

Summary
Glucose transporter 4 (GLUT4) is the major insulin regulated glucose transporter expressed mainly in muscle and adipose tissue. GLUT4 is stored in a poorly characterized intracellular vesicular compartment and translocates to the cell surface in response to insulin stimulation resulting in increase glucose uptake. This process is essential for the maintenance of normal glucose homeostasis and involves a complex interplay of trafficking events and intracellular signaling cascades. Recent studies have identified sortilin as an essential element for the formation of GLUT4 storage vesicles during adipogenesis and GGA (Golgi-localized γ-ear-containing Arf-binding protein) as a key coat adaptor for the entry of newly synthesized GLUT4 into the specialized compartment. Insulin-stimulated GLUT4 translocation from this compartment to the plasma membrane appears to require the Akt/protein kinase B substrate, termed AS160 (Akt Substrate of 160 kDa). In addition, the VPS9 domain-containing protein Gapex-5 in complex with CIP4 appears to function as a Rab31 guanylnucleotide exchange factor that is necessary for insulin-stimulated GLUT4 translocation. Here we attempt to summaries recent advances in GLUT4 vesicle biogenesis, intracellular trafficking and membrane fusion.
Keywords: GLUT4, translocation, insulin signaling, exocytosis, endocytosis
Introduction
GLUT4, the major insulin-responsive glucose transporter, belongs to a large family of 12 membrane spanning domain proteins called the facilitative glucose transporter proteins. GLUT4 is highly expressed in striated muscle and adipose tissue, and is responsible for the postprandial removal of glucose from the circulation [1-3]. In the basal state, GLUT4 undergoes a slow but continuous recycling between the plasma membrane and several intracellular compartments, with only 5% of the total GLUT4 protein pool localized to the plasma membrane. In response to acute insulin stimulation (2-3 min), however, the rate of GLUT4 exocytosis markedly increases concomitant with a small decrease in endocytosis, so that approximately 50% of the GLUT4 protein are relocated to the cell surface [1,4,5]. The rate of exocytosis subsequently decreases following the removal of the insulin signal, and this trafficking system returns back to the basal condition.
Significant achievements have been made to elucidate the mechanisms responsible for insulin-stimulated glucose uptake and new findings have changed our understanding of GLUT4 trafficking over the past years. In the present review, we mainly focus on the past two-year advances of GLUT4 vesicle biogenesis, intracellular trafficking and plasma membrane docking and fusion.
GLUT4 vesicle biogenesis, formation of the insulin-responsive storage compartment
Upon differentiation from fibroblasts, both adipocytes and striated muscle induce the expression of endogenous GLUT4 and dramatically acquire insulin responsiveness and glucose uptake. Small glucose transporter-containing vesicles represent the major insulin-responsive compartments (IRC) in these cell types. Although the molecular identification of these insulin-responsive vesicle storage compartments remains poorly characterized, biochemical data suggest that they can best be classified as enriched for GLUT4, IRAP and VAMP2 while negative for the small vesicle transport marker cellugyrin and endosome markers EEA1 and transferrin receptor [6-8]
Similarly, the mechanisms responsible for the sorting of GLUT4 into the IRC during muscle development and adipogenesis is also poorly understood. Recent studies support a general model of vesicle trafficking that include the recruitment of coat and adaptor proteins that drive the formation of budding compartments to specific sites on donor membranes [9]. In turn, the coat and adaptors associates with the donor membranes through multiple interactions with the cytoplasmic tails of cargo proteins, Arf and phosphatidylinositol phosphates [9]. The adaptor protein AP1 or AP3 (less likely) have been suggested in participating the formation of GLUT4 compartments on intracellular donor membranes [10-13].
Recently, several lines of evidence have indicated a requirement for the Golgi-localized γ-ear-containing Arf-binding proteins (GGA) of coat adaptor proteins in the biosynthetic sorting of GLUT4 from the TGN into the IRC. For example, expression of a dominant-interfering GGA mutant inhibited insulin-stimulated GLUT4 translocation and GST-GGA fusion proteins were found to bind to GLUT4 containing transport vesicles but did not directly bind to GLUT4 itself [9]. Moreover, using IRAP as a surrogate to study GLUT4 translocation, it was demonstrated that siRNA mediated knockdown of GGA prior to GLUT4 expression completely prevented insulin-stimulated translocation [14]. However, if GLUT4 was expressed first and then GGA protein was reduced, then insulin-stimulated GLUT4 translocation was completely unaffected. These data defined GGA function at a processing step, which sort the GLUT4 protein from the Golgi to the IRC, but not in the insulin-stimulated trafficking from the IRC to the plasma membrae (Figure 1).
Figure 1. Insulin signaling pathway regulates GLUT4 intracellular trafficking.
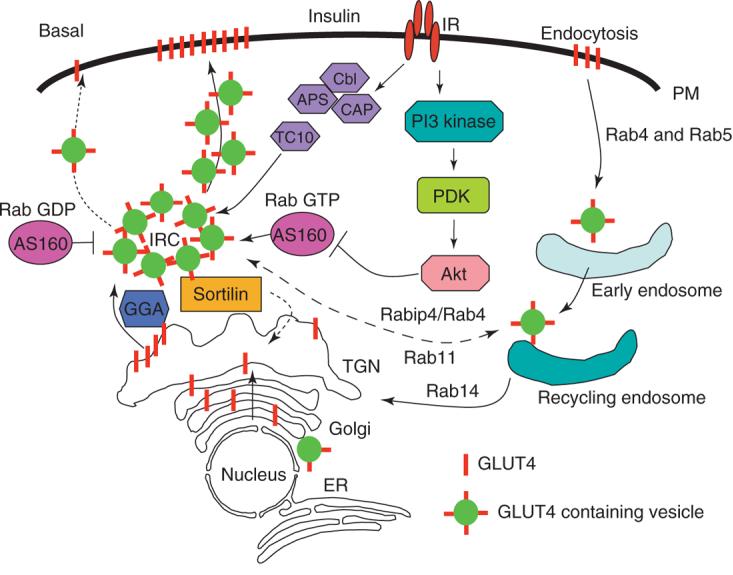
Newly synthesized GLUT4 traffic through a GGA-dependent and trans-Golgi Network (TGN) sorting into the insulin-responsive storage compartment (IRC). In the basal state, GLUT4 is mainly sequestered into the IRC and translocates to the plasma membrane (PM) after insulin stimulation. Exit from the IRC is prevented by AS160 that function to maintain a putative Rab in an inactive state (left). However, following insulin stimulation, AS160 is phosphorylated and inactivated by Akt-dependent phosphorylation (right). Several Rab proteins and effectors that have been implicated endocytosis (Rab4 and 5), recycling back and forth between endosome and TGN (Rabip4/Rab4, Rab11 and 14) are indicated.
Although the cargo-binding domain of GGA (VHS) does not appear to directly associate with GLUT4, there are several other well-established cargo proteins that contain VHS consensus binding motifs (DxxLL) that have shown to directly interact. In particular, sortilin has recently been shown to play an important role in the formation of the specialized GLUT4 IRC in adipocytes [15]. During 3T3L1 differentiation from the fibroblast to adipocyte state, the IRC is formed concomitant with the induction of sortilin expression prior to the expression of GLUT4 [15]. Moreover, when GLUT4 is over-expressed prior to the induction of sortilin, the GLUT4 protein undergoes rapid degradation, whereas over expression of sortilin stabilizes the GLUT4 protein, increases the formation of the IRC and promotes insulin-stimulated glucose uptake [15]. Conversely siRNA-mediated sortilin knockdown decreases both the formation of the IRC and insulin-regulated glucose uptake. In support of a critical role of sortilin in the sorting of GLUT4, chemical cross-linking and yeast two-hybrid studies indicated a direct interaction of the luminal domains of GLUT4 with sortilin [16]. Together, these data suggest a model in which sortilin serves as the cargo adaptor linking GLUT4 to GGA coated transport vesicles that are then sorted to the IRC.
GLUT4 intracellular retention and exocytosis
Despite intensive investigation, there is considerable uncertainty with regard to the precise intracellular localization and trafficking pathways of the GLUT4-containing vesicles in the intracellular vesicular compartment. In part this is because at steady-state GLUT4 is distributed at varying levels throughout most of the endomembrane system [17-21]. Indeed, following insulin-stimulated translocation, GLUT4 is retrieved from the plasma membrane by endocytosis and routed through a complex trafficking itinerary that appears to include endosome compartments and perhaps the trans-Golgi network, before returning to the IRC. This complex itinerary has made it difficult to reliably distinguish between IRC and the general endosome recycling compartments occupied by GLUT4. For example, immunoelectron microscopy has demonstrated the predominant localization of GLUT4 protein (∼60%) in tubulo-vesicular elements beneath the plasma membrane with the remaining protein localized to the trans-Golgi network (TGN), clathrin-coated vesicles and to endosome structures.[18-21]. The content of GLUT4 in all these compartments was found to decrease in response to insulin concomitant with an increased distribution to the plasma membrane.
Since these are primarily static measurements and GLUT4 undergoes continuous dynamic trafficking, several studies took advantage of chemical ablation using HRP-transferrin to selectively inactive the general endosome recycling pathway, that is the endosome compartments containing the transferrin receptor [6,8,17,21]. These studies suggested the presence of two GLUT4 populations: one that overlaps with the transferrin receptor consistent with the endosome recycling compartments and a second separate population that represents the IRC.
With the advent of fluorescent proteins numerous studies have expressed GLUT4-GFP constructs in culture adipocytes. These studies have suggested that insulin-regulated GLUT4 pool is peri-nuclear localized that undergoes enhanced exocytosis and trafficking to the plasma membrane [8,22]. However, more recent studies using Total Internal Reflectance Fluorescent (TIRF) microscopy, suggests that GLUT4 vesicles are continuously trafficking to and from the plasma membrane in an insulin-independent manner [23]. In the presence of insulin, however, these transport vesicles have increased pausing (tethering) at the plasma membrane with a higher probability of plasma membrane fusion. Kinetic analysis and the development of a novel in vitro plasma membrane fusion assay are consistent with GLUT4 vesicle plasma membrane fusion as the key insulin-regulated step in the translocation process [24,25].
GLUT4 vesicle docking and plasma membrane fusion
In several systems, the actin cytoskeleton is required for the trafficking of transport vesicles to specific docking sites at the plasma membrane[23,27]. In the case of GLUT4, myosin Ic appears to be function as the essential actin motor driving GLUT4 vesicle to their docking sites [28]. However, our general knowledge of the vesicle docking, tethering and fusion process itself is poorly understood process (Figure 2). The docking of GLUT4 vesicles with the plasma membrane appears to require the Exocyst complex that is formed from the assembly of 8 distinct proteins [29-32]. In the case of GLUT4, the Exo70, Sec6 and Sec8 protein component of this complex redistribute to the plasma membrane in response to insulin [29] These events are triggered by the insulin activation of a small GTP binding protein TC10 that recruits Exo70 with Sec6 and Sec8 to lipid raft microdomains in the plasma membrane [33,34]. Moreover, Sec8 associates with the PDZ domain of SAP97 a MAGUK (membrane-associated guanylate kinase) family member that is recruited along with Sec8 [33]. Interestingly, expression of a dominant-interfering Exo70 protein or siRNA-mediated knockdowns did not prevent GLUT4 vesicle trafficking to the plasma membrane but did inhibit GLUT4 plasma membrane fusion whereas over expression of Sec6 and Sec8 enhanced GLUT4 translocation to the plasma membrane[33,35]. Although these data indicate that the Exocyst complex plays an important role in the insulin-regulated plasma membrane docking/tethering of GLUT4 vesicles, it is well established the physiologic minimal fusion machinery is dependent upon the assembly of the SNARE complex [36-38]
Figure 2. GLUT4 vesicle docking, tethering and fusion.
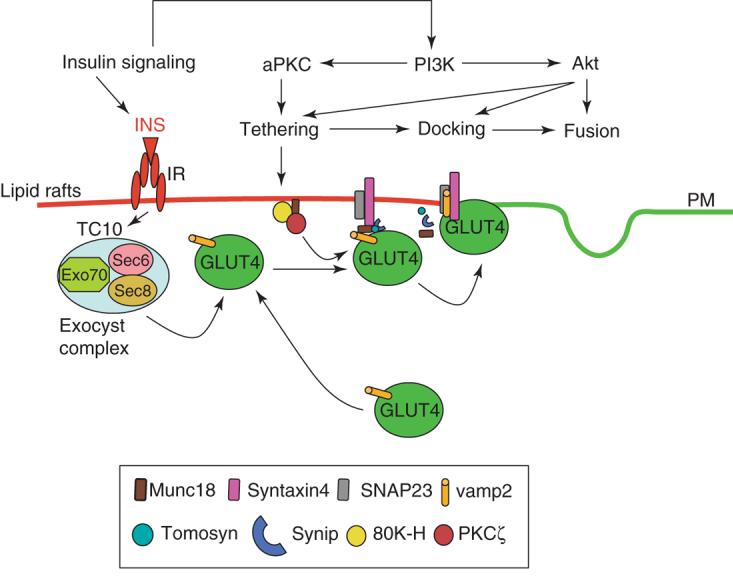
This schematic model shown that the docking of GLUT4 vesicles with the plasma membrane requires the Exocyst complex that is formed from the assembly of 8 distinct proteins. Insulin activates a small GTP binding protein TC10 that recruits Exo70 with Sec6 and Sec8 to lipid raft microdomains in the plasma membrane. The SNARE dependent fusion involves VAMP2, Syntaxin4 and SNAP23. Several syntaxin 4 binding partners have been identified including one member of the Munc18 family (Munc18c), Synip and Tomosyn. Munc18 proteins induces a closed conformational state of syntaxin thereby rendering it unable to bind to VAMP or SNAP. More over, insulin activation of PKC induces the formation of a complex with 80K-H and Munc18c to promote VAMP2 binding to syntaxin 4 that enhances GLUT4 fusion with the plasma membrane.
SNARE proteins can be generally classified as v-SNAREs (or R-SNAREs) that possess a single coil-coiled domain and a transmembrane domain that are localized in vesicle compartment [39-42]. The second family, t-SNAREs (or Q-SNAREs) are dimeric, with one partner belongs to the family of syntaxin isoforms and the second partner belonging to the family of SNAP isoform. Syntaxin isoforms possess a single SNARE domain (coiled-coil), a transmembrane domain and a N-terminal extension involved in the binding of regulator factors. The SNAP isoforms possess 2 coiled-coil domains and are usually link to the plasma membrane by palmitoylation. These t-SNAREs are localized on the acceptor or target compartment [24,43,44]. The assembly of a four helical bundle composed of one helix from the v-SNARE and three helices from the t-SNAREs provide the minimal machinery to drive vesicle fusion both in reconstituted and cellular systems [24,25].
The use of various proteolytic toxins and dominant-interfering mutants has demonstrated that VAMP2 is the predominant v-SNARE required for insulin-stimulated GLUT4 translocation [45,46]. Interestingly, although VAMP2 is essential for insulin-stimulated GLUT4 translocation is completely dispensable for other signals inducing GLUT4 translocation, such as osmotic shock. In this pathway TI-VAMP (VAMP7) appears to be the primary v-SNARE required, consistent with the presence of at least two-independent GLUT4 storage vesicle populations [26]. The cognate plasma membrane t-SNAREs for insulin-stimulated GLUT4 translocation appears to be the syntaxin 4 and the SNAP23 isoforms, based upon antibody blocking studies and expression of dominant-interfering mutants and blocking peptides [47-49]. However, involvement of these t-SNAREs has not been as extensively investigated and further studies are still required to determine the specific function steps of these isoforms in the translocation process. In any case, since the Exocyst complex itself is non-fusogenic some mechanism also must be present to convert the docked GLUT4 vesicles (Exocyst complex) to that of a SNARE complex capable of inducing the fusion reaction.
To address this issue, several investigators have been attempting to identify regulatory partners that display insulin-dependence. Several syntaxin 4 binding partners have been identified including one member of the Munc18 family (Munc18c), Synip and Tomosyn [47,50,51]. Although the potential role of Tomosyn has not been well studied, the evidence supporting a critical role for either Munc18c and/or Synip has remained controversial. Munc18c was originally identified as one of three mammalian isoforms of the sec1 protein in yeast [52]. In all systems to date, over expression of the appropriate Munc18 protein isoform was found to inhibit membrane vesicle fusion, suggesting that this protein function as a fusogenic inhibitor [48,53-56]. Structural studies indicated that the Munc18 proteins induces a closed conformational state of syntaxin thereby rendering it unable to bind to VAMP or SNAP consistent with in vitro binding assays [57,58]. However, genetic studies in yeast, worms, flies and in the mammalian nervous system have also found that the loss of Munc18 expression also prevents vesicle fusion suggesting that Munc18 proteins play a required positive fusogenic role [56,59]. In this regard, recent in vitro studies have suggested that the Munc18 proteins function to anchor VAMP to syntaxin thereby promoting fusion if added during the fusion reaction [58].
To address the in vivo role of Munc18c in GLUT4 translocation, two laboratories have generated conventional Munc18c knockout mice. One study found that homozygotic knockouts of Munc18c results in early embryonic lethality whereas that the heterozygotic mice had no significant phenotype on a normal diet but displayed mild glucose intolerance on a high fat diet [60]. These data are consistent with studies of neural Munc18 knockouts and that of lower organisms [61,62]. However another study, found that homozygous Munc18c knockout mice were viable and had increased insulin sensitivity consistent with Munc18c actin as a fusogenic inhibitor [63]. Insulin has also been observed to induce the association of PKC and Munc18c concomitant with a dissociation of the syntaxin4/Munc18c complex [50]. More over, insulin triggers PKC forms a complex with 80K-H and munc18c to promote VAMP2 binding to syntaxin4 that enhanced the GLUT4 fusion with the plasma membrane.[64]. These findings are consistent with studies suggesting that PKC is an important component of insulin signaling pathways leading to GLUt4 translocation [65,66]. At present, the basis for the divergent genetic, biochemical and cell biological data concerning the functional role of the Munc18 proteins and particularly Munc18c in GLUT4 trafficking remains an open question.
Another potential syntaxin4 binding regulatory factor, Synip has also been observed to prevent the formation of a ternary SNARE complex by inhibiting the binding of VAMP2 and SNAP23 to syntaxin4 [67]. Further studies have suggested that insulin induces a specific Akt 2 dependent Synip phosphorylation on serine 99 is required for the dissociation of Synip from syntaxin4 and thereby allowing for the assembly of the ternary SNARE-complex [51]. However, this model has been recently challenged as another group has reported that mutation of serine 99 had no significant effect on insulin-stimulated GLUT4 translocation [68]. As with Munc18c, the basis for these different results is not readily apparent and further studies are necessary to determine the specific regulatory steps controlling GLUT4 vesicle fusion with the plasma membrane.
Insulin signaling leading to GLUT4 translocation
It is well established that insulin stimulation initiates a cascade of signaling events that regulates a complex set of anabolic processes including glucose uptake, lipogenesis, glycogen, protein and DNA synthesis. The initiation step is the tyrosine autophosphorylation and substrate kinase activation of the insulin receptor itself following insulin binding (Figure 1). This occurs through an intramolecular trans-autophosphorylation mechanism in which one β subunit tyrosine kinase domain phosphorylates the adjacent β subunit resulting in the activation of the intrinsic substrate kinase activity of the insulin receptor [69,70]. The most studied immediate downstream substrate target of the insulin receptor tyrosine kinase is the insulin receptor substrate (IRS) family members', IRS1 and IRS2 [71]. Tyrosine phosphorylation of these substrates generates multiple phosphotyrosine docking sites for a variety of effectors, in particular the p85 regulatory subunits of the Type 1A phosphatidylinositol 3-kinase (PI3K). The primary product of this lipid kinase is phosphatidylinositol-3,4,5-trisphosphate (PI(3,4,5)P3) that is formed at the plasma membrane. In turn, PI(3,4,5)P3 functions to recruit and acivate atypical protein kinase C isoforms (PKCζ/λ) and the Akt protein kinases (sometimes referred to as (PKB) protein kinase B). Akt activation occurs through phosphorylation on threonine 308 by phosphoinositide-dependent protein kinase 1 (PDK1) and serine 473 by PDK2 [71-73]. Although there are three Akt isoforms, Akt2 function appears to be specifically required for insulin-stimulated GLUT4 translocation [73-76]. Akt can phosphorylate numerous downstream target proteins, including Synip as previously described. However, recently another Akt target that has received substantial attention is a Rab-GTPase-activating protein (GAP) of 160 kDa, termed AS160 [77]. AS160 appears to be active GAP for Rab2, Rab8, Rab10 and/or Rab14 [78], but when phosphorylated by Akt the GAP activity is inhibited. Similarly, siRNA mediated knockdowns of AS160 result in a basal state translocation of GLUT4 to the cell surface [79]. Thus, it has been proposed that insulin induces phosphorylation of AS160, resulting in the inhibition of GAP activity, thereby converting a putative Rab protein to the active GTP-bound form. The active Rab would then promote vesicle trafficking in an as of yet undefined manner. Alternatively, it has also been proposed that AS160 directly associates with another IRC cargo protein IRAP, and that insulin-dependent Akt phosphorylation results in the dissociation of AS160 from GLUT4 compartment and would also lead to Rab activation [80]. Although these studies have suggested that AS160 serves as the insulin-regulated rate-limiting step in GLUT4 translocation, both an in vitro plasma membrane reconstitution fusion system and in vivo TIRF microscopy measurements indicate that the critical site of insulin regulation is the plasma membrane fusion process downstream of AS160 [23,25].
In addition to IRS proteins serving as important targets to scaffold various signaling effectors, the insulin receptor is also well established to induce the tyrosine phosphorylation of the Cbl proteins [81]. Cbl proteins containing several protein modulatory regions and most notably a ring finger domain that functions as an E3 ligase in the ubiquitation and rapid down-regulation of various receptors including the EGF, PDGF, T and B cell receptors [82-85]. However, the insulin receptor does not undergo a rapid down-regulation process following ligand engagement in contrast to these other receptors. In this case, Cbl phosphorylation has been suggested to provide a pathway for the activation of a small GTP binding protein TC10 that is localized to lipid raft microdomains [86]. Using a constitutively active TC10 protein as bait, several interacting partners were identified including Exo70, as previously described, and CIP4/2 [35,87]. CIP4/2 (Cdc42-interacting protein 4/2) is a modular domain protein containing an FCH domain, which has been recently recognized as part of a larger structural domain called EFC or F-BAR. This domain is similar to the BAR domain that is involved in regulating membrane curvature [88,89]. In addition, CIP4/2 also contain two coiled-coil domains, and an SH3 domain that is recruited to the plasma membrane in response to insulin activation of TC10. Expression of a dominant-interfering CIP4/2 mutant inhibited insulin-stimulated GLUT4 translocation. Further studies using CIP4 as bait, identified Gapex-5, a RasGAP and VPS9 domain-containing protein that functions as a guanine nucleotide exchange factor (GEF) for Rab31, a Rab5 subfamily GTPase that involved in trans-Golgi network (TGN)-to-endosome trafficking [34]. Insulin recruited the CIP4/Gapex-5 complex to the plasma membrane, thereby decreasing Rab31 activity. These data suggest that Rab31 may function in the dynamic retention of GLUT4 vesicles and the inhibition of Rab31 activity allows for release of this GLUT4 vesicles population.
More recently, several investigators have been to elucidate another regulatory aspect of membrane fusion events that are dependent on the lipid composition of donor and acceptor membranes. For example, product of phospholipase D (PLD), phosphatidic acid (PA) appears to play an important role as diminished PA production was found to inhibit GLUT4 plasma membrane fusion without affecting trafficking or docking/tethering [90]. Moreover, in vitro reconstitution studies demonstrated that PA in the syntaxin4/SNAP23 acceptor membrane markedly accelerated the rate of fusion whereas PA in the VAMP2 donor vesicles was inhibitory [24]. As it is well established that insulin alters the lipid composition of various intracellular membranes, further studies are still need to address the specific roles of the donor and acceptor lipid membrane environments as the potential target events responsible for the control of GLUT4 vesicle plasma membrane fusion.
GLUT4 endocytosis
In all trafficking systems, the rate of anterograde traffic is balanced by retrograde traffic to achieve a dynamic equilibrium. As outlined above, activation of the insulin receptor triggers a large increase in the rate of GLUT4 vesicle plasma membrane exocytosis, that is anterograde transport. The insulin-mediated increase in exocytosis is probably the major step for GLUT4 translocation, since a complete inhibition of GLUT4 endocytosis (retrograde transport from the plasma membrane) results in a very slow time-dependent increase in plasma membrane associated GLUT4 protein without affecting the extent of insulin-stimulated GLUT4 translocation at normal physiologic response times [91]. Nevertheless, insulin does appear to induce a smaller decrease in the rate of plasma membrane internalization by endocytosis [92-95]. Similar to most proteins, GLUT4 endocytosis primarily occurs through a clathrin-coated pit, dynamin-dependent mechanism [96,97]. However, caeveolin-enriched lipid raft microdomains may also play a role in this process. For example, disruption of caveolin organization by expression of dominant-interfering mutants partially inhibits GLUT4 endocytosis [98]. More recently, the dependence of caveolin versus clathrin-dependent endocytosis appears to be different in the basal versus insulin-stimulated state [99].
Once GLUT4 internalized, GLUT4 must necessarily undergo multiple sorting steps en route back to the IRC. Immediately following internalization, GLUT4 co-localizes with the early endosome antigen marker EEA1 and the transferrin receptor [100,101]. Subsequently GLUT4 becomes sequestered from these compartments and equilibrates with the remaining GLUT4 protein in the IRC [38]. It has also been suggested that GLUT4 may not necessary directly traffic back to the IRC but may re-enter into the Golgi/TGN secretory system and then undergo sorting to the IRC [102]. Although the sorting proteins involved in these processes have only been poorly defined, the adaptor protein 2 (AP2) complex is required for the plasma membrane sorting into clathrin-coated pits [103,104]. In particular, Rab5 has been reported to function in the initial trafficking of GLUT4 from small early endocytotic vesicles into the early endosome recycling system through dynein and microtubule-dependent process [105]. Similarly, the small GTPases Rab4 and the Rab4 interacting protein Rabip4 have been implicated in endocytotic GLUT4 sorting back to the IRC as disrupting Rabip4 function prevents internalized GLUT4 from populating the IRC [106].
Concluding remarks
The biosynthetic sorting, intracellular trafficking and insulin regulatory pathways controlling glucose uptake in skeletal muscle and adipose tissues remain a very complicated puzzle that as inspired numerous studies over the past several decades. As GLUT4 translocation is the essential step in post-prandial glucose clearance, defects in this pathway results in states of impaired glucose tolerance and insulin resistance. In this brief review, we have highlighted several of the new findings but also the complexities, inconsistency and controversies that are continually evolving around the mechanisms accounting for GLUT4 trafficking and glucose uptake. Although substantial progress has been made in several of these areas there remains a remarkable amount that remains to be learned. Whether or not developing experimental or therapeutic strategies that increase glucose uptake by increasing GLUT4 at the cell surface is a viable approach, understanding the intricacies of insulin regulated trafficking will have numerous applications for other cell biological processes.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bryant NJ, Govers R, James DE. Regulated transport of the glucose transporter GLUT4. Nat Rev Mol Cell Biol. 2002;3:267–277. doi: 10.1038/nrm782. [DOI] [PubMed] [Google Scholar]
- 2.Ducluzeau PH, Fletcher LM, Vidal H, Laville M, Tavare JM. Molecular mechanisms of insulin-stimulated glucose uptake in adipocytes. Diabetes Metab. 2002;28:85–92. [PubMed] [Google Scholar]
- 3.Watson RT, Kanzaki M, Pessin JE. Regulated membrane trafficking of the insulin-responsive glucose transporter 4 in adipocytes. Endocr Rev. 2004;25:177–204. doi: 10.1210/er.2003-0011. [DOI] [PubMed] [Google Scholar]
- 4.Holman GD, Sandoval IV. Moving the insulin-regulated glucose transporter GLUT4 into and out of storage. Trends Cell Biol. 2001;11:173–179. doi: 10.1016/s0962-8924(01)01953-5. [DOI] [PubMed] [Google Scholar]
- 5.Pessin JE, Thurmond DC, Elmendorf JS, Coker KJ, Okada S. Molecular basis of insulin-stimulated GLUT4 vesicle trafficking. Location! Location! Location! J Biol Chem. 1999;274:2593–2596. doi: 10.1074/jbc.274.5.2593. [DOI] [PubMed] [Google Scholar]
- 6.Martin S, Tellam J, Livingstone C, Slot JW, Gould GW, James DE. The glucose transporter (GLUT-4) and vesicle-associated membrane protein-2 (VAMP-2) are segregated from recycling endosomes in insulin-sensitive cells. J Cell Biol. 1996;134:625–635. doi: 10.1083/jcb.134.3.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hah JS, Ryu JW, Lee W, Kim BS, Lachaal M, Spangler RA, Jung CY. Transient changes in four GLUT4 compartments in rat adipocytes during the transition, insulin-stimulated to basal: implications for the GLUT4 trafficking pathway. Biochemistry. 2002;41:14364–14371. doi: 10.1021/bi026474n. [DOI] [PubMed] [Google Scholar]
- 8.Zeigerer A, Lampson MA, Karylowski O, Sabatini DD, Adesnik M, Ren M, McGraw TE. GLUT4 Retention in Adipocytes Requires Two Intracellular Insulin- regulated Transport Steps. Mol Biol Cell. 2002;13:2421–2435. doi: 10.1091/mbc.E02-02-0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li LV, Kandror KV. Golgi-localized, gamma-ear-containing, Arf-binding protein adaptors mediate insulin-responsive trafficking of glucose transporter 4 in 3T3-L1 adipocytes. Mol Endocrinol. 2005;19:2145–2153. doi: 10.1210/me.2005-0032. [DOI] [PubMed] [Google Scholar]
- 10.Marsh BJ, Martin S, Melvin DR, Martin LB, Alm RA, Gould GW, James DE. Mutational analysis of the carboxy-terminal phosphorylation site of GLUT-4 in 3T3-L1 adipocytes. Am J Physiol. 1998;275:E412–422. doi: 10.1152/ajpendo.1998.275.3.E412. [DOI] [PubMed] [Google Scholar]
- 11.Gillingham AK, Koumanov F, Pryor PR, Reaves BJ, Holman GD. Association of AP1 adaptor complexes with GLUT4 vesicles. J Cell Sci. 1999;112:4793–4800. doi: 10.1242/jcs.112.24.4793. [DOI] [PubMed] [Google Scholar]
- 12.Al-Hasani H, Kunamneni RK, Dawson K, Hinck CS, Muller-Wieland D, Cushman SW. Roles of the N- and C-termini of GLUT4 in endocytosis. J Cell Sci. 2002;115:131–140. doi: 10.1242/jcs.115.1.131. [DOI] [PubMed] [Google Scholar]
- 13.Martin S, Ramm G, Lyttle CT, Meerloo T, Stoorvogel W, James DE. Biogenesis of insulin-responsive GLUT4 vesicles is independent of brefeldin A-sensitive trafficking. Traffic. 2000;1:652–660. doi: 10.1034/j.1600-0854.2000.010809.x. [DOI] [PubMed] [Google Scholar]
- 14.Hou JC, Suzuki N, Pessin JE, Watson RT. A Specific Dileucine Motif Is Required for the GGA-dependent Entry of Newly Synthesized Insulin-responsive Aminopeptidase into the Insulin-responsive Compartment. J Biol Chem. 2006;281:33457–33466. doi: 10.1074/jbc.M601583200. [DOI] [PubMed] [Google Scholar]
- 15.Shi J, Kandror KV. Sortilin is essential and sufficient for the formation of Glut4 storage vesicles in 3T3-L1 adipocytes. Dev Cell. 2005;9:99–108. doi: 10.1016/j.devcel.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 16.Shi J, Kandror KV. The luminal Vps10p domain of sortilin plays the predominant role in targeting to insulin-responsive Glut4-containing vesicles. J Biol Chem. 2007 doi: 10.1074/jbc.M608971200. [DOI] [PubMed] [Google Scholar]
- 17.Lim SN, Bonzelius F, Low SH, Wille H, Weimbs T, Herman GA. Identification of discrete classes of endosome-derived small vesicles as a major cellular pool for recycling membrane proteins. Mol Biol Cell. 2001;12:981–995. doi: 10.1091/mbc.12.4.981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Piper RC, Hess LJ, James DE. Differential sorting of two glucose transporters expressed in insulin-sensitive cells. Am J Physiol. 1991;260:C570–C580. doi: 10.1152/ajpcell.1991.260.3.C570. [DOI] [PubMed] [Google Scholar]
- 19.Slot JW, Geuze HJ, Gigengack S, Lienhard GE, James DE. Immuno-localization of the insulin regulatable glucose transporter in brown adipose tissue of the rat. J Cell Biol. 1991;113:123–135. doi: 10.1083/jcb.113.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marette A, Burdett E, Douen A, Vranic M, Klip A. Insulin induces the translocation of GLUT4 from a unique intracellular organelle to transverse tubules in rat skeletal muscle. Diabetes. 1992;41:1562–1569. doi: 10.2337/diab.41.12.1562. [DOI] [PubMed] [Google Scholar]
- 21.Coster AC, Govers R, James DE. Insulin stimulates the entry of GLUT4 into the endosomal recycling pathway by a quantal mechanism. Traffic. 2004;5:763–771. doi: 10.1111/j.1600-0854.2004.00218.x. [DOI] [PubMed] [Google Scholar]
- 22.Karylowski O, Zeigerer A, Cohen A, McGraw TE. GLUT4 is retained by an intracellular cycle of vesicle formation and fusion with endosomes. Mol Biol Cell. 2004;15:870–882. doi: 10.1091/mbc.E03-07-0517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lizunov VA, Matsumoto H, Zimmerberg J, Cushman SW, Frolov VA. Insulin stimulates the halting, tethering, and fusion of mobile GLUT4 vesicles in rat adipose cells. J Cell Biol. 2005;169:481–489. doi: 10.1083/jcb.200412069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vicogne J, Vollenweider D, Smith JR, Huang P, Frohman MA, Pessin JE. Asymmetric phospholipid distribution drives in vitro reconstituted SNARE-dependent membrane fusion. Proc Natl Acad Sci U S A. 2006;103:14761–14766. doi: 10.1073/pnas.0606881103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koumanov F, Jin B, Yang J, Holman GD. Insulin signaling meets vesicle traffic of GLUT4 at a plasma-membrane-activated fusion step. Cell Metab. 2005;2:179–189. doi: 10.1016/j.cmet.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 26.Randhawa VK, Thong FS, Lim DY, Li D, Garg RR, Rudge R, Galli T, Rudich A, Klip A. Insulin and hypertonicity recruit GLUT4 to the plasma membrane of muscle cells by using N-ethylmaleimide-sensitive factor-dependent SNARE mechanisms but different v-SNAREs: role of TI-VAMP. Mol Biol Cell. 2004;15:5565–5573. doi: 10.1091/mbc.E04-03-0266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu L, Jedrychowski MP, Gygi S, Pilch PF. Role of insulin-dependent cortical fodrin/spectrin remodeling in glucose transporter 4 translocation in rat adipocytes. Mol Biol Cell. 2006;17:4249–4256. doi: 10.1091/mbc.E06-04-0278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bose A, Robida S, Furcinitti PS, Chawla A, Fogarty K, Corvera S, Czech MP. Unconventional myosin Myo1c promotes membrane fusion in a regulated exocytic pathway. Mol Cell Biol. 2004;24:5447–5458. doi: 10.1128/MCB.24.12.5447-5458.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ewart MA, Clarke M, Kane S, Chamberlain LH, Gould GW. Evidence for a role of the exocyst in insulin-stimulated Glut4 trafficking in 3T3-L1 adipocytes. J Biol Chem. 2005;280:3812–3816. doi: 10.1074/jbc.M409928200. [DOI] [PubMed] [Google Scholar]
- 30.Bowser R, Muller H, Govindan B, Novick P. Sec8p and Sec15p are components of a plasma membrane-associated 19.5S particle that may function downstream of Sec4p to control exocytosis. J Cell Biol. 1992;118:1041–1056. doi: 10.1083/jcb.118.5.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.TerBush DR, Novick P. Sec6, Sec8, and Sec15 are components of a multisubunit complex which localizes to small bud tips in Saccharomyces cerevisiae. J Cell Biol. 1995;130:299–312. doi: 10.1083/jcb.130.2.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.TerBush DR, Maurice T, Roth D, Novick P. The Exocyst is a multiprotein complex required for exocytosis in Saccharomyces cerevisiae. Embo J. 1996;15:6483–6494. [PMC free article] [PubMed] [Google Scholar]
- 33.Inoue M, Chiang SH, Chang L, Chen XW, Saltiel AR. Compartmentalization of the exocyst complex in lipid rafts controls Glut4 vesicle tethering. Mol Biol Cell. 2006;17:2303–2311. doi: 10.1091/mbc.E06-01-0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lodhi IJ, Chiang SH, Chang L, Vollenweider D, Watson RT, Inoue M, Pessin JE, Saltiel AR. Gapex-5, a Rab31 guanine nucleotide exchange factor that regulates Glut4 trafficking in adipocytes. Cell Metab. 2007;5:59–72. doi: 10.1016/j.cmet.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Inoue M, Chang L, Hwang J, Chiang SH, Saltiel AR. The exocyst complex is required for targeting of Glut4 to the plasma membrane by insulin. Nature. 2003;422:629–633. doi: 10.1038/nature01533. [DOI] [PubMed] [Google Scholar]
- 36.Cheatham B, Volchuk A, Kahn CR, Wang L, Rhodes CJ, Klip A. Insulin-stimulated translocation of GLUT4 glucose transporters requires SNARE-complex proteins. Proc Natl Acad Sci USA. 1996;93:15169–15173. doi: 10.1073/pnas.93.26.15169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Foran PG, Fletcher LM, Oatey PB, Mohammed N, Dolly JO, Tavare JM. Protein kinase B stimulates the translocation of GLUT4 but not GLUT1 or transferrin receptors in 3T3-L1 adipocytes by a pathway involving SNAP- 23, synaptobrevin-2, and/or cellubrevin. J Biol Chem. 1999;274:28087–28095. doi: 10.1074/jbc.274.40.28087. [DOI] [PubMed] [Google Scholar]
- 38.Bryant NJ, Govers R, James DE. Regulated transport of the glucose transporter GLUT4. Nat Rev Mol Cell Biol. 2002;3:267–277. doi: 10.1038/nrm782. [DOI] [PubMed] [Google Scholar]
- 39.Hong W. SNAREs and traffic. Biochim Biophys Acta. 2005;1744:493–517. [PubMed] [Google Scholar]
- 40.Bonifacino JS, Glick BS. The mechanisms of vesicle budding and fusion. Cell. 2004;116:153–166. doi: 10.1016/s0092-8674(03)01079-1. [DOI] [PubMed] [Google Scholar]
- 41.Fasshauer D, Sutton RB, Brunger AT, Jahn R. Conserved structural features of the synaptic fusion complex: SNARE proteins reclassified as Q- and R-SNAREs. Proc Natl Acad Sci U S A. 1998;95:15781–15786. doi: 10.1073/pnas.95.26.15781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bock JB, Matern HT, Peden AA, Scheller RH. A genomic perspective on membrane compartment organization. Nature. 2001;409:839–841. doi: 10.1038/35057024. [DOI] [PubMed] [Google Scholar]
- 43.Sutton RB, Fasshauer D, Jahn R, Brunger AT. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature. 1998;395:347–353. doi: 10.1038/26412. [DOI] [PubMed] [Google Scholar]
- 44.Antonin W, Fasshauer D, Becker S, Jahn R, Schneider TR. Crystal structure of the endosomal SNARE complex reveals common structural principles of all SNAREs. Nat Struct Biol. 2002;9:107–111. doi: 10.1038/nsb746. [DOI] [PubMed] [Google Scholar]
- 45.Randhawa VK, Bilan PJ, Khayat ZA, Daneman N, Liu Z, Ramlal T, Volchuk A, Peng XR, Coppola T, Regazzi R, et al. VAMP2, but not VAMP3/cellubrevin, mediates insulin-dependent incorporation of GLUT4 into the plasma membrane of L6 myoblasts. Mol Biol Cell. 2000;11:2403–2417. doi: 10.1091/mbc.11.7.2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Olson AL, Knight JB, Pessin JE. Syntaxin 4, VAMP2, and/or VAMP3/cellubrevin are functional target membrane and vesicle SNAP receptors for insulin-stimulated GLUT4 translocation in adipocytes. Mol Cell Biol. 1997;17:2425–2435. doi: 10.1128/mcb.17.5.2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Widberg CH, Bryant NJ, Girotti M, Rea S, James DE. Tomosyn interacts with the t-SNAREs syntaxin4 and SNAP23 and plays a role in insulin-stimulated GLUT4 translocation. J Biol Chem. 2003;278:35093–35101. doi: 10.1074/jbc.M304261200. [DOI] [PubMed] [Google Scholar]
- 48.Tamori Y, Kawanishi M, Niki T, Shinoda H, Araki S, Okazawa H, Kasuga M. Inhibition of insulin-induced GLUT4 translocation by Munc18c through interaction with syntaxin4 in 3T3-L1 adipocytes. J Biol Chem. 1998;273:19740–19746. doi: 10.1074/jbc.273.31.19740. [DOI] [PubMed] [Google Scholar]
- 49.Tellam JT, Macaulay SL, McIntosh S, Hewish DR, Ward CW, James DE. Characterization of Munc-18c and syntaxin-4 in 3T3-L1 adipocytes. Putative role in insulin-dependent movement of GLUT-4. J Biol Chem. 1997;272:6179–6186. doi: 10.1074/jbc.272.10.6179. [DOI] [PubMed] [Google Scholar]
- 50.Hodgkinson CP, Mander A, Sale GJ. Protein kinase-zeta interacts with munc18c: role in GLUT4 trafficking. Diabetologia. 2005;48:1627–1636. doi: 10.1007/s00125-005-1819-y. [DOI] [PubMed] [Google Scholar]
- 51.Yamada E, Okada S, Saito T, Ohshima K, Sato M, Tsuchiya T, Uehara Y, Shimizu H, Mori M. Akt2 phosphorylates Synip to regulate docking and fusion of GLUT4-containing vesicles. J Cell Biol. 2005;168:921–928. doi: 10.1083/jcb.200408182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Egerton M, Zueco J, Boyd A. Molecular characterization of the SEC1 gene of Saccharomyces cerevisiae: subcellular distribution of a protein required for yeast protein secretion. Yeast. 1993;9:703–713. doi: 10.1002/yea.320090704. [DOI] [PubMed] [Google Scholar]
- 53.Riento K, Galli T, Jansson S, Ehnholm C, Lehtonen E, Olkkonen VM. Interaction of Munc-18-2 with syntaxin 3 controls the association of apical SNAREs in epithelial cells. J Cell Sci. 1998;111(Pt 17):2681–2688. doi: 10.1242/jcs.111.17.2681. [DOI] [PubMed] [Google Scholar]
- 54.Latham CF, Lopez JA, Hu SH, Gee CL, Westbury E, Blair DH, Armishaw CJ, Alewood PF, Bryant NJ, James DE, et al. Molecular dissection of the Munc18c/syntaxin4 interaction: implications for regulation of membrane trafficking. Traffic. 2006;7:1408–1419. doi: 10.1111/j.1600-0854.2006.00474.x. [DOI] [PubMed] [Google Scholar]
- 55.Thurmond DC, Ceresa BP, Okada S, Elmendorf JS, Coker K, Pessin JE. Regulation of insulin-stimulated GLUT4 translocation by munc18c in 3T3L1 adipocytes. J Biol Chem. 1998;273:33876–33883. doi: 10.1074/jbc.273.50.33876. [DOI] [PubMed] [Google Scholar]
- 56.Toonen RF, Verhage M. Vesicle trafficking: pleasure and pain from SM genes. Trends Cell Biol. 2003;13:177–186. doi: 10.1016/s0962-8924(03)00031-x. [DOI] [PubMed] [Google Scholar]
- 57.Misura KM, Scheller RH, Weis WI. Three-dimensional structure of the neuronal-Sec1-syntaxin 1a complex. Nature. 2000;404:355–362. doi: 10.1038/35006120. [DOI] [PubMed] [Google Scholar]
- 58.Shen J, Tareste DC, Paumet F, Rothman JE, Melia TJ. Selective activation of cognate SNAREpins by Sec1/Munc18 proteins. Cell. 2007;128:183–195. doi: 10.1016/j.cell.2006.12.016. [DOI] [PubMed] [Google Scholar]
- 59.Oh E, Thurmond DC. The stimulus-induced tyrosine phosphorylation of Munc18c facilitates vesicle exocytosis. J Biol Chem. 2006;281:17624–17634. doi: 10.1074/jbc.M601581200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Oh E, Spurlin BA, Pessin JE, Thurmond DC. Munc18c heterozygous knockout mice display increased susceptibility for severe glucose intolerance. Diabetes. 2005;54:638–647. doi: 10.2337/diabetes.54.3.638. [DOI] [PubMed] [Google Scholar]
- 61.Weimer RM, Richmond JE, Davis WS, Hadwiger G, Nonet ML, Jorgensen EM. Defects in synaptic vesicle docking in unc-18 mutants. Nat Neurosci. 2003;6:1023–1030. doi: 10.1038/nn1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Heeroma JH, Roelandse M, Wierda K, van Aerde KI, Toonen RF, Hensbroek RA, Brussaard A, Matus A, Verhage M. Trophic support delays but does not prevent cell-intrinsic degeneration of neurons deficient for munc18-1. Eur J Neurosci. 2004;20:623–634. doi: 10.1111/j.1460-9568.2004.03503.x. [DOI] [PubMed] [Google Scholar]
- 63.Kanda H, Tamori Y, Shinoda H, Yoshikawa M, Sakaue M, Udagawa J, Otani H, Tashiro F, Miyazaki J, Kasuga M. Adipocytes from Munc18c-null mice show increased sensitivity to insulin-stimulated GLUT4 externalization. J Clin Invest. 2005;115:291–301. doi: 10.1172/JCI22681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hodgkinson CP, Mander A, Sale GJ. Identification of 80K-H as a protein involved in GLUT4 vesicle trafficking. Biochem J. 2005;388:785–793. doi: 10.1042/BJ20041845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu LZ, Zhao HL, Zuo J, Ho SK, Chan JC, Meng Y, Fang FD, Tong PC. Protein kinase Czeta mediates insulin-induced glucose transport through actin remodeling in L6 muscle cells. Mol Biol Cell. 2006;17:2322–2330. doi: 10.1091/mbc.E05-10-0969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kanzaki M, Mora S, Hwang JB, Saltiel AR, Pessin JE. Atypical protein kinase C (PKCzeta/lambda) is a convergent downstream target of the insulin-stimulated phosphatidylinositol 3-kinase and TC10 signaling pathways. J Cell Biol. 2004;164:279–290. doi: 10.1083/jcb.200306152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Min J, Okada S, Coker K, Ceresa BP, Elmendorf JS, Syu L-J, Noda Y, Saltiel AR, Pessin JE. Synip: A novel insulin-regulated syntaxin 4 binding protein mediating GLUT4 translocation in adipocytes. Mol Cell. 1999;3:751–760. doi: 10.1016/s1097-2765(01)80007-1. [DOI] [PubMed] [Google Scholar]
- 68.Sano H, Kane S, Sano E, Lienhard GE. Synip phosphorylation does not regulate insulin-stimulated GLUT4 translocation. Biochem Biophys Res Commun. 2005;332:880–884. doi: 10.1016/j.bbrc.2005.05.027. [DOI] [PubMed] [Google Scholar]
- 69.Ullrich A, Schlessinger J. Signal transduction by receptors with tyrosine kinase activity. Cell. 1990;61:203–212. doi: 10.1016/0092-8674(90)90801-k. [DOI] [PubMed] [Google Scholar]
- 70.Czech MP, Corvera S. Signaling mechanisms that regulate glucose transport. J Biol Chem. 1999;274:1865–1868. doi: 10.1074/jbc.274.4.1865. [DOI] [PubMed] [Google Scholar]
- 71.White MF. IRS proteins and the common path to diabetes. Am J Physiol Endocrinol Metab. 2002;283:E413–422. doi: 10.1152/ajpendo.00514.2001. [DOI] [PubMed] [Google Scholar]
- 72.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 73.Welsh GI, Hers I, Berwick DC, Dell G, Wherlock M, Birkin R, Leney S, Tavare JM. Role of protein kinase B in insulin-regulated glucose uptake. Biochem Soc Trans. 2005;33:346–349. doi: 10.1042/BST0330346. [DOI] [PubMed] [Google Scholar]
- 74.Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw EB, 3rd, Kaestner KH, Bartolomei MS, Shulman GI, Birnbaum MJ. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta) Science. 2001;292:1728–1731. doi: 10.1126/science.292.5522.1728. [DOI] [PubMed] [Google Scholar]
- 75.Jiang ZY, Zhou QL, Coleman KA, Chouinard M, Boese Q, Czech MP. Insulin signaling through Akt/protein kinase B analyzed by small interfering RNA-mediated gene silencing. Proc Natl Acad Sci U S A. 2003;100:7569–7574. doi: 10.1073/pnas.1332633100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.George S, Rochford JJ, Wolfrum C, Gray SL, Schinner S, Wilson JC, Soos MA, Murgatroyd PR, Williams RM, Acerini CL, et al. A family with severe insulin resistance and diabetes due to a mutation in AKT2. Science. 2004;304:1325–1328. doi: 10.1126/science.1096706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sano H, Kane S, Sano E, Miinea CP, Asara JM, Lane WS, Garner CW, Lienhard GE. Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J Biol Chem. 2003;278:14599–14602. doi: 10.1074/jbc.C300063200. [DOI] [PubMed] [Google Scholar]
- 78.Miinea CP, Sano H, Kane S, Sano E, Fukuda M, Peranen J, Lane WS, Lienhard GE. AS160, the Akt substrate regulating GLUT4 translocation, has a functional Rab GTPase-activating protein domain. Biochem J. 2005;391:87–93. doi: 10.1042/BJ20050887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Eguez L, Lee A, Chavez JA, Miinea CP, Kane S, Lienhard GE, McGraw TE. Full intracellular retention of GLUT4 requires AS160 Rab GTPase activating protein. Cell Metab. 2005;2:263–272. doi: 10.1016/j.cmet.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 80.Peck GR, Ye S, Pham V, Fernando RN, Macaulay SL, Chai SY, Albiston AL. Interaction of the Akt substrate, AS160, with the glucose transporter 4 vesicle marker protein, insulin-regulated aminopeptidase. Mol Endocrinol. 2006;20:2576–2583. doi: 10.1210/me.2005-0476. [DOI] [PubMed] [Google Scholar]
- 81.Ribon V, Saltiel AR. Insulin stimulates tyrosine phosphorylation of the proto-oncogene product of c-Cbl in 3T3-L1 adipocytes. Biochem J. 1997;324:839–845. doi: 10.1042/bj3240839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Molero JC, Turner N, Thien CB, Langdon WY, James DE, Cooney GJ. Genetic ablation of the c-Cbl ubiquitin ligase domain results in increased energy expenditure and improved insulin action. Diabetes. 2006;55:3411–3417. doi: 10.2337/db06-0955. [DOI] [PubMed] [Google Scholar]
- 83.Vantler M, Huntgeburth M, Caglayan E, Ten Freyhaus H, Schnabel P, Rosenkranz S. PI3-kinase/Akt-dependent antiapoptotic signaling by the PDGF alpha receptor is negatively regulated by Src family kinases. FEBS Lett. 2006;580:6769–6776. doi: 10.1016/j.febslet.2006.11.034. [DOI] [PubMed] [Google Scholar]
- 84.Dragone LL, Myers MD, White C, Gadwal S, Sosinowski T, Gu H, Weiss A. Src-like adaptor protein (SLAP) regulates B cell receptor levels in a c-Cbl-dependent manner. Proc Natl Acad Sci U S A. 2006;103:18202–18207. doi: 10.1073/pnas.0608965103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Iwaki S, Jensen BM, Gilfillan AM. Ntal/Lab/Lat2. Int J Biochem Cell Biol. 2006 doi: 10.1016/j.biocel.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Saltiel AR, Pessin JE. Insulin signaling pathways in time and space. Trends Cell Biol. 2002;12:65–71. doi: 10.1016/s0962-8924(01)02207-3. [DOI] [PubMed] [Google Scholar]
- 87.Chang L, Adams RD, Saltiel AR. The TC10-interacting protein CIP4/2 is required for insulin-stimulated Glut4 translocation in 3T3L1 adipocytes. Proc Natl Acad Sci U S A. 2002;99:12835–12840. doi: 10.1073/pnas.202495599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tsujita K, Suetsugu S, Sasaki N, Furutani M, Oikawa T, Takenawa T. Coordination between the actin cytoskeleton and membrane deformation by a novel membrane tubulation domain of PCH proteins is involved in endocytosis. J Cell Biol. 2006;172:269–279. doi: 10.1083/jcb.200508091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Itoh T, Erdmann KS, Roux A, Habermann B, Werner H, De Camilli P. Dynamin and the actin cytoskeleton cooperatively regulate plasma membrane invagination by BAR and F-BAR proteins. Dev Cell. 2005;9:791–804. doi: 10.1016/j.devcel.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 90.Huang P, Altshuller YM, Hou JC, Pessin JE, Frohman MA. Insulin-stimulated plasma membrane fusion of Glut4 glucose transporter-containing vesicles is regulated by phospholipase D1. Mol Biol Cell. 2005;16:2614–2623. doi: 10.1091/mbc.E04-12-1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Watson RT, Khan AH, Furukawa M, Hou JC, Li L, Kanzaki M, Okada S, Kandror KV, Pessin JE. Entry of newly synthesized GLUT4 into the insulin-responsive storage compartment is GGA dependent. Embo J. 2004 doi: 10.1038/sj.emboj.7600159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Czech MP. Molecular actions of insulin on glucose transport. Annu Rev Nutr. 1995;15:441–471. doi: 10.1146/annurev.nu.15.070195.002301. [DOI] [PubMed] [Google Scholar]
- 93.Holman GD, Cushman SW. Subcellular localization and trafficking of the GLUT4 glucose transporter isoform in insulin-responsive cells. Bioessays. 1994;16:753–759. doi: 10.1002/bies.950161010. [DOI] [PubMed] [Google Scholar]
- 94.Kandror KV, Pilch PF. Compartmentalization of protein traffic in insulin-sensitive cells. Am J Physiol. 1996;271:E1–E14. doi: 10.1152/ajpendo.1996.271.1.E1. [DOI] [PubMed] [Google Scholar]
- 95.Martin OJ, Lee A, McGraw TE. GLUT4 distribution between the plasma membrane and the intracellular compartments is maintained by an insulin-modulated bipartite dynamic mechanism. J Biol Chem. 2006;281:484–490. doi: 10.1074/jbc.M505944200. [DOI] [PubMed] [Google Scholar]
- 96.Kao AW, Ceresa BP, Santeler SR, Pessin JE. Expression of a dominant interfering dynamin mutant in 3T3L1 adipocytes inhibits GLUT4 endocytosis without affecting insulin signaling. J Biol Chem. 1998;273:25450–25457. doi: 10.1074/jbc.273.39.25450. [DOI] [PubMed] [Google Scholar]
- 97.Al-Hasani H, Hinck CS, Cushman SW. Endocytosis of the glucose transporter GLUT4 is mediated by the GTPase dynamin. J Biol Chem. 1998;273:17504–17510. doi: 10.1074/jbc.273.28.17504. [DOI] [PubMed] [Google Scholar]
- 98.Cohen AW, Combs TP, Scherer PE, Lisanti MP. Role of caveolin and caveolae in insulin signaling and diabetes. Am J Physiol Endocrinol Metab. 2003;285:E1151–1160. doi: 10.1152/ajpendo.00324.2003. [DOI] [PubMed] [Google Scholar]
- 99.Shigematsu S, Watson RT, Khan AH, Pessin JE. The adipocyte plasma membrane caveolin functional/structural organization is necessary for the efficient endocytosis of GLUT4. J Biol Chem. 2003;278:10683–10690. doi: 10.1074/jbc.M208563200. [DOI] [PubMed] [Google Scholar]
- 100.Ramm G, Slot JW, James DE, Stoorvogel W. Insulin recruits GLUT4 from specialized VAMP2-carrying vesicles as well as from the dynamic endosomal/trans-Golgi network in rat adipocytes. Mol Biol Cell. 2000;11:4079–4091. doi: 10.1091/mbc.11.12.4079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kandror KV, Pilch PF. Multiple endosomal recycling pathways in rat adipose cells. Biochem J. 1998;331:829–835. doi: 10.1042/bj3310829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Shewan AM, van Dam EM, Martin S, Luen TB, Hong W, Bryant NJ, James DE. GLUT4 recycles via a trans-Golgi network (TGN) subdomain enriched in Syntaxins 6 and 16 but not TGN38: involvement of an acidic targeting motif. Mol Biol Cell. 2003;14:973–986. doi: 10.1091/mbc.E02-06-0315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Robinson LJ, Pang S, Harris DS, Heuser J, James DE. Translocation of the glucose transporter (GLUT4) to the cell surface in permeabilized 3T3-L1 adipocytes: effects of ATP insulin, and GTP gamma S and localization of GLUT4 to clathrin lattices. J Cell Biol. 1992;117:1181–1196. doi: 10.1083/jcb.117.6.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Blot V, McGraw TE. GLUT4 is internalized by a cholesterol-dependent nystatin-sensitive mechanism inhibited by insulin. Embo J. 2006;25:5648–5658. doi: 10.1038/sj.emboj.7601462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Huang J, Imamura T, Olefsky JM. Insulin can regulate GLUT4 internalization by signaling to Rab5 and the motor protein dynein. Proc Natl Acad Sci U S A. 2001;98:13084–13089. doi: 10.1073/pnas.241368698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mari M, Monzo P, Kaddai V, Keslair F, Gonzalez T, Le Marchand-Brustel Y, Cormont M. The Rab4 effector Rabip4 plays a role in the endocytotic trafficking of Glut 4 in 3T3-L1 adipocytes. J Cell Sci. 2006;119:1297–1306. doi: 10.1242/jcs.02850. [DOI] [PubMed] [Google Scholar]