

Abstract
Human immunodeficiency virus type 1 (HIV-1) infection is known to cause neuronal injury and dementia in a significant proportion of patients. However, the mechanism by which HIV-1 mediates its deleterious effects in the brain is poorly defined. The present study was undertaken to investigate the effect of the HIV-1 tat gene on the expression of inducible nitric-oxide synthase (iNOS) in human U373MG astroglial cells and primary astroglia. Expression of the tat gene as RSV-tat but not that of the CAT gene as RSV-CAT in U373MG astroglial cells led to the induction of NO production and the expression of iNOS protein and mRNA. Induction of NO production by recombinant HIV-1 Tat protein and inhibition of RSV-tat-induced NO production by anti-Tat antibodies suggest that RSV-tat-induced production of NO is dependent on Tat and that Tat is secreted from RSV-tat-transfected astroglia. Similar to U373MG astroglial cells, RSV-tat also induced the production of NO in human primary astroglia. The induction of human iNOS promoter-derived luciferase activity by the expression of RSV-tat suggests that RSV-tat induces the transcription of iNOS. To understand the mechanism of induction of iNOS, we investigated the role of NF-κB and C/EBPβ, transcription factors responsible for the induction of iNOS. Activation of NF-κB as well as C/EBPβ by RSV-tat, stimulation of RSV-tat-induced production of NO by the wild type of p65 and C/EBPβ, and inhibition of RSV-tat-induced production of NO by Δp65, a dominant-negative mutant of p65, and ΔC/EBPβ, a dominant-negative mutant of C/EBPβ, suggest that RSV-tat induces iNOS through the activation of NF-κB and C/EBPβ. In addition, we show that extracellular signal-regulated kinase (ERK) but not that p38 mitogen-activated protein kinase (MAPK) is involved in RSV-tat induced production of NO. Interestingly, PD98059, an inhibitor of the ERK pathway, and ΔERK2, a dominant-negative mutant of ERK2, inhibited RSV-tat-induced production of NO through the inhibition of C/EBPβ but not that of NF-κB. This study illustrates a novel role for HIV-1 tat in inducing the expression of iNOS in human astrocytes that may participate in the pathogenesis of HIV-associated dementia.
HIV-1-associated dementia (HAD)1 is a severe form of neurological disability, observed in 20–30% of patients with acquired immunodeficiency syndrome (AIDS) (1). The histopathological signs of HAD include infiltration of inflammatory cells, astrogliosis, pallor of myelin sheaths, abnormalities of dendritic processes, and neuronal apoptotic death. Productive HIV-1 infection in the brain occurs predominantly in macrophages, microglia, and multinucleated giant cells (2, 3). Infection of astrocytes may also occur with restricted virus replication, affirming that the effects of HIV on astrocytes may be indirect (4–6). Neurons also are not infected. The correlation between the disease severity and the viral load is unconvincing, and the neurotoxicity of the virus itself is controversial (4, 7, 8). Furthermore, little or no virus has been found in AIDS-related vacuolar myelopathy (9). Taken together, these findings suggest that indirect mechanisms possibly play an important role in the observed neuronal loss in HAD.
One means by which indirect effects may be exerted upon neural cells is via nitric oxide (NO) production. NO, a diffusible gas, plays an important role in many physiological and diverse pathophysiological conditions (10, 11). At low concentration, NO has been shown to play a unique role in neurotransmission and vasodilation, whereas at higher concentrations it is neurotoxic (10, 11). Consistently, NO, derived in excessive amount from the activation of inducible nitric-oxide synthase (iNOS) in glial cells (astroglia and microglia) and macrophages, is assumed to contribute to neuronal abnormalities in HAD (12–14). By immunocytochemical analysis, Zhao et al. (15) have shown that iNOS expression is present in all of the HAD cases tested and that iNOS immunoreactivity is localized primarily to reactive astrocytes. Analysis of cerebrospinal fluid and serum from HAD patients has shown increased levels of nitrite and nitrate compared with non-HIV infected patients (16). The reaction of NO with forms peroxynitrite, ONOO–, a strong nitrosating agent capable of nitrosating tyrosine residues of a protein to nitrotyrosine. Consistently increasing levels of nitrotyrosine have been found in brains of demented, but not in nondemented, AIDS patients (17). Subsequently, reverse transcription-PCR and Western blot analysis of normal and HAD brains also show markedly higher expression of iNOS mRNA and protein in HAD brains than in normal brains (13).
However, the mechanism by which NO is produced in the brains of HAD patients is unclear. The HIV-1 regulatory protein, Tat, is a potent transactivator of viral and cellular gene expression that is produced in the early phase of infection and actively secreted into the extracellular environment, from where it can act in an autocrine or a paracrine manner (18). We report herein that the HIV-1 tat gene induces the production of NO and the expression of iNOS through the activation of NF-κB and C/EBPβ in human astrocytes.
Materials and Methods
Reagents
Fetal bovine serum, Hanks' balanced salt solution, and Dulbecco's modified Eagle's medium/F-12 were from Invitrogen. l-NG-Monomethylarginine (l-NMA), d-NG-monomethylarginine (d-NMA), PD98059, and SB203580 were purchased from Biomol. Arginase was purchased from Sigma. Antibodies against mouse macrophage iNOS were obtained from Calbiochem. Recombinant Tat protein and anti-Tat monoclonal antibodies were obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, National Institutes of Health. HIV-Tat was from Dr. J. Brady, Tat monoclonal antibodies from Dr. K. Krohn. 125I-labeled protein A, [α-32P]dCTP, and [γ-32P]ATP were obtained from PerkinElmer Life Sciences. Dominant-negative mutants of ERK1, ERK2, and p38 were kindly provided by Dr. Jawed Alam (Alton Ochsner Medical Foundation, New Orleans, LA). The wild type p65, the wild type C/EBPβ, and the dominant-negative mutant of C/EBPβ were kindly provided by Dr. Sankar Ghosh (Yale University School of Medicine), Dr. Ormond A. Macdougald (University of Michigan Medical School), and Dr. Steve Smale (University of California at Los Angeles), respectively.
Preparation of Human Astrocytes
Human CNS tissue was obtained from the Human Embryology Laboratory, University of Washington, Seattle. The CNS tissue from each specimen was processed separately and independently, as were subsequent cell cultures. There was no pooling of CNS tissue from distinct specimens. All of the experimental protocols were reviewed and approved by the Institutional Review Board (IRB 224-01-FB) of the University of Nebraska Medical Center. These cells were grown in a serum-free, defined medium (B16) enriched with 5 ng of basic fibroblast growth factor/ml for optimal growth of astrocytes and for the suppression of fibroblast growth (19). By immunofluorescence assay, these cultures homogeneously expressed glial fibrillary acidic protein (GFAP). Cells were trypsinized, subcultured, and stimulated with different cytokines in serum-free Dulbecco's modified Eagle's medium/F-12 medium.
Human U373MG astrocytoma cells obtained from American Type Culture Collection (ATCC) were also maintained and induced with different stimuli as indicated above.
Preparation of RSV-CAT and RSV-tat Constructs
The plasmid RSV-CAT expressing the chloramphenicol acetyltransferase enzyme under the control of the Rous sarcoma virus promoter was constructed as described (20). The recombinant plasmid RSV-tat was constructed by replacing the CAT gene in the RSV-CAT construct with the SalI-KpnI fragment of HIV-1, which contains the exon I of the HIV tat gene. The construction of this plasmid was described in detail previously (21).
Expression of RSV-CAT and RSV-tat in Human U373MG Astroglial Cells and Primary Astrocytes
Cells at 50–60% confluence were transfected with different amounts of either RSV-CAT or RSV-tat construct by LipofectAMINE Plus (Invitrogen) following the manufacturer's protocol (22–24). Twenty-four hours after transfection, cells were incubated under serum-free conditions. After 24 h of incubation, culture supernatants were transferred to measure NO production.
Assay for NO Synthesis
Synthesis of NO was determined by assay of culture supernatants for nitrite, a stable reaction product of NO with molecular oxygen. Briefly, 400 μl of culture supernatant was allowed to react with 200 μl of Griess reagent (25–28) and incubated at room temperature for 15 min. The optical density of the assay samples was measured spectrophotometrically at 570 nm. Fresh culture medium served as the blank in all experiments. Nitrite concentrations were calculated from a standard curve derived from the reaction of NaNO2 the assay. Protein was measured by the procedure of Bradford (29).
Immunoblot Analysis for iNOS
Immunoblot analysis for iNOS was carried out as described earlier (26–28). Briefly, cells were detached by scraping, washed with Hanks' buffer, and homogenized in 50 mm Tris-HCl (pH 7.4) containing protease inhibitors (1 mm phenylmethylsulfonyl fluoride, 5 μg/ml aprotinin, 5 μg/ml pepstatin A, and 5 μg/ml leupeptin). After electrophoresis the proteins were transferred onto a nitrocellulose membrane, and the iNOS band was visualized by immunoblotting with antibodies against mouse macrophage iNOS and 125I-labeled protein A.
RNA Isolation and Northern Blot Analysis
Cells were taken out of the culture dishes directly by adding Ultraspec-II RNA reagent (Biotecx Laboratories, Inc.), and total RNA was isolated according to the manufacturer's protocol. For Northern blot analyses, 20 μg of total RNA was electrophoresed on 1.2% denaturing formaldehyde-agarose gels, electrotransferred to Hybond nylon membrane (Amersham Biosciences), and hybridized at 68 °C with 32P-labeled cDNA probe using Express Hyb hybridization solution (Clontech) as described by the manufacturer. The cDNA probe was made by polymerase chain reaction amplification using two primers (forward primer, 5′-CTC CTT CAA AGA GGC AAA AAT A-3′; reverse primer, 5′-CAC TTC CTC CAG GAT GTT GT-3′) (26–28). After hybridization, the filters were washed two or three times in solution I (2× SSC, 0.05% SDS) for 1 h at room temperature followed by solution II (0.1× SSC, 0.1% SDS) at 50 °C for another hour. The membranes were then dried and exposed to x-ray films (Kodak). The same amount of RNA was hybridized with probe for glyceraldehyde 3-phosphate dehydrogenase.
Assay of iNOS Promoter-derived Reporter Activity
Cells plated at 50–60% confluence in 6-well plates were cotransfected with 0.5 μg of phiNOS(7.2)Luc2 and different amounts of either RSV-tat or RSV-CAT by LipofectAMINE Plus (Invitrogen) following the manufacturer's protocol (22–24). All transfections also included 50 ng of pRL-TK (a plasmid encoding Renilla luciferase, used as transfection efficiency control; Promega)/μg of total DNA. Twenty-four hours after transfection, cells were incubated with serum-free medium for 24 h. Firefly and Renilla luciferase activities were obtained by analyzing total cell extract according to standard instructions provided in the Dual Luciferase Kit (Promega) in a TD-20/20 luminometer (Turner Designs). Relative luciferase activity of cell extracts was typically represented as the ratio of firefly luciferase value/Renilla luciferase value × 103.
Assay of Transcriptional Activities of NF-κB and C/EBPβ
To assay the transcriptional activities of NF-κB and C/EBPβ, cells at 50 –60% confluence were transfected with either pBIIX-Luc, an NF-κB-dependent reporter construct (31), or pC/EBPβ-Luc using the LipofectAMINE Plus method (Invitrogen) (22–24). Construction of pC/EBPβ-Luc has been described earlier (31). This C/EBPβ-sensitive promoter contains four consensus C/EBPβ-binding sites. All transfections included 50 ng/μg total DNA of pRL-TK (a plasmid encoding Renilla luciferase, used as transfection efficiency control; Promega). After 24 h of transfection, cells were treated with different stimuli for 6 h. Firefly and Renilla luciferase activities were analyzed as described above.
Assay of ERK and p38 MAPK
U373MG astroglial cells (50–60% confluent) were transfected with different concentrations of either RSV-tat or RSV-CAT. Twenty-four hours after transfection, cells were incubated with serum-free medium. After 18 h of incubation, ERK and p38 MAPK activities were measured using assay kits obtained from Cell Signaling Technology. Briefly, cells were harvested under nondenaturing conditions at different time intervals, and cell lysates were prepared. Activated forms of ERK and p38 MAPK were pulled down from the cell lysate by immunoprecipitation using immobilized phospho-ERK and phospho-p38 MAPK monoclonal antibodies, respectively. The pellets were washed twice with kinase buffer and finally resuspended with 50 μl kinase buffer supplemented with 200 μm ATP and either ELK-1 fusion protein (for ERK) or ATF-2 fusion protein (for p38 MAPK). Following incubation at 30 °C for 30 min, samples were analyzed by Western blot using antibodies against phospho-ELK-1 or phospho-ATF-2.
Results
Expression of RSV-tat but Not RSV-CAT Induces the Expression of iNOS in Human U373MG Astroglial Cells
To study the effect of HIV-1 Tat on the expression of iNOS, we transfected human astroglial cells transiently with HIV-1 tat gene. The plasmid RSV-tat expressing the exon I of the HIV-1 tat gene under the control of the Rous sarcoma virus promoter was used to transfect human U373MG astroglial cells. Expression of RSV-tat but not of RSV-CAT induced the production of NO (Table I). The inhibition of NO production by arginase, an enzyme that degrades the substrate (l-arginine) of NOS, and l-NMA, a competitive inhibitor of NOS, but not by d-NMA, a negative control of l-NMA, suggests that RSV-tat induced the production of NO in U373MG astroglial cells through NOS-mediated arginine metabolism (Table I). To study the dose dependence of RSV-tat on the induction of NO production, cells plated in 6-well plates were transfected with RSV-tat at different doses ranging from 0.05 to 0.5 μg. The induction of NO production started at 0.05 μg of RSV-tat, reached the maximum at 0.2 μg of RSV-tat, and decreased at higher doses (Fig. 1A). This decrease in NO production was due to the increase in astroglial cell death when transfected at higher doses of RSV-tat (data not shown). In contrast, cells transfected with different doses of RSV-CAT were unable to induce the production of NO (Fig. 1A) suggesting that the induction of NO production is due to the expression of the tat gene. To understand the mechanism of NO production, we examined the effect of RSV-tat and RSV-CAT on protein and mRNA levels of iNOS. For Northern blot analysis, cell plated in 100-mm dishes were transfected with different doses of either RSV-CAT or RSV-tat (Fig. 1C). Consistent with the induction of NO production, Western blot analysis with antibodies against murine macrophage iNOS and Northern blot analysis for iNOS mRNA clearly showed that expression of RSV-CAT alone did not induce the expression of iNOS protein and mRNA. However, marked expression of iNOS protein (Fig. 1B) and mRNA (Fig. 1C) was observed in cells transfected with different doses of RSV-tat. Similar to the induction of NO production, the expression of iNOS protein and mRNA by RSV-tat was also dose-dependent. The induction of iNOS protein was maximal in cells transfected with 0.2 μg (per well of 6-well plate) of RSV-tat (Fig. 1B), whereas the expression of iNOS mRNA was maximal in cells transfected with 0.8 μg (100-mm dish) of RSV-tat (Fig. 1C).
Table I. Expression of RSV-tat induces the production of NO in human U373MG astroglial cells.
Cells plated at 50–60% confluence in 6-well plates were transfected with 0.2 μg of either RSV-CAT or RSV-tat using LipofectAMINE Plus (Invitrogen) as described under “Materials and Methods.” After 24 h of transfection, cells were incubated under serum-free conditions in the presence or absence of l-NMA (0.1 mm), d-NMA (0.1 mm), and arginase (100 units/ml). After 24 h of incubation, the concentrations of nitrite were measured in the supernatants as described under “Materials and Methods.” Data are mean ± S.D. of three different experiments.
| Transfections and treatments | Nitrite |
|---|---|
| μg/mg protein/24 h | |
| RSV-CAT (0.2 μg) | 4.9 ± 0.5 |
| RSV-tat (0.2 μg) | 36.2 ± 4.1 |
| RSV-tat (0.2 μg) + arginase | 6.7 ± 0.4 |
| RSV-tat (0.2 μg) + l-NMA | 6.2 ± 0.8 |
| RSV-tat (0.2 μg) + d-NMA | 35.7 ± 4.5 |
Fig. 1. Expression of RSV-tat induces the production of NO and the expression of iNOS protein in human U373MG astroglial cells.
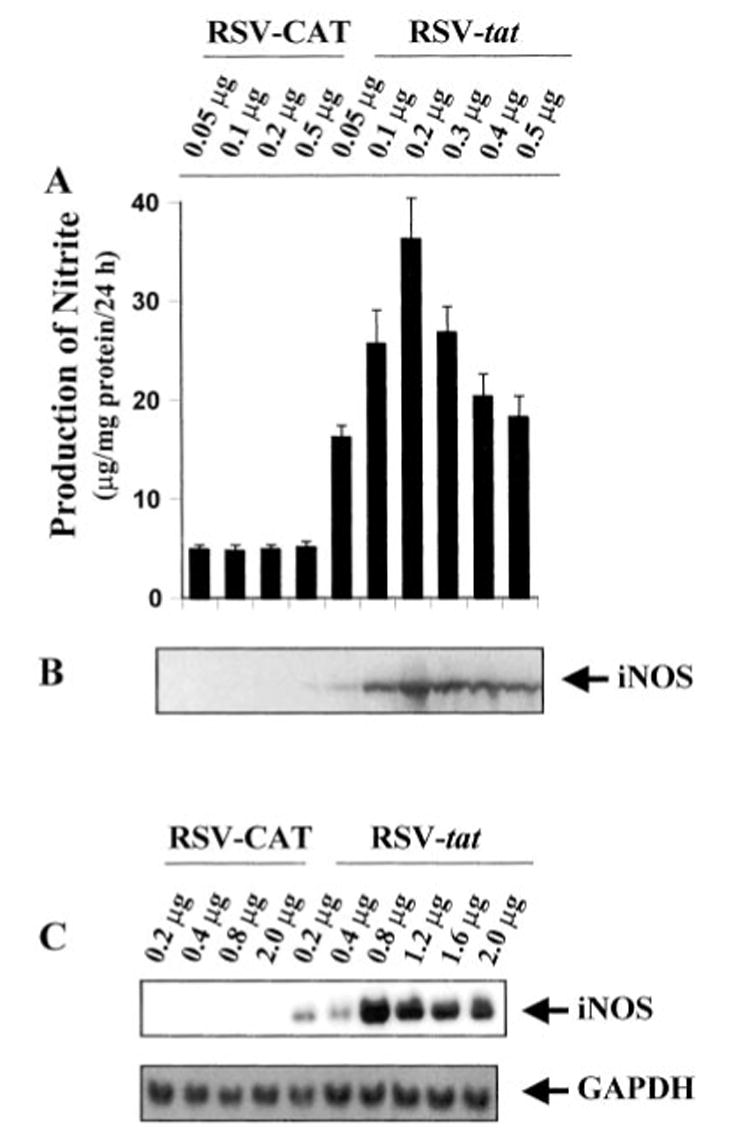
Cells plated at 50–60% confluence in 6-well plates were transfected with different amounts of either RSV-CAT or RSV-tat using LipofectAMINE Plus (Invitrogen) as described under “Materials and Methods.” After 24 h of transfection, cells were incubated under serum-free conditions. A, after 24 h of incubation, supernatants were used for nitrite assay. Data are the mean ± S.D. of three different experiments. B, cell homogenates were electrophoresed, transferred onto nitrocellulose membrane, and immunoblotted with antibodies against mouse macrophage iNOS as mentioned under “Materials and Methods.” C, cells plated at 50–60% confluence in 100-mm dishes were transfected with different amounts of either RSV-CAT or RSV-tat. After 24 h of transfection, cells were incubated under serum-free conditions. After 24 h of incubation, cells were taken out directly by adding Ultraspec-II RNA reagent (Biotecx Laboratories Inc.) to the plates for isolation of total RNA, and Northern blot analysis for iNOS mRNA was carried out as described under “Materials and Methods.”
In the CNS, only microglia is known to be infected productively by HIV-1 and to secrete the HIV-1 regulatory protein, Tat (2, 3, 32, 33) which in turn may act on astroglia in a paracrine fashion (18). Therefore, to understand whether Tat protein is secreted from RSV-tat-transfected astroglial cells and whether Tat protein is in fact responsible for the induction of iNOS, we incubated RSV-tat-transfected cells with anti-Tat antibodies. Although anti-Tat antibodies at a dose of 1 μg/ml was not very effective in blocking the induction of NO production, about 50% inhibition of NO production was observed when RSV-tat-transfected cells were incubated with 5 μg/ml anti-Tat antibodies (Fig. 2A). In contrast, control IgG had no effect on the induction of NO production. We next examined whether exogenously added recombinant HIV-1 Tat protein is able to induce the production of NO. Consistent with the induction of NO production by RSV-tat, recombinant Tat protein also dose-dependently induced the production of NO with the maximum induction observed at 100 ng/ml (Fig. 2B). However, in contrast to the 8–9-fold induction of NO production by the expression of RSV-tat (Fig. 1A), recombinant Tat protein induced the production of NO by about 4-fold (Fig. 2B). Taken together, these observations suggest that the induction of iNOS in RSV-tat-transfected astroglial cells is due to Tat and that Tat protein is secreted from RSV-tat-transfected astroglial cells.
Fig. 2. Expression of RSV-tat induces the production of NO through the secretion of tat in human U373MG astroglial cells.
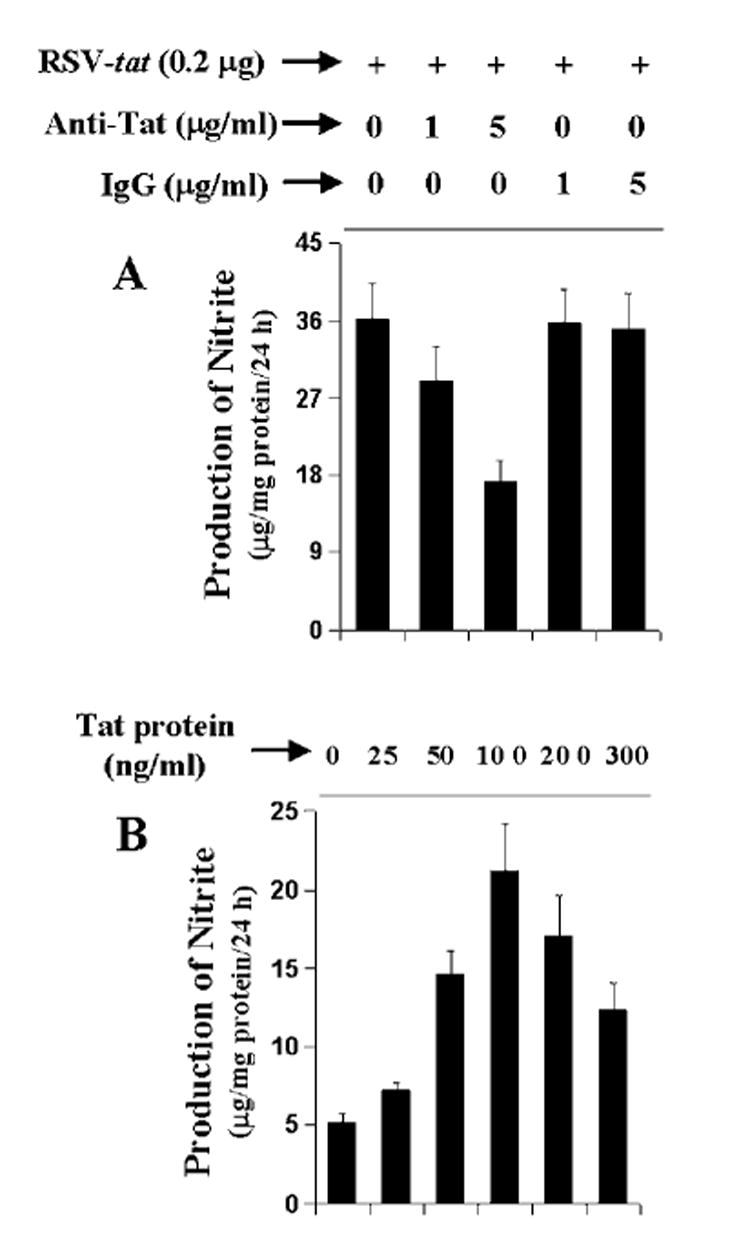
A, cells plated in 6-well plates were transfected with 0.2 μg of RSV-tat. After 24 h of transfection, cells were incubated under serum-free conditions in the presence of different concentrations of anti-Tat antibodies and control IgG. After 24 h of incubation, supernatants were used for nitrite assay. Data are the mean ± S.D. of three different experiments. B, cells were treated with different concentrations of recombinant HIV-1 Tat protein under serum-free conditions. After 24 h, supernatants were used for nitrite assay. Data are the mean ± S.D. of three different experiments.
Expression of RSV-tat Induces the Production of NO in Human Primary Astroglia
Human primary astrocytes have been shown to induce the expression of iNOS in the presence of different proinflammatory cytokines (23, 24).2 Because HIV-1 tat gene induced the production of NO in human U373MG astroglial cells, we examined whether HIV-1 tat gene was also able to induce the production of NO in human primary astrocytes (Fig. 3). Consistent with the induction of NO production in human U373MG astroglial cells, expression of RSV-tat but not RSV-CAT (the control plasmid) dose-dependently induced the production of NO in human primary astroglia. The induction of NO was maximum at 0.2 μg of RSV-tat and decreased at higher concentration (Fig. 3).
Fig. 3. Expression of RSV-tat induces the production of NO in human primary astroglia.
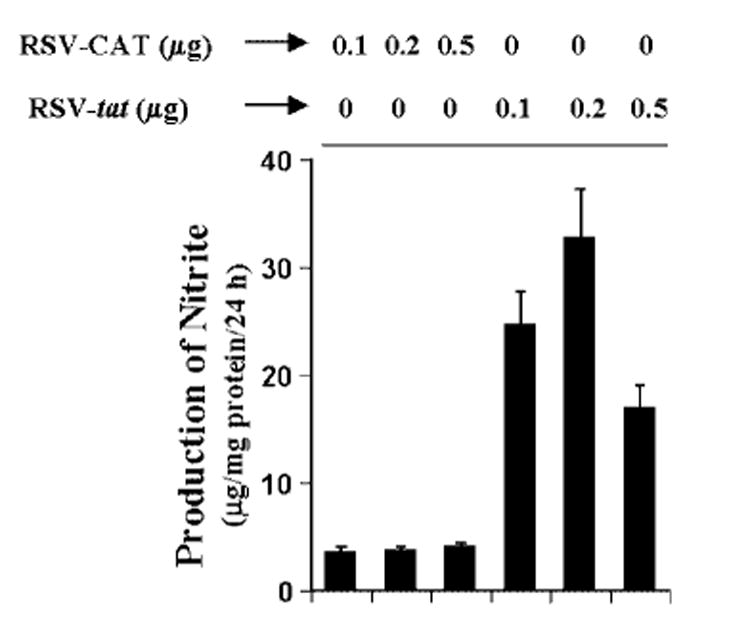
Cells plated at 50–60% confluence in 6-well plates were transfected with different amounts of either RSV-CAT or RSV-tat. After 24 h of transfection, cells were incubated under serum-free conditions. After 24 h of incubation, supernatants were used for nitrite assay. Data are the mean ± S.D. of three different experiments.
RSV-tat Induces Human iNOS Promoter-derived Luciferase Activity in Human U373MG Astroglial Cells
To understand the effect of the tat gene on the transcription of iNOS, U373MG glial cells were cotransfected with phiNOS(7.2)Luc, a construct containing the human iNOS promoter fused to the luciferase gene,2 and either RSV-tat or RSV-CAT. Activation of the iNOS promoter was measured after incubating the cells with serum-free media. It is evident from Fig. 4 that transfection of cells with different amounts of RSV-tat but not RSV-CAT led to the induction of iNOS promoter-derived luciferase activity. About 3.7-fold activation of iNOS promoter-derived luciferase activity was observed in cells transfected with 0.2 μg of RSV-tat (Fig. 4). These results suggest that the induction rate of the human iNOS promoter construct is much lower than the induction rate of human iNOS mRNA expression (Fig. 1C). We used a 7.2-kb human iNOS promoter for this study. Earlier, Taylor et al. (34) showed a 4.1-fold induction of this human iNOS promoter in human AKN-1 liver cells. They have also shown that transfection of a 16-kb human iNOS promoter construct produced a 9-fold increase in luciferase activity following cytokine stimulation, suggesting that there are additional functional elements even further upstream from 7.2 kb. Consistently, Marks-Konczalik et al. (35) have also shown the presence of extra NF-κB-and AP-1-binding sites within 7.2–8.3-kb human iNOS promoter, which is responsible for higher promoter activity in response to cytokines. Therefore, the observed low induction of 7.2-kb human iNOS promoter by RSV-tat in human U373MG astroglial cells is probably due to the absence of extra NF-κB-and AP-1-binding sites.
Fig. 4. Expression of RSV-tat induces iNOS promoter-derived luciferase activity in human U373MG astroglial cells.
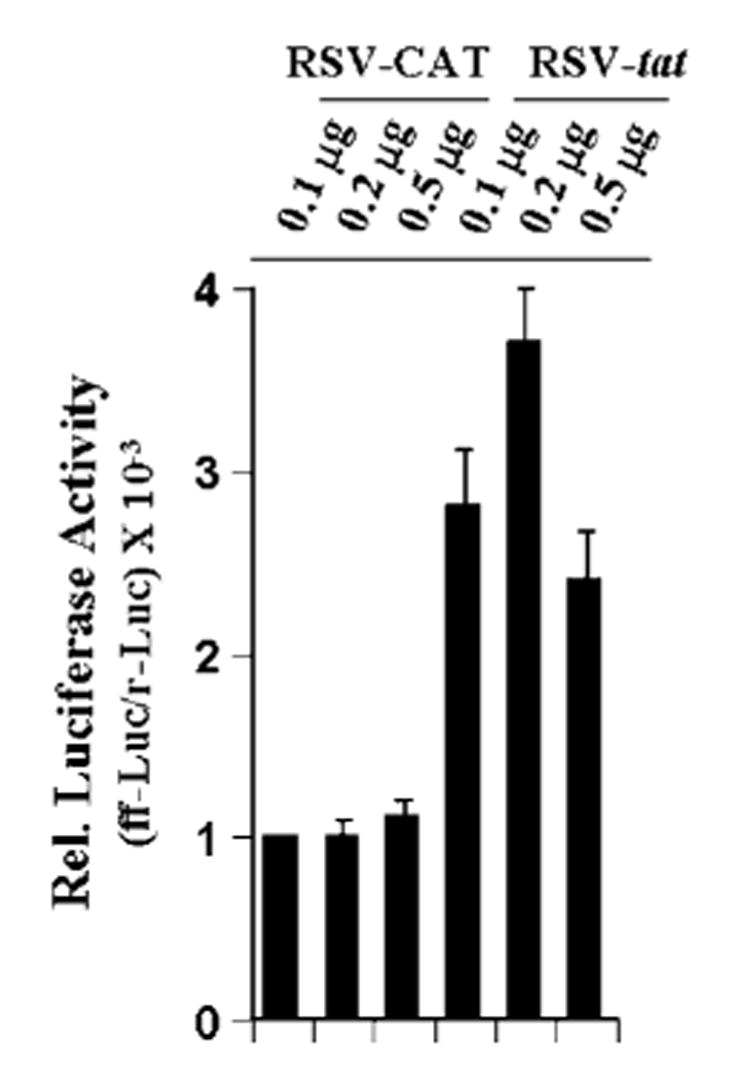
Cells plated in 6-well plates were cotransfected with 0.5 μg of phiNOS(7.2)Luc (a construct containing the human iNOS promoter fused to the luciferase gene) and different amounts of either RSV-CAT or RSV-tat. All transfections also included 50 ng/μg pRL-TK (as transfection efficiency control). After 24 h of transfection, cells were incubated under serum-free conditions for 18 h. Firefly (ff-Luc) and Renilla (r-Luc) luciferase activities were obtained by analyzing total cell extract as described under “Materials and Methods.” Data are the mean ± S.D. of three different experiments.
Role of NF-κB and C/EBPβ in RSV-tat-mediated Induction of iNOS in U373MG Astroglial Cells
The presence of NF-κB DNA-binding sites in the promoter of iNOS (34–36) and the inhibition of expression of iNOS by the inhibitors of NF-κB suggests that NF-κB plays an important role in the expression of iNOS (22–24, 26–28, 31, 36). Recent studies also have shown that activation of C/EBPβ is important for the induction of mouse as well as human iNOS (31).2 These findings prompted us to ask whether activation of NF-κB and C/EBPβ may be responsible for the induction of iNOS following HIV-1 tat gene expression in U373MG astroglial cells. First, we considered whether these two transcription factors are activated by RSV-tat. Activation of these transcription factors was monitored by transcriptional activities using the expression of luciferase from reporter constructs like pBIIX-Luc (for NF-κB) and pC/ EBPβ-Luc (for C/EBPβ) as an assay. Expression of RSV-tat but not RSV-CAT induced the activation of both NF-κB (Fig. 5A) and C/EBPβ(Fig. 5B) in a dose-dependent fashion. The maximum activation (∼9 –12-fold) of these transcription factors occurred in cells transfected with 0.2 μg of RSV-tat.
Fig. 5. Expression of RSV-tat induces activation of NF-κB and C/EBPβ in human U373MG astroglial cells.
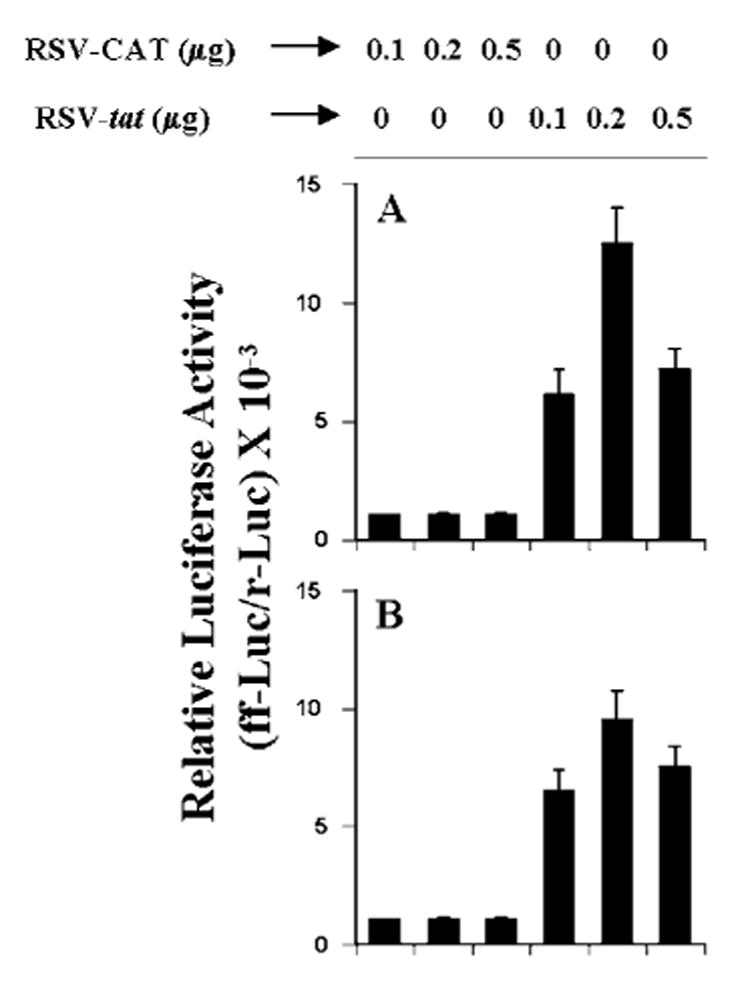
Cells plated in 6-well plates were cotransfected with 0.5 μg of either pBIIX-Luc (A) or pC/ EBPβ-Luc (B) and different amounts of either RSV-CAT or RSV-tat. All transfections also included 50 ng/μg pRL-TK. After 24 h of transfection, cells were incubated under serum-free conditions for 18 h. Firefly (ff-Luc) and Renilla (r-Luc) luciferase activities were obtained by analyzing total cell extract. Data are the mean ± S.D. of three different experiments.
Next we examined whether activation of both NF-κB and C/EBPβ is important for RSV-tat-induced production of NO. Overexpression of dominant-negative molecules provides an effective tool with which to investigate the in vivo functions of different transcription factors and signaling molecules. NF-κB was inhibited by a dominant-negative mutant of p65 (Δp65) (31). A naturally occurring alternate C/EBPβ translation product, known as LIP, lacks an “activation domain” and yet retains the ability to inhibit the function of C/EBPβ (37). LIP therefore acts as a dominant-negative mutant of C/EBPβ (37). Expression of Δp65 as well as ΔC/EBPβ, but not the empty vector, inhibited the production of NO (Fig. 6) in RSV-tat-transfected cells. In contrast, expression of wild type p65 and C/EBPβ stimulated RSV-tat-induced production of NO (Fig. 6). These studies suggest that activation of both NF-κB and C/EBPβ is important for RSV-tat-induced production of NO in human astroglial cells.
Fig. 6. Effect of wild-type p65 and C/EBPβ and dominant-negative mutants of p65 (Δp65) and C/EBPβ (ΔC/EBPβ) on RSV-tat-induced production of NO in human U373MG astroglial cells.
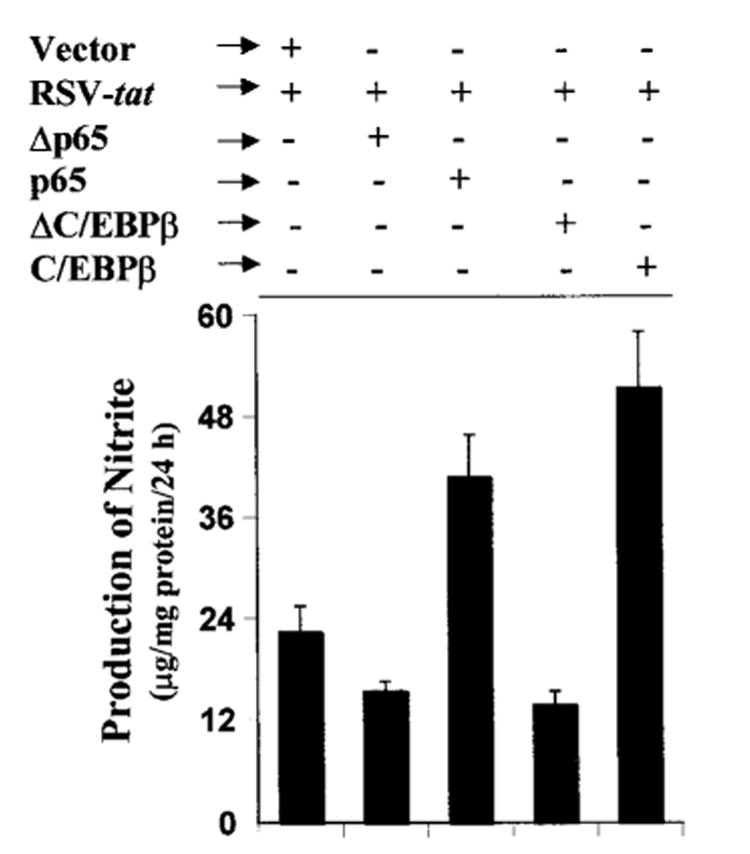
Cells plated in 6-well plates were cotransfected with 0.2 μg of RSV-tat and 0.5 μg of an empty vector, p65, C/EBPβ, Δp65, or ΔC/EBPβ. After 24 h of transfection, cells were incubated under serum-free conditions for another 24 h. Supernatants were used for the nitrite assay. Data are the mean ± S.D. of three different experiments.
Role of ERK and p38 MAPK in RSV-tat-induced Production of NO in Human U373MG Astroglial Cells
In eukaryotic cells, an important group of signaling pathways is the mitogen-activated protein kinase (MAPK) signaling cascades (38). Because the activation of MAPK pathways such as ERK and p38 MAPK by lipopolysaccharide and cytokines represents a potential signaling mechanism for NO production during the inflammatory response (27, 39, 40), we investigated the role of ERK and p38 MAPK in RSV-tat-induced production of NO. Therefore, we first examined the effect of RSV-tat on the activation of these kinases in U373MG astroglial cells. Interestingly, expression of RSV-tat induced the activation of ERK (Fig. 7). Consistent with the effect of RSV-tat on the induction of NO production and the activation of NF-κB and C/EBPβ, there was an inhibition of ERK activation in cells transfected with higher amount (0.5 or 1.0 μg) of RSV-tat (Fig. 7). In contrast, under the same conditions, the activation of p38 MAPK was not detected, suggesting that ERK but not p38 MAPK may play an important role in RSV-tat-induced production of NO in human astroglial cells.
Fig. 7. Expression of RSV-tat induces activation of ERK in human U373MG astroglial cells.

Cells were transfected with different amounts of RSV-tat. After 24 h of transfection, cells were incubated under serum-free conditions. After 18 h of incubation, activities of ERK and p38 MAPK were assayed as described under “Materials and Methods.”
Therefore, we investigated the role of ERK and p38 MAPK in RSV-tat-induced production of NO using specific pharmacological inhibitors of MEK-ERK (PD98059) and p38 MAPK (SB203580). Twenty-four hours after transfection with 0.2 μg of RSV-tat, cells were incubated with different concentrations of PD98059 or SB203580 under serum-free conditions. After 24 h of incubation, the concentration of nitrite was measured in supernatants. Both PD98059 and SB203580 were dissolved in Me2SO. These drugs were added to the cell culture at a final Me2SO concentration of 0.02– 0.06%. Me2SO (0.06%) was used as a control. Consistent with the activation of ERK but not p38 MAPK by RSV-tat, PD98059 but not SB203580 dose-dependently inhibited the production of NO in RSV-tat-transfected cells (Fig. 8A), suggesting that ERK but not p38 MAPK is involved in RSV-tat-induced production of NO. The inhibition by PD98059 was not from Me2SO because Me2SO alone did not exhibit any inhibitory effect at the highest concentration used in our study (data not shown). To further confirm the involvement of ERK but not p38 MAPK in RSV-tat-induced production of NO, we studied the effect of dominant-negative mutants of ERK1 (ΔERK1), ERK2 (ΔERK2), and p38 (Δp38) on RSV-tat-induced production of NO. Interestingly, RSV-tat-induced production of NO was inhibited by ΔERK2 but not by ΔERK1, suggesting that ERK2 but not ERK1 is involved in RSV-tat-induced production of NO (Fig. 8B). In addition, consistent with the inability of RSV-tat to induce the activation of p38 MAPK and the inability of SB203580 to inhibit RSV-tat-induced production of NO, Δp38 had no effect on RSV-tat-induced production of NO (Fig. 8B).
Fig. 8. Role of ERK and p38 MAPK in RSV-tat-induced NO production in human U373MG astroglial cells.
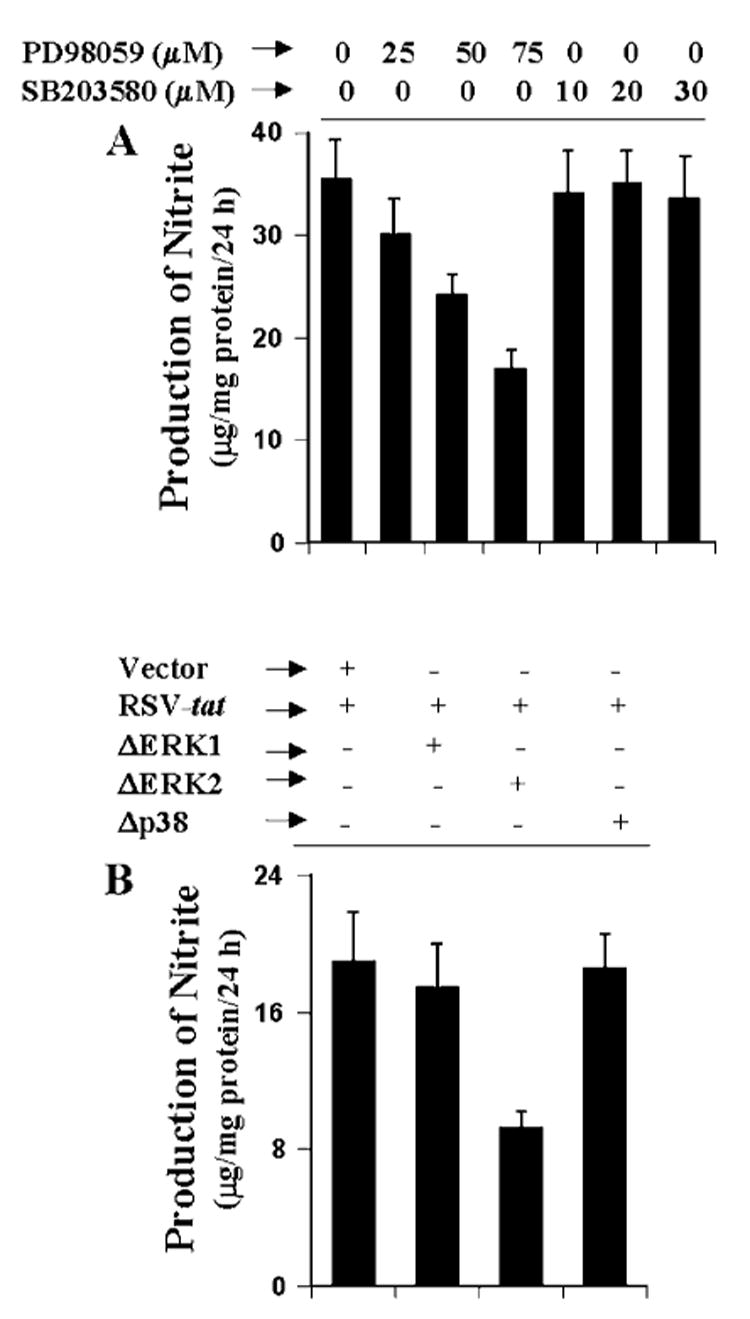
A, cells were transfected with 0.2 μg of RSV-tat. After 24 h of transfection, cells were incubated under serum-free conditions in the presence or absence of different concentrations of PD98059 and SB203580. After 24 h of incubation, supernatants were used for the nitrite assay. Data are the mean ± S.D. of three different experiments. B, cells were cotransfected with 0.2 μg of RSV-tat and 0.5 μg of either an empty vector or ΔERK1, ΔERK2, or Δp38. After 24 h of transfection, cells were incubated under serum-free conditions for another 24 h. Supernatants were used for the nitrite assay. Data are the mean ± S.D. of three different experiments.
Is ERK2 Involved in RSV-tat-induced Activation of NF-κB and C/EBPβ in Human U373MG Astroglial Cells?
The results presented in Fig. 8 show that RSV-tat induces the production of NO through ERK2. Because proinflammatory transcription factors NF-κB and C/EBPβ are also involved in RSV-tat-induced production of NO, we next examined the effect of PD98059 and ΔERK2 on RSV-tat-induced activation of NF-κB and C/EBPβ. Interestingly, PD98059 at different tested doses had no inhibitory effect on RSV-tat-induced activation of NF-κB (Fig. 9A). Even PD98059 at a dose of 25 μm stimulated RSV-tat-induced activation of NF-κB (Fig. 9A). In sharp contrast, PD98059 dose-dependently inhibited RSV-tat-induced activation of C/EBPβ (Fig. 9B). Because PD98059 inhibits the MEK-ERK pathway, these results suggest that ERK2 is probably involved in RSV-tat-induced activation of C/EBPβ but not of NF-κB. To further confirm this observation, we tested the effect of ΔERK2 on RSV-tat-induced activation of NF-κB and C/EBPβ. Consistent with the effect of PD98059 on RSV-tat-induced activation of NF-κB and C/EBPβ, ΔERK2 specifically inhibited the activation of C/EBPβ (Fig. 10B) but not of NF-κB (Fig. 10A). These experiments suggest that PD98059 and ΔERK2 inhibit RSV-tat-induced production of NO by inhibiting the activation of C/EBPβ but not NF-κB.
Fig. 9. Effect of PD98059 on RSV-tat-induced activation of NF-κB and C/EBPβ in human U373MG astroglial cells.
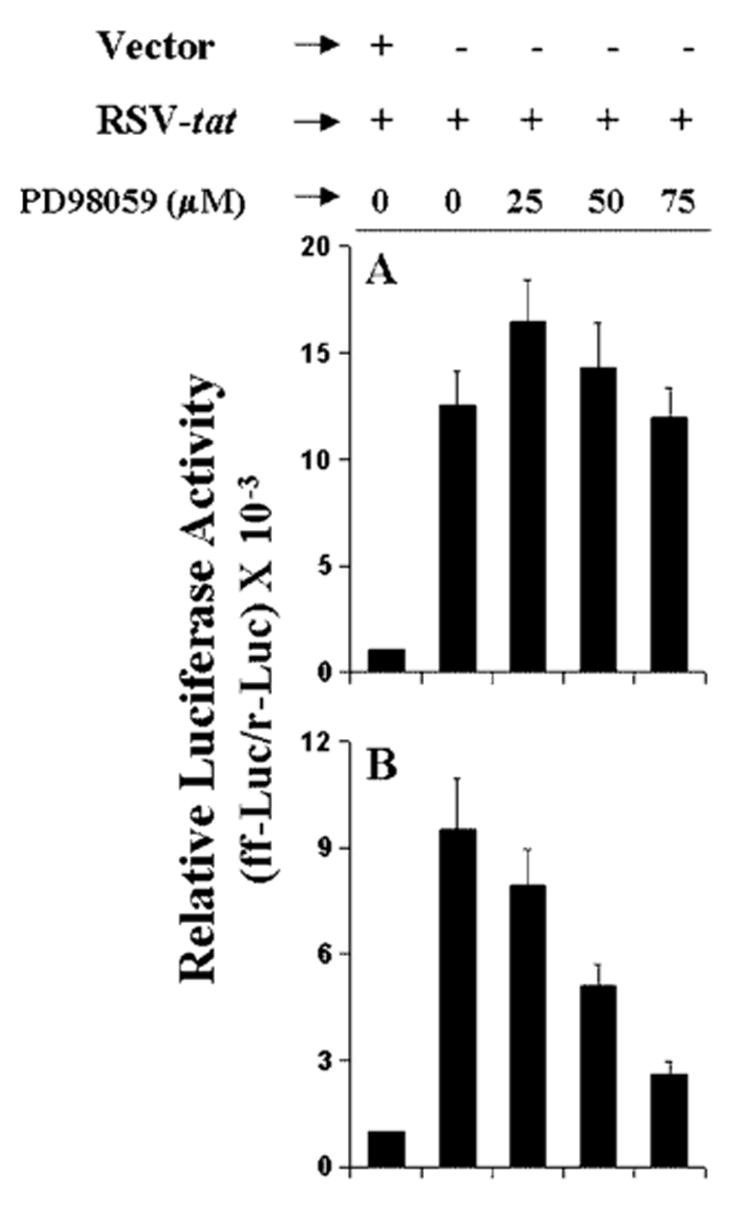
Cells plated in 6-well plates were cotransfected with 0.2 μg of RSV-tat and 0.5 μg of either pBIIX-Luc (A) or pC/EBPβ-Luc (B). All transfections also included 50 ng/μg pRL-TK. After 24 h of transfection, cells were incubated in the presence or absence of different concentrations of PD98059 under serum-free conditions for 18 h. Firefly (ff-Luc) and Renilla (r-Luc) luciferase activities were obtained by analyzing total cell extract. Data are the mean ± S.D. of three different experiments.
Fig. 10. Effect of ΔERK2, the dominant-negative mutant of ERK2, on RSV-tat-induced activation of NF-κB and C/EBPβ in human U373MG astroglial cells.
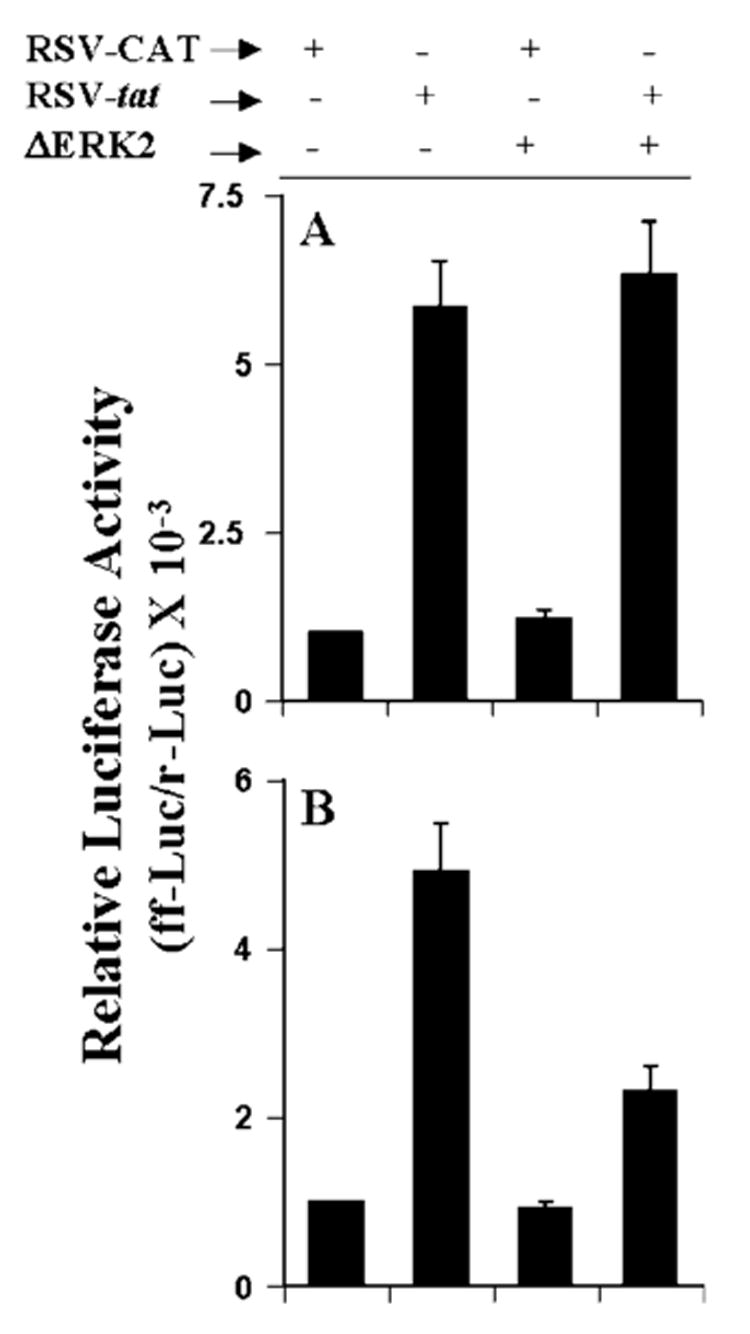
Cells were cotransfected with 0.2 μg of RSV-tat, 0.5 μg of either an empty vector or ΔERK2, and 0.5 μg of either pBIIX-Luc (A) or pC/EBPβ-Luc (B). After 24 h of transfection, cells were incubated under serum-free conditions for 18 h. Firefly (ff-Luc) and Renilla (r-Luc) luciferase activities were obtained by analyzing total cell extract. Data are the mean ± S.D. of three different experiments.
Discussion
The pathogenesis of dementia associated with HIV-1 infection involves the complex interactions between viral products and host immune system, which eventually result in neuronal dysfunction and cell loss. Several findings suggest that extracellular Tat plays an important role in the pathogenesis of HAD. First, Tat is actively secreted by infected cells of HAD patients (32, 33). Second, anti-Tat antibodies are frequently detected in the serum of HAD patients (41). Third, Tat mRNA is elevated in patients with HAD (42, 43). Fourth, a single intraventricular injection of Tat leads to macrophage infiltration, progressive glial activation, and neuronal death, pathologies that are also observed in HAD brains (44). However, the mechanism by which Tat causes neuronal death in HAD patients is poorly understood. Astroglia are the major glial cells in the CNS, and reactive astroglia have been found in large numbers in the brains of HAD patients. The evidence presented in this manuscript that RSV-tat but not RSV-CAT (the control plasmid) induced the production of NO and the expression of iNOS, that anti-Tat antibodies blocked RSV-tat-induced production of NO, and that recombinant Tat protein also induced the production of NO clearly support the conclusion that Tat induces the expression of iNOS in human astrocytes. The ability of Tat to induce the production of NO in humanU373MG astroglial cells and primary astroglia suggests that neurons in the vicinity of Tat-activated astroglia in brains of HAD patients could be subjected to NO-induced damage.
In vitro iNOS has been shown to be induced in glial cells following exposure to HIV-1 or HIV-1 envelope glycoproteins such as gp120 and gp41 (45–47). Although earlier studies reported iNOS expression in monocyte-derived macrophages, microglia, and astrocytes (45–46), more recent studies have demonstrated an inability of macrophages or microglia to express iNOS following stimulation with HIV or gp41 (47) at least in vitro. Particularly, gp41 has been shown to induce iNOS in astrocytes but not in the absence of monocyte-derived macrophages (14). Recently Polazzi et al. (48) reported that HIV-1 Tat protein induces the production of NO in mouse microglial cells. However, immunocytochemical analysis of HAD brains has shown that iNOS is primarily expressed in astrocytes (15). In contrast, the majority of macrophages or microglia (including multinucleated giant cells) remain iNOS-negative (15). In light of these findings, our observations suggest that Tat may also contribute to the expression of iNOS observed in astroglia of HAD brains. However, Barton et al. (49) have reported that HIV-1 Tat inhibits interferon-γ-induced iNOS activity in murine macrophages. There are several instances in which the induction of iNOS is differentially regulated in astrocytes and macrophages. First, increasing cAMP or protein kinase A inhibits iNOS in astrocytes (26, 50) but stimulates iNOS macrophages (26). Second, inhibitors of protein phosphatase 1/2A stimulate iNOS in astrocytes (51) but inhibit iNOS in macrophages (51, 52). Third, ceramide stimulates iNOS in astrocytes (26) but inhibits iNOS in macrophages (53). Therefore, Tat may regulate the expression of iNOS differentially in astrocytes and macrophages.
The signaling events in the induction of iNOS have not been completely established so far. Proinflammatory cytokines (tumor necrosis factor-α, interleukin-1β, or interferon-γ) and lipopolysaccharide bind to their respective receptors and induce the expression of iNOS via NF-κB activation (22–24, 26–28, 31, 36). The presence of a consensus sequence in the promoter region of iNOS for the binding of NF-κB (34–36) and the inhibition of iNOS expression with the inhibition of NF-κB activation establish an essential role for NF-κB activation in the induction of iNOS. Activation of NF-κB by various cellular stimuli involves the proteolytic degradation of IκB, the inhibitory subunit of the NF-κB complex, and the concomitant nuclear translocation of the liberated NF-κB heterodimer. Although the biochemical mechanism underlying the degradation of IκB remains unclear, it appears that degradation of IκB induced by various mitogens and cytokines occurs in association with the transient phosphorylation of IκB on serines 32 and 36. Consistently, two closely related kinases (IKKα and IKKβ) that directly phosphorylate IκBαhave also been described (reviewed in Ref. 54). Upon phosphorylation, IκB that is still bound to NF-κB apparently becomes a high affinity substrate for an ubiquitin-conjugating enzyme. After phosphorylation-dependent ubiquitination, the IκB is rapidly and completely degraded by the 20 or 26 S proteasome, the NF-κB heterodimer enters into the nucleus (reviewed in Ref. 54) and binds to the consensus DNA-binding site present in the promoter region of iNOS. Our results have shown clearly that the activation of NF-κB is important for the expression of iNOS in RSV-tat-transfected astrocytes. First, RSV-tat but not RSV-CAT induced the activation of NF-κB. Second, Δp65, a dominant-negative mutant of p65, but not the empty vector inhibited RSV-tat-induced production of NO. Third, overexpression of the wild type p65 stimulated RSV-tat-induced production of NO.
CCAAT/enhancer-binding protein (C/EBP) is a member of the basic region-leucine zipper family of transcription factors that controls the transcription of a number of genes through protein-protein interactions at the gene level (37). In particular, C/EBPβ not only can bind to its own family members such as C/EBP proteins, Fos, and Jun (55), but also forms complexes with other transcription factors such as NF-κB, estrogen receptor, and an Sp1 factor (56). In addition, C/EBPβ may also alter transcription by complex interactions with coactivators and basal transcription factors such as TFIIB and p300 (57). The 8.3-kb human iNOS promoter contains 15 nucleotide sequences (58) that conform to the consensus C/EBP box TKNNGYAAK (37). Recently we have found that the dominant-negative mutant of C/EBPβ inhibits cytokine-induced production of NO in human astroglial cells2 suggesting the involvement of C/EBPβ in cytokine-induced expression of iNOS. The results presented in this article clearly demonstrate that the activation of C/EBPβ is also essential for RSV-tat-mediated induction of iNOS in human astrocytes. First, RSV-tat induced the activation of C/EBPβ. Second, ΔC/EBPβ, a truncated alternate C/EBPβ translation product, LIP, which acts as a dominant-negative inhibitor of C/EBPβ activity (37), inhibited RSV-tat-induced production of NO. Third, overexpression of wild type C/EBPβ stimulated RSV-tat-induced production of NO.
At present, it is unclear how Tat activates NF-κB and C/EBPβ to induce the production of NO in astroglial cells. MAPKs are Ser-Thr kinases that have been shown to activate a number of transcription factors in different cell types in response to various stimuli (38). We have found that the expression of RSV-tat induces the activation of ERK only and not p38 MAPK. Consistently, using pharmacological inhibitors and dominant-negative mutants, we have elucidated that ERK but not p38 MAPK is involved in RSV-tat-induced production of NO. There are two isoforms of ERK (ERK1 and ERK2) (38). Interestingly, ERK2 but not ERK1 is involved in RSV-tat-induced production of NO in human astroglial cells. In addition, RSV-tat-induced ERK2 couples with C/EBPβ only and not with NF-κB. Taken together, our studies suggest that Tat activates ERK2, which ultimately couples to the activation of C/EBPβ but not to NF-κB for the induction of iNOS. Further studies are under way to delineate the Tat-induced signaling pathway(s) that couples with NF-κB for the induction of iNOS.
NO, a diffusible free radical, plays many roles as a signaling and an effector molecule in diverse biological systems, including neurotransmission, vasodilation, and antimicrobial and antitumor activities (10, 11). In the nervous system small amounts of NO produced by neuronal NO synthase act as a neurotransmitter. In sharp contrast, excessive amounts of NO produced from the activation of iNOS is directly or indirectly neurotoxic via stimulation of N-methyl-d-aspartate receptors, leading to necrosis and apoptosis (59). In addition, NO and peroxynitrite (reaction product of NO and ) may mediate toxicity in neurons and oligodendrocytes through the formation of iron-NO complexes of iron-containing enzyme systems, oxidation of protein sulfhydryl groups, nitration of proteins, and nitrosylation of nucleic acids and DNA strand breaks (11). NO can also modulate macrophage apoptosis, lymphocyte proliferation, and chemokine induction (11, 60, 61) and thus could regulate the overall inflammatory responses in the CNS. iNOS expressed in astrocytes could contribute to the breach of the blood-brain barrier due to its potent vasorelaxing property, allowing extravasation of the harmful molecules to the CNS. Such an abnormality in the blood-brain barrier could contribute to the diffuse myelin damage that is often associated with HAD (62). On the other hand, NO also exhibits antiviral activity against a wide range of viruses in rodents (e.g. herpes simplex, vaccinia, ectromelia, vesicular stomatitis, Friend leukemia, coxsackievirus, Japanese encephalitis viruses, and coronavirus) (reviewed in Ref. 10). Therefore, the existence of NO-mediated antiviral activity against HIV-1 is probably responsible for the observed low levels of virus detectable in vivo even in the presence of significant neurologic damage.
In the CNS, iNOS is expressed mainly by activated astroglia and microglia, the two glial cell types involved in intracerebral immune regulation. Astroglia are the major glial cell population in the CNS, and therefore, induction of iNOS in astroglia by Tat may be an important source of NO in HAD associated with astrogliosis and neuronal death.
Acknowledgments
We thank Tom Dunn and associates for help in preparing this manuscript.
Footnotes
This study was supported by Public Health Service Grants NS39940 (to K. P.) and P20-RR15635 and CA76958 (to C. W.). from the National Institutes of Health.
The abbreviations used are: HAD, HIV-1-associated dementia; HIV, human immunodeficiency virus; iNOS, inducible nitric-oxide synthase; NMA, l-NG-monomethylarginine; d-NMA, d-NG-monomethylarginine; ERK, extracellular signal-regulated kinase; MAPK, mitogen-activated protein kinase; CNS, central nervous system; CAT, chloramphenicol acetyltransferase; RSV, Rous sarcoma virus; C/EBP, CCAAT enhancerbinding protein; IKK, IκB kinase; LIP, liver inhibitory protein.
Contributor Information
Xiaojuan Liu, Department of Oral Biology, University of Nebraska Medical Center, Lincoln, Nebraska 68583.
Malabendu Jana, Department of Oral Biology, University of Nebraska Medical Center, Lincoln, Nebraska 68583.
Subhajit Dasgupta, Department of Oral Biology, University of Nebraska Medical Center, Lincoln, Nebraska 68583.
Sreenivas Koka, Department of Oral Biology, University of Nebraska Medical Center, Lincoln, Nebraska 68583.
Jun He, Nebraska Center for Virology and School of Biological Sciences, University of Nebraska, Lincoln, Nebraska 68588.
Charles Wood, Nebraska Center for Virology and School of Biological Sciences, University of Nebraska, Lincoln, Nebraska 68588.
Kalipada Pahan, Department of Oral Biology, University of Nebraska Medical Center, Lincoln, Nebraska 68583.
References
- 1.Kolson DL, Gonzalez-Scarano F. J Clin Invest. 2000;106:11–13. doi: 10.1172/JCI10553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bagasra O, Lavi E, Bobroski L, Khalili K, Pestaner JB, Pomerantz RJ. AIDS. 1996;10:573–585. doi: 10.1097/00002030-199606000-00002. [DOI] [PubMed] [Google Scholar]
- 3.Wiley CA, Schrier RD, Nelson JA, Lampert PW, Oldstone MBA. Proc Natl Acad Sci U S A. 1986;83:7089–7093. doi: 10.1073/pnas.83.18.7089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Glass JD, Johnson RT. Annu Rev Neurosci. 1996;19:1–26. doi: 10.1146/annurev.ne.19.030196.000245. [DOI] [PubMed] [Google Scholar]
- 5.Tornatore C, Chandra R, Berger JR, Major EO. Neurology. 1994;44:481–487. doi: 10.1212/wnl.44.3_part_1.481. [DOI] [PubMed] [Google Scholar]
- 6.Ranki A, Nyberg M, Ovod V, Haltia M, Elovaara I, Raininko R, Haapasalo H, Krohn K. AIDS. 1995;9:1001–1008. doi: 10.1097/00002030-199509000-00004. [DOI] [PubMed] [Google Scholar]
- 7.Bernton EW, Bryant HU, Decoster MA, Orenstein JM, Ribas JL, Meltzer MS, Gendelman HE. AIDS Res Hum Retroviruses. 1992;8:495–503. doi: 10.1089/aid.1992.8.495. [DOI] [PubMed] [Google Scholar]
- 8.Johnson RT, Glass JD, McArthur JC, Chesebro BW. Ann Neurol. 1996;39:392–395. doi: 10.1002/ana.410390319. [DOI] [PubMed] [Google Scholar]
- 9.Petito CK, Vecchio D, Chen YT. J Neuropathol Exp Neurol. 1994;53:86–94. doi: 10.1097/00005072-199401000-00011. [DOI] [PubMed] [Google Scholar]
- 10.Nathan C. FASEB J. 1992;6:3051–3064. [PubMed] [Google Scholar]
- 11.Jaffrey SR, Snyder SH. Annu Rev Cell Dev Biol. 1995;11:417–440. doi: 10.1146/annurev.cb.11.110195.002221. [DOI] [PubMed] [Google Scholar]
- 12.Adamson DC, McArthur JC, Dawson TM, Dawson VL. Mol Med. 1999;5:98–109. [PMC free article] [PubMed] [Google Scholar]
- 13.Adamson DC, Wildemann B, Sasaki M, Glass JD, McArthur JC, Christov VI, Dawson TM, Dawson VL. Science. 1996;274:1917–1921. doi: 10.1126/science.274.5294.1917. [DOI] [PubMed] [Google Scholar]
- 14.Hori K, Burd PR, Furuke K, Kutza J, Weih KA, Clouse KA. Blood. 1999;93:1843–1850. [PubMed] [Google Scholar]
- 15.Zhao ML, Kim MO, Morgello S, Lee SC. J Neuroimmunol. 2001;115:182–191. doi: 10.1016/s0165-5728(00)00463-x. [DOI] [PubMed] [Google Scholar]
- 16.Giovannoni G, Miller RF, Heales SJ, Land JM, Harrison MJ, Thompson EJ. J Neurol Sci. 1998;156:53–58. doi: 10.1016/s0022-510x(98)00021-5. [DOI] [PubMed] [Google Scholar]
- 17.Boven LA, Gomes L, Hery C, Gray F, Verhoef J, Portegies P, Tardieu M, Nottet HS. J Immunol. 1999;162:4319–4327. [PubMed] [Google Scholar]
- 18.Chang HK, Gallo RC, Ensoli B. J Biomed Sci. 1995;2:189–202. doi: 10.1007/BF02253380. [DOI] [PubMed] [Google Scholar]
- 19.McCarthy M, Wood C, Fedoseyeva L, Whittemore S. J Neurovirol. 1995;1:275–285. doi: 10.3109/13550289509114024. [DOI] [PubMed] [Google Scholar]
- 20.Yamamoto T, Jay G, Pastan I. Proc Natl Acad Sci U S A. 1980;77:176–180. doi: 10.1073/pnas.77.1.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jung M, Wood C. Life Sci Adv. 1991;10:77–88. [Google Scholar]
- 22.Pahan K, Sheikh FG, Liu X, Hilger S, McKinney M, Petro TM. J Biol Chem. 2001;276:7899–7905. doi: 10.1074/jbc.M008262200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pahan K, Liu X, Wood C, Raymond JR. FEBS Lett. 2000;472:203–207. doi: 10.1016/s0014-5793(00)01465-4. [DOI] [PubMed] [Google Scholar]
- 24.Pahan K, Liu X, McKinney MJ, Wood C, Sheikh FG, Raymond JR. J Neurochem. 2000;74:2288–2295. doi: 10.1046/j.1471-4159.2000.0742288.x. [DOI] [PubMed] [Google Scholar]
- 25.Feinstein DL, Galea E, Roberts S, Berquist H, Wang H, Reis DJ. J Neurochem. 1994;62:315–321. doi: 10.1046/j.1471-4159.1994.62010315.x. [DOI] [PubMed] [Google Scholar]
- 26.Pahan K, Namboodiri AMS, Sheikh FG, Smith BT, Singh I. J Biol Chem. 1997;272:7786–7791. doi: 10.1074/jbc.272.12.7786. [DOI] [PubMed] [Google Scholar]
- 27.Pahan K, Sheikh FG, Khan M, Namboodiri AMS, Singh I. J Biol Chem. 1998;273:2591–2600. doi: 10.1074/jbc.273.5.2591. [DOI] [PubMed] [Google Scholar]
- 28.Pahan K, Raymond JR, Singh I. J Biol Chem. 1999;274:7528–7536. doi: 10.1074/jbc.274.11.7528. [DOI] [PubMed] [Google Scholar]
- 29.Bradford M. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 30.Deleted in proof
- 31.Jana M, Liu X, Koka S, Ghosh S, Petro TM, Pahan K. J Biol Chem. 2001;276:44527–44533. doi: 10.1074/jbc.M106771200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tardieu M, Hery C, Peudenier S, Boespflug O, Montagnier L. Ann Neurol. 1992;132:11–17. doi: 10.1002/ana.410320104. [DOI] [PubMed] [Google Scholar]
- 33.Ensoli B, Buonaguro L, Barillari G, Fiorelli V, Gendelman R, Morgan R, Wingfield P, Gallo RC. J Virol. 1993;67:277–287. doi: 10.1128/jvi.67.1.277-287.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taylor BS, de Vera ME, Ganster RW, Wang Q, Shapiro RA, Morris SM, Billiar TR, Geller DA. J Biol Chem. 1998;273:15148–15156. doi: 10.1074/jbc.273.24.15148. [DOI] [PubMed] [Google Scholar]
- 35.Marks-Konczalik J, Chu SC, Moss J. J Biol Chem. 1998;273:22201–22208. doi: 10.1074/jbc.273.35.22201. [DOI] [PubMed] [Google Scholar]
- 36.Xie Q, Kashiwabara Y, Nathan C. J Biol Chem. 1994;269:4705–4708. [PubMed] [Google Scholar]
- 37.Descombes P, Schibler U. Cell. 1991;67:569–579. doi: 10.1016/0092-8674(91)90531-3. [DOI] [PubMed] [Google Scholar]
- 38.Dong C, Davis RJ, Flavell RA. Annu Rev Immunol. 2002;20:55–72. doi: 10.1146/annurev.immunol.20.091301.131133. [DOI] [PubMed] [Google Scholar]
- 39.Bhat NR, Zhang P, Lee JC, Hogan EL. J Neurosci. 1998;18:1633–1641. doi: 10.1523/JNEUROSCI.18-05-01633.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Silva JD, Pierrat B, Mary JL, Lesslauer W. J Biol Chem. 1997;272:28373–28380. doi: 10.1074/jbc.272.45.28373. [DOI] [PubMed] [Google Scholar]
- 41.Aldovini A, Debouck C, Feinberg MB, Rosenberg M, Arya SK, Wong-Staal F. Proc Natl Acad Sci U S A. 1986;83:6672–6676. doi: 10.1073/pnas.83.18.6672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wesselingh SL, Power C, Glass JD, Tyor WR, McArthur J, Farber JM, Griffin JW, Griffin DE. Ann Neurol. 1993;33:576–582. doi: 10.1002/ana.410330604. [DOI] [PubMed] [Google Scholar]
- 43.Wiley CA, Baldwin M, Achim CL. AIDS. 1996;10:843–847. doi: 10.1097/00002030-199607000-00007. [DOI] [PubMed] [Google Scholar]
- 44.Jones M, Olafson K, Del Bigio MR, Peeling J, Nath A. J Neuropathol Exp Neurol. 1998;57:563–570. doi: 10.1097/00005072-199806000-00004. [DOI] [PubMed] [Google Scholar]
- 45.Kong LY, Wilson BC, McMillian MK, Bing G, Hudson PM, Hong JS. Cell Immunol. 1996;172:77–83. doi: 10.1006/cimm.1996.0217. [DOI] [PubMed] [Google Scholar]
- 46.Koka P, He K, Zack JA, Kitchen S, Peacock W, Fried I, Tran T, Yashar SS, Merrill JE. J Exp Med. 1995;182:941–951. doi: 10.1084/jem.182.4.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hu S, Sheng WS, Ehrlich LC, Peterson PK, Chao CC. J Neurosci. 1999;19:6468–6474. doi: 10.1523/JNEUROSCI.19-15-06468.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Polazzi E, Levi G, Minghetti L. J Neuropathol Exp Neurol. 1999;58:825–831. doi: 10.1097/00005072-199908000-00005. [DOI] [PubMed] [Google Scholar]
- 49.Barton CH, Biggs TE, Mee TR, Mann DA. J Gen Virol. 1996;77:1643–1647. doi: 10.1099/0022-1317-77-8-1643. [DOI] [PubMed] [Google Scholar]
- 50.Galea E, Feinstein DL. FASEB J. 1999;13:2125–2137. doi: 10.1096/fasebj.13.15.2125. [DOI] [PubMed] [Google Scholar]
- 51.Pahan K, Sheikh FG, Namboodiri AM, Singh I. J Biol Chem. 1998;273:12219–12226. doi: 10.1074/jbc.273.20.12219. [DOI] [PubMed] [Google Scholar]
- 52.Dong Z, Yang X, Xie K, Juang SH, Llansa N, Fidler IJ. J Leukocyte Biol. 1995;58:725–732. doi: 10.1002/jlb.58.6.725. [DOI] [PubMed] [Google Scholar]
- 53.Hsu YW, Chi KH, Huang WC, Lin WW. J Immunol. 2001;166:5388–5397. doi: 10.4049/jimmunol.166.9.5388. [DOI] [PubMed] [Google Scholar]
- 54.Ghosh S, May MJ, Kopp EB. Ann Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 55.Hsu W, Kerppola TK, Chen PL, Chen-Kiang S. Mol Cell Biol. 1994;14:268–276. doi: 10.1128/mcb.14.1.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stein B, Yang MX. Mol Cell Biol. 1995;15:4971–4979. doi: 10.1128/mcb.15.9.4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mink S, Haenig B, Klempnauer KH. Mol Cell Biol. 1997;17:6609–6617. doi: 10.1128/mcb.17.11.6609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chu SC, Marks-Konczalik J, Wu HP, Banks TC, Moss J. Biochem Biophys Res Commun. 1998;248:871–878. doi: 10.1006/bbrc.1998.9062. [DOI] [PubMed] [Google Scholar]
- 59.Hewett SJ, Csernansky CA, Choi DW. Neuron. 1994;13:487–494. doi: 10.1016/0896-6273(94)90362-x. [DOI] [PubMed] [Google Scholar]
- 60.Albina JE, Cui S, Mateo RB, Reichner JS. J Immunol. 1993;150:5080–5085. [PubMed] [Google Scholar]
- 61.Muhl H, Dinarello CA. J Immunol. 1997;159:5063–5069. [PubMed] [Google Scholar]
- 62.Strelow LI, Janigro D, Nelson JA. Adv Virus Res. 2001;56:355–388. doi: 10.1016/s0065-3527(01)56033-9. [DOI] [PubMed] [Google Scholar]