

Abstract
Networks synthesized through thiol-acrylate photopolymerization or Michael-type addition step growth reactions contain esters with neighboring sulfide groups. Previous work has demonstrated that these esters are readily hydrolyzable at physiological pH. Here, the influence of the distance between the sulfide and ester, as well as the water concentration, on ester hydrolysis was characterized. These preliminary results indicate that reducing the number of carbons between the sulfide and the ester from 2 to 1 increased the rate of ester hydrolysis from 0.022 ± 0.001 to 0.08 ± 0.015 days-1. Increases in ester hydrolysis rates were also observed as hydrophilicity increased for oligomers prepared from a trithiol, tetrathiol and dithiol monomer (0.012 ± 0.003, 0.032 ± 0.004, and 0.091 ± 0.003 days-1, respectively). Additionally, in bulk-eroding polymeric biomaterials, variations in pH impacted the ester hydrolysis rate. This work confirms that small variations in buffer pH predictably alter the mass loss profile of a thiol-acrylate photopolymer. More specifically, as buffer pH was changed from 7.4 to 8.0, the rate of ester hydrolysis increased from 0.074 ± 0.003 to 0.28 ± 0.005 days-1. The magnitude of this observed change in ester hydrolysis rate was correlated to the increase in hydroxide ion concentration that accompanied this pH change.
1. Introduction
Degradable polymeric hydrogels are commonly used in tissue engineering and drug delivery research. Numerous advantages make this class of materials well suited for these types of applications. For example, the high water content of most hydrogels enables successful cell and protein encapsulation [1-24]. Proper selection of the macromolecular precursors used to synthesize the gels allows the initial mechanical properties, degradation behavior and biological activity of the network to be selectively tuned for a variety of applications. Previous work has demonstrated that network degradation can be used to control drug release temporally and influence tissue formation [1,2,6,9,11,14,20,21,25-29]. Degradation in covalently crosslinked polymeric biomaterials has been achieved through hydrolytic cleavage of esters and anhydrides and also through enzymatically induced cleavage of select peptide sequences that have been incorporated into the material [2,22,27,30-32].
Much of the work involving hydrolytically degradable hydrogels has focused on materials formed from triblock oligomers containing poly(lactic acid) (PLA) segments flanking a hydrophilic, water-stable block [4,10,19,24,33-37]. These segments contain multiple ester linkages which degrade when exposed to basic or acidic environments. The number of ester bonds that the PLA segments contain directly influences the probability that one PLA linkage in the block is cleaved, thereby breaking the crosslink. Recently, researchers have found that networks formed from Michael-type addition reactions between thiol and acrylate monomers also degrade hydrolytically at physiological pH through degradation of single ester groups located near sulfide bonds [1,2,38-40]. Furthermore, the number of carbon atoms between the ester and sulfide groups was qualitatively shown to influence the rate of ester hydrolysis [39].
Here, the quantitative investigation of how the carbon spacer length between the ester and sulfide and the monomer hydrophobicity impact ester hydrolysis rates is presented. To this end, thiol-acrylate oligomers were formed through Michael-type addition of different multifunctional thiols with a monoacrylated poly(ethylene glycol) (PEG). These oligomers were incubated in pH 8.0 phosphate buffer that contained 10% D2O. Changes in 1H NMR spectra were directly used to quantify ester degradation.
2. Materials and Methods
2.1. Comparing ester hydrolysis at pH 7.4 and pH 8.0
Crosslinked thiol-acrylate photopolymers were made from monomer solutions containing a PLA-PEG2000-PLA diacrylate [41-43] and a monofunctional thiol (butyl-3-mercaptopropionate, Aldrich). The monomers were mixed in a 7:3 ratio of acrylate to thiol functional groups before 50 wt.% dimethyl sulfoxide and 0.1 wt.% 1--2-hydroxy-2-methyl-1-propane-1-one (Irgacure 2959, I2959, Ciba) were added, and the mixture was photopolymerized into disks 10 mm in diameter and 1 mm thick using 5 mW cm-2 of 365 nm light. These disks were then degraded in either pH 7.4 or 8.0 phosphate buffer and their mass loss profiles compared.
2.2. Preparation of oligomers from thiols and acrylates
Mixtures of PEG monoacrylate ( , Aldrich) and two different trithiols, a dithiol or a tetrathiol (1 trimethylolpropane tris(2-mercaptoacetate), 2 trimethylolpropane tris(3-mercaptopropionate), 3 2,2′-(ethylenedioxy) diethanethiol and 4 pentaerythritol tetrakis(3-mercaptopropionate), Fig. 1, Aldrich) were prepared such that stoichiometric ratios of acrylate and thiol functional groups were achieved. Triethylamine (2 wt.%, Fisher Scientific) was added to catalyze the Michael-type addition reactions, and each mixture was stirred for 1 h.
Fig. 1.
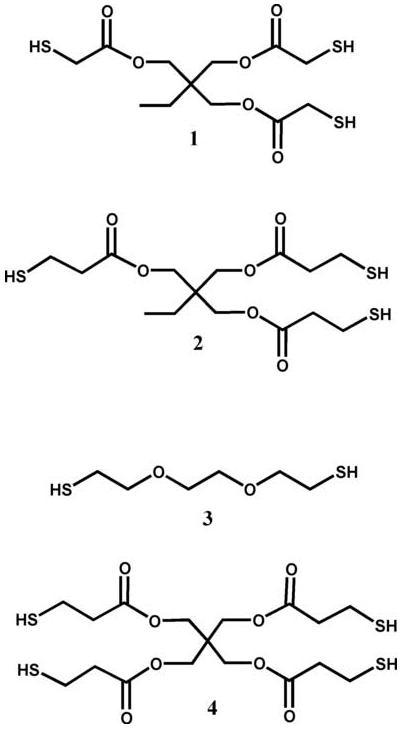
Thiol monomers used to make the thiol-acrylate oligomers with hydrolyzable esters: 1, trimethylolpropane tris(2-mercaptoacetate); 2, trimethylolpropane tris(3-mercaptopropionate); 3, 2,2′-(ethylenedioxy) diethanethiol; and 4, pentaerythritol tetrakis(3-mercaptopropionate).
2.3. Ester hydrolysis of oligomers made from thiols and acrylates
Approximately 0.6 g of each mixture was combined with 1 ml of 0.1 M pH 8.0 phosphate buffer that contained 10 wt.% D2O. The samples were incubated at 37 °C and periodically their 1H-NMR spectra were determined (Varian 500 MHz, NTR probe). NMR was used to quantify ester hydrolysis in these materials even though it is less sensitive than chromatographic techniques because it allowed the cleavage of different types of ester groups to be monitored independently in the same sample. For the dithiol, the tetrathiol and the mercaptopropionate trithiol samples, the disappearance of the peaks at 2.76 and 4.19 ppm and the appearance of the peak at 2.36 ppm were used to determine the extent of hydrolysis from the acrylate-based ester. The chemical structure proximate to the esters in these thiol-acrylate oligomers is shown in Fig. 2, as are the degradation products that result following ester hydrolysis. The 1H-NMR peaks at 4.2-4.0, 2.76 and 2.36 ppm correspond to the protons in the figure that are labeled “a”, “b” and “c”, respectively. The protons neighboring the acrylate-based and thiol-based esters were not distinguishable from each other for these oligomers using this technique. They are distinguishable, however, for the mercaptoacetate trithiol, where the hydrolysis of the acrylate-based ester corresponds to the disappearance of the peaks between 4.1 and 4.2 ppm and the appearance of the peak at 2.36 ppm, while hydrolysis of the thiol-based ester corresponds to the disappearance of the 3.33 and 2.8 ppm peaks and the appearance of the 3.13 ppm peak. The NMR data for each peak was normalized by dividing the peak areas by the ideal number of protons for that peak (e.g. 8 protons for the 2.36 ppm peak for the tetrathiol oligomers), and the data for the peaks at 2.36 and 3.13 were further manipulated by subtracting their value from 1 so that their appearance could be compared with the disappearance of the other peaks in each spectra.
Fig. 2.
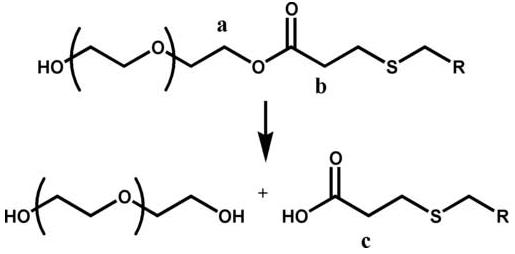
Protons corresponding to 1H-NMR shifts of (a) 4.19, (b) 2.76 and (c) 2.36 ppm.
3. Results and discussion
3.1. Impact of buffer pH on the rate of ester hydrolysis
Often, hydrolytic degradation of crosslinked polymeric biomaterials is investigated at physiological pH (7.4). To decrease the time required for this study, experiments were conducted at pH 8.0. To confirm that the mechanism by which ester hydrolysis occurs in these systems is not significantly impacted by this change in pH, an experiment comparing mass loss profiles of a thiol-acrylate photopolymer degraded in phosphate buffers at pH 7.4 and 8.0 was performed.
Fig. 3(a) shows the mass loss extent as a function of degradation time for thiol-acrylate samples that were degraded in either pH 7.4 or 8.0 buffer. This result reveals that as the buffer pH is increased, so is the rate of mass loss. When this observation was quantified by comparing the observed hydrolysis rate constants (khyd, predicted from experimental fits of the data using a mass loss model [43]), a similar trend was observed: khyd was 0.074 ± 0.003 days-1 for samples degraded at pH 7.4 and 0.28 ± 0.005 days-1 at pH 8.0.
Fig. 3.


(a) Mass loss profiles of thiol-acrylate photopolymers at pH 7.4 (◆) and pH 8.0 (■). (b) Same data as in (a) but with the time axis of the pH 8.0 data adjusted to account for in the increase in OH- concentration. The polymer was made from a PLA-PEG-PLA diacrylate and a monothiol (butyl 3-mercaptopropionate) where 30 mol.% of the functional groups were thiol.
The rate of ester hydrolysis in these types of crosslinked polymer networks was first modeled by Metters et al. [35] in degradable acrylate hydrogels using Eq. (1), where a lumped, overall kinetic parameter, khyd, combines the ester hydrolysis rate constant, k, the network’s water concentration, [H2O], and the hydroxide ion concentration, [OH-]. When the difference in OH- concentration between the pH 7.4 and 8.0 buffers is accounted for (2.5×10-7 and 1×10-6 M, respectively), similar k values are observed for the two systems (i.e., k is 5.4 ± 0.2 109 days-1 for samples degraded at pH 7.4 and 5.1 ± 0.09 109 days-1 at pH 8.0).
| (1) |
In Fig. 3(b) this analysis was taken one step further when the degradation times for the samples degraded in pH 8.0 buffer were multiplied by a factor of 4 and plotted against unmodified data for the samples degraded at pH 7.4 (e.g. the initial time to reach 43% mass loss at pH 8.0 is 1.8 ± 0.05 days and the modified time for the same sample is 7.1 ± 0.2 days). The time adjustment (i.e. modified time = 4 × original time) corresponds to the fourfold increase in hydroxide ions associated with the increased pH. When the pH 8.0 data are adjusted in this manner, the two mass loss profiles are nearly identical up to the point where reverse gelation occurs. The extended mass loss profile for the samples degraded at pH 8.0 highlights the fact that the transition from loosely crosslinked networks to entanglements of highly branched polymer chains which occurs at reverse gelation is hard to capture using mass loss data because a change in mass loss is dependent on ester hydrolysis and diffusion of the degraded material out of the rest of the network. This limitation is exaggerated when the time axis is multiplied by a factor of 4. Despite these shortcomings regarding the time where reverse gelation occurs, it was concluded that while an increase in pH from 7.4 to 8.0 increases the mass loss rate, the degradation mechanism is not significantly affected and both systems are degraded by a bulk-erosion mechanism. Additionally, the polymer networks that were utilized for this set of experiments contain two types of ester groups: esters that are clustered in PLA blocks and esters near sulfides that form when the thiol and acrylate groups react. Importantly, while the hydrolysis data in Fig. 3 do not differentiate between these two types of esters, the scaling of the reaction rate with OH- concentration that is depicted in Fig. 3(b) is strongly indicative of the degradation mechanism’s dependence on buffer pH being the same for the two ester types. Thus, the influence of various factors on the ester hydrolysis rate can be studied in a pH 8.0 buffer and directly correlated to physiological conditions at pH 7.4 for esters with neighboring sulfide groups.
3.2. Studying hydrolysis of esters with neighboring sulfides using thiol-acrylate oligomers
Four different thiol monomers were used to study the ester hydrolysis with neighboring sulfides: a hydrophilic dithiol that did not contain any esters, two trithiols that have either one or two carbons between their ester and the sulfide, and a tetrathiol with two carbons separating the ester and the sulfide (Fig. 1). Each of the oligomers formed from the reaction of these thiols with PEG monoacrylate contains an additional degradable ester for every attached acrylate.
As the esters in these oligomers were hydrolyzed, the extent of ester hydrolysis was quantified by NMR. An example of how 1H-NMR was used to study hydrolysis rates using these oligomers is shown in Fig. 4. In this figure the 1H-NMR spectra of the dithiol-based oligomer is shown initially (part a) and after 11 days in pH 8.0 buffer (part b). The decrease in the areas under the proton peaks at 4.2 and 2.76 ppm and the increase in peak area at 2.36 ppm are associated with the hydrolysis of the neighboring ester group. The changes in peak areas were used to calculate the khyd for each type of oligomer.
Fig. 4.
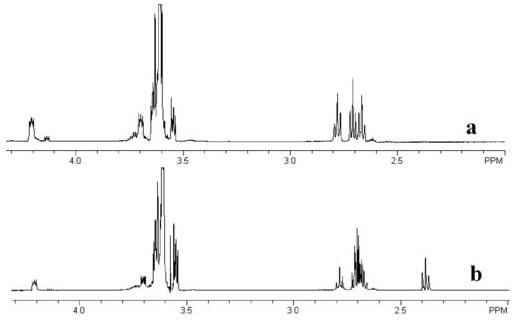
(a) Initial 1H NMR spectra and (b) a spectra after 11 days in pH 8.0 buffer for the dithiol-monoacrylate oligomer. Ester peaks at 4.2 and 2.76 ppm decrease with time while the peak associated with the ester hydrolysis appears at 2.36 ppm.
3.2.1. Influence of carbon spacer length on ester hydrolysis
Examining the data for the mercaptoacetate trithiol (1) oligomers provides information about how the number of carbons between the sulfide and ester groups influences ester hydrolysis. Fig. 5 shows the chemical structure surrounding the degradable esters in both the mercaptoacetate- and mercaptopropionate-based oligomers (a and b, respectively). Each type of oligomer has two degradable esters surrounding the sulfide: one originating from the thiol monomer and another from the acrylate monomer. In the mercaptopropionate-based oligomer (b), there are two carbon atoms between the ester and sulfide for both of the esters. In the mercaptoacetate oligomer (1), the thiol-based ester only has a single carbon spacer between the ester and the sulfide while the acrylate-based ester still has two carbons between the ester and the sulfide.
Fig. 5.

Chemical structures of the (a) mercaptoacetate-based and (b) mercaptopropionate-based oligomers highlighting the number of carbons between the sulfide and esters for the (1) thiol-based and (2) acrylate-based esters.
Analysis of the changes in the 1H-NMR spectra during degradation of this oligomer provides insights into how the ester-sulfide carbon spacer length influences the ester hydrolysis rate. Fig. 6 plots the change in peak area as a function of time for the protons that neighbor both the thiol-based and acrylate-based esters in the mercaptoacetate-based oligomer’s NMR spectra. The protons that correspond to each curve in the plot are labeled with respect to the chemical structures of both the initial oligomer and its degradation products. From the data in this figure, khyd values were calculated as 0.08 ± 0.015 and 0.022 ± 0.001 days-1 for the thiol- and acrylate-based esters, respectively. These observations support earlier claims made by Schoenmakers et al. [39] that the closer the ester is to the sulfide, the more rapidly it hydrolyzes.
Fig. 6.


Changes in peak area due to ester hydrolysis for thiol-acrylate oligomers formed through Michael-type addition between a PEG375 monoacrylate and trimethylolpropane tris(2-mercaptoacetate). Changes in peak area that occur when the thiol-based ester hydrolyzes are the filled symbols, while changes in peak area due to hydrolysis of the acrylate-based ester are the open symbols: 4.2 (◇), 3.33 (■), 3.13 (▲), 2.8 (●) and 2.36 (○) ppm.
3.2.2. Influence of substrate hydrophobicity on ester hydrolysis
Comparing the ester hydrolysis rate data for the oligomers made from the trithiol (2), the dithiol (3) and the tetrathiol (4) provides information about how the ester’s local chemical environment influences hydrolytic degradation. Changes in the peak areas of the protons neighboring the degradable esters are show in Fig. 7 as a function of degradation time for these three different oligomers. The khyd values calculated from the data in each of these plots are 0.012 ± 0.003, 0.032 ± 0.004 and 0.091 ±0.003 days-1 for the oligomers made from the trithiol, tetrathiol and dithiol monomers, respectively. Of these three oligomers, the one made from the dithiol is the most hydrophilic, followed by the tetrathiol and then the trithiol. This decrease in hydrophilicity associated with the thiol monomer chemistries decreases the local water concentration and the ester hydrolysis rate as evidenced by the plots of the NMR data shown in Fig. 7.
Fig. 7.



Changes in peak area due to ester hydrolysis for thiol-acrylate oligomers formed through Michael-type addition between a PEG375 monoacrylate and a tri- (a) or tetrafunctional (b) mercaptopropionate or a difunctional thiol (c) that does not contain esters. 4.2 (◇), 4.15 (◀), 4.1 (□), 2.76 (△) and 2.36 (○) ppm.
4. Conclusions
Small deviations in pH predictably alter the rate of ester hydrolysis without impacting the network degradation mechanism. This allows mass loss in pH 8.0 degradation buffer to be correlated to mass loss at physiological pH (7.4) by accounting for the fourfold increase in hydroxide ion concentration. Additionally, this work confirmed that esters with neighboring sulfide groups readily hydrolyze. The ester hydrolysis rate in these systems is influenced both by the number of carbons between the sulfide and ester and by the network’s water concentration. The khyd values were observed to shift from 0.08 ± 0.015 to 0.02 ± 0.001 days-1 when the number of carbons between the sulfide and ester was increased from 1 to 2. Changing oligomer hydrophobicity through changes in thiol monomer chemistry and functionality allowed khyd to vary from 0.01 ± 0.003 to 0.09 ± 0.003 days-1.
Acknowledgements
The authors thank their funding sources for this work: a grant from the NIH (R01 DE12998), a Department of Education GAANN fellowship and a University of Colorado Beverly Sears Graduate Student Grant to AER.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Elbert DL, Pratt AB, Lutolf MP, Halstenberg S, Hubbell JA. Protein delivery from materials formed by self-selective conjugate addition reactions. J. Control. Release. 2001;76:11. doi: 10.1016/s0168-3659(01)00398-4. [DOI] [PubMed] [Google Scholar]
- [2].Van de Wetering P, Metters AT, Schoenmakers RG, Hubbell JA. Poly(ethylene glycol) hydrogels formed by conjugate addition with controllable swelling, degradation, and release of pharmaceutically active proteins. J. Control. Release. 2005;102:619. doi: 10.1016/j.jconrel.2004.10.029. [DOI] [PubMed] [Google Scholar]
- [3].Masters KS, Shah DN, Leinwand LA, Anseth KS. Crosslinked hyaluronan scaffolds as a biologically active carrier for valvular interstitial cells. Biomaterials. 2005;26:2517. doi: 10.1016/j.biomaterials.2004.07.018. [DOI] [PubMed] [Google Scholar]
- [4].Nuttelman CR, Tripodi MC, Anseth KS. Synthetic hydrogel niches that promote hMSC viability. Matrix Biology. 2005;24:208. doi: 10.1016/j.matbio.2005.03.004. [DOI] [PubMed] [Google Scholar]
- [5].Quick DJ, Macdonald KK, Anseth KS. Delivering DNA from photocrosslinked, surface eroding polyanhydrides. Journal of Controlled Release. 2004;97:333. doi: 10.1016/j.jconrel.2004.03.001. [DOI] [PubMed] [Google Scholar]
- [6].Bryant SJ, Bender RJ, Durand KL, Anseth KS. Encapsulating chondrocytes in degrading PEG hydrogels with high modulus: engineering gel structural changes to facilitate cartilaginous tissue production. Biotechnology and Bioengineering. 2004;86:747. doi: 10.1002/bit.20160. [DOI] [PubMed] [Google Scholar]
- [7].Nuttelman CR, Tripodi MC, Anseth KS. In vitro osteogenic differentiation of human mesenchymal stem cells photoencapsulated in PEG hydrogels. Journal of Biomedical Materials Research Part A. 2004;68A:773. doi: 10.1002/jbm.a.20112. [DOI] [PubMed] [Google Scholar]
- [8].Rice MA, Martens P, Bryant SJ, Mahoney MJ, Bowman CN, Anseth KS. Photopolymerization of synthetic hydrogel niches for 3D cell culture and tissue regeneration. Abstr. Pap. Am. Chem. Soc. 2004;228:U391. [Google Scholar]
- [9].Bryant SJ, Anseth KS, Lee DA, Bader DL. Crosslinking density influences the morphology of chondrocytes photoencapsulated in PEG hydrogels during the application of compressive strain. Journal of Orthopaedic Research. 2004;22:1143. doi: 10.1016/j.orthres.2004.02.001. [DOI] [PubMed] [Google Scholar]
- [10].Quick DJ, Anseth KS. Gene delivery in tissue engineering: a photopolymer platform to coencapsulate cells and plasmid DNA. Pharmaceutical Research. 2003;20:1730. doi: 10.1023/b:pham.0000003368.66471.6a. [DOI] [PubMed] [Google Scholar]
- [11].Anseth KS, Bryant SJ, Martens P. In situ forming cell gel constructs: monitoring gel degradation to control extracellular matrix evolution. Abstr. Pap. Am. Chem. Soc. 2003;225:U683. [Google Scholar]
- [12].Bryant SJ, Durand KL, Anseth KS. Manipulations in hydrogel chemistry control photoencapsulated chondrocyte behavior and their extracellular matrix production. Journal of Biomedical Materials Research Part A. 2003;67A:1430. doi: 10.1002/jbm.a.20003. [DOI] [PubMed] [Google Scholar]
- [13].Poshusta AK, Burdick JA, Mortisen DJ, Padera RF, Ruehlman D, Yaszemski MJ, Anseth KS. Histocompatibility of photocrosslinked polyanhydrides: a novel in situ forming orthopaedic biomaterial. Journal of Biomedical Materials Research Part A. 2003;64A:62. doi: 10.1002/jbm.a.10274. [DOI] [PubMed] [Google Scholar]
- [14].Anseth KS, Metters AT, Bryant SJ, Martens PJ, Elisseeff JH, Bowman CN. In situ forming degradable networks and their application in tissue engineering and drug delivery. J. Control. Release. 2002;78:199. doi: 10.1016/s0168-3659(01)00500-4. [DOI] [PubMed] [Google Scholar]
- [15].Burdick JA, Anseth KS. Photoencapsulation of osteoblasts in injectable RGD-modified PEG hydrogels for bone tissue engineering. Biomaterials. 2002;23:4315. doi: 10.1016/s0142-9612(02)00176-x. [DOI] [PubMed] [Google Scholar]
- [16].Bryant SJ, Anseth KS. Hydrogel properties influence ECM production by chondrocytes photoencapsulated in poly(ethylene glycol) hydrogels. Journal of Biomedical Materials Research. 2002;59:63. doi: 10.1002/jbm.1217. [DOI] [PubMed] [Google Scholar]
- [17].Burdick JA, Mason MN, Hinman AD, Thorne K, Anseth KS. Delivery of osteoinductive growth factors from degradable PEG hydrogels influences osteoblast differentiation and mineralization. J. Control. Release. 2002;83:53. doi: 10.1016/s0168-3659(02)00181-5. [DOI] [PubMed] [Google Scholar]
- [18].Burdick JA, Padera RF, Huang JV, Anseth KS. An investigation of the cytotoxicity and histocompatibility of in situ forming lactic acid based orthopedic biomaterials. Journal of Biomedical Materials Research. 2002;63:484. doi: 10.1002/jbm.10298. [DOI] [PubMed] [Google Scholar]
- [19].Mason MN, Metters AT, Bowman CN, Anseth KS. Predicting controlled-release behavior of degradable PLA-b-PEG-b-PLA hydrogels. Macromolecules. 2001;34:4630. [Google Scholar]
- [20].Bryant SJ, Anseth KS. The effects of scaffold thickness on tissue engineered cartilage in photocrosslinked poly(ethylene oxide) hydrogels. Biomaterials. 2001;22:619. doi: 10.1016/s0142-9612(00)00225-8. [DOI] [PubMed] [Google Scholar]
- [21].Ratner BD, Bryant SJ. Biomaterials: where we have been and where we are going. Annual Review of Biomedical Engineering. 2004;6:41. doi: 10.1146/annurev.bioeng.6.040803.140027. [DOI] [PubMed] [Google Scholar]
- [22].West JL, Hubbell JA. Polymeric biomaterials with degradation sites for proteases involved in cell migration. Macromolecules. 1999;32:241. [Google Scholar]
- [23].Langer R, Peppas NA. Advances in biomaterials, drug delivery, and bionanotechnology. Aiche Journal. 2003;49:2990. [Google Scholar]
- [24].Peppas NA, Langer R. New challenges in biomaterials. Science. 1994;263:1715. doi: 10.1126/science.8134835. [DOI] [PubMed] [Google Scholar]
- [25].Martens PJ, Bryant SJ, Anseth KS. Tailoring the degradation of hydrogels formed from multivinyl poly(ethylene glycol) and poly(vinyl alcohol) macromers for cartilage tissue engineering. Biomacromolecules. 2003;4:283. doi: 10.1021/bm025666v. [DOI] [PubMed] [Google Scholar]
- [26].Bryant SJ, Chowdhury TT, Lee DA, Bader DL, Anseth KS. Crosslinking density influences chondrocyte metabolism in dynamically loaded photocrosslinked poly(ethylene glycol) hydrogels. Annals of Biomedical Engineering. 2004;32:407. doi: 10.1023/b:abme.0000017535.00602.ca. [DOI] [PubMed] [Google Scholar]
- [27].Lutolf MP, Weber FE, Schmoekel HG, Schense JC, Kohler T, Muller R, Hubbell JA. Repair of bone defects using synthetic mimetics of collagenous extracellular matrices. Nature Biotechnology. 2003;21:513. doi: 10.1038/nbt818. [DOI] [PubMed] [Google Scholar]
- [28].Burdick JA, Lovestead TM, Anseth KS. Kinetic chain lengths in highly crosslinked networks formed by the photoinitiated polymerization of divinyl monomers: a gel permeation chromatography investigation. Biomacromolecules. 2003;4:149. doi: 10.1021/bm025677o. [DOI] [PubMed] [Google Scholar]
- [29].Burdick JA, Frankel D, Dernell WS, Anseth KS. An initial investigation of photocurable three-dimensional lactic acid based scaffolds in a critical-sized cranial defect. Biomaterials. 2003;24:1613. doi: 10.1016/s0142-9612(02)00538-0. [DOI] [PubMed] [Google Scholar]
- [30].Lutolf MP, Lauer-Fields JL, Schmoekel HG, Metters AT, Weber FE, Fields GB, Hubbell JA. Synthetic matrix metalloproteinase-sensitive hydrogels for the conduction of tissue regeneration: engineering cell-invasion characteristics. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:5413. doi: 10.1073/pnas.0737381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lutolf MP, Raeber GP, Zisch AH, Tirelli N, Hubbell JA. Cell-responsive synthetic hydrogels. Advanced Materials. 2003;15:888. [Google Scholar]
- [32].Sawhney AS, Pathak CP, Hubbell JA. Bioerodible hydrogels based on photopolymerized poly(ethylene glycol)-co-poly(alpha-hydroxy acid) diacrylate macromers. Macromolecules. 1993;26:581. [Google Scholar]
- [33].Davis KA, Burdick JA, Anseth KS. Photoinitiated crosslinked degradable copolymer networks for tissue engineering applications. Biomaterials. 2003;24:2485. doi: 10.1016/s0142-9612(02)00582-3. [DOI] [PubMed] [Google Scholar]
- [34].Blaine G. The uses of plastics in surgery. Lancet. 1946;248:525. doi: 10.1016/s0140-6736(46)91802-8. [DOI] [PubMed] [Google Scholar]
- [35].Metters AT, Anseth KS, Bowman CN. Fundamental studies of a novel, biodegradable PEG-b-PLA hydrogel. Polymer. 2000;41:3993. [Google Scholar]
- [36].Metters AT, Anseth KS, Bowman CN. A statistical kinetic model for the bulk degradation of PLA-b-PEG-b-PLA hydrogel networks. Journal of Physical Chemistry B. 2000;104:7043. [Google Scholar]
- [37].Metters AT, Anseth KS, Bowman CN. A statistical kinetic model for the bulk degradation of PLA-b-PEG-b-PLA hydrogel networks: Incorporating network non-idealities. Journal of Physical Chemistry B. 2001;105:8069. [Google Scholar]
- [38].Elbert DL, Hubbell JA. Conjugate addition reactions combined with free-radical cross-linking for the design of materials for tissue engineering. Biomacromolecules. 2001;2:430. doi: 10.1021/bm0056299. [DOI] [PubMed] [Google Scholar]
- [39].Schoenmakers RG, van de Wetering P, Elbert DL, Hubbell JA. The effect of the linker on the hydrolysis rate of drug-linked ester bonds. Jounal of Controlled Release. 2004;95:291. doi: 10.1016/j.jconrel.2003.12.009. [DOI] [PubMed] [Google Scholar]
- [40].DuBose JW, Cutshall C, Metters AT. Controlled release of tethered molecules via engineered hydrogel degradation: model development and validation. Journal of Biomedical Materials Research. 2005;74A:104. doi: 10.1002/jbm.a.30307. [DOI] [PubMed] [Google Scholar]
- [41].Rydholm AE, Bowman CN, Anseth KS. Degradable thiol-acrylate photopolymers: polymerization and degradation behavior of an in situ forming biomaterial. Biomaterials. 2005;26:4495. doi: 10.1016/j.biomaterials.2004.11.046. [DOI] [PubMed] [Google Scholar]
- [42].Rydholm AE, Held NL, Bowman CN, Anseth KS. Gel Permeation chromatography characterization of the kinetic chain length distributions in thiol-acrylate photopolymers. Macromolecules. 2006;39(23):7882–7888. doi: 10.1021/ma060858u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Rydholm AE, Reddy SK, Anseth KS, Bowman CN. Controlling network structure in degradable thiol-acrylate biomaterials to tune mass loss behavior. Biomacromolecules. 2006;7(10):2827–2836. doi: 10.1021/bm0603793. [DOI] [PubMed] [Google Scholar]