuclear translocation of the liberated NF-κB heterodimer (44, 45). Although the biochemical mechanism underlying the degradation of IκB remains unclear, it appears that degradation of IκB induced by various mitogens and cytokines occurs in association with the transient phosphorylation of IκB on serines 32 and 36 (46). Consistently, two closely related kinases (IKKα and IKKβ) that directly phosphorylate IκBβ have also been described (47-48). Upon phosphorylation, IκB that is still bound to NF-κB apparently becomes a high affinity substrate for an ubiquitin-conjugating enzyme (49). After phosphorylation-dependent ubiquitination, the IκB is rapidly and completely degraded by the 20 or 26 S proteosome; the NF-κB heterodimer enters into the nucleus (50) and binds to the consensus DNA-binding site present in the promoter region of iNOS. Our results have clearly shown that the activation of NF-κB is important for the expression of iNOS and that CD40 ligation stimulates the expression of iNOS in microglial cells through the stimulation of NF-κB activation. First, CD40 ligation alone induced the activation of NF-κB and stimulated IFN-γ-induced activation of NF-κB. Second, Δp65, a dominant-negative mutant of p65, but not the empty vector inhibited the production of NO and the expression of iNOS mRNA in BV-2 glial cells that were stimulated with the combination of IFN-γ and anti-CD40. However, despite the ability of CD40 ligation alone to induce the activation of NF-κB, it was unable to induce the expression of iNOS, suggesting that the activation of NF-κB is not sufficient for the induction of iNOS and that the activation of other transcription factors is also essential for the induction of iNOS.
C/EBP is a member of the basic region-leucine zipper family of transcription factors that controls transcription of a number of genes through protein-protein interactions at the gene level (43). In particular, C/EBPβ can not only bind to its own family members, such as the C/EBP proteins and Fos and Jun (51), but also forms complexes with other transcription factors such as NF-κB, the estrogen receptor, and the Sp1 factor (52). In addition, C/EBPβ may also alter transcription through complex interactions with coactivators and basal transcription factors such as TFIIB and p300 (15). The murine iNOS promoter contains 12 nucleotide sequences that conform to the consensus C/EBPβ box TKNNGYAAK (43). Using footprinting analysis of the 5′ -flanking region of the iNOS gene in activated RAW 264.7 cells, Goldring et al. (41) have also demonstrated protection of guanine 146 within the C/EBPβ box, suggesting that C/EBPβ binds to this site. In addition, correlative changes in the activation of C/EBPβ and the expression of the iNOS gene in cultured cardiac myocytes and vascular smooth muscle cells (40, 42) provided evidence to suggest a role for C/EBPβ in the expression of iNOS. Results presented in this manuscript clearly demonstrate that the activation of C/EBPβ is also essential for the induction of iNOS in mouse microglial cells. First, IFN-γ, which alone induced iNOS, also induced the activation of both NF-κB and C/EBPβ. Second, CD40 ligation that alone was unable to induce iNOS induced the activation of only NF-κB but not of C/EBPβ. Third, overexpression of ΔC/EBPβ, the truncated alternate C/EBPβ translation product LIP that acts as a dominant-negative inhibitor of C/EBPβ activity (43), inhibited the production of NO and the expression of iNOS mRNA in BV-2 glial cells that were stimulated with the combination of IFN-γ and anti-CD40.
NO, a diffusible free radical, plays many roles as a signaling and effector molecule in diverse biological systems; it is a neuronal messenger and is involved in vasodilation as well as in antimicrobial and antitumor activities (1, 2). In the nervous system, NO appears to have both neurotoxic and neuroprotec-tive effects and may have a role in the pathogenesis of stroke and other neurodegenerative diseases and demyelinating conditions (e.g. MS and experimental allergic encephalopathy) associated with infiltrating cells and the production of proinflammatory cytokines (2). NO and peroxynitrite (reaction product of NO and ) are molecules potentially toxic to neurons and oligodendrocytes that may mediate toxicity through the formation of iron-NO complexes of iron-containing enzyme systems, the oxidation of protein sulfhydryl groups, the nitration of proteins, and the nitrosylation of nucleic acids and DNA strand breaks (2). Although monocytes/macrophages are the primary source of iNOS in inflammation, proinflammatory cytokines induce a similar response in microglia (10, 24, 37). NO derived from microglia has also been implicated in the damage of myelin-producing oligodendrocytes in demyelinating disorders like multiple sclerosis and neuronal death during Alzheimer’s disease and brain trauma (7). Therefore, the identification of the stimulatory effect of CD40 ligation on the expression of iNOS defines a novel role for CD40 ligation in the regulation of neural injury in the inflamed central nervous system through NO production.
Acknowledgments
We thank Tom Dunn and associates for help in preparing this manuscript.
Footnotes
This study was supported by National Institutes of Health Grant NS39940 (to K. P.) and National Multiple Sclerosis Society Grant PP077 (to T. M. P.).
The abbreviations used are: iNOS, inducible nitric-oxide synthase; MS, multiple sclerosis; EAE, experimental allergic encephalomyelitis;PCR, polymerase chain reaction; IFN-γ, interferon-γ; GAPDH, glyceraldehyde- 3-phosphate dehydrogenase; C/EBPβ, CCAAT/enhancerbindingprotein β; CD40L, CD40 ligand; NOS, nitric-oxide synthase; NMA, NG-monomethylarginine.
References
- 1.Nathan C. FASEB J. 1992;6:3051–3064. [PubMed] [Google Scholar]
- 2.Jaffrey SR, Snyder SH. Annu Rev Cell Dev Biol. 1995;11:417–440. doi: 10.1146/annurev.cb.11.110195.002221. [DOI] [PubMed] [Google Scholar]
- 3.Galea E, Feinstein DL, Reis DJ. Proc Natl Acad Sci U S A. 1992;89:10945–10949. doi: 10.1073/pnas.89.22.10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koprowski H, Zheng YM, Heber-Katz E, Fraser N, Rorke L, Fu ZF, Hanlon C, Dietzshold B. Proc Natl Acad Sci U S A. 1993;90:3024–3027. doi: 10.1073/pnas.90.7.3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mitrovic B, Ignarro LJ, Montestruque S, Smoll A, Merril JE. Neuroscience. 1994;61:575–585. doi: 10.1016/0306-4522(94)90435-9. [DOI] [PubMed] [Google Scholar]
- 6.Bo L, Dawson TM, Wesselingh S, Mork S, Choi S, Kong PA, Hanley D, Trapp BD. Ann Neurol. 1994;36:778–786. doi: 10.1002/ana.410360515. [DOI] [PubMed] [Google Scholar]
- 7.Merrill JE, Ignarro LJ, Sherman MP, Melinek J, Lane TE. J Immunol. 1993;151:2132–2141. [PubMed] [Google Scholar]
- 8.Kolb H, Kolb-Bachofen V. Immunol Today. 1992;13:157–160. doi: 10.1016/0167-5699(92)90118-Q. [DOI] [PubMed] [Google Scholar]
- 9.McCatney-Francis N, Allen JB, Mizel DE, Albina JE, Xie Q-W, Nathan CF, Wahl SM. J Exp Med. 1993;178:749–754. doi: 10.1084/jem.178.2.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu SX, Sheng WS, Peterson PK, Chao CC. Glia. 1995;15:491–494. doi: 10.1002/glia.440150412. [DOI] [PubMed] [Google Scholar]
- 11.Johnson AW, Land JM, Thompson EJ, Bolanos JP, Clark JB, Heales SJR. J Neurol Neurosurg Psychiatry. 1995;58:107–115. doi: 10.1136/jnnp.58.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brenner T, Brocke S, Szafer F, Sobel RA, Parkinson JF, Perez DH, Steinman L. J Immunol. 1997;158:2940–2946. [PubMed] [Google Scholar]
- 13.Hooper DC, Scott GS, Zborek A, Mikheeva T, Kean RB, Koprowski H, Spitsin SV. FASEB J. 2000;14:691–698. doi: 10.1096/fasebj.14.5.691. [DOI] [PubMed] [Google Scholar]
- 14.Brosan CF, Battistini L, Raine CS, Dickson DW, Casadevall A, Lee SC. Dev Neurosci. 1994;16:152–161. doi: 10.1159/000112102. [DOI] [PubMed] [Google Scholar]
- 15.Mink S, Haenig B, Klempnauer KH. Mol Cell Biol. 1997;17:6609–6617. doi: 10.1128/mcb.17.11.6609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Kooten C, Banchereau J. Curr Opin Immunol. 1997;9:330–337. doi: 10.1016/s0952-7915(97)80078-7. [DOI] [PubMed] [Google Scholar]
- 17.Foy TM, Aruffo A, Bajorath J, Buhlmann JE, Noelle RJ. Annu Rev Immunol. 1996;14:591–617. doi: 10.1146/annurev.immunol.14.1.591. [DOI] [PubMed] [Google Scholar]
- 18.Grewal IS, Flavell RA. Immunol Today. 1996;17:410–414. doi: 10.1016/0167-5699(96)10030-x. [DOI] [PubMed] [Google Scholar]
- 19.Grewal IS, Foellmer HG, Grewal KD, Xu J, Hardardottir F, Baron JL, Janeway CA, Flavell RA. Science. 1996;273:1864–1867. doi: 10.1126/science.273.5283.1864. [DOI] [PubMed] [Google Scholar]
- 20.Gerritse K, Laman JD, Noelle RJ, Aruffo A, Ledbetter JA, Boersma WJ, Claassen E. Proc Natl Acad Sci U S A. 1996;93:2499–2504. doi: 10.1073/pnas.93.6.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Samoilova EB, Horton JL, Zhang H, Chen Y. J Mol Med (Berl) 1997;75:603–608. doi: 10.1007/s001090050145. [DOI] [PubMed] [Google Scholar]
- 22.Giulian D, Baker TJ. J Neurosci. 1986;6:2163–2178. doi: 10.1523/JNEUROSCI.06-08-02163.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pahan K, Sheikh FG, Namboodiri AMS, Singh I. Free Radic Biol Med. 1998;24:39–48. doi: 10.1016/s0891-5849(97)00137-8. [DOI] [PubMed] [Google Scholar]
- 24.Pahan K, Sheikh FG, Liu X, Hilger S, McKinney M, Petro TM. J Biol Chem. 2001;276:7899–7905. doi: 10.1074/jbc.M008262200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pahan K, Liu X, Wood C, Raymond JR. FEBS Lett. 2000;472:203–207. doi: 10.1016/s0014-5793(00)01465-4. [DOI] [PubMed] [Google Scholar]
- 26.Pahan K, Liu X, McKinney MJ, Wood C, Sheikh FG, Raymond JR. J Neurochem. 2000;74:2288–2295. doi: 10.1046/j.1471-4159.2000.0742288.x. [DOI] [PubMed] [Google Scholar]
- 27.Feinstein DL, Galea E, Roberts S, Berquist H, Wang H, Reis DJ. J Neurochem. 1994;62:315–321. doi: 10.1046/j.1471-4159.1994.62010315.x. [DOI] [PubMed] [Google Scholar]
- 28.Pahan K, Namboodiri AMS, Sheikh FG, Smith BT, Singh I. J Biol Chem. 1997;272:7786–7791. doi: 10.1074/jbc.272.12.7786. [DOI] [PubMed] [Google Scholar]
- 29.Pahan K, Sheikh FG, Khan M, Namboodiri AMS, Singh I. J Biol Chem. 1998;273:2591–2600. doi: 10.1074/jbc.273.5.2591. [DOI] [PubMed] [Google Scholar]
- 30.Pahan K, Raymond JR, Singh I. J Biol Chem. 1999;274:7528–7536. doi: 10.1074/jbc.274.11.7528. [DOI] [PubMed] [Google Scholar]
- 31.Bradford M. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 32.Zhong H, SuYang H, Erdjument-Bromage H, Tempst P, Ghosh S. Cell. 1997;89:413–424. doi: 10.1016/s0092-8674(00)80222-6. [DOI] [PubMed] [Google Scholar]
- 33.Tan J, Town T, Paris D, Placzek A, Parker T, Crawford F, Yu H, Humphrey J, Mullan M. J Neuroimmunol. 1999;97:77–85. doi: 10.1016/s0165-5728(99)00053-3. [DOI] [PubMed] [Google Scholar]
- 34.Nguyen VT, Walker WS, Benveniste EN. Eur J Immunol. 1998;28:2537–2548. doi: 10.1002/(SICI)1521-4141(199808)28:08<2537::AID-IMMU2537>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 35.Xie Q, Kashiwabara Y, Nathan C. J Biol Chem. 1994;269:4705–4708. [PubMed] [Google Scholar]
- 36.Taylor BS, de Vera ME, Ganster RW, Wang Q, Shapiro RA, Morris SM, Billiar TR, Geller DA. J Biol Chem. 1998;273:15148–15156. doi: 10.1074/jbc.273.24.15148. [DOI] [PubMed] [Google Scholar]
- 37.Pahan K, Sheikh FG, Namboodiri AMS, Singh I. J Clin Invest. 1997;100:2671–2679. doi: 10.1172/JCI119812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pahan K, Sheikh FG, Namboodiri AMS, Singh I. J Biol Chem. 1998;273:12219–12226. doi: 10.1074/jbc.273.20.12219. [DOI] [PubMed] [Google Scholar]
- 39.Eberhardt W, Pluss C, Hummel R, Pfeilschifter J. J Immunol. 1998;160:4961–4969. [PubMed] [Google Scholar]
- 40.Gupta AK, Kone BC. Am J Physiol. 1999;276:F599–F605. doi: 10.1152/ajprenal.1999.276.4.F599. [DOI] [PubMed] [Google Scholar]
- 41.Goldring CE, Reveneau S, Algarte M, Jeannin JF. Nucleic Acids Res. 1996;24:1682–1687. doi: 10.1093/nar/24.9.1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hecker M, Preiss C, Schini Kerth VB. Br J Pharmacol. 1997;120:1067–1074. doi: 10.1038/sj.bjp.0701026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Descombes P, Schibler U. Cell. 1991;67:569–579. doi: 10.1016/0092-8674(91)90531-3. [DOI] [PubMed] [Google Scholar]
- 44.Stefanova I, Corcoran ML, Horak EM, Wahl LM, Bolen JB, Horak ID. J Biol Chem. 1993;268:20725–20728. [PubMed] [Google Scholar]
- 45.Salkowski CA, Detore G, McNally R, van Rooijen N, Vogel SN. J Immunol. 1997;158:905–912. [PubMed] [Google Scholar]
- 46.Beg AA, Ruben SM, Scheinman RI, Haskil S, Rosen CA, Baldwin AS., Jr Genes Dev. 1992;6:1899–1913. doi: 10.1101/gad.6.10.1899. [DOI] [PubMed] [Google Scholar]
- 47.DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M. Nature. 1997;388:548–554. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- 48.Maniatis T. Science. 1997;278:818–819. doi: 10.1126/science.278.5339.818. [DOI] [PubMed] [Google Scholar]
- 49.Sun S-C, Ganchi PA, Ballard DW, Greene WC. Science. 1993;259:1912–1915. doi: 10.1126/science.8096091. [DOI] [PubMed] [Google Scholar]
- 50.Brown K, Gerstberger S, Carlson L, Franzoso G, Siebenlist U. Science. 1995;267:1485–1488. doi: 10.1126/science.7878466. [DOI] [PubMed] [Google Scholar]
- 51.Hsu W, Kerppola TK, Chen PL, Chen-Kiang S. Mol Cell Biol. 1994;14:268–276. doi: 10.1128/mcb.14.1.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stein B, Yang MX. Mol Cell Biol. 1995;15:4971–4979. doi: 10.1128/mcb.15.9.4971. [DOI] [PMC free article] [PubMed] [Google Scholar]