

Abstract
Interleukin-12 (IL-12) is composed of two different subunits, p40 and p35. Expression of p40 mRNA but not that of p35 mRNA in excessive amount in the central nervous system of patients with multiple sclerosis (MS) suggests that IL-12 p40 may have a role in the pathogenesis of the disease. However, the mode of action of p40 is completely unknown. Because nitric oxide produced from the induction of nitric-oxide synthase (iNOS) also plays a vital role in the pathophysiology of MS, the present study was undertaken to explore the role of p40 in the induction of NO production and the expression of iNOS in microglia. Both IL-12 and p402, the p40 homodimer, dose-dependently induced the production of NO in BV-2 microglial cells. This induction of NO production was accompanied by an induction of iNOS protein and mRNA. Induction of NO production by the expression of mouse p40 cDNA but not that of the mouse p35 cDNA suggests that the p40 but not the p35 subunit of IL-12 is involved in the expression of iNOS. In addition to BV-2 glial cells, p402 also induced the production of NO in mouse primary microglia and peritoneal macrophages. However, both IL-12 and p402 were unable to induce the production of NO in mouse primary astrocytes. Because activation of NF-κB is important for the expression of iNOS, we investigated the effect of p402 on the activation of NF-κB. Induction of the DNA binding as well as the transcriptional activity of NF-κB by p402 and inhibition of p402-induced expression of iNOS by SN50, a cell-permeable peptide carrying the nuclear localization sequence of p50 NF-κB, but not by SN50M, a nonfunctional peptide mutant, suggests that p402 induces the expression of iNOS through the activation of NF-κB. This study delineates a novel role of IL-12 p40 in inducing the expression of iNOS in microglial cells, which may participate in the pathogenesis of neuroinflammatory diseases.
Nitric oxide (NO), derived in excessive amount from the activation of inducible nitric-oxide synthase (iNOS)1 in glial cells (microglia and astrocytes), is assumed to contribute to oligodendrocyte degeneration in demyelinating diseases and neuronal death during neurodegenerative diseases (1–5). Evidence from several laboratories emphasizes the involvement of NO in the pathophysiology of multiple sclerosis (MS) and experimental allergic encephalomyelitis (EAE), the animal model of MS (2, 6–7). Analysis of CSF from MS patients has shown increased levels of nitrite and nitrate compared with normal control (9). The reaction of NO with forms peroxynitrite, ONOO–, a strong nitrosating agent capable of nitrosating tyrosine residues of a protein to nitrotyrosine. Increased levels of nitrotyrosine have been found in demyelinating lesions of MS brains as well as in spinal cords of mice with EAE (10, 11). Subsequently, semiquantitative reverse transcriptase-polymerase chain reaction for iNOS mRNA in MS brains also shows markedly higher expression of iNOS mRNA in MS brains than in normal brains (12, 4).
On the other hand, interleukin-12 (IL-12) plays a critical role in the early inflammatory response to infection and in the generation of T helper type 1 Th-1 cells, which favor cell-mediated immunity (14). Recently, it has been found that overproduction of IL-12 can be dangerous to the host as it is involved in the pathogenesis of a number of autoimmune inflammatory diseases (e.g. multiple sclerosis, arthritis, insulin-dependent diabetes, Refs. 15, 16). IL-12 consists of a heavy chain (p40) and a light chain (p35) linked covalently by disulfide bonds to give rise to a heterodimeric (p70) molecule (17, 18). It is known that the heterodimeric p70 molecule is the bioactive IL-12 cytokine, and both subunits must be coexpressed in the same cell to generate the bioactive form (19). However, the level of p40 is much higher than that of p35 in IL-12 producing cells (19). Again, several reports (15, 19–21) indicate that the level of p40 mRNA in the central nervous system (CNS) of patients with MS is much higher than the CNS of control subjects whereas the level of p35 mRNA is about the same or decreases compared with controls. Similarly, in mice with experimental allergic encephalomyelitis (EAE), an animal model of MS, the expression of p40 mRNA but not that of p35 mRNA increases in brain and spinal cord (22). However, the functional significance of marked overexpression of IL-12 p40 subunit in neural tissues of MS patients and EAE animals has not been delineated so far.
We herein report the first evidence that p402, the IL-12 p40 homodimer, markedly induces the production of NO and the expression of iNOS through the activation of NF-κB in mouse microglia.
Materials and Methods
Reagents
Fetal bovine serum, Hank's balanced salt solution and DMEM/F-12 were from Life Technologies, Inc. L-NG-Monomethylarginine (L-NMMA) and D-NG-monomethylarginine (D-NMMA), NF-κB SN50, and NF-κB SN50 M were purchased from Biomol. LPS (Escherichia coli, serotype 0111:B4) and arginase were from Sigma. Antibodies against mouse macrophage iNOS were obtained from CalBiochem. Recombinant mouse IL-12 and p40 homodimer were obtained from Pharmingen. Recombinant mouse IFN-γ, TNF-α, and IL-1β were obtained from R&D.
Isolation of Mouse Microglia and Astrocytes
Astrocytes were prepared from mouse cerebral tissue as described by McCarthy and DeVellis (23). Cells were maintained in DMEM/F-12 medium containing 10% fetal bovine serum. After 10 days of culture, astrocytes were separated from microglia and oligodendrocytes by shaking for 24 h in an orbital shaker at 240 rpm. To ensure the removal of oligodendrocytes and microglia, the shaking was repeated twice after a gap of 1 or 2 days. Astrocyte cultures were >95% positive for glial fibrillary acidic protein, a specific marker for astrocytes. Cells were trypsinized, subcultured, and stimulated with IL-12 p70, IL-12 p402, and other cytokines in serum-free DMEM/F-12.
Microglial cells were isolated from mixed glial cultures according to the procedure of Guilian and Baker (24). Briefly, on day 7–9, the mixed glial cultures were washed three times with DMEM/F-12 and were shaken at 240 rpm for 2 h at 37 °C on a rotary shaker. The floating cells were washed and seeded onto plastic tissue culture flasks and incubated at 37 °C for 2 h. The attached cells were removed by trypsinization and seeded onto new plates for further studies. Ninety to ninety-five percent of this preparation was found to be positive for Mac-1 surface antigen. For the induction of NO production, cells were stimulated with IL-12 p70, IL-12 p402, and other cytokines in serum-free DMEM/F-12.
Mouse BV-2 microglial cells (kind gift from Virginia Bocchini of the University of Perugia) were also maintained and induced with different stimuli as indicated above.
Isolation of Mouse Macrophages and Induction of NO Production
Resident macrophages were obtained from mouse by peritoneal lavage with sterile RPMI 1640 medium containing 1% fetal bovine serum and 100 μg/ml gentamicin (25). Cells were washed three times with RPMI 1640 at 4 °C and were maintained at 37 °C in a humidified incubator containing 5% CO2 in air. Macrophages at a concentration of 2 × 106/ml in RPMI 1640 medium containing L-glutamine and gentamicin were added in volumes of 1 ml to a 35-mm plate. After 1 h, nonadherent cells were removed by washing, and 1 ml of serum-free RPMI 1640 medium with various stimuli was added to the adherent cells. After 24 h, the culture supernatants were transferred to measure NO production.
Construction of Mouse p40 and p35 cDNA Expression Constructs
Recombinant plasmids containing cDNA for mouse p40 (ATCC 87595) and p35 (ATCC 87596) in pBluescript SK+ were obtained from the ATCC. The p35 and p40 cDNA were cut out of the plasmids utilizing the restriction enzymes XhoI and NotI. The enzyme reaction products were size fractionated on 0.8% agarose/0.5× Tris-acetate-EDTA and visualized by ethidium bromide fluorescence. Bands of ∼750 base pairs and 1050 base pairs corresponding to p35 and p40 cDNA, respectively, were isolated from the gel using Qiagen gel miniprep kit according to the manufacturer's specifications. The isolated cDNA was ligated into XhoI/NotI-cut pCIneo mammalian expression vector (Promega, Madison, WI) utilizing T4 DNA ligase according to the manufacturer's specifications. The cloned cDNA was used to transform competent E. coli JM109. Several transformed clones were isolated and plasmids were prepared from each. The sequence of the inserted DNA in several plasmid constructs was confirmed at the core facilities of the Beadle Center for Biotechnology, University of Nebraska.
Expression of Mouse p40 and p35 cDNAs in BV-2 Glial Cells
Cells at 50–60% confluence were transfected with 1 μg each of p40 and p35 cDNAs by LipofectAMINE Plus (Life Technologies, Inc.) following the manufacturer's protocol (26, 27). Twenty-four hours after transfection, cells were incubated with serum-free media. To exclude the influence of bioactive IL-12 p70 on this experiment, we also added anti-IL-12 p70 (1 μg /ml) to the serum-free media. After 24 h of incubation, culture supernatants were transferred to measure NO production.
Assay for NO Synthesis
Synthesis of NO was determined by assay of culture supernatants for nitrite, a stable reaction product of NO with molecular oxygen. Briefly, 400 μl of culture supernatant was allowed to react with 200 μl of Griess reagent (28–31) and incubated at room temperature for 15 min. The optical density of the assay samples was measured spectrophotometrically at 570 nm. Fresh culture medium served as the blank in all experiments. Nitrite concentrations were calculated from a standard curve derived from the reaction of NaNO2 in the assay. Protein was measured by the procedure of Bradford (32).
Immunoblot Analysis for iNOS
Immunoblot analysis for iNOS was carried out as described earlier (28–30). Briefly, cells were scraped off, washed with Hank's buffer, and homogenized in 50 mM Tris-HCl, pH 7.4 containing protease inhibitors (1 mM phenylmethylsulfonyl fluoride, 5 μg/ml aprotinin, 5 μg/ml pepstatin A, and 5 μg/ml leupeptin). After electrophoresis, the proteins were transferred onto a nitrocellulose membrane, and the iNOS band was visualized by immunoblotting with antibodies against mouse macrophage iNOS and by chemiluminescence assay.
RNA Isolation and Northern Blot Analysis
Cells were taken out of the culture dishes directly by adding Ultraspec-II RNA reagent (Biotecx Laboratories Inc.), and total RNA was isolated according to the manufacturer’s protocol. For Northern blot analyses, 20 μg of total RNA was electrophoresed on 1.2% denaturing formaldehyde-agarose gels, electrotransferred to Hybond nylon membrane (Amersham Pharmacia Bio-tech) and hybridized at 68 °C with 32P-labeled cDNA probe using Express Hyb hybridization solution (CLONTECH) as described by the manufacturer. The cDNA probe was made by polymerase chain reaction amplification using two primers (forward primer, 5′-CTCCTTCAAAGAGGCAAAAATA-3′; reverse primer, 5′-CACTTCCTCCAGGATGTTGT-3′; Refs. 28–30). After hybridization, the filters were washed two or three times in solution I (2× SSC, 0.05% SDS) for 1 h at room temperature followed by solution II (0.1× SSC, 0.1% SDS) at 50 °C for another hour. The membranes were then dried and exposed to x-ray films (Kodak). The same amount of RNA was hybridized with probe for glyceraldehyde-3-phosphate dehydrogenase.
Preparation of Nuclear Extracts and Electrophoretic Mobility Shift Assay
Nuclear extracts from p402-stimulated or unstimulated cells (1 × 107 cells) were prepared using the method of Dignam et al. (33) with slight modifications. Cells were harvested, washed twice with ice-cold phosphate-buffered saline, and lysed in 400 μl of buffer A (10 mM HEPES, pH 7.9, 10 mM KCl, 2 mM MgCl2, 0.5 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 5 μg /ml aprotinin, 5 μg /ml pepstatin A, and 5 μg/ml leupeptin) containing 0.1% Nonidet P-40 for 15 min on ice, vortexed vigorously for 15 s, and centrifuged at 14,000 rpm for 30 s. The pelleted nuclei were resuspended in 40 μl of buffer B (20 mM HEPES, pH 7.9, 25% (v/v) glycerol, 0.42 M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 5 μg/ml aprotinin, 5 μg/ml pepstatin A, and 5 μg/ml leupeptin). After 30 min on ice, lysates were centrifuged at 14,000 rpm for 10 min. Supernatants containing the nuclear proteins were diluted with 20 μl of modified buffer C (20 mM HEPES, pH 7.9, 20% (v/v) glycerol, 0.05 M KCl, 0.2 mM EDTA, 0.5 mM dithiothreitol, and 0.5 mM phenylmethylsulfonyl fluoride) and stored at –70 °C until use. Nuclear extracts were used for the electrophoretic mobility shift assay using the NF-κB DNA-binding protein detection system kit (Life Technologies, Inc./Life Technologies, Inc.) according to the manufacturer's protocol.
Assay of Transcriptional Activity of NF-κB
To assay the transcriptional activity of NF-κB, cells at 50–60% confluence were transfected with pNF-κB-Luc, an NF-κB-dependent reporter construct (obtained from Stratagene), using the LipofectAMINE Plus method (Life Technologies, Inc.) (26, 27). All transfections included 50 ng/μg total DNA of pRL-TK (a plasmid encoding Renilla luciferase, used as transfection efficiency control; Promega). After 24 h of transfection, cells were treated with different stimuli for 6 h. Firefly and Renilla luciferase activities were obtained by analyzing total cell extract according to standard instructions provided in the Dual Luciferase Kit (Promega) in a TD-20/20 Luminometer (Turner Designs). Relative luciferase activity of cell extracts was typically represented as (firefly luciferase value/Renilla luciferase value) × 10–3.
Results
IL-12 Induces the Production of NO and the Expression of iNOS in BV-2 Microglial Cells
IL-12 is a potent regulator of cell-mediated immune responses (14, 19). To understand the role of IL-12 in the induction of iNOS, we examined the effect of IL-12 p70 on the production of NO in mouse BV-2 microglial cells. Results in Table I show that mouse IL-12 markedly induced the production of NO. The inhibition of NO production by arginase, an enzyme that degrades the substrate (L-arginine) of NOS and L-NMA, a competitive inhibitor of NOS, but not by D-NMA, a negative control of L-NMA, suggest that IL-12-induced NO production in BV-2 glial cells is dependent on NOS-mediated arginine metabolism (Table I). To understand the mechanism of NO production in IL-12-stimulated BV-2 cells, we examined the effect of IL-12 on the protein level of iNOS. Western blot analysis with antibodies against murine macrophage iNOS of IL-12-stimulated BV-2 cells clearly showed that IL-12 significantly induced the expression of iNOS protein (Fig. 1B). To understand the specificity of induction of iNOS, BV-2 glial cells were stimulated with different cytokines and LPS. Among all the inducers tested, LPS, IFN-γ, and IL-12 efficiently induced the production of NO and the expression of iNOS protein whereas IL-1β was less efficient in inducing the expression of iNOS (Fig. 1). However, there was no induction of iNOS by TNF-α and IL-6 (Fig. 1). Similarly, TNF-α and IL-6 were also ineffective in inducing the production of NO in mouse primary microglia (data not shown).
Table I. Induction of NO production by IL-12 in BV-2 glial cells.
BV-2 glial cells were cultured for 24 h in serum-free DMEM/F-12 with the listed reagents, and nitrite concentration in the supernatants was measured as described under “Materials and Methods.” Arginase (100 units/ml), L-NMA (0.1 mM), and D-NMA (0.1 mM) were added to the cells together with IL-12. Data are mean ± S.D. of three different experiments.
| Treatments | Nitrite |
|---|---|
| nmol/mg/24 h | |
| Control | 4.3 ± 0.5 |
| IL-12 (1 ng/ml) | 35.6 ± 4.1 |
| IL-12 (2 ng/ml) | 66.3 ± 7.1 |
| IL-12 (5 ng/ml) | 85.5 ± 9.8 |
| IL-12 (10 ng/ml) | 86.3 ± 8.7 |
| IL-12 (5 ng/ml) + Arginase | 8.4 ± 1.0 |
| IL-12 (5 ng/ml) + L-NMA | 12.9 ± 1.5 |
| IL-12 (5 ng/ml) + D-NMA | 83.7 ± 7.5 |
Fig. 1. Induction of NO production and expression of iNOS by different cytokines in BV-2 glial cells.
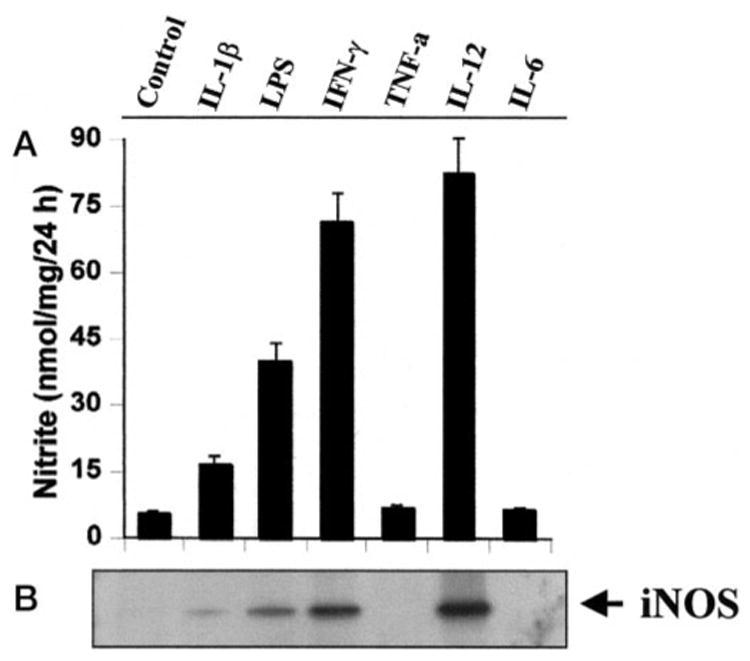
Cells were cultured with different cytokines under serum-free conditions. A, after 24 h, supernatants were used for nitrite assay as described under “Materials and Methods.” Data are mean ± S.D. of three different experiments. B, cell homogenates were electrophoresed, transferred onto nitrocellulose membranes, and immunoblotted with antibodies against mouse macrophage iNOS as described under “Materials and Methods.” Concentrations of different stimuli were: LPS, 1.0 μg/ml; TNF-α, 50 ng/ml; IL-1β, 10 ng/ml; IFN-γ, 25 units/ml; IL-12, 5 ng/ml; IL-6, 20 ng/ml.
IL-12 p40 Induces the Production of NO and the Expression of iNOS in BV-2 Microglial Cells
It is known that biologically active IL-12 is a 70-kDa heterodimeric glycoprotein comprised of disulfide-bonded 35-kDa (p35) and 40-kDa (p40) subunits (17–19). However, the p40 but not the p35 mRNA is expressed in excessive amount in neural tissues of MS and EAE (15, 19–21). Therefore, we examined the effect of p402 on the expression of iNOS. Fig. 2A shows that p402 dose-dependently induced the production of NO. About 18–20-fold induction of NO production was observed when p402 was used at a concentration of 5 or 10 ng/ml (Fig. 2A). This recombinant p402 (Pharmingen) was pure, and it showed a single 40-kDa protein band with SDS-polyacrylamide gel electrophoresis (data not shown). The induction of NO production by p402 was also inhibited by anti-mouse p40 but not by anti-mouse p70 (data not shown) suggesting that bioactive IL-12 p70 is not involved in p402-mediated induction of NO production. Moreover, recombinant mouse p402 obtained from a different source (R&D) also induced the production of NO in BV-2 glial cells (data not shown). Taken together, these observations clearly show that p402 is capable of inducing the production of NO in BV-2 glial cells. To understand the mechanism of induction of NO production, we examined the effect of p402 on protein and mRNA levels of iNOS. Western blot analysis with antibodies against murine macrophage iNOS and Northern blot analysis for iNOS mRNA of p402-stimulated BV-2 glial cells clearly showed that p402 induced the expression of iNOS protein (Fig. 2B) and iNOS mRNA (Fig. 2C). Under physiological conditions, IL-12 p40 exists as both monomer and dimer (19). To understand the role of p40 monomer in the induction of iNOS, we examined the effect of human p40 monomer (obtained from R&D) on the induction of NO production in human THP1 monocytic cells. In contrast to the effect of mouse p402 on NO production in mouse BV-2 glial cells, the human p40 monomer alone did not induce the production of NO in THP1 cells. However, the human p40 monomer markedly stimulated the production of NO in IFN-γ-treated THP1 cells.
Fig. 2. Dose-dependent induction of NO production and expression of iNOS by IL-12 p40 in BV-2 glial cells.
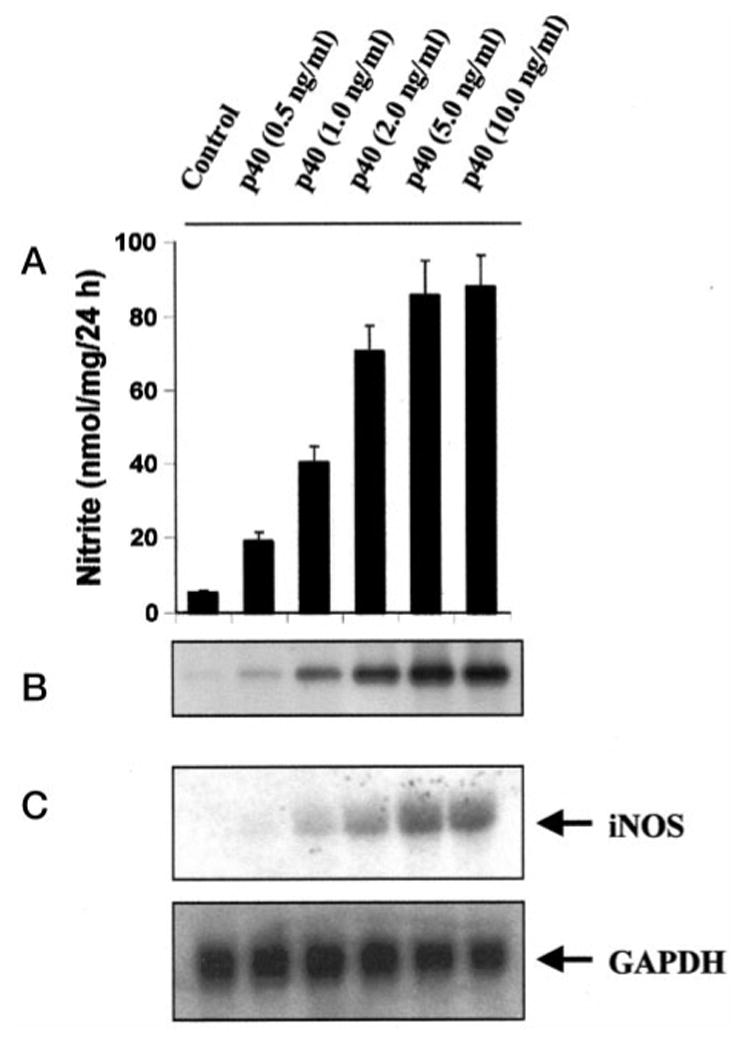
Cells were cultured with different p402 concentrations under serum-free conditions. A, after 24 h, supernatants were used for nitrite assay. Data are mean ± S.D. of three different experiments. B, cell homogenates were electrophoresed, transferred onto nitrocellulose membranes, and immunoblotted with antibodies against mouse macrophage iNOS. C, after a 6-h incubation, cells were taken out directly by adding Ultraspec-II RNA reagent (Biotecx Laboratories Inc.) to the plates for isolation of total RNA, and Northern blot analysis for iNOS mRNA was carried out as described under “Materials and Methods.”
Expression of Mouse p40 cDNA Induces the Production of NO and the Expression of iNOS in BV-2 Glial Cells
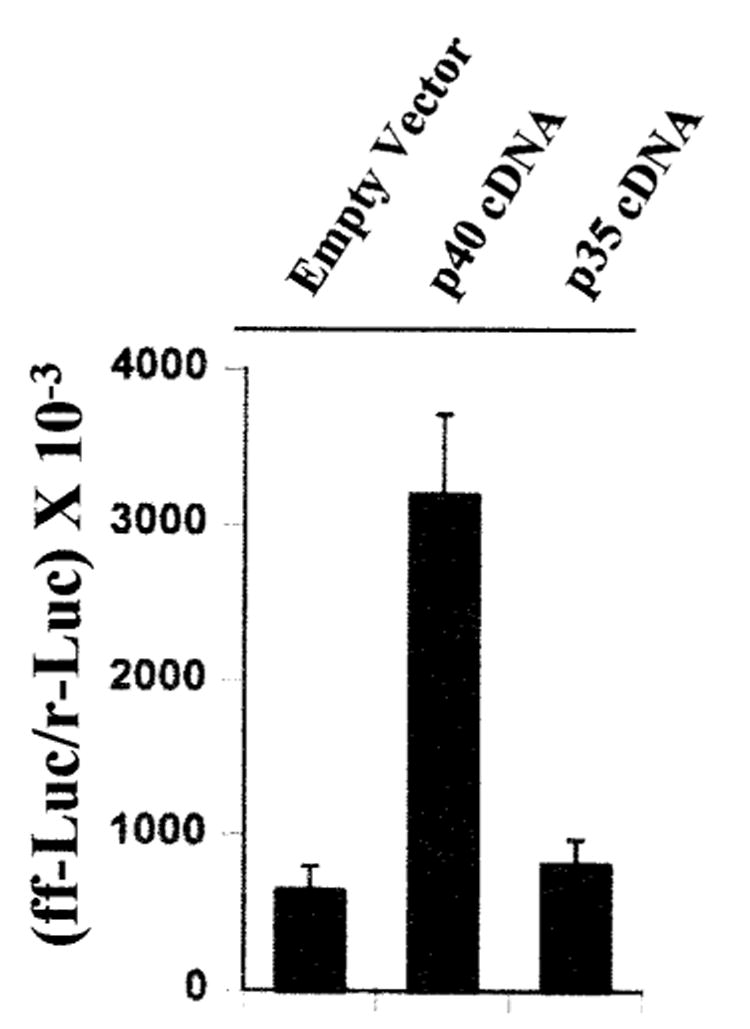
To further confirm the induction of iNOS by p40, we examined the effect of transient expression of mouse p40 and p35 cDNAs on the production of NO and the expression of iNOS in BV-2 glial cells. Similar to macrophages, microglial cells are also known to produce IL-12 (34, 35). Mouse p40 or p35 protein generated from transfected p40 or p35 cDNA may react with endogenous p35 or p40 protein to produce the bioactive IL-12 p70 heterodimer, which in turn may influence the effect of transfected p40 or p35 cDNA. Therefore, to exclude the possible influence by IL-12 p70, we also added anti-mouse p70 to the serum-free media. Consistent to the induction of iNOS by p402, expression of p40 cDNA but not that of p35 cDNA markedly induced the production of NO (Fig. 3A) and the expression of iNOS protein (Fig. 3B) suggesting that p40 but not the p35 subunit of IL-12 is involved in the induction of iNOS in microglial cells and that p40 induces iNOS independent of the so-called bioactive IL-12 p70.
Fig. 3. Expression of p40 cDNA but not that of p35 cDNA induces the expression of iNOS in BV-2 glial cells.
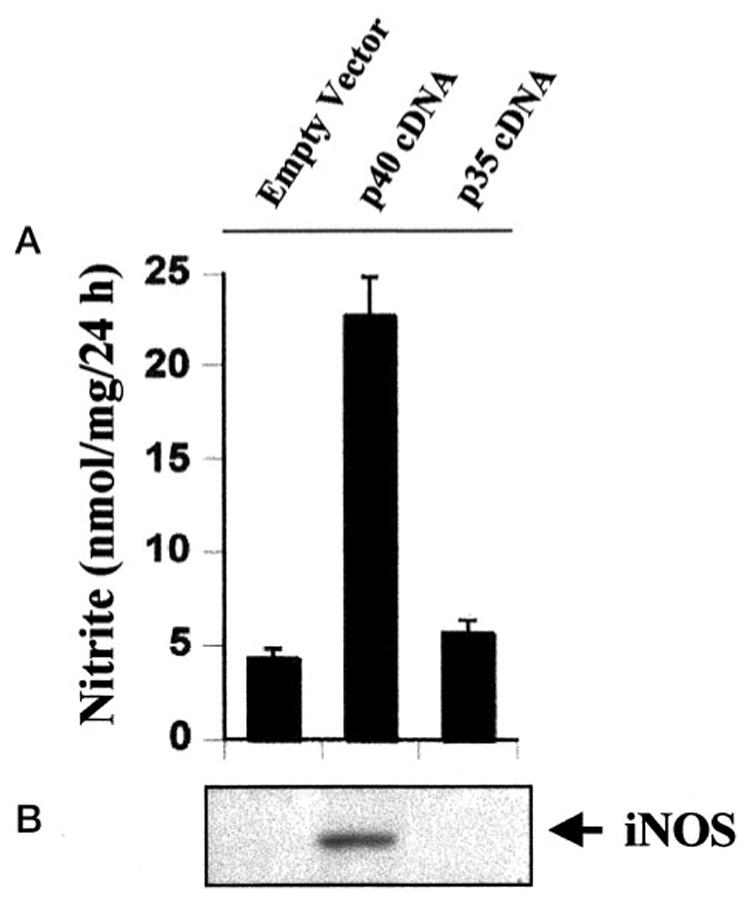
Cells plated at 50–60% confluence in 6-well plates were transfected with 1 μg each of p40 and p35 cDNAs by LipofectAMINE Plus (Life Technologies, Inc.) as described under “Materials and Methods.” Twenty-four hours after transfection, cells were incubated with serum-free medium in the presence of 1 μg/ml of anti-mouse IL-12 p70. A, after 24 h, supernatants were used for nitrite assay. Data are mean ± S.D. of three different experiments. B, cell homogenates were electrophoresed, transferred onto nitrocellulose membranes, and immunoblotted with antibodies against mouse macrophage iNOS.
IL-12 p70 and IL-12 p40 Induce the Production of NO in Mouse Peritoneal Macrophages and Primary Microglia but Not in Primary Astrocytes
To understand whether p402 induces the production of NO in primary cells, we examined the effect of p402 on the production of NO in mouse primary glial cells (astrocytes and microglia) and peritoneal macrophages (Table II). Consistent to the induction of iNOS in BV-2 glial cells, both IL-12 p70 and p402 markedly induced the production of NO in mouse primary microglia and peritoneal macrophages. However, both IL-12 p70 and p402 were unable to induce the production of NO in mouse primary astrocytes (Table 2) suggesting that p402 specifically induces iNOS in mouse microglia and macrophages but not in astrocytes.
Table II. Induction of NO production by IL-12 p70 and IL-12 p402 in mouse primary astrocytes, microglia, and macrophages.
Mouse peritoneal macrophages and primary astrocytes and microglia were cultured for 24 h in serum-free DMEM/F-12 with the listed reagents. Nitrite concentration in the supernatants was measured as described under “Materials and Methods.” Data are mean ± S.D. of three different experiments.
| Nitrite, nmol/mg/24 h | |||
|---|---|---|---|
| Treatments | |||
| Macrophages | Astrocytes | Microglia | |
| Control | 9.2 ± 1.2 | 3.4 ± 0.3 | 6.7 ± 0.8 |
| IL-12 (5 ng/ml) | 112 ± 13.5 | 3.5 ± 0.4 | 92.5 ± 11.2 |
| p402 (5 ng/ml) | 120 ± 15 | 3.4 ± 0.4 | 95.2 ± 10.8 |
IL-12 p40 Induces the Expression of iNOS through the Activation of NF-κB in BV-2 Glial Cells
Because activation of NF-κB is necessary for induction of iNOS (26–30, 36, 37), to understand the basis of expression of iNOS, we examined the effect of p402 on the activation of NF-κB in BV-2 glial cells. Activation of NF-κB was monitored by both DNA binding and transcriptional activity of NF-κB (26, 27, 31). DNA binding activity of NF-κB was evaluated by the formation of a distinct and specific complex in a gel shift DNA binding assay. Treatment of BV-2 glial cells with different concentrations of p402 resulted in dose-dependent induction of DNA binding activity of NF-κB with the maximum induction observed at 5 ng/ml (Fig. 4A). This gel shift assay detected a specific band in response to p402 that was competed off by an unlabeled probe suggesting that p402 induces the DNA binding activity of NF-κB. We then tested the effect of p402 on NF-κB-dependent transcription of luciferase in BV-2 glial cells, using the expression of luciferase from a reporter construct, pNF-κB-Luc (Stratagene), as an assay. Consistent to the effect of p402 on the DNA binding activity of NF-κB, p402 also induced NF-κB-dependent transcription of luciferase in a dose-dependent manner (Fig. 4B). To further confirm the activation of NF-κB by p402, we expressed mouse p40 and p35 cDNAs in BV-2 glial cells and examined the transcriptional activity of NF-κB. It is evident from Fig. 4C that expression of mouse p40 but not that of p35 cDNA induced NF-κB-dependent expression of luciferase, suggesting that the p40 but not the p35 subunit of IL-12 is involved in the activation of NF-κB. Because the activation of NF-κB is important for the induction of iNOS, these results also suggest that unlike p40, p35 is unable to induce iNOS because of its inability to induce the activation of NF-κB (Fig. 5). To investigate further that p402-induced expression of iNOS in BV-2 microglial cells depends on the activation of NF-κB, we examined the effect of SN50 on p402-mediated expression of iNOS. SN50 is a synthetic peptide containing signal sequences of Kaposi’s fibroblast growth factor and the nuclear localization sequence of NF-κB p50 (38). It has been reported to have the capacity to specifically block the nuclear translocation of activated NF-κB (38). Inhibition of p402-mediated activation of NF-κB (Fig. 6A), induction of NO production (Fig. 6B), and expression of iNOS mRNA (Fig. 6C) by SN50 but not by SN50M, a nonfunctional mutant of SN50, suggests that activation of NF-κB is necessary for the expression of iNOS in p402-stimulated BV-2 cells. However, p402 was unable to activate NF-κB in mouse primary astrocytes (data not shown), suggesting that p402 is unable to induce iNOS in mouse astrocytes (Table 2) because of its inability to induce the activation of NF-κB.
Fig. 4. IL-12 p40 induces the activation of NF-κB in BV-2 glial cells.
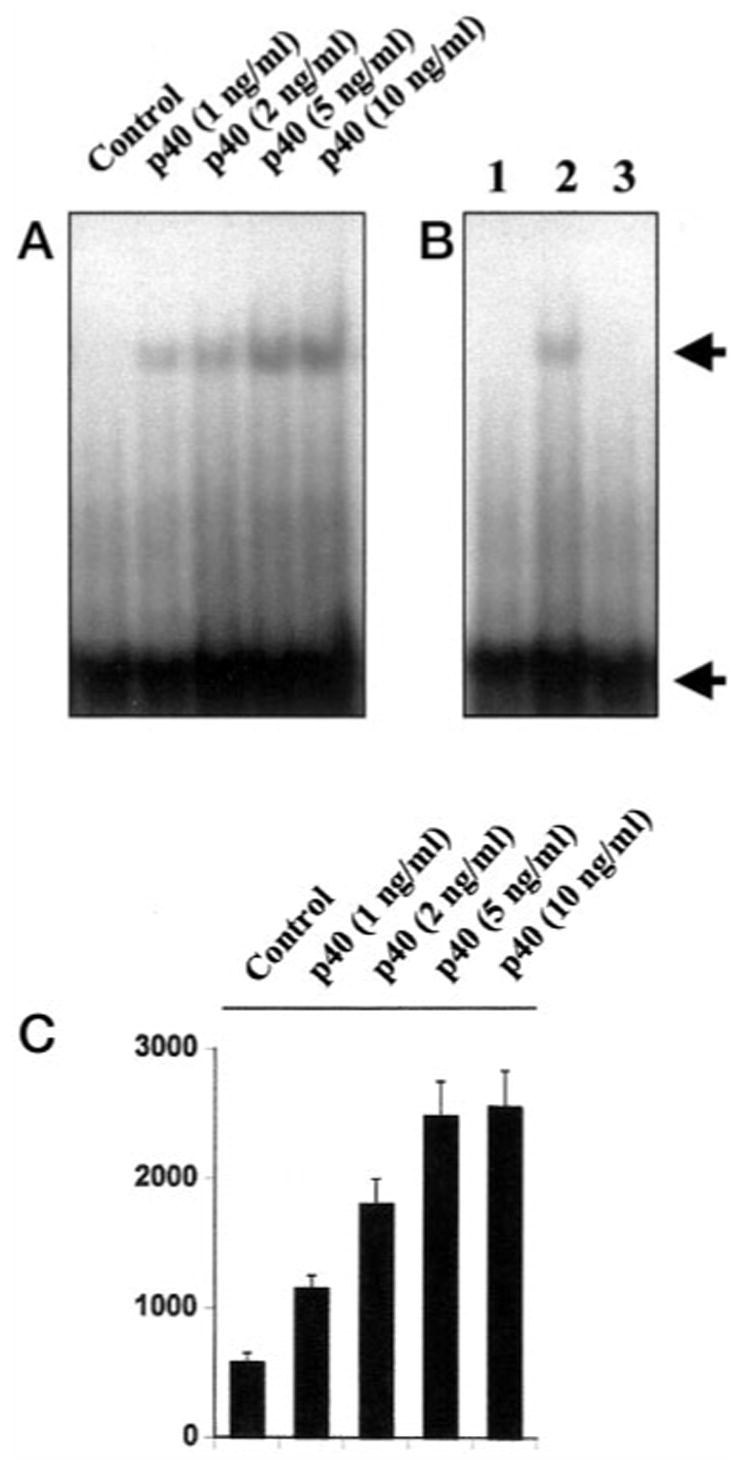
A, cells incubated in serum-free DMEM/F-12 were treated with different concentrations of p402. After 1 h of incubation, cells were taken out to prepare nuclear extracts, and nuclear proteins were used for the electrophoretic mobility shift assay as described under “Materials and Methods.” B, lanes 1–3 represent nuclear extract of control cells, nuclear extract of p402-treated cells, and nuclear extract of p402-treated cells incubated with a 100-fold excess of unlabeled oligonucleotide. The concentration of p402 used in this experiment was 5 ng/ml. The upper arrow indicates the induced NF-κB band, and the lower arrow indicates the unbound probe. C, cells plated at 50–60% confluence in 6-well plates were cotransfected with 1 μg of pNF-κB-Luc (an NF-κB-dependent reporter construct) and 50 ng of pRL-TK (a plasmid encoding Renilla luciferase, used as transfection efficiency control) using LipofectAMINE Plus as described under “Materials and Methods.” After 24 h of transfection, cells were stimulated with different concentrations of p402 for 6 h under serum-free conditions. Firefly and Renilla luciferase activities were obtained by analyzing total cell extract as described under “Materials and Methods.”
Fig. 5. Expression of p40 cDNA but not that p35 cDNA induces the activation of NF-κB in BV-2 glial cells.
Cells plated at 50–60% confluence in 6-well plates were cotransfected with 1 μg of pNF-κB-Luc (an NF-κB-dependent reporter construct) and 1 µg of p40, p35, or the empty vector using the LipofectAMINE Plus method. All transfections included 50 ng/µg total DNA of pRL-TK. After 24 h of transfection, cells were incubated with serum-free medium for 24 h in the presence of 1 µg/ml of anti-mouse IL-12 p70. Firefly and Renilla luciferase activities were obtained by analyzing total cell extract as described under “Materials and Methods.”
Fig. 6. SN50, a specific cell-permeable peptide inhibitor of NF-κB nuclear translocation, inhibits p402-mediated activation of NF-κB and expression of iNOS in BV-2 glial cells.
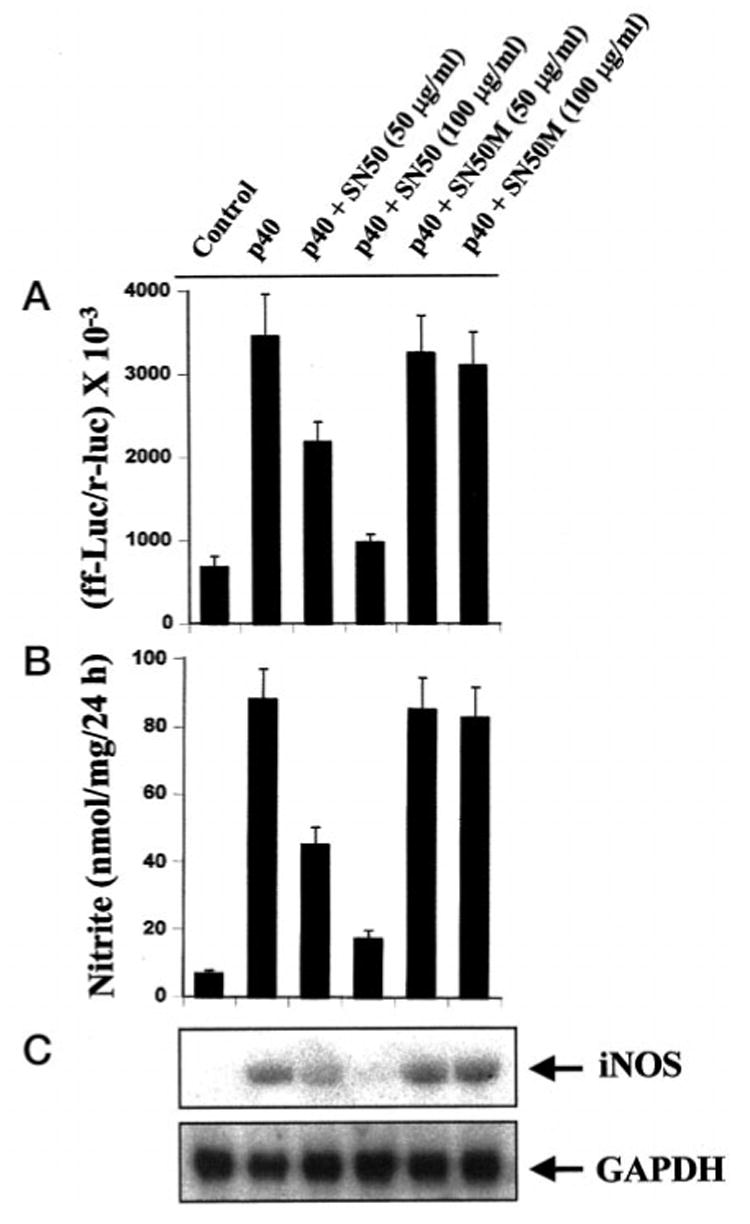
A, cells plated at 50–60% confluence in 6-well plates were cotransfected with 1 μg of pNF-κB-Luc and 50 ng of pRL-TK using LipofectAMINE Plus. After 24 h of transfection, cells were incubated with different concentrations of SN50 or SN50M (a nonfunctional mutant of SN50) for 1 h followed by stimulation with 5 ng/ml of p402 for 6 h under serum-free conditions. Firefly and Renilla luciferase activities were obtained by analyzing total cell extract as described above. B, cells preincubated with different concentrations of SN50 or SN50M for 1 h received 5 ng/ml of p402 under serum-free conditions. After 24 h, supernatants were used for nitrite assay. Data are mean ± S.D. of three different experiments. C, after 6 h of incubation, cells were taken out directly by adding Ultraspec-II RNA reagent (Biotecx Laboratories Inc.) to the plates for isolation of total RNA, and Northern blot analysis for iNOS mRNA was carried out as described above.
The cytokines (TNF-α, IL-1β, IFN-γ, IL-12 p70, IL-12 p40, IL-6) or drugs (SN50 or SN50M) used under these experimental conditions had no effect on the viability of BV-2 glial cells, measured by trypan blue exclusion. Therefore, the conclusions drawn in this study are not due to any change in viability of the cells.
Discussion
IL-12, a heterodimeric cytokine, is most noted for its ability to regulate the balance between type 1 and type 2 helper T cells (14, 19). Neither IL-12 subunits (p35 or p40) alone was found to display significant biological activity over a large range of concentrations (17–19). However, several lines of evidence indicate that p40 is expressed in excessive amount in the CNS of different demyelinating diseases such as multiple sclerosis (MS), Guillain-Barre syndrome, and animal models experimental autoimmune encephalomyelitis and neuritis (15, 16, 19–21). On the other hand, the expression of p35 remains almost constant or decreased to some extent in the CNS of these demyelinating diseases compared with the CNS of control subjects (19–21). However, the biological significance of this over-expression of p40 in the CNS of patients with demyelinating diseases is not known.
Several lines of evidence presented in this manuscript clearly support the conclusion that IL-12 p40 homodimer, p402, induces the expression of iNOS in mouse microglia and macrophages. This conclusion was based on the following observations. First, p402 induces the production of NO, which is inhibited by arginase, the enzyme which degrades the substrate of NOS, and by L-NMA, an inhibitor of NOS. Second, p402-mediated production of NO and expression of iNOS is inhibited by anti-mouse p40 but not by anti-mouse p70. Third, the expression of mouse p40 cDNA but not the mouse p35 cDNA induces the production of NO and the expression of iNOS, suggesting that the p40 but not the p35 subunit of IL-12 is involved in the induction of iNOS. Because NO produced from the activation of iNOS in the CNS participates in the pathophysiology of MS (4, 6–12), the overexpression of p40 mRNA in the CNS of MS patients (15, 19–21) and the induction of iNOS by p40 suggest that p40 may participate in the pathophysiology of MS through the induction of iNOS.
The level of p40 is much higher (5- to 500-fold) than that of the heterodimeric p70 in IL-12-producing cells (19). This excess p40 produced either in vitro in activated cells or in vivo in serum of endotoxin-treated mice exists as both dimer (20–40%) and monomer (the remainder, Ref. 19). Although the biological role of the monomeric as well as the dimeric form of p40 is not known, it has been suggested that p402 may act as a physiologic regulator of bioactive IL-12, because p402 possesses IL-12 antagonist activity (19, 39). Therefore, the induction of iNOS by p402 suggests that p402 exhibits IL-12 antagonist activity possibly through the activation of iNOS. However, our observation that both p402 and the so-called bioactive IL-12 (heterodimeric p70) induce the production of NO and the expression of iNOS precludes this possibility. If iNOS-derived NO mediates the IL-12 antagonist activity of p402 then IL-12 itself can regulate its own function through the activation of iNOS. Apart from the IL-12 antagonist activity of p40, experiments on Listeria monocytogenes infection in p40- and p35-deficient mice have shown that p40-deficient mice were susceptible to infection, but p35-deficient mice were able to eliminate bacteria despite the mouse's inability to produce biologically active heterodimeric IL-12 (19). Interestingly, it has also been found that the p35-deficient mouse produces normal levels of p40 (19). Taken together, these observations suggest that p40 alone may carry out some of the biological functions of heterodimeric IL-12. Here we present the first evidence that similar to IL-12, p402 can also induce the expression of iNOS and that iNOS-derived NO may account for the bacteria-eliminating property of both IL-12 and p40.
The signaling events in cytokine-mediated induction of iNOS are not completely established so far. Proinflammatory cytokines (TNF-α, IL-1β, or IFN-γ) bind to their respective receptors and induce the expression of iNOS via NF-κB activation (26–30, 36, 37). The presence of a consensus sequence in the promoter region of iNOS for the binding of NF-κB (36) and the inhibition of iNOS expression with the inhibition of NF-κB activation establishes an essential role of NF-κB activation in the induction of iNOS (26–30, 36, 37). Activation of NF-κB by various cellular stimuli involves the proteolytic degradation of IkB, the inhibitory subunit of NF-κB complex, and the concomitant nuclear translocation of the liberated NF-κB heterodimer (40, 41). Although the biochemical mechanism underlying the degradation of IkB remains unclear, it appears that degradation of IkB induced by various mitogens and cytokines occurs in association with the transient phosphorylation of IkB on serines 32 and 36 (42). Consistently, two closely related kinases (IKKα and IKKβ) that directly phosphorylate IkBα have also been described (43–45). Upon phosphorylation, IkB that is still bound to NF-κB apparently becomes a high affinity substrate for an ubiquitin-conjugating enzyme (46). After phosphorylation-controlled ubiquitination, the IkB is rapidly and completely degraded by the 20 S or 26 S proteosome, and the NF-κB heterodimer enters into the nucleus (47) and binds to the consensus DNA-binding site present in the promoter region of iNOS.
Our results have clearly shown that p402 induces the expression of iNOS through the activation of NF-κB. First, p402 induces the DNA-binding as well as the transcriptional activity of NF-κB. Second, expression of the mouse p40 cDNA but not the mouse p35 cDNA induces the activation of NF-κB and the expression of iNOS. Third, SN50, a ell-permeable peptide carrying the nuclear localization sequence of p50 NF-κB, but not mutant SN50 (SN50M) inhibits p402-mediated activation of NF-κB and expression of iNOS. It has been demonstrated that SN50 specifically blocks the nuclear translocation of NF-κB, but does not affect the activity of AP-1, SP-1 factor, and OCT-1 transcriptional factors (48) suggesting that SN50 inhibits the expression of iNOS in p402-stimulated microglial cells by inhibiting the activation of NF-κB. In addition, these results also suggest that IL-12 p402 is biologically active and that p402 alone can activate microglial cells.
At present, it is unclear how p402 activates NF-κB and induces iNOS in microglial cells. IL-12 p402 has been shown to antagonize bioactive IL-12 heterodimer by binding to the IL-12 receptor complex (19). The high affinity IL-12 receptor is composed of a low affinity IL-12Rβ1 combined with a low affinity IL-12Rβ2, which are responsible for Tyk2/Jak2 activation, respectively, and STAT4 activation (19, 49). It appears that p402 binds to IL-12Rβ1 rather than IL-12Rβ2, whereas bioactive IL-12 binds the receptor complex with high affinity (50). Therefore, it is possible that p402 activates NF-κB and induces the expression of iNOS through the IL-12Rβ1.
NO, a diffusible free radical, plays many roles as a signaling and as a effector molecule in diverse biological systems including neuronal messenger, vasodilation, and antimicrobial and antitumor activities (51, 52). In the nervous system the NO appears to have both neurotoxic and neuroprotective effects and may have a role in the pathogenesis of stroke and other neurodegenerative diseases and in demyelinating conditions (e.g. multiple sclerosis, experimental allergic encephalopathy, X-adrenoleukodystrophy) associated with infiltrating macrophages and the production of proinflammatory cytokines (53). NO and peroxynitrite (reaction product of NO and ) are potentially toxic molecules to neurons and oligodendrocytes that may mediate toxicity through the formation of iron-NO complexes of iron containing enzyme systems (54), oxidation of protein sulfhydryl groups (55), nitration of proteins, and nitrosylation of nucleic acids and DNA strand breaks (56). Although monocytes/macrophages are the primary source of iNOS in inflammation, LPS and proinflammatory cytokines induce a similar response in microglia (35, 8, 13). NO derived from microglia has also been implicated in the damage of myelin-producing oligodendrocytes in demyelinating disorders like multiple sclerosis and neuronal death during Alzheimer's disease and brain trauma (2–5).
Because IL-12 p40 is overexpressed in the CNS of the neuroinflammatory diseases, the induction of iNOS expression by IL-12 p40 in microglia and macrophages suggests that expression of p40 may induce/potentiate the neural injury in the inflamed CNS through the induction of NO production.
Footnotes
This study was supported by Grants NS39940 from the National Institutes of Health, 0692 from Smokeless Tobacco Research Council and 00–09 from the UNMC College of Dentistry.
The abbreviations used are: iNOS, inducible nitric-oxide synthase; IL-12, interleukin 12; DMEM, Dulbecco's modified Eagle's medium; LPS, lipopolysaccharide.
Contributor Information
Kalipada Pahan, Department of Oral Biology, University of Nebraska Medical Center, Lincoln, Nebraska 68583.
Faruk G. Sheikh, Department of Oral Biology, University of Nebraska Medical Center, Lincoln, Nebraska 68583.
Xiaojuan Liu, Department of Oral Biology, University of Nebraska Medical Center, Lincoln, Nebraska 68583.
Shilo Hilger, Department of Oral Biology, University of Nebraska Medical Center, Lincoln, Nebraska 68583.
Michael McKinney, Department of Pharmacology, Mayo Clinic at Jacksonville, Jacksonville, Florida 32224.
Thomas M. Petro, Department of Oral Biology, University of Nebraska Medical Center, Lincoln, Nebraska 68583
References
- 1.Galea E, Feinstein DL, Reis DJ. Proc Natl Acad Sci U S A. 1992;89:10945–10949. doi: 10.1073/pnas.89.22.10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koprowski H, Zheng YM, Heber-Katz E, Fraser N, Rorke L, Fu ZF, Hanlon C, Dietzshold B. Proc Natl Acad Sci U S A. 1993;90:3024–3027. doi: 10.1073/pnas.90.7.3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mitrovic B, Ignarro LJ, Montestruque S, Smoll A, Merril JE. Neurosci. 1994;61:575–585. doi: 10.1016/0306-4522(94)90435-9. [DOI] [PubMed] [Google Scholar]
- 4.Bo L, Dawson TM, Wesselingh S, Mork S, Choi S, Kong PA, Hanley D, Trapp BD. Ann Neurol. 1994;36:778–786. doi: 10.1002/ana.410360515. [DOI] [PubMed] [Google Scholar]
- 5.Merrill JE, Ignarro LJ, Sherman MP, Melinek J, Lane TE. J Immunol. 1993;151:2132–2141. [PubMed] [Google Scholar]
- 6.Kolb H, Kolb-Bachofen V. Immunol Today. 1992;13:157–160. doi: 10.1016/0167-5699(92)90118-Q. [DOI] [PubMed] [Google Scholar]
- 7.McCatney-Francis N, Allen JB, Mizel DE, Albina JE, Xie QW, Nathan CF, Wahl SM. J Ex Med. 1993;178:749–754. doi: 10.1084/jem.178.2.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pahan K, Sheikh FG, Namboodiri AMS, Singh I. J Biol Chem. 1998;273:12219–12226. doi: 10.1074/jbc.273.20.12219. [DOI] [PubMed] [Google Scholar]
- 9.Johnson AW, Land JM, Thompson EJ, Bolanos JP, Clark JB, Heales SJR. J Neurol Neurosurg Psychiatry. 1995;58:107–115. doi: 10.1136/jnnp.58.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brenner T, Brocke S, Szafer F, Sobel RA, Parkinson JF, Perez DH, Steinman L. J Immunol. 1997;158:2940–2946. [PubMed] [Google Scholar]
- 11.Hooper DC, Scott GS, Zborek A, Mikheeva T, Kean RB, Koprowski H, Spitsin SV. FASEB J. 2000;14:691–698. doi: 10.1096/fasebj.14.5.691. [DOI] [PubMed] [Google Scholar]
- 12.Brosan CF, Battistini L, Raine CS, Dickson DW, Casadevall A, Lee SC. Dev Neurosci. 1994;16:152–161. doi: 10.1159/000112102. [DOI] [PubMed] [Google Scholar]
- 13.Hu SX, Sheng WS, Peterson PK, Chao CC. Glia. 1995;15:491–494. doi: 10.1002/glia.440150412. [DOI] [PubMed] [Google Scholar]
- 14.Hsieh CS, Macatonia SE, Tripp CS, Wolf SF, O'Garra A, Murphy KM. Science. 1993;260:547–549. doi: 10.1126/science.8097338. [DOI] [PubMed] [Google Scholar]
- 15.Constantinescu CS, Goodman DB, Hilliard B, Wysocka M, Cohen JA. Neurosci Lett. 2000;287:171–174. doi: 10.1016/s0304-3940(00)01184-8. [DOI] [PubMed] [Google Scholar]
- 16.Zipris D, Greiner DL, Malkani S, Whalen B, Mordes JP, Rossini AA. J Immunol. 1996;156:1315–1321. [PubMed] [Google Scholar]
- 17.Wolf SF, Temple PA, Kobayashi M, Young D, Dicig M, Lowe L, Dzialo R, Fitz L, Ferenz C, Hewick RM. J Immunol. 1991;146:3074–3081. [PubMed] [Google Scholar]
- 18.Schoenhaut DS, Chua AO, et al. J Immunol. 1992;148:3433–3440. [PubMed] [Google Scholar]
- 19.Gately MK, Renzetti LM, Magram J, Stern AS, Adorini L, Gubler U, Presky DH. Annu Rev Immunol. 1998;16:495–521. doi: 10.1146/annurev.immunol.16.1.495. [DOI] [PubMed] [Google Scholar]
- 20.van Boxel-Dezaire AH, Hoff SC, et al. Ann Neurol. 1999;45:695–703. doi: 10.1002/1531-8249(199906)45:6<695::aid-ana3>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 21.Fassbender K, Ragoschke A, Rossol S, Schwartz A, Mielke O, Paulig A, Hennerici M. Neurology. 1998;51:753–758. doi: 10.1212/wnl.51.3.753. [DOI] [PubMed] [Google Scholar]
- 22.Bright JJ, Musuro BF, Du C, Sriram S. J Neuroimmunol. 1998;82:22–30. doi: 10.1016/S0165-5728(97)00184-7. [DOI] [PubMed] [Google Scholar]
- 23.McCarthy K, DeVellis J. J Cell Biol. 1980;85:890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Giulian D, Baker TJ. J Neurosci. 1986;6:2163–2178. doi: 10.1523/JNEUROSCI.06-08-02163.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pahan K, Sheikh FG, Namboodiri AMS, Singh I. Free Rad Biol Med. 1998;24:39–48. doi: 10.1016/s0891-5849(97)00137-8. [DOI] [PubMed] [Google Scholar]
- 26.Pahan K, Liu X, Wood C, Raymond JR. FEBS Lett. 2000;472:203–207. doi: 10.1016/s0014-5793(00)01465-4. [DOI] [PubMed] [Google Scholar]
- 27.Pahan K, Liu X, McKinney MJ, Wood C, Sheikh FG, Raymond JR. J Neurochem. 2000;74:2288–2295. doi: 10.1046/j.1471-4159.2000.0742288.x. [DOI] [PubMed] [Google Scholar]
- 28.Feinstein DL, Galea E, Roberts S, Berquist H, Wang H, Reis DJ. J Neurochem. 1994;62:315–321. doi: 10.1046/j.1471-4159.1994.62010315.x. [DOI] [PubMed] [Google Scholar]
- 29.Pahan K, Namboodiri AMS, Sheikh FG, Smith BT, Singh I. J Biol Chem. 1997;272:7786–7791. doi: 10.1074/jbc.272.12.7786. [DOI] [PubMed] [Google Scholar]
- 30.Pahan K, Sheikh FG, Khan M, Namboodiri AMS, Singh I. J Biol Chem. 1998;273:2591–2600. doi: 10.1074/jbc.273.5.2591. [DOI] [PubMed] [Google Scholar]
- 31.Pahan K, Raymond JR, Singh I. J Biol Chem. 1999;274:7528–7536. doi: 10.1074/jbc.274.11.7528. [DOI] [PubMed] [Google Scholar]
- 32.Bradford M. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 33.Dignam JD, Lebovitz RM, Roeder RG. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aloisi F, Penna G, Cerase J, Menendez Iglesias B, Adorini L. J Immunol. 1997;159:1604–1612. [PubMed] [Google Scholar]
- 35.Suzumura A, Sawada M, Takayanagi T. Brain Res. 1998;787:139–142. doi: 10.1016/s0006-8993(97)01166-9. [DOI] [PubMed] [Google Scholar]
- 36.Xie Q, Kashiwabara Y, Nathan C. J Biol Chem. 1994;269:4705–4708. [PubMed] [Google Scholar]
- 37.Pahan K, Sheikh FG, Namboodiri AMS, Singh I. J Clin Invest. 1997;100:2671–2679. doi: 10.1172/JCI119812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin YZ, Yao SY, Veach RA, Torgerson TR, Hawiger J. J Biol Chem. 1995;270:14255–14258. doi: 10.1074/jbc.270.24.14255. [DOI] [PubMed] [Google Scholar]
- 39.Germann T, Rude E, Mattner F, Gately MK. Immunol Today. 1995;16:500–501. doi: 10.1016/0167-5699(95)80035-2. [DOI] [PubMed] [Google Scholar]
- 40.Stefanova I, Corcoran ML, Horak EM, Wahl LM, Bolen JB, Horak ID. J Biol Chem. 1993;268:20725–20728. [PubMed] [Google Scholar]
- 41.Salkowski CA, Detore G, McNally R, van Rooijen N, Vogel SN. J Immunol. 1997;158:905–912. [PubMed] [Google Scholar]
- 42.Beg AA, Ruben SM, Scheinman RI, Haskil S, Rosen CA, Baldwin AS., Jr Genes Dev. 1992;6:1899–1913. doi: 10.1101/gad.6.10.1899. [DOI] [PubMed] [Google Scholar]
- 43.DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M. Nature. 1997;388:548–554. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- 44.Maniatis T. Science. 1997;278:818–819. doi: 10.1126/science.278.5339.818. [DOI] [PubMed] [Google Scholar]
- 45.Mercurio F, Zhu H, Murray BW, et al. Science. 1997;278:860–866. doi: 10.1126/science.278.5339.860. [DOI] [PubMed] [Google Scholar]
- 46.Sun SC, Ganchi PA, Ballard DW, Greene WC. Science. 1993;259:1912–1915. doi: 10.1126/science.8096091. [DOI] [PubMed] [Google Scholar]
- 47.Brown K, Gerstberger S, Carlson L, Franzoso G, Siebenlist U. Science. 1995;267:1485–1488. doi: 10.1126/science.7878466. [DOI] [PubMed] [Google Scholar]
- 48.Qin ZH, Wang Y, Nakai M, Chase TN. Mol Pharmacol. 1998;53:33–42. doi: 10.1124/mol.53.1.33. [DOI] [PubMed] [Google Scholar]
- 49.Wang X, Wilkinson VL, Podlaski FJ, Wu C, Stern AS, Presky DH, Magram J. Eur J Immunol. 1999;29:2007–2013. doi: 10.1002/(SICI)1521-4141(199906)29:06<2007::AID-IMMU2007>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 50.Presky DH, Yang H, Minetti LJ, Chua AO, Nabavi N, Wu CY, Gately MK, Gubler U. Proc Natl Acad Sci U S A. 1996;93:14002–14007. doi: 10.1073/pnas.93.24.14002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nathan C. FASEB J. 1992;6:3051–3064. [PubMed] [Google Scholar]
- 52.Jaffrey SR, Snyder SH. Annu Rev Cell Dev Biol. 1995;11:417–440. doi: 10.1146/annurev.cb.11.110195.002221. [DOI] [PubMed] [Google Scholar]
- 53.Dawson VL, Dawson TM, London ED, Bredt DT, Snyder SH. Proc Natl Acad Sci U S A. 1991;88:6368–6371. doi: 10.1073/pnas.88.14.6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Drapier JC, Hibbs JB. J Immunol. 1988;140:2829–2838. [PubMed] [Google Scholar]
- 55.Radi R, Beckman JS, Bush KM, Freeman BA. J Biol Chem. 1991;266:4244–4250. [PubMed] [Google Scholar]
- 56.Wink DA, Kasprazak KS, et al. Science. 1991;254:1001–1003. doi: 10.1126/science.1948068. [DOI] [PubMed] [Google Scholar]