

Summary
Alpha-11 giardin is a member of the multi-gene alpha giardin family in the intestinal protozoan, Giardia lamblia. This gene family shares an ancestry with the annexin super family, whose common characteristic is calcium-dependent binding to membranes that contain acidic phospholipids. Several alpha giardins are highly expressed during parasite-induced diarrhea in humans. Despite being a member of a large family of proteins, little is known about the function and cellular localization of alpha-11 giardin, although giardins are often associated with the cytoskeleton. It has been shown that Giardia exhibits high levels of alpha-11 giardin mRNA transcript throughout its life cycle; however, constitutive expression of this protein is lethal to the parasite. Determining the three-dimensional structure of an alpha giardin is essential to identifying functional domains shared in the alpha giardin family. Here we report the crystal structures of the apo and Ca2+-bound forms of alpha-11 giardin, the first alpha giardin to be characterized structurally. Crystals of apo and Ca2+-bound alpha-11 giardin diffracted to 1.1 Å and 2.93 Å, respectively. The crystal structure of selenium-substituted apo alpha-11 giardin reveals a planar array of four tandem repeats of predominantly α-helical domains, reminiscent of previously determined annexin structures, making this the highest-resolution structure of an annexin to date. The apo alpha-11 giardin structure also reveals a hydrophobic core formed between repeats I/IV and II/III, a region typically hydrophilic in other annexins. Surprisingly, the Ca2+-bound structure contains only a single calcium ion, located in the DE loop of repeat I and coordinated differently from the two types of calcium sites observed in previous annexin structures. The apo and Ca2+-bound alpha-11 giardin structures assume overall similar conformations; however, calcium-bound alpha-11 giardin crystallized in a lower-symmetry spacegroup with four molecules in the asymmetric unit. Vesicle-binding studies suggest that alpha-11 giardin, unlike most other annexins, does not bind to vesicles composed of acidic phospholipids in a calcium-dependent manner.
Keywords: Giardia, annexin, intestinal protozoan, calcium-binding, cytoskeleton
Introduction
The protozoan parasite Giardia lamblia (syn. G. intestinalis, G. duodenalis) can infect a wide range of vertebrates including humans, pets and livestock.1 Giardia lamblia has a global distribution and it is one of the most frequent intestinal parasites in humans, although infection outcomes vary from asymptomatic to acute or chronic diarrhea (giardiasis).2 About 200 million people worldwide have symptoms of intestinal Giardia infection and around 500,000 new cases occur each year.3
Despite rapid progress in understanding the biology of Giardia, few virulence factors have been identified and the cause of disease is still unknown. However, there is a strong link between the cytoskeleton and virulence since the disease-causing form of the parasite, the trophozoite, uses its cytoskeleton to move in the intestine, to divide and to attach to the epithelium. The cytoskeleton of Giardia is composed of both classical cytoskeleton structures (microtubules and microfilaments) and Giardia-specific proteins like the alpha-giardins.4 The alpha-giardins are related to the annexin super family, whose common characteristic is calcium-dependent phospholipid binding.5 Several annexins have also been shown to bind F-actin and to link the cytoskeleton to the membrane.6 The microfilament system in Giardia is highly reduced but the annexin-related alpha-giardin gene family with 21 members is instead highly expanded.4,7 The alpha-giardins are highly expressed proteins that interact with the cytoskeleton and different trophozoite membrane structures.7 Many of the alpha-giardins are also highly immunoreactive during acute human giardiasis.8
The overall three-dimensional structure of the C-terminal core domain of annexins is a curved disk structure with a convex and a concave face.9 The calcium- and membrane-binding sites are located on the convex face but additional phospholipid-binding sites have been described for the N-terminal domain located on the concave face (annexin A1).10,11 The N-terminal domains of annexins are highly variable and confer specific properties to each particular annexin. The alpha-giardins have been classified as representatives of a new annexin subfamily (annexin E family) due to their low but significant sequence identity with annexins from other organisms, but to date no three dimensional structure has been determined for any alpha-giardin.12 Determining the three dimensional structure of an alpha-giardin is essential to identify functional domains that are specific for the alpha-giardin family and this can be important in the development of drugs that interfere with the cytoskeleton of this intestinal parasite.
In the present work our goal was to solve the structure of the most highly expressed protein in the alpha-giardin family, alpha-11 giardin.7 The overall three-dimensional fold of alpha-11 giardin is similar to the general annexin fold, but significant local differences were detected.
Results and Discussion
Structure of alpha-11 giardin
The crystal structure of alpha-11 giardin was solved by multi-wavelength anomalous dispersion (MAD) phasing using selenomethionine-derivatized protein. The native protein as well as the selenium-labeled protein crystallized in the orthorhombic spacegroup P212121 with one monomer in the asymmetric unit. Attempts to solve the structure of alpha-11 giardin using the structure of the most closely related annexin, annexin A4 (sequence identity 26%, PDB code: 1ANN), were unsuccessful. Native crystals diffracted to 1.1 Å and selenium-substituted crystals to 1.92 Å. All data collection statistics are shown in Table 1. Three-wavelengths MAD phasing with the programs SOLVE and RESOLVE produced a 2.5 Å electron density map of excellent quality (Figure 1). After careful crystallographic refinement and extension of the resolution to 1.1 Å (Figure 2), the entire amino acid sequence of alpha-11 giardin (306 amino acids) was built into electron density maps, with the exception of terminal residues Ser1 and Lys306, which were not visible in the electron density maps. The final model consists of 304 amino acids (eight with alternate conformations), 620 waters (five with alternate locations) and two sulfate ions. Alternative conformations of the amino acid side chains were identified for Glu19, Lys54, Cys71, Asn214, His237, Ser238, Ser282 and Gln285. The final model of apo alpha-11 giardin exhibits excellent stereochemistry with Rwork and Rfree values of 19.5% and 22.5%, respectively (Table 2).
Table 1.
Data collection and MAD phasing statistics.
| Ca2+ bound | Native (apo) | Edge | SeMet MAD inflection | remote | |
|---|---|---|---|---|---|
| Data collection | |||||
| Wavelength (Å)/(eV) | 0.979 | 0.979 | 0.979/12,661 | 0.979/12,658 | 0.905/13,700 |
| f′/f″ | -- | -- | −9.31/7.76 | −12.5/4.07 | −1.6/3.3 |
| Resolution | 50.0–2.93 | 42.74–1.10 | 50.0–1.92 | 50.0–1.92 | 50.0–1.92 |
| range (Å)a | (3.01-2.93) | (1.14-1.10) | (1.95-1.92) | (1.95-1.92) | (1.95-1.92) |
| Spacegroup | P21 | P212121 | P212121 | P212121 | P212121 |
| Unit-cell parameters | |||||
| a (Å) | 129.30 | 45.95 | 45.27 | 45.27 | 45.33 |
| b (Å) | 45.29 | 60.95 | 61.26 | 61.27 | 61.34 |
| c (Å) | 131.90 | 119.91 | 119.88 | 119.89 | 120.05 |
| β (º) | 116.39 | 90.00 | 90.00 | 90.00 | 90.00 |
| Total observations | 730,522 | 1,203,126 | 736,908 | 733,668 | 907,860 |
| Unique reflections | 30,834 | 133,460 | 26,366 | 26,251 | 26,661 |
| Completeness (%)a | 99.6 (99.4) | 97.2 (94.2) | 98.5 (79.7) | 98.9 (87.0) | 99.6 (99.4) |
| Rmerge#(%)a | 10.4 (49.3) | 12.1 (48.2) | 7.5 (31.2) | 7.3 (30.4) | 7.5 (34.3) |
| Average I/σ(I)a | 13.7 (2.6) | 6.4 (2.0) | 16.6 (3.4) | 6.1 (3.6) | 17.4 (3.3) |
| Redundancya | 3.7 (3.6) | 9.0 (5.98) | 5.2 (4.7) | 5.3 (4.7) | 5.2 (4.9) |
| Mosaicity (º) | 0.72 | 0.36 | 0.47 | 0.51 | 0.48 |
| MAD Phasing | |||||
| Resolution range (Å) | 30.0–2.5 | ||||
| No. of selenomethionines per ASU | 5 | ||||
| Solve peaks (peak heights/σ) | S1=24.0, S2=25.3, S3=24.7, S4=25.0, S5=20.5 | ||||
| Overall SOLVE Z score | 51.8 | ||||
| Figure of merit before density modification | 0.71 | ||||
| Figure of merit after density modification (RESOLVE) | 0.86 | ||||
Rmerge = Σhkl Σj |Ij(hkl)−<I(hkl)>|/Σhkl Σj<I(hkl)>, where Ij(hkl) and <I(hkl)> are the intensity of measurement j and the mean intensity for the reflection with indices hkl, respectively
Values in parentheses are for the highest resolution shell.
Figure 1.

Experimental electron density map at 2.5 Å and contoured at 1.0 σ obtained from selenomethionine MAD phasing using the program RESOLVE36 is shown with the final model of alpha-11 giardin. The figure was prepared using the program Coot.39
Figure 2.
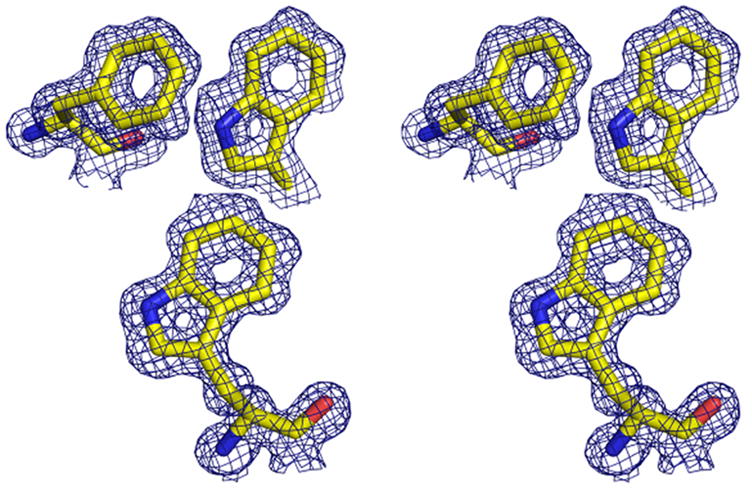
Stereo image of the 2Fo-Fc electron density contoured at 1.0σ after phase extension and refinement to 1.1 Å. Amino acids Phe24 (top left), Trp69 (top right) and Trp28 (bottom left) are shown in gold. The stereo image was created with the molecular graphics program Pymol [http://pymol.sourceforge.net/].
Table 2.
Refinement statistics.
| Alpha-11 giardin (Apo) | Ca2+-bound Alpha-11 giardin | |
|---|---|---|
| Refinement | ||
| Resolution range (Å) | 42.72–1.10 | 40.00–2.93 |
| Number of reflections (working set/test set) | 126,647/6,722 | 27,068/3,012 |
| Rwork/Rfreea,b (%) | 19.5/22.5 | 26.3/30.7 |
| Average B factor (Å2) | 13.8 | 49.6 |
| Ramachandran plotc | ||
| Most favorable regions (%) | 94.7 | 86.5 |
| Additionally allowed regions (%) | 5.3 | 12.3 |
| Generously allowed regions (%) | 0.0 | 1.1 |
| Disallowed regions (%) | 0.0 | 0.0 |
| R.m.s. deviations from ideality | ||
| Bond lengths (Å) | 0.010 | 0.010 |
| Bond angles (º) | 1.256 | 1.426 |
| Model details | ||
| Protein residues/waters | 304/620 | 1,208/111 |
| No. of protein atoms | 3,150 | 9,819 |
| No. of calcium ions | 0 | 4 |
| No. of sulfate ions | 2 | 0 |
Rwork= Σ|Fobs−Fcalc|/(Σ|Fobs|).
The Rfree is the R-factor based on 5%–10% of the data excluded from refinement.
Determined by PROCHECK39
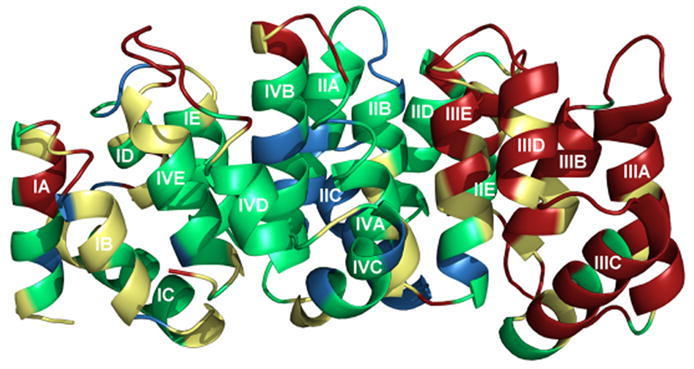
The crystal structure of alpha-11 giardin from Giardia lamblia is the first from the alpha giardin family. The overall structure displays the same topology as the core domains of numerous other members of the annexin family and thus the alpha-11 giardin structure represents the highest-resolution annexin structure to date.13 Alpha-11 giardin folds into a predominantly α-helical curved disk with a concave face and a convex face and short loops connecting the helices (Figure 3(a) and (b)). The disk is formed by a planar packing of four tandem repeats (I–IV) with each repeat consisting of five α-helices (A–E) that form a small hydrophobic core. Another small hydrophobic core located between repeats I/IV and II/III on the concave face is formed by a cluster of seven aliphatic hydrophobic residues (Leu67, Val79, Val96, Ile99, Ile220, Val232 and Leu258) and one aromatic hydrophobic residue (Phe259) (Figure 3(c)). There are significant differences between the residues forming the hydrophobic core in alpha-11 giardin and homologous residues in other annexin structures.14 In annexins A1, A2, A4, A5, A6 and A7 this region is generally hydrophilic and only contains three to five hydrophobic residues in contrast to the eight hydrophobic residues forming the hydrophobic core in alpha-11 giardin (Figure 4).
Figure 3.
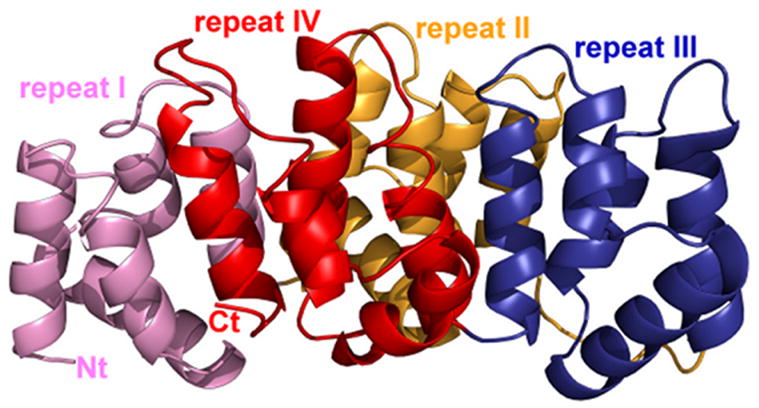
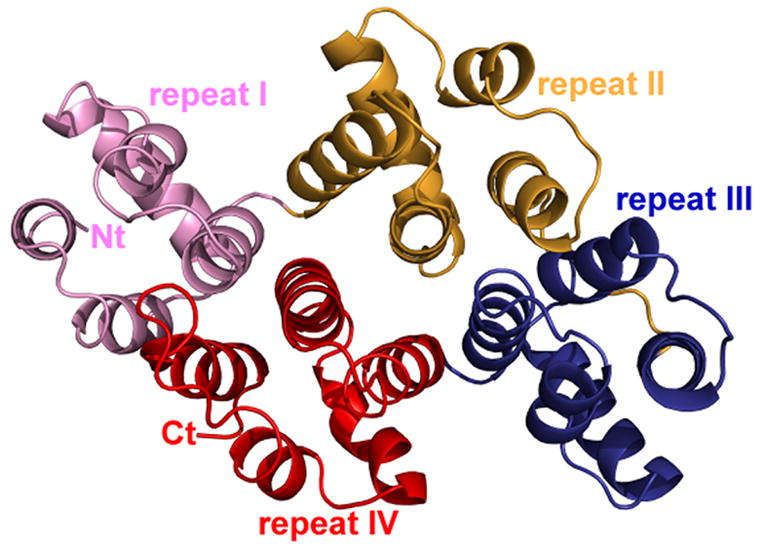
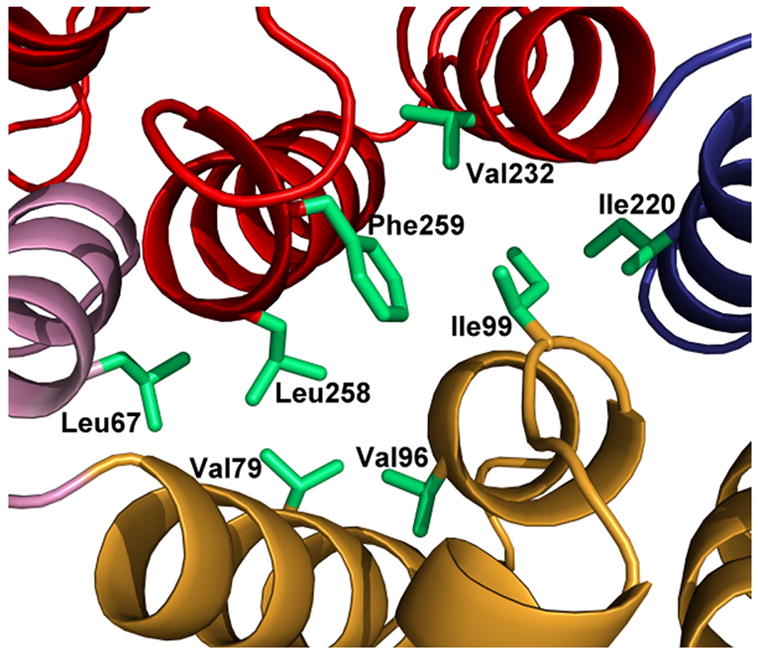
Ribbon diagram of alpha-11 giardin. (a) Side view of the structure with the concave face located at the bottom and the convex face at the top and (b) top view from the convex side. Repeat I is shown in pink, repeat II in gold, repeat III in blue and repeat IV in red. (c) bottom view from the concave side showing the hydrophobic core formed by residues Leu67, Val79, Val96, Ile99, Ile220, Val232, Leu258 and Phe259 (side chains shown in light green). The N-terminus and C-terminus are labeled as Nt and Ct, respectively. The figure was prepared in the molecular graphics program Pymol [http://pymol.sourceforge.net/].
Figure 4.
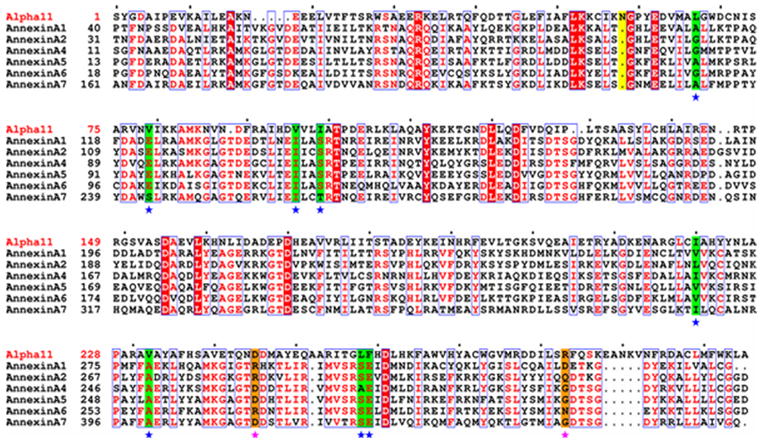
Sequence alignment of alpha-11 giardin and annexins A1, A2, A4, A5, A6 and A7. Note that the first amino acid is a serine residue in the alpha-11 giardin sequence since the first methionine is absent due to the presence of an N-terminal glutathione-S-transferase (GST) tag that was introduced during cloning. Residues denoted as white letters on red background are strictly conserved. Residues boxed in blue and denoted in red are homologous, while residues denoted in black are non-homologous in this group. Highlighted in green and indicated below with a blue star are the residues that line the hydrophobic core located between repeats I/IV and II/III. Highlighted in orange and indicated below with a pink star are residues Asp245 and Arg283 (alpha-11) that form a salt bridge in the AB loop of repeat IV preventing a calcium ion from binding and homologues of these alpha-11 residues in the annexin sequences are highlighted in orange. Highlighted in yellow is Asn58 of alpha-11 giardin which is involved in the type IIIb calcium coordinating site in the DE loop of repeat I. The sequence alignment was performed using CLUSTALW45,46 and the figure was prepared with the program ESPript47.
Unlike most other annexin structures, alpha-11 giardin lacks an amino-terminal domain on the concave side of the core domain. The N-terminal domain, which commonly precedes the annexin core domain and is typically located on the concave side of the disk, is unique for a given member of the annexin family and can range from as few as twelve residues to as many as 169 residues.15 In the alpha-11 giardin structure there are only two N-terminal amino acids preceding the core domain. The absence of an N-terminal domain is not an unusual feature of the alpha giardin family. Out of the 21 members of the alpha giardin family, only six members (alpha 7.1, 7.2, 10, 14, 16 and 19 giardins) have N-terminal domains preceding their respective core domains that range from eleven (alpha 14 giardin) to 93 amino acids (alpha 7.1 and 7.2 giardins). Alpha 7.1, 7.2 and 16 which have long N-terminal domains ranging from 71–93 amino acids share 15.1% sequence identity and 34.5% sequence homology with each other. Whether the long N-terminal domains of these alpha giardins form repeats in addition to repeats I–IV in the core domain remains unclear.
A sequence alignment of alpha-11 giardin with ten other alpha giardin members (alpha-1, 2, 5, 6, 7.1, 7.2, 7.3, 8, 9 and 10 giardins) that share approximately 25–29% sequence identity with alpha-11 giardin reveals several distinct features of alpha-11 giardin (Figure 5). In helix E of repeat II alpha-11 giardin has two alanine residues (Ala134 and Ala141) instead of the conserved tryptophan residues present in all the other alpha giardin sequences. And while there is a conserved lysine present in all other alpha giardin sequences, alpha-11 giardin has a tryptophan residue (Trp267) in helix C of repeat IV. Furthermore, instead of a conserved aromatic residue in helix C of repeat II present in the other alpha giardin sequences, alpha-11 has a charged amino acid (Arg105). In addition, alpha-11 giardin has a longer DE loop in repeat IV in comparison to the length of the DE loops in repeat IV in the other alpha giardins in Figure 5. Whether these differences within the alpha giardin family are functionally and/or structurally significant is yet undetermined and will only be understood once more structural and biochemical studies have been performed on other alpha giardins. Despite these key differences between alpha-11 giardin and the other members of the giardin family, there are several regions in the amino acid sequence that are homologous. Repeats I, II, III and IV are approximately 3.0%, 3.8%, 3.9% and 6.5% identical and 20.9%, 17.6%, 18.0% and 10.8% homologous, respectively, among the alpha giardins aligned in Figure 5. There are a total of thirteen strictly conserved residues (six located in repeat IV) throughout the amino acid sequence, including a conserved tryptophan located in helix E of repeat IV (Trp302) and two conserved arginines (Arg146 and Arg149) located in the connector loop between repeats II and III. Most of the α-helices located in each of the repeats show homology except for helix A in repeat III.
Figure 5.
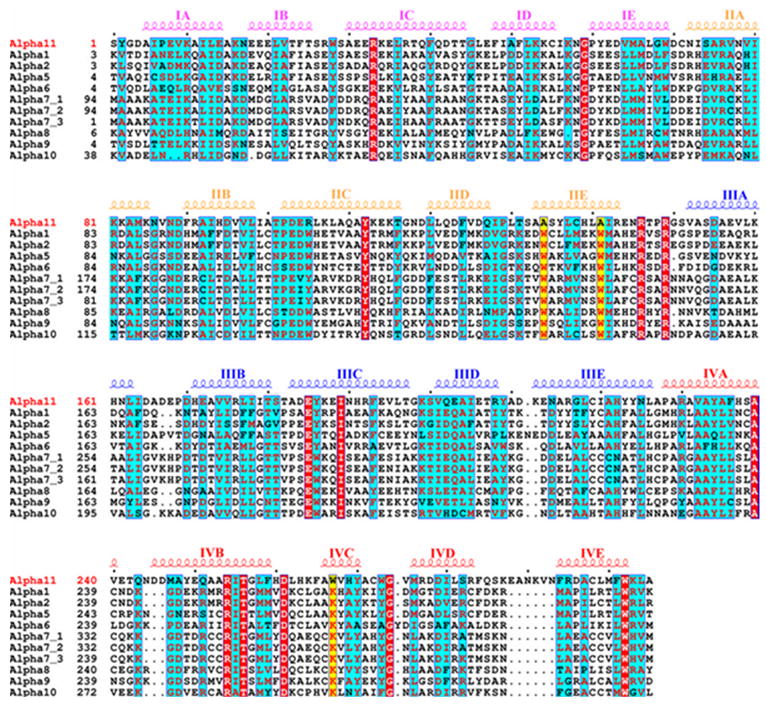
Sequence alignment of alpha-11 giardin and ten other members of the alpha giardin family, which possess 25–29% sequence identity. The secondary structural elements for alpha-11 giardin are indicated above the sequence alignment and colored according to repeats I–IV. Note that the first amino acid is a serine residue in the alpha-11 giardin sequence since the first methionine is absent due to the presence of an N-terminal GST tag that was introduced during cloning. Residues denoted as white letters on red background are strictly conserved. Residues denoted in red on a cyan background are homologous while residues denoted in black on a cyan background are non-homologous in this group. Ala134, Ala141 and Trp267 in the alpha-11 giardin sequence are highlighted in yellow background to stress location in the alpha-11 giardin sequence where it differs from all other alpha giardins. The sequence alignment was performed using CLUSTALW45,46 and the figure was prepared with the program ESPript47.
Although the overall topology and structure of alpha-11 giardin are similar to those of the previously reported annexin core domains, molecular replacement trials with the closest homologue with a known structure, annexin A4, as a search model failed. Even when searching with individual repeats or different combinations of the four repeats of annexin A4, molecular replacement trials were unsuccessful, despite a sequence identity of 26%. After solving the structure of alpha-11 giardin using MAD, it became clear why the previous molecular replacement trials using annexin A4 as the search model had been unfruitful. The r.m.s.d. of the Cα atoms between alpha-11 giardin and annexin A4 is 1.69 Å, indicating that there are significant differences between these two structures despite the same overall fold. The largest differences occur in repeat III where a majority of the Cα atoms change position by more than 3.0 Å (Figure 6). Most of these large changes occur between residues 151–182 in helices A and B. In addition to the differences in the helical regions of the protein there are significant conformational differences around the loop regions of alpha-11 giardin. The largest difference in the position of Cα atoms occurs in the following six loop regions, which deviate more than 3.0 Å: AB loop of repeat I, AB and DE loops of repeat III, the connector loop between repeats II and III on the concave side of the core domain and the AB and DE loops in repeat IV. A superposition of the alpha-11 giardin and annexin A4 structures indicates that there is a difference in the relative orientation of the helices and domains (Figure 7(a) and (b)). The hydrophobic core located between repeats I/IV and repeats II/III in alpha-11 giardin is more shielded from the bulk solvent on the concave side of the core domain in comparison to annexin A4.
Figure 6.
Alpha-11 giardin colored according to its r.m.s.d. from annexin A4, the non-giardin annexin with the highest homology. Regions colored in blue deviate < 1.0 Å, in green 1.0–2.0 Å, in yellow 2.0–3.0 Å and in red are residues that could not be aligned or deviate > 3.0 Å. Repeats and helices are labeled for clarification. The r.m.s.d. was calculated using RMSDcalc [http://www.igs.cnrs-mrs.fr/Caspr2/RMSDcalc.cgi]. The figure was prepared with the molecular graphics program Pymol [http://pymol.sourceforge.net/].
Figure 7.
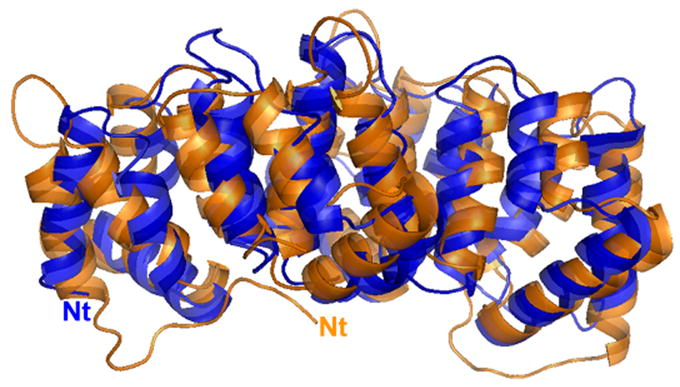
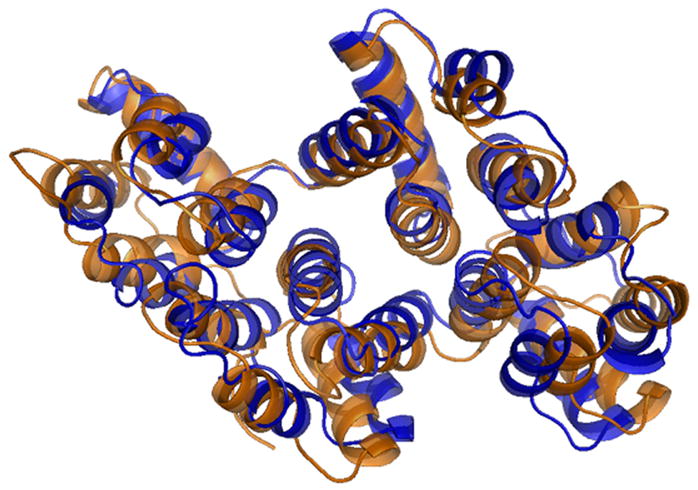
Overlay of alpha-11 giardin (blue) and annexin A4 (orange) crystal structures. (a) Side view of the structure and (b) top view from the convex side. The N-termini (Nt) of alpha-11 giardin and annexin A4 are labeled in their respective colors. Superposition of the alpha-11 giardin and annexin A4 structures was performed in Coot39 and the figures were produced with the molecular graphics program Pymol [http://pymol.sourceforge.net/].
Structure of Ca2+ bound alpha-11 giardin
The crystals of apo and Ca2+-bound alpha-11 giardin were grown under two different conditions and in two different spacegroups with different unit cell parameters, suggesting that alpha-11 giardin is capable of binding calcium. The X-ray crystal structure of Ca2+-bound alpha-11 giardin was solved by molecular replacement using apo alpha-11 giardin as the search model. The possibility that alpha giardins may bind calcium is not a surprising feature since nearly all members of the annexin family have been shown to bind calcium, and numerous calcium ions are present in many annexin crystal structures.9,16 Calcium coordination in annexins can occur at the AB loop (type II binding site) and at the DE loop (type III binding site). The calcium-binding loops are located on the convex face of the core domain, and calcium coordination occurs in a pentagonal bipyramidal or octa-coordination sphere. In type II binding sites, calcium is coordinated by three backbone carbonyls within the (M,L)-X-G-(X)-G sequence (coordinating residues are highlighted in bold and X is a variable residue), a bidentate carboxylate ligand from either an aspartic acid or glutamic acid residue (“cap residue”) located approximately 44 residues downstream in sequence and one to two water molecules. In the traditional type III binding site, the calcium ion is coordinated by two backbone carbonyl oxygens, one acidic side chain located approximately eight residues downstream in sequence and one to three water molecules.17 In contrast to other annexins, in the Ca2+-bound alpha-11 giardin structure, only one calcium ion was found, namely in the DE loop of repeat I in each of the four independent monomers that comprise the asymmetric unit (for details see section on calcium coordination below).
Ca2+-bound alpha-11 giardin crystallized in a spacegroup with four monomers in the asymmetric unit. The four monomers are arranged such that monomers B and D are situated side-by-side and monomers A and C are tilted by 180° relative to monomers B and D (not interacting) and shifted such that the convex portion of the core domains of monomers A and C are slightly face-to-face with the convex portions of monomers B and D (Figure 8). Even though Ca2+-bound alpha-11 giardin crystallized with four monomers in the asymmetric unit, from the observed packing it appears unlikely that alpha-11 giardin forms multimers in solution. The total buried surface area between monomers A and D is 308 Å2 (two hydrogen bonds and no salt bridges) and the total buried surface area between monomers B and C is 392 Å2 (no salt bridges or hydrogen bonds). The total buried surface area between monomers A and B is 152 Å2, while the total buried surface between the other pairs is negligible. There are thus no significant intermolecular contacts between the monomers in this crystal form to suggest alpha-11 giardin oligomerization in solution. Interestingly, the only intermolecular contact between monomers B and C occurs through the coordination of the calcium ion located in the DE loop of repeat I of monomer C. This calcium ion is coordinated in a hexagonal geometry by the backbone carbonyls of Lys53 and Ile56, the side chain carbonyl of Asn58, the unidentate carboxylate of Glu62 from molecule C, one water molecule and the backbone carbonyl of Asp126 from molecule B (Figure 9(a)). The coordination of a calcium ion by an amino acid residue in a neighboring molecule in the DE loop of repeat I has been reported previously in the Ca2+-bound crystal structures of annexin A1 and annexin A2.18,19 Surprisingly, for alpha-11 giardin the coordination of a calcium ion from an adjacent molecule is only seen for monomer C. The backbone carbonyl oxygen of the equivalent Asp126 residues in the neighboring molecules are each approximately 7 Å away from the calcium ion located in the nearby monomers (A, B and D), placing those residues well out of reach for calcium coordination. The calcium ions in monomers A, B and D are coordinated by the backbone carbonyls of Lys53 and Ile56, the side chain carbonyl of Asn58, the unidentate carboxylate of Glu62 and one or two water molecules (Figure 9(b)), in either a hexagonal (monomer D) or pentagonal coordination (monomers A and B).
Figure 8.
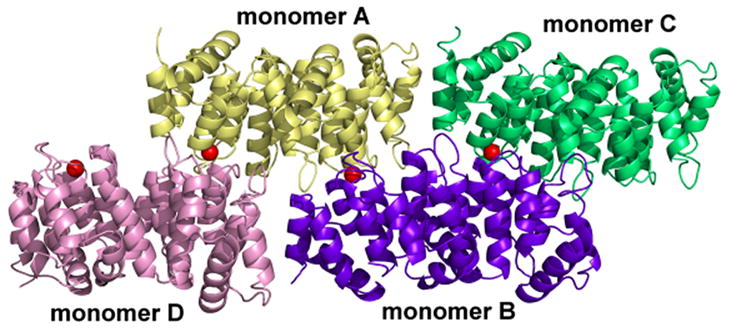
Arrangement of the four monomers in the asymmetric unit of Ca2+-bound alpha-11 giardin. Monomer A is shown in yellow, monomer B in purple, monomer C in green and monomer D in teal. Calcium ions bound to the DE loop of repeat I in each of the four monomers are illustrated as red spheres. The figure was prepared with the molecular graphics program Pymol [http://pymol.sourceforge.net/].
Figure 9.
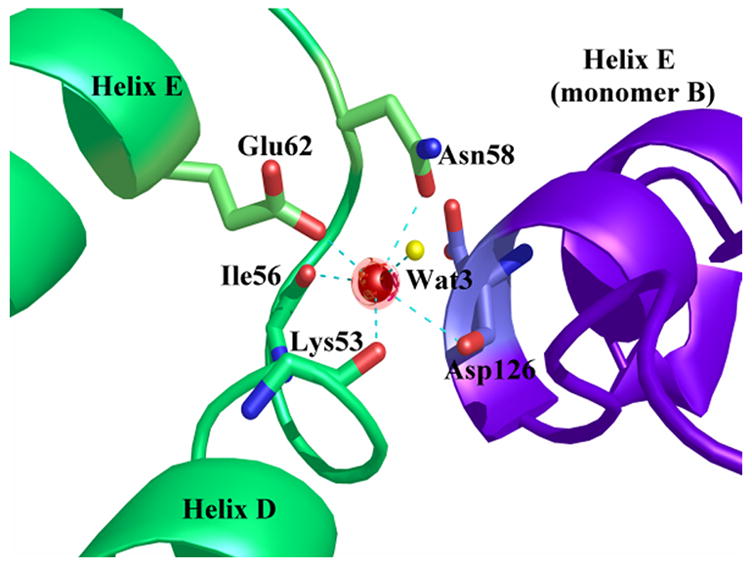
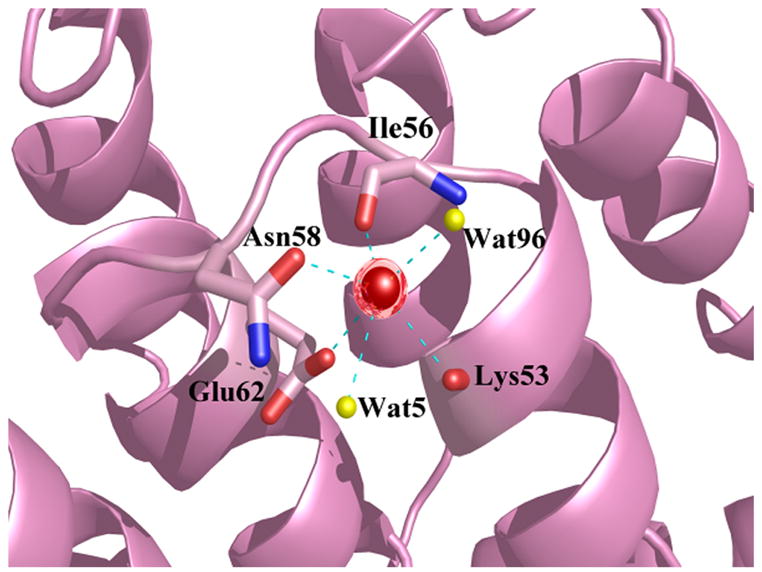
Coordination of the calcium ion in the DE loop of repeat in Ca2+-bound alpha-11 giardin. Calcium ions are represented as red spheres and water molecules as yellow spheres and the coordination between the calcium ion and oxygen atoms is shown as cyan dotted lines. (a) On the left hand side are the two helices (D and E) from monomer C and on the right hand side is helix E from monomer B (in purple). The coordination distances between the calcium ion in monomer C and the coordinating oxygen atoms (Ca-O) are as follows: Ca-O Lys53 2.83 Å, Ca-O Ile56 2.52 Å, Ca-OD1 Asn58 2.84 Å, Ca-OE1 Glu62 2.71 Å, Ca-O HOH3 2.63 Å and Ca-O Asp126 (monomer B) 2.98 Å. (b) Helices and loops from monomer D are shown in pink. The bond distances between the calcium ion in monomer D and the coordinating oxygen atoms (Ca-O) are as follows: Ca-O Lys53 2.76 Å, Ca-O Ile56 2.75 Å, Ca-OD1 Asn58 2.73 Å, Ca-OE1 Glu62 2.56 Å, Ca-O HOH5 2.89 Å and Ca-O HOH96 2.78 Å. Figures were produced with the programs Pymol [http://pymol.sourceforge.net/] and POVRAY [http://www.povray.org/].
Superposition of the individual monomers A, B, C and D of the Ca2+-bound alpha-11 structure with apo alpha-11 giardin results in r.m.s.d. values of 0.60 Å, 0.61 Å, 0.60 Å and 0.61 Å, respectively. This indicates that there are no significant overall conformational changes upon binding of calcium. The largest difference in the positions of Cα atoms occurs in repeat I, namely between residues 7–16 in helix A and residues 44–55 in helices C and D, which move by approximately 1–2 Å.
Different coordination of calcium in alpha-11 giardin and in annexins
There is a clear difference when comparing the calcium coordination in the DE loop of repeat I in alpha-11 giardin with those reported for the traditional type III sites in the DE loops for various other annexin structures. In the crystal structures of annexins A1, A2, A5 and A6 (PDB codes: 1MCX,18 1XJL,19 1AVR20 and 1AVC,16 respectively), the calcium is coordinated (traditional type III binding site) by the backbone carbonyl oxygens of lysine and leucine residues, the carboxylate of a glutamic acid and one to three waters (Table 3). In alpha-11 giardin the calcium ion is coordinated by the backbone carbonyl oxygens of lysine and isoleucine residues, the carboxylate of a glutamic acid, one or two waters and the side-chain carbonyl of an asparagine residue. The presence of the side chain of an asparagine residue in the calcium coordination sphere is highly unusual and has not been reported for any of the calcium sites of previously published annexin structures. In addition, the asparagine residue (Asn58) in alpha-11 giardin appears to be an insertion occurring only in alpha giardins as this residue is not present in annexins A1, A2, A4, A5, A6 and A7 (Figure 4). We thus propose the term type IIIb calcium binding site for the arrangement observed in DE loop of repeat I of alpha-11 giardin.
Table 3.
A comparison of the residues involved in calcium coordination in the DE loops of repeat I in alpha-11 giardin and other annexin structures.
| Structure | Residues | Amino Acid Sequence | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Alpha-11 giardin | 53–62, 126 (monomer B)† | K | K | C | I | K | N | G | P | Y | E,* | D‡* |
| Annexin A1 | 97–106, 196 (monomer B) | K | K | A | L | T | G | H | L | E* | E, | D‡* |
| Annexin A2 | 88–97, 36 (monomer B) | K | S | A | L | S | G | H | L | E* | T, | E‡* |
| Annexin A5 | 75–84 | K | S | E | L | T | G | K | F | E** | K | |
| Annexin A6 | 70–79 | K | Y | E | L | T | G | K | F | E | R | |
Residues involved in calcium coordination in the DE loop of repeat I are high-lighted in bold and colored red (backbone carbonyl oxygens), orange (polar side chain) and blue (acidic side chain: unidentate (*) and bidentate carboxylate (**)). Annexin PDB codes: A1 - 1MCX,18 A2 - 1XJL,19 A5 - 1AVR20 and A6 - 1AVC.16
Calcium coordination from a neighboring molecule is only seen for monomer C.
Residues from a neighboring molecule.
Another unusual feature of the Ca2+-bound alpha-11 structure is the absence of a calcium ion in the AB loop of repeat IV. The type II calcium binding site in the AB loop of repeat IV is conserved in annexins A1, A2, A4, A5 and A6. The calcium ion present in this loop is coordinated by the backbone carbonyl oxygens in the following sequence M-X-G-X-G (coordinating residues are shown in bold and X is a variable residue), a downstream aspartic or glutamic acid residue (located approximately 44 residues downstream of the methionine) and one or two water molecules. In the AB loop of repeat IV of alpha-11 giardin, there is a potential calcium binding site that could be coordinated by the backbone carbonyl oxygens of Thr242 and Gln243 and the side chain of Asp245; however, the side chain of Arg283 located in the DE loop obstructs a calcium ion from binding in this area since it forms a salt bridge with Asp245. In place of a charged, basic arginine residue (Arg283 in alpha-11 giardin), alpha-1, 2, 5, 6, 7.1, 7.2, 7.3, 8, 9 and 10 giardins have either a hydrophobic aliphatic or a polar residue (cysteine, alanine, threonine, valine or leucine), while the acidic aspartic residue (Asp245 in alpha-11 giardin) is absent in all alpha giardin sequences mentioned above, indicating that a salt bridge is absent at this position and does not obstruct a calcium ion from binding in the AB loop of repeat IV in the other alpha giardins aligned in Figure 5. Annexins A1, A2, A4, A5, A6 and A7 either have an aspartic acid or arginine homologue (Asp245 in alpha-11 giardin) and either an aspartic acid, glutamine, asparagine or glycine homologue (Arg283 in alpha-11 giardin) (Figure 4). The only possible salt bridge that could form in the annexin structures at the alpha-11 homologous residues in the AB loop of repeat IV is in annexin A1 between an arginine residue (Asp245 in alpha-11 giardin) and an aspartic acid residue (Arg283 in alpha-11 giardin). However, in the crystal structure of Ca2+-bound annexin A1, the side chains of the arginine (Arg292 in annexin A1) and aspartic acid (Asp329 in annexin A1) residues are over 15.0 Å apart and can not form a salt bridge. Thus it appears that the AB loop in repeat IV is devoid of a calcium ion due to a salt bridge that is only present in alpha-11 giardin. It remains to be seen if the other Ca2+-bound alpha giardin structures have a bound calcium ion in the AB loop of repeat IV and in what type of calcium coordination.
The absence of type II calcium coordination sites in alpha giardins may be a common feature. According to Morgan and coworkers, a hidden Markov model (HMM) profile of the Giardia family revealed a general absence of type II calcium coordination sites.21 The alpha-11 giardin and annexin alignment in Figure 4 suggests the absence of type II coordinating residues in the AB loop of repeat I. In particular the glycine residues in the type II calcium coordination sequence conserved in annexins (M-X-G-X-G -- coordinating residues are shown in bold and X is a variable residue) are absent in the alpha-11 giardin sequence. It has been shown that alpha-1 giardin associates with multilamellar phosphatidylserine-containing vesicles in a calcium-dependent manner; however, molecular modeling of the amino acid sequence of alpha-1 giardin based on the X-ray structure of annexin A5 suggested that the calcium coordination sites in alpha-1 giardin are low affinity type III, in contrast to the type II calcium coordination sites that are thought to be primarily responsible for membrane binding.22 Four potential type III calcium-binding sites on the convex face of alpha-1 giardin were predicted at residues Glu20 and Glu63 (repeat I), Glu134 (repeat II) and Glu245 (repeat IV). The homologues of these alpha-1 giardin glutamic acid residues in alpha-11 giardin are Glu18 and Tyr61 (repeat I), Ser132 (repeat II) and Tyr249 (repeat IV). This suggests only one potential type III calcium coordination site in repeat I of alpha-11 giardin which would be involving residue Glu18; however, our crystal structure in the presence of calcium reveals that this site is not occupied by an ordered calcium ion. It is unclear why this potential type III calcium coordination site is not occupied in the alpha-11 giardin structure.
Calcium-dependent phospholipid binding
To test whether alpha-11 giardin exhibits the calcium-dependent binding to acidic phospholipids, generally regarded as a property of the annexins, we tested its calcium-dependent binding to small unilamellar phosphatidylserine (PS)- and phosphatidylcholine (PC)-containing vesicles (see materials and methods section).23,24 To our surprise, alpha-11 giardin did not bind to the POPS/POPC [1-palmitoyl-2-pleoyl-sn-glycero-3-[phospho-L-serine] (POPS) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC)] containing vesicles regardless of the calcium concentration (0.05–10 mM) used in each set-up (results not shown). Even though our phospholipid binding studies demonstrate that alpha-11 giardin does not bind to 20% PS/80% PC-containing vesicles in a calcium-dependent manner, this result does not completely rule out the possibility that alpha-11 giardin binds to membranes under other circumstances. In order to test whether alpha-11 giardin exhibits some of the calcium-dependent phospholipid binding features observed for most annexins, additional phospholipid binding studies will need to be performed with different lipids such as cardiolipin, cholesterol, phosphoinositides and/or higher percentages of PS. One reason why alpha-11 giardin did not associate with acidic phospholipid vesicles in our assay may be due to the fact that alpha-11 giardin binds only one calcium ion, in a type IIIb site with presumably weak calcium affinity. Even though the crystal structure of alpha-11 giardin reported herein is overall similar to that of other annexins, it is still unclear what the exact role of alpha-11 giardin is in the intestinal protozoan parasite, Giardia lamblia. One possibility is that alpha-11 giardin may have extracellular localization where it could interact with glycosaminoglycans (GAGs) on the epithelial cell lining, initiating the first contact with the microvilli in the small intestine.25 Several in vitro and crystallographic studies have shown that some annexins can bind to heparin in a calcium-dependent and independent manner.26–30 Crystallographic and kinetic studies performed on rat annexin A5 revealed the presence of two heparin-derived tetrasaccharides (HTS) binding sites, one that is calcium dependent and located on the convex face (HTS1 near Arg207 and Lys208) and the other that is calcium independent and located on the concave face (HTS2 near Arg285-Lys286-X-X-Arg289-Lys290, where X is a variable residue).31 The amino acid sequence alignment of alpha-11 and annexin A5 reveals that neither HTS binding site in annexin A5 is conserved in alpha-11 giardin. However, a closer look at the alpha-11 giardin amino acid sequence reveals that there may be two heparin binding sites located in repeat I, one that is calcium independent (concave face) and the other that is calcium-dependent (convex face). The calcium-independent heparin binding site would be located on the concave face off alpha-11 giardin and formed by residues Arg33-Lys34-X-X-Arg37, which is very similar to the R-K-X-X-R-K motif of HTS1 in annexin A5. The other possible heparin binding site could be calcium-dependent and formed by residues Lys53 and Lys54 located in the calcium binding site in the DE loop of repeat I, which may be the reason why alpha-11 giardin binds calcium. Further biochemical experiments will have to be carried out to determine if alpha-11 giardin functions like lectins and where it localizes in the parasite.
Conclusions
We have solved the first three-dimensional structure of a member of the alpha giardin family in the apo and Ca2+-bound forms. The crystal structure of alpha-11 giardin reveals a highly α-helical, tightly packed core domain similar to the conserved annexin core domain.13 The apo structure of alpha-11 giardin was solved to a resolution of 1.1 Å, making this the highest resolution for an annexin structure to date. The overall fold consists of four repeats (approximately 70 amino acids each), with each repeat comprised of five α-helices. In contrast to nearly all published annexin structures, the structure of alpha-11 giardin lacks an amino terminal domain on the concave side of the protein.15 Despite having the same overall fold, the alpha-11 giardin structure reveals different orientations of the helices within each repeat and conformational differences in the loop regions in comparison to the annexin closest in primary sequence, annexin A4. The structure of the Ca2+-bound form of alpha-11 giardin reveals only one calcium ion bound in a newly described calcium binding site in the DE loop of repeat I. In comparison to previously determined Ca2+-bound annexin structures, the alpha-11 giardin structure shows significant differences in the coordination of the calcium ion in the DE loop of repeat I, and we thus propose the term type IIIb binding site, in contrast to the traditional type III binding site in previous annexin structures.16,18–20 In addition to the conserved amino acids coordinating the calcium ion in type III sites, the side chain of an asparagine residue was found in the coordination sphere. Another unusual feature of alpha-11 giardin was revealed in the vesicle pull-down assay, which showed that alpha-11 giardin does not associate with 20% PS/80% PC-containing vesicles under a wide range of calcium concentrations. Even though our results suggest that alpha-11 giardin does not bind to vesicles in a calcium-dependent manner, further phospholipid-binding studies will be performed to determine if alpha-11 giardin does indeed lack the landmark feature exhibited by most annexins. Although alpha-11 giardin is one of only two members of the alpha giardin family that show high levels of mRNA transcription (alpha-1 giardin being the other one) and presumably corresponding protein expression, it has yet to be revealed what the exact function of alpha-11 giardin is in Giardia lamblia and whether this member of the alpha giardin family associates with the cytoskeleton. The crystal structures of apo and Ca2+-bound alpha-11 giardin reported herein are an important step in deciphering the role and function of alpha-11 giardin.
Materials and Methods
Protein expression and purification
Expression and purification of recombinant native alpha-11 giardin and Ca2+-bound alpha-11 giardin was described previously.32 The recombinant plasmid pGEX-6P3-alpha-11 was transformed into BL21-CodonPlus(DE3)-RIL-X (Stratagene) methionine auxotrophic Escherichia coli strain to express selenomethionine-substituted alpha-11 giardin. 20 ml of cells grown from an overnight pre-culture incubated at 37 °C were inoculated in 2 l of M9 minimal media [100 ml/l of 10x M9 salts (68 g/l Na2HPO4, 30 g/l KH2PO4, 5 g/l NaCl), 1 ml/l 1M MgSO4, 1 g/l NH4Cl, 1 ml/l 100 mM CaCl2, 15 ml/l, 20% (v/v) glucose, 10 ml GIBCO MEM Vitamin Solution (Fisher Scientific)] supplemented with 50 μg/ml of ampicillin and 50 μg/ml of L-methionine. The culture was grown to an A600 of 0.8 at 37 °C and then harvested by centrifugation, washed twice in M9 minimal media and finally resuspended in M9 minimal media supplemented with 50 μg/ml of ampicillin. Cells were grown at 37 °C for 1 h to exhaust any remaining L-methionine in the media. After 1 h 100 mg/l of L-selenomethionine was added and incubated for 30 min before the culture was induced with 1 mM isopropyl β-D-thiogalactopyranoside (IPTG) and incubated overnight at 37 °C. Cells were harvested by centrifugation at 6000g for 15 min and suspended in 50 mM Tris-HCl pH 7.4, 1 mM EDTA, 100 mM NaCl, 1% (v/v) Nonidet P40 (NP40), 10% (v/v) glycerol, 1 mM dithiothreitol (DTT) and 1 mM phenylmethylsulfonyl fluoride (PMSF) containing a protease inhibitor cocktail (20 μg/ml aprotinin, 2 μg/ml leupeptin, 2 μg/ml pepstatin, 0.5 mM benzamidine and 10 μM trans-epoxysuccinyl-L-leucylamido(4-guanidino)butane (E-64)) and 0.5 mg/ml lysozyme. After the cells were lysed in a French Press, the lysate was centrifuged at 40,000g for 1 h and filtered through a 0.45 μm filter to remove any cell debris.
N-terminal glutathione-S-transferase (GST) tagged alpha-11 giardin was purified on a pre-equilibrated (50 mM Tris-HCl pH 7.4, 1 mM EDTA, 100 mM NaCl, 1% (v/v) NP40, 10% (v/v) glycerol) 20 ml bed volume GSTrap Fast Flow column (Amersham). The cell lysate was incubated on the GST column for 1 ½ h, washed with ten column volumes of 1 M NaCl and ten column volumes of 50 mM Tris-HCl pH 8.0 and 150 mM NaCl to remove any unbound protein and impurities. The GST moiety was cleaved on the column in protease cleavage buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1 mM EDTA and 1 mM DTT) with 10 units of PreScission Protease (Amersham) per mg of GST fusion protein overnight at 4 °C. Alpha-11 giardin was eluted in protease cleavage buffer and fractions were collected. In the final purification step, the PreScission Protease (GST tag) was separated from the alpha-11 giardin by applying fractions onto a GSTrap Fast Flow column equilibrated with protease cleavage buffer. During this purification step, the PreScission Protease remained bound to the GSTrap Fast Flow column while the alpha-11 giardin eluted in the flow-through in protease cleavage buffer. Fractions containing alpha-11 giardin were collected and the purity was verified by SDS-PAGE before the protein was concentrated in a Centricon 10 kDa cutoff concentrator (Millipore) to 11 mg/ml for crystallization.
Crystallization and data collection
Apo, Ca2+-bound and selenomethionyl-derivatized alpha-11 giardin crystals were obtained at 4 °C using the sitting-drop vapor-diffusion method in the JCSG suite from Nextal Biotechnologies (native and selenomethionine derivatized crystals) and the Index Screen from Hampton Research (Ca2+-bound crystals). 2 μl of protein solution were mixed with 2 μl of reservoir solution over wells containing 200 μl of reservoir solution. Native and selenomethionyl derivatized alpha-11 giardin crystals were obtained in 200 mM lithium sulfate, 100 mM MES pH 6.0, and 25% (w/v) PEG 3350 and grew in about one week. Ca2+-bound alpha-11 giardin crystals were obtained in 50 mM calcium chloride, 100 mM Bis-Tris pH 6.5, 24% (w/v) PEG 550 MME and appeared after two weeks. Crystals were transferred to a solution containing the reservoir solution and 30% (v/v) glycerol as a cryo-protectant before being flashed-cooled in liquid nitrogen for data collection.
Diffraction data on apo, Ca2+-bound and selenomethionyl-derivatized alpha-11 giardin were collected at beamline 9-1 at the Stanford Synchrotron Radiation Laboratory (SSRL), CA, U.S.A. using remote robotic data collection. The Q315 CCD area detector from Area Detector Systems Corporation (ADSC) was used for data collection. 180 frames were collected with an oscillation angle of 1° for native and Ca2+-bound alpha-11 giardin crystals. Two data sets from a single native alpha-11 giardin crystal were collected to 1.1 Å and 1.4 Å resolution (5 and 30 seconds exposure time, respectively) and one data set was collected on a Ca2+-bound alpha-11 giardin crystal to 2.93 Å (30 seconds exposure time). An X-ray absorption scan of a selenomethionyl derivatized crystal was collected near the Se K absorption edge to select the appropriate wavelengths for multi-wavelength anomalous dispersion (MAD) experiments. MAD diffraction data were collected on a single crystal at three wavelengths corresponding to the inflection point, peak and high energy remote wavelengths with a crystal-to-image plate distance of 280 mm, an oscillation angle of 1° and 8 seconds exposure time. For each wavelength, 360° of data were collected in 30° wedges to 1.92 Å. The two data sets collected on the native alpha-11 giardin crystal were indexed and integrated separately and later merged and scaled together with the program d*TREK33 in the primitive orthorhombic spacegroup P222. The data sets collected on the Ca2+ bound alpha-11 giardin crystal and the selenomethionyl derivatized crystal were processed with HKL200034 in the primitive monoclinic spacegroup P2 and primitive orthorhombic spacegroup P222, respectively.
Structure determination by MAD phasing and refinement
Initially, molecular replacement trials were carried out to solve the structure of alpha-11 giardin with the program PHASER35 using bovine annexin A4 (PDB code: 1ANN) either in its entirety or broken up into separate repeats as the search model. Since all attempts to solve the structure of alpha-11 giardin via molecular replacement failed, MAD phasing was the next approach. Crystals of selenomethionine-labeled alpha-11 giardin were isomorphous to native crystals allowing the determination of initial experimental phases. The structure of apo alpha-11 giardin was solved in the spacegroup P212121 using the data sets collected at the inflection point, peak and high energy remote wavelengths. All five selenium sites were located with the program SOLVE14 using data to a maximum resolution of 2.5 Å. The peak heights for all selenium sites in the SOLVE map were greater than 20σ and the overall Z score after running SOLVE was 51.8. The initial electron density map from SOLVE was improved using the density modification program RESOLVE36 which increased the figure of merit (FOM) from 0.71 to 0.86 at 2.5 Å. Phases were extended to 1.1 Å using the high-resolution native data set and the automated protein model building program ARP/wARP.37 After ten cycles of auto-building and 10 cycles of REFMAC38 refinement between each auto-building cycle, the final model contained seven peptide chains with a connectivity index of 0.95. ARP/wARP built 281 amino acid residues out of 306 amino acid residues and 79 water molecules with a crystallographic R-factor of 26.1 % and an Rfree of 33.7%. Manual model building was performed with the modeling program Coot39 and anisotropic B-factor refinement was carried out in REFMAC5.40
For the Ca2+-bound form of alpha-11 giardin the Matthew’s coefficient was calculated to be 2.47 Å3 Da−1 (corresponding to a solvent content of 50.2%) when assuming four molecules in the asymmetric unit.41 A self-rotation function plot showed the presence of two non-crystallographic 2-fold peaks in addition to the crystallographic 2-fold peak, confirming the presence of non-crystallographic symmetry.42 All four chains of Ca2+-bound alpha-11 giardin were located using the molecular replacement program PHASER and apo alpha-11 giardin as the search model. Manual model building was performed with the program Coot after three rounds of rigid body refinement to a maximum resolution of 3.0 Å in CNS.43 Preliminary rounds of refinement were carried out to a maximum resolution of 3.0 Å with cycles of energy minimization, torsion-angle simulated annealing and temperature-factor optimization using the maximum-likelihood target function while keeping the atomic coordinates restrained by non-crystallographic symmetry (NCS) in CNS. Four calcium ions and ordered solvent water molecules were added using water pick in CNS and by visual inspection. Several rounds of re-building were continued based on 3Fo-2Fc, 2Fo-Fc, Fo-Fc, NCS-averaged and 2Fo-Fc simulated annealing omit maps, and subsequent rounds of refinement were carried out with the program CNS using data to the maximum resolution of 2.93 Å.
The final model of apo alpha-11 giardin includes one monomer per asymmetric unit (residues 2–305), 620 water molecules and two sulfate ions. The final model of Ca2+-bound alpha-11 giardin includes four monomers, 111 water molecules and four calcium ions. Good stereochemistry for the apo alpha-11 giardin and Ca2+-bound alpha-11 giardin models was confirmed using the program PROCHECK.44 Final refinement statistics for apo alpha-11 giardin and Ca2+ bound alpha-11 giardin are listed in Table 2.
Vesicle pull-down assay
Phospholipid vesicles composed of 1-palmitoyl-2-oleoyl-sn-glycero-3-[phospho-L-serine] (POPS) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) were prepared in a 1:4 molar ratio.24 25 mg of POPS and 97.1 mg of POPC (Avanti Polar Lipids) were dissolved in 4 ml of chloroform and 2 ml of methanol. The organic solvents were evaporated under a stream of nitrogen and dried overnight. The lipids were dissolved in phospholipid buffer (50 mM imidazole-HCl pH 7.4 and 100 mM NaCl) and then subjected to four freeze-thaw cycles. Small unilamellar vesicles 0.2 μm in size were prepared by filtering through an extruder (15 times) at 37 °C. The association of alpha-11 giardin with the vesicles was assessed with a co-pelleting assay using annexin A2 as a positive control and ovalbumin as a negative control.19 For each reaction (200 μl), a total of 20 nmoles of vesicles were incubated with 0.2 nmoles of protein in phospholipid buffer with varying amounts of calcium chloride (0.05 mM–10mM). All samples were incubated for 1 h at room temperature and then centrifuged for 10 min at 14,000g to separate vesicles with protein bound (pellet) from soluble protein (supernatant). Pellet and supernatant samples were analyzed on an SDS-PAGE gel and subsequently stained with Coomassie Brilliant Blue (results not shown).
Protein Data Bank accession codes
The atomic coordinates and structure factors for apo and Ca2+ bound alpha-11 giardin have been deposited in the RCSB Protein Data Bank under the accession codes 2II2 and 2IIC, respectively.
Acknowledgments
We would like to thank Lutz Vogeley and Jason Stagno for assistance with data processing and Dr. Anja Rosengarth for assistance in the vesicle pull-down assay. This research was supported by NIH grant R01-GM067808.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hunter PR, Thompson RC. The zoonotic transmission of Giardia and Cryptosporidium. Int J Parasitol. 2005;35:1181–1190. doi: 10.1016/j.ijpara.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 2.Gardner TB, Hill DR. Treatment of giardiasis. Clin Microbiol Rev. 2001;14:114–128. doi: 10.1128/CMR.14.1.114-128.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.WHO. The World Health Report 1996. 68 Organization WH; Geneva: 1996. [Google Scholar]
- 4.Elmendorf HG, Dawson SC, McCaffery JM. The cytoskeleton of Giardia lamblia. Int J Parasitol. 2003;33:3–28. doi: 10.1016/s0020-7519(02)00228-x. [DOI] [PubMed] [Google Scholar]
- 5.Rescher U, Gerke V. Annexins--unique membrane binding proteins with diverse functions. J Cell Sci. 2004;117:2631–2639. doi: 10.1242/jcs.01245. [DOI] [PubMed] [Google Scholar]
- 6.Hayes MJ, Rescher U, Gerke V, Moss SE. Annexin-actin interactions. Traffic. 2004;5:571–576. doi: 10.1111/j.1600-0854.2004.00210.x. [DOI] [PubMed] [Google Scholar]
- 7.Weiland ME, McArthur AG, Morrison HG, Sogin ML, Svard SG. Annexin-like alpha giardins: a new cytoskeletal gene family in Giardia lamblia. Int J Parasitol. 2005;35:617–626. doi: 10.1016/j.ijpara.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 8.Palm JE, Weiland ME, Griffiths WJ, Ljungstrom I, Svard SG. Identification of immunoreactive proteins during acute human giardiasis. J Infect Dis. 2003;187:1849–1859. doi: 10.1086/375356. [DOI] [PubMed] [Google Scholar]
- 9.Gerke V, Creutz CE, Moss SE. Annexins: linking Ca2+ signalling to membrane dynamics. Nat Rev Mol Cell Biol. 2005;6:449–461. doi: 10.1038/nrm1661. [DOI] [PubMed] [Google Scholar]
- 10.Hoekstra D, Buist-Arkema R, Klappe K, Reutelingsperger CP. Interaction of annexins with membranes: the N-terminus as a governing parameter as revealed with a chimeric annexin. Biochemistry. 1993;32:14194–14202. doi: 10.1021/bi00214a019. [DOI] [PubMed] [Google Scholar]
- 11.de la Fuente M, Ossa CG. Binding to phosphatidyl serine membranes causes a conformational change in the concave face of annexin I. Biophys J. 1997;72:383–387. doi: 10.1016/S0006-3495(97)78677-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moss SE, Morgan RO. The annexins. Genome Biol. 2004;5:219. doi: 10.1186/gb-2004-5-4-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gerke V, Moss SE. Annexins: from structure to function. Physiol Rev. 2002;82:331–371. doi: 10.1152/physrev.00030.2001. [DOI] [PubMed] [Google Scholar]
- 14.Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr D Biol Crystallogr. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seaton BA, Dedman JR. Annexins. Biometals. 1998;11:399–404. doi: 10.1023/a:1009205925714. [DOI] [PubMed] [Google Scholar]
- 16.Avila-Sakar AJ, Creutz CE, Kretsinger RH. Crystal structure of bovine annexin VI in a calcium-bound state. Biochim Biophys Acta. 1998;1387:103–116. doi: 10.1016/s0167-4838(98)00111-3. [DOI] [PubMed] [Google Scholar]
- 17.Huber R, Schneider M, Mayr I, Romisch J, Paques EP. The calcium binding sites in human annexin V by crystal structure analysis at 2.0 Å resolution. Implications for membrane binding and calcium channel activity. FEBS Lett. 1990;275:15–21. doi: 10.1016/0014-5793(90)81428-q. [DOI] [PubMed] [Google Scholar]
- 18.Rosengarth A, Luecke H. A calcium-driven conformational switch of the N-terminal and core domains of annexin A1. J Mol Biol. 2003;326:1317–1325. doi: 10.1016/s0022-2836(03)00027-5. [DOI] [PubMed] [Google Scholar]
- 19.Rosengarth A, Luecke H. Annexin A2: Does it induce membrane aggregation by a new multimeric state of the protein? Annexins. 2004;1:129–136. [Google Scholar]
- 20.Huber R, Berendes R, Burger A, Schneider M, Karshikov A, Luecke H, et al. Crystal and molecular structure of human annexin V after refinement. Implications for structure, membrane binding and ion channel formation of the annexin family of proteins. J Mol Biol. 1992;223:683–704. doi: 10.1016/0022-2836(92)90984-r. [DOI] [PubMed] [Google Scholar]
- 21.Morgan RO, Martin-Almedina S, Garcia M, Jhoncon-Kooyip J, Fernandez MP. Deciphering function and mechanism of calcium-binding proteins from their evolutionary imprint. Biochim Biophys Acta. 2006;1763:1238–1249. doi: 10.1016/j.bbamcr.2006.09.028. [DOI] [PubMed] [Google Scholar]
- 22.Bauer B, Engelbrecht S, Bakker-Grunwald T, Scholze H. Functional identification of alpha 1-giardin as an annexin of Giardia lamblia. FEMS Microbiol Lett. 1999;173:147–153. doi: 10.1111/j.1574-6968.1999.tb13496.x. [DOI] [PubMed] [Google Scholar]
- 23.Hofmann A, Huber R. Liposomes in assessment of annexin-membrane interactions. Methods Enzymol. 2003;372:186–216. doi: 10.1016/S0076-6879(03)72011-4. [DOI] [PubMed] [Google Scholar]
- 24.Reeves JP, Dowben RM. Formation and properties of thin-walled phospholipid vesicles. J Cell Physiol. 1969;73:49–60. doi: 10.1002/jcp.1040730108. [DOI] [PubMed] [Google Scholar]
- 25.Weiland ME, Palm JE, Griffiths WJ, McCaffery JM, Svard SG. Characterisation of alpha-1 giardin: an immunodominant Giardia lamblia annexin with glycosaminoglycan-binding activity. Int J Parasitol. 2003;33:1341–1351. doi: 10.1016/s0020-7519(03)00201-7. [DOI] [PubMed] [Google Scholar]
- 26.Kojima K, Yamamoto K, Irimura T, Osawa T, Ogawa H, Matsumoto I. Characterization of carbohydrate-binding protein p33/41: relation with annexin IV, molecular basis of the doublet forms (p33 and p41), and modulation of the carbohydrate binding activity by phospholipids. J Biol Chem. 1996;271:7679–7685. doi: 10.1074/jbc.271.13.7679. [DOI] [PubMed] [Google Scholar]
- 27.Kassam G, Manro A, Braat CE, Louie P, Fitzpatrick SL, Waisman DM. Characterization of the heparin binding properties of annexin II tetramer. J Biol Chem. 1997;272:15093–15100. doi: 10.1074/jbc.272.24.15093. [DOI] [PubMed] [Google Scholar]
- 28.Ishitsuka R, Kojima K, Utsumi H, Ogawa H, Matsumoto I. Glycosaminoglycan binding properties of annexin IV, V, and VI. J Biol Chem. 1998;273:9935–9941. doi: 10.1074/jbc.273.16.9935. [DOI] [PubMed] [Google Scholar]
- 29.Capila I, VanderNoot VA, Mealy TR, Seaton BA, Linhardt RJ. Interaction of heparin with annexin V. FEBS Lett. 1999;446:327–330. doi: 10.1016/s0014-5793(99)00245-8. [DOI] [PubMed] [Google Scholar]
- 30.Fitzpatrick SL, Kassam G, Manro A, Braat CE, Louie P, Waisman DM. Fucoidan-dependent conformational changes in annexin II tetramer. Biochemistry. 2000;39:2140–2148. doi: 10.1021/bi992180z. [DOI] [PubMed] [Google Scholar]
- 31.Capila I, Hernaiz MJ, Mo YD, Mealy TR, Campos B, Dedman JR, et al. Annexin V--heparin oligosaccharide complex suggests heparin sulfate--mediated assembly on cell surfaces. Structure. 2001;9:57–64. doi: 10.1016/s0969-2126(00)00549-9. [DOI] [PubMed] [Google Scholar]
- 32.Pathuri P, Nguyen ET, Luecke H. Expression, purification, crystallization and preliminary X-ray diffraction analysis of alpha-11 giardin from Giardia lamblia. Acta Crystallograph Sect F Struct Biol Cryst Commun. 2006;62:1108–1112. doi: 10.1107/S1744309106039650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pflugrath JW. The finer things in X-ray diffraction data collection. Acta Crystallogr D Biol Crystallogr. 1999;55:1718–1725. doi: 10.1107/s090744499900935x. [DOI] [PubMed] [Google Scholar]
- 34.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 35.Storoni LC, McCoy AJ, Read RJ. Likelihood-enhanced fast rotation functions. Acta Crystallogr D Biol Crystallogr. 2004;60:432–438. doi: 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- 36.Terwilliger TC. Maximum-likelihood density modification. Acta Crystallogr D Biol Crystallogr. 2000;56:965–972. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lamzin VS, Wilson KS. Automated refinement of protein models. Acta Crystallogr D Biol Crystallogr. 1993;49:129–147. doi: 10.1107/S0907444992008886. [DOI] [PubMed] [Google Scholar]
- 38.Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nat Struct Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 39.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 40.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 41.Matthews BW. Solvent content of protein crystals. J Mol Biol. 1968;33:491–497. doi: 10.1016/0022-2836(68)90205-2. [DOI] [PubMed] [Google Scholar]
- 42.Vagin A, Teplyakov A. A translation-function approach for heavy-atom location in macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 1998;54:400–402. doi: 10.1107/s0907444997014923. [DOI] [PubMed] [Google Scholar]
- 43.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 44.Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 45.Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, et al. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003;31:3497–3500. doi: 10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gouet P, Courcelle E, Stuart DI, Metoz F. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics. 1999;15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]