

Summary
Aminoacyl-tRNA synthetases are an ancient class of enzymes responsible for the matching of amino acids with anticodon sequences of tRNAs. Eukaryotic tRNA synthetases are often larger than their bacterial counterparts, and several mammalian enzymes use the additional domains to facilitate assembly into a multi-synthetase complex. Human cysteinyl-tRNA synthetase (CysRS) does not associate with the multi-synthetase complex, yet contains a eukaryotic-specific C-terminal extension that follows the tRNA anticodon-binding domain. Here we show by mutational and kinetic analysis that the C-terminal extension of human CysRS is used to selectively improve recognition and binding of the anticodon sequence, such that the specificity of anticodon recognition by human CysRS is higher than that of its bacterial counterparts. However, the improved anticodon recognition is achieved at the expense of a significantly slower rate in the aminoacylation reaction, suggesting a previously unrecognized kinetic quality control mechanism. This kinetic quality control reflects an evolutionary adaptation of some tRNA synthetases to improve the anticodon specificity of tRNA aminoacylation from bacteria to humans, possibly to accommodate concomitant changes in codon usage.
Keywords: cysteinyl-tRNA synthetase, transient kinetics, burst kinetics, product release, induced fit
Introduction
Aminoacyl-tRNA synthetases (aaRSs) provide the first level of accuracy in protein synthesis, matching amino acids with specific transfer RNAs with an error rate less than one per 104–105 codons 1. The matching is achieved in two steps 2. In the first step, an aaRS activates an amino acid with ATP to generate an enzyme-bound aminoacyl-adenylate. Next, the enzyme transfers the activated aminoacyl group to the 3′ end of a cognate tRNA to synthesize an aminoacyl-tRNA (aa-tRNA). Although subsequent steps following synthesis of aa-tRNAs also provide some selectivity 3, the very high specificity of protein synthesis that is necessary in vivo is provided primarily by the aaRSs. In cases where ambiguities are difficult to prevent, some aaRSs possess editing quality control mechanisms to remove incorrectly activated amino acids or non-cognate aa-tRNA 4. Here we report studies of a eukaryotic aaRS that suggest an alternative kinetic quality control mechanism for achieving very high levels of specificity.
Eukaryotic aaRSs differ from their bacterial counterparts by containing additional domains that are usually present as extensions to the N- or C-termini. Previous studies have focused on nine mammalian aaRSs (specifying Asp, Lys, Glu-Pro, Gln, Arg, Met, Leu, and Ile) that form a multi-synthetase complex in vivo 5; 6; 7. It is thought that the function of this complex may be to enhance tRNA channeling during protein synthesis 8. Extension domains of the synthetases in the multi-synthetase complex serve two known functions. First, they enhance tRNA binding in a sequence (and sometimes structure)-independent manner. Examples include the lysine-rich domain at the N-terminus of LysRS 9, the repeated R domains found in the linker region of the bi-functional Glu-ProRS 10, and similar repeated R motifs at the N- and C-termini of MetRS 11. Second, the extension domains promote protein-protein contacts within the multi-synthetase complex. Examples of tRNA synthetase domains functioning in this capacity include the leucine-rich motif at the N-terminus of ArgRS 12; 13; 14, the GST-like motif at the N-termini of MetRS and Glu-ProRS 15, and the imperfect repeats in the C-terminal region of IleRS 16. However, little is known of the extension domains found in mammalian tRNA synthetases that are not present in the multi-synthetase complex. In one such case, repeated motifs found in the N-terminal extension of human HisRS were shown to contain the major epitope that is recognized by auto-antibodies in the myositis disease 17. Few other mammalian tRNA synthetases not found in the multi-synthetase complex have been studied, and it is unknown whether the additional domains present in these enzymes might play a role in enhancing the specificity of aa-tRNA formation.
Human CysRS is an example of a mammalian tRNA synthetase not found in the multi-synthetase complex, that contains extension domains as compared with its prokaryotic homologs. The enzyme has three such extensions: (i) a short N-terminal peptide (~30 aa), (ii) an insertion domain found between the two pseudo-symmetrical halves of the active-site Rossmann fold (~100 aa), and (iii) a long C-terminal extension (~100 aa) (Fig 1A). None of these three additional peptides possesses the repeated motifs present in other eukaryotic enzymes that are found either within or apart from the multi-synthetase complex. Among the three extensions in human CysRS, only the C-terminal polypeptide is found broadly among homologs in the eukaryotic domain.
Fig. 1.
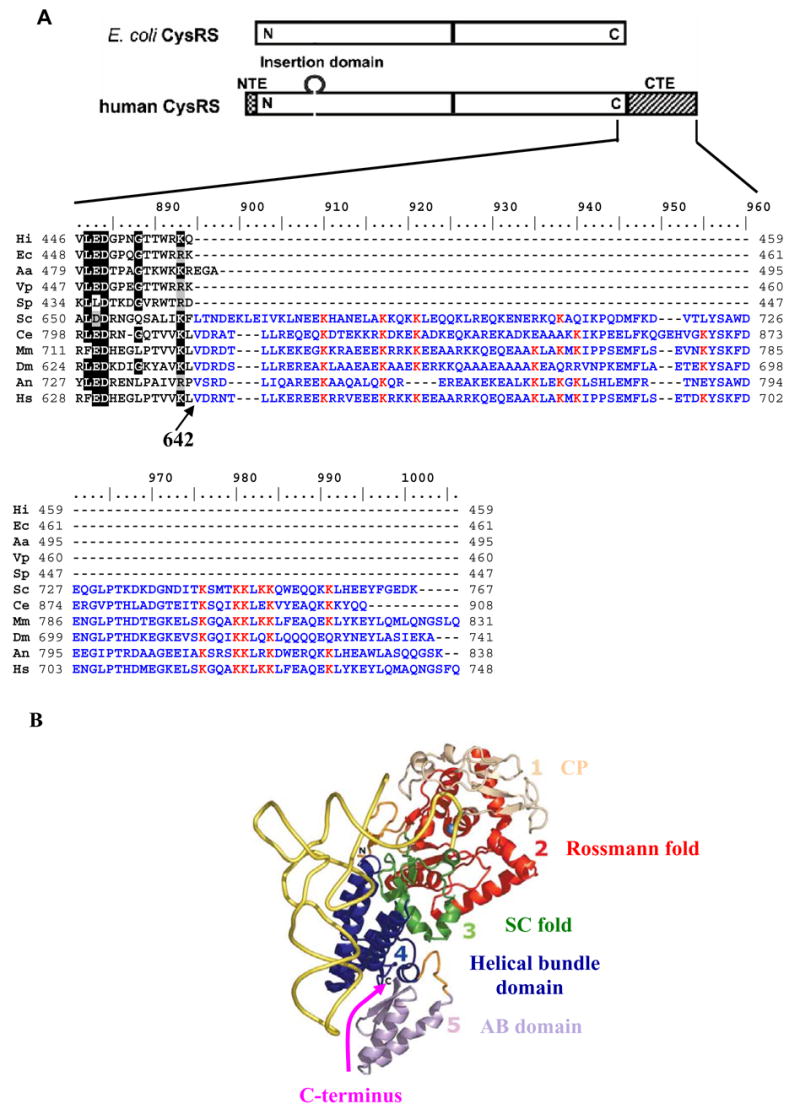
(A) Domain alignment (top) of human and E. coli CysRSs, showing the N- and C-extensions and an insertion peptide in the human enzyme. The sequence of the eukaryotic extension is shown in detail (bottom), where residues conserved among eukaryotic CysRSs are boxed. (B) The crystal structure of the E. coli CysRS-tRNACys binary complex 20, showing the four domains of the enzyme and highlighting the C-terminus by an arrow. Sequences in the alignment include those of H. influenzae (Hi), E. coli (Ec), A. aeolicus (Aa), V. parahaemolyticus (Vp), S. pneumoniae (Sp), S. cerevisiae (Sc), C. elegans (Ce), M. musculus (Mm), D. melanogaster (Dm), A. nidulans (An), and H. sapiens (Hs).
Human CysRS shares high sequence identity with its bacterial counterparts in the regions of the enzyme that are conserved among all the homologs 18. The crystal structure of E. coli CysRS reveals the typical catalytic Rossmann-fold of class I synthetases at the N-terminus, a small connective polypeptide insertion (the CP domain) within the Rossmann fold, a bridging stem-contact fold (the SC fold), a helical bundle domain conserved in all subclass 1a tRNA synthetases, and a unique mixed α/β anticodon-binding domain that provides the specificity determinants for all three anticodon nucleotides (Fig. 1B) 19; 20. The C-terminus of E. coli CysRS corresponds to Val642 in human CysRS, and the eukaryotic-specific C-terminal extension emanates from this point.
The proximity of the eukaryotic-specific C-terminal extension to the anticodon-binding domain suggests the possibility that it might play an additional role in anticodon recognition. However, the C-terminal extension could in principle also affect recognition of other parts of the tRNA molecule. The landscape of tRNA recognition by both E. coli and human CysRSs is dominated by four nucleotides: the 5′-GCA-3′ anticodon 21, and U73 in the acceptor stem 22; 23, all of which are strictly conserved among tRNACys species. However, tRNA recognition by human CysRS differs from that of E. coli CysRS in at least two respects. First, while E. coli CysRS recognizes an unusual 15–48 base pair (G15-G48) in the tertiary core of E. coli tRNACys through an indirect readout mechanism 20; 24, human CysRS does not depend on the nature of the 15–48 base pair as a selective determinant for aminoacylation 22. Thus the human enzyme places less emphasis on an indirect readout at the 15–48 position for tRNA recognition. Second, while E. coli CysRS functions as a monomer, all known eukaryotic CysRS enzymes exist as dimers 18; 25; 26. The functional significance of the dimeric structure in eukaryotes is unknown.
To elucidate the role of the C-terminal extension of eukaryotic CysRS enzymes, we performed an extensive comparative kinetic analysis of recombinant full-length human CysRS and a mutant enzyme in which the eukaryotic-specific C-terminal domain is deleted. We find that the C-terminal extension plays a major role in anticodon recognition, but that it does not affect the dimeric structure or recognition pattern of the tRNA tertiary core. Unexpectedly, while the C-terminal extension enhances stable binding of human CysRS to its cognate tRNA during aminoacylation, and promotes efficient enzyme turnover in the steady state, its presence appears to slow the rate of an induced-fit conformational rearrangement that occurs en route to the Michaelis complex formation. Further, we show that human CysRS exhibit an overall higher specificity for anticodon recognition than E. coli CysRS, suggesting that the C-terminal extension allows the human enzyme to exploit the increased affinity for tRNACys to allow a closer inspection of the anticodon. Because the improved specificity occurs concomitantly with a decrease in the aminoacylation rate, it appears that the C-terminal extension provides a kinetic “quality control” function. This kinetic quality control is conceptually distinct from the previously characterized editing quality control of aaRS that removes incorrectly synthesized aa-tRNA. The ability of a eukaryote-specific extension domain to exert quality control of tRNA recognition may be a specific mechanism that permits adaptation to changes in codon usage along the evolutionary trajectory from bacteria to humans.
Results
A stable mutant of human CysRS lacking the C-terminal extension
To determine the role of the C-terminal extension in human CysRS, we constructed a ΔC mutant of the enzyme that terminates at V642, removing residues 643 to 748 from the full-length enzyme (Fig. 1A). The ΔC mutant was made by expression of a truncated cDNA of the human cysS gene, under the control of the inducible T7 RNA polymerase promoter. As with the full-length enzyme, the ΔC mutant was stably produced in E. coli with a C-terminal His-tag, and remained stable throughout the entire purification procedure. Both the full-length enzyme and ΔC mutant were purified to homogeneity as examined by SDS-PAGE; each migrated as expected with monomer molecular masses of 82 and 71 kDa, respectively.
The stable ΔC mutant was active in the first step of aminoacylation: the ATP-dependent synthesis of cysteinyl-adenylate. This activity was assayed by using TLC to monitor the forward reaction by which γ-32P-ATP is converted to 32P-PPi upon synthesis of Cys-AMP 27. To increase the sensitivity of detection, the labeled ATP substrate was kept at near saturating concentrations. The steady-state rates of Cys-AMP synthesis catalyzed by the ΔC mutant exhibited classical Michaelis-Menten kinetics with Km of 10.3 μM for cysteine and kcat of 18.1 s−1 (Table 1). Remarkably, both kcat and Km are improved compared with the full-length enzyme, with an overall increase in kcat/Km of 3.5-fold (Table 1).
Table I.
Parameters of Cysteine Activation Observed by ATP-PPi Exchange and of Aminoacylation with Respect to Human tRNACys Transcript.
| A: Apparent Parameters for Cysteine in the Activation Step
| ||||||
|---|---|---|---|---|---|---|
| Km (μM) | kcat (s−1) | kcat/Km (s−1M−1) | Relative Activity | |||
| Full-length | 20.3 ± 1.3 | 10.3 ± 1.6 | 0.51 × 106 | 1.0 | ||
| ΔC | 10.3 ± 0.3 | 18.1 ± 0.4 | 1.76 × 106 | 3.5 | ||
|
| ||||||
| B: Kinetic Parameters for tRNA in the Aminoacylation Step
| ||||||
| Steady-State
|
Single Turnover
|
Multiple turnover rate from burst kinetics
|
||||
| Km (μM) | kcat (s−1) | kcat/Km (s−1M−1) | Relative activity | kapp* (s−1) | kcat (s−1) | |
|
| ||||||
| Full-length | 0.9 ± 0.1 | 2.3 ± 0.1 | 2.6 × 106 | 1.0 | 14 (kconf) | 2.6 |
| ΔC | 4.5 ± 0.7 | 0.20 ± 0.01 | 4.4 × 104 | 1.7 × 10−2 | ~120 (kchem) | 0.2 |
kapp is the saturating rate constant of the overall two-step aminoacylation reaction. It refers to the conformational change rate for the wild type enzyme and the chemical rate for the ΔC mutant. Both the WT human CysRS and the ΔC mutant exhibit burst kinetics.
The C-terminal extension is not involved in dimer formation
To determine if the C-terminal extension of human CysRS is responsible for dimerization, the full-length enzyme and the ΔC mutant were examined by gel filtration using a calibrated Superose 12 column under native conditions (Fig. 2A). The full-length enzyme eluted at fractions corresponding to an apparent mass of 207 kDa, while the ΔC mutant eluted at fractions corresponding to 148 kDa. Each of these determinations is most consistent with a dimeric structure. Additionally, size-exclusion chromatography light scattering (SEC-LS) returned molecular weights of 177 kDa for the full-length CysRS and 147 kDa for the truncation mutant, each again closely approximating the calculated molecular weight of a dimer (Fig. 2C). It appears then that the C-terminal extension of human CysRS does not function as a dimerization domain.
Fig. 2.
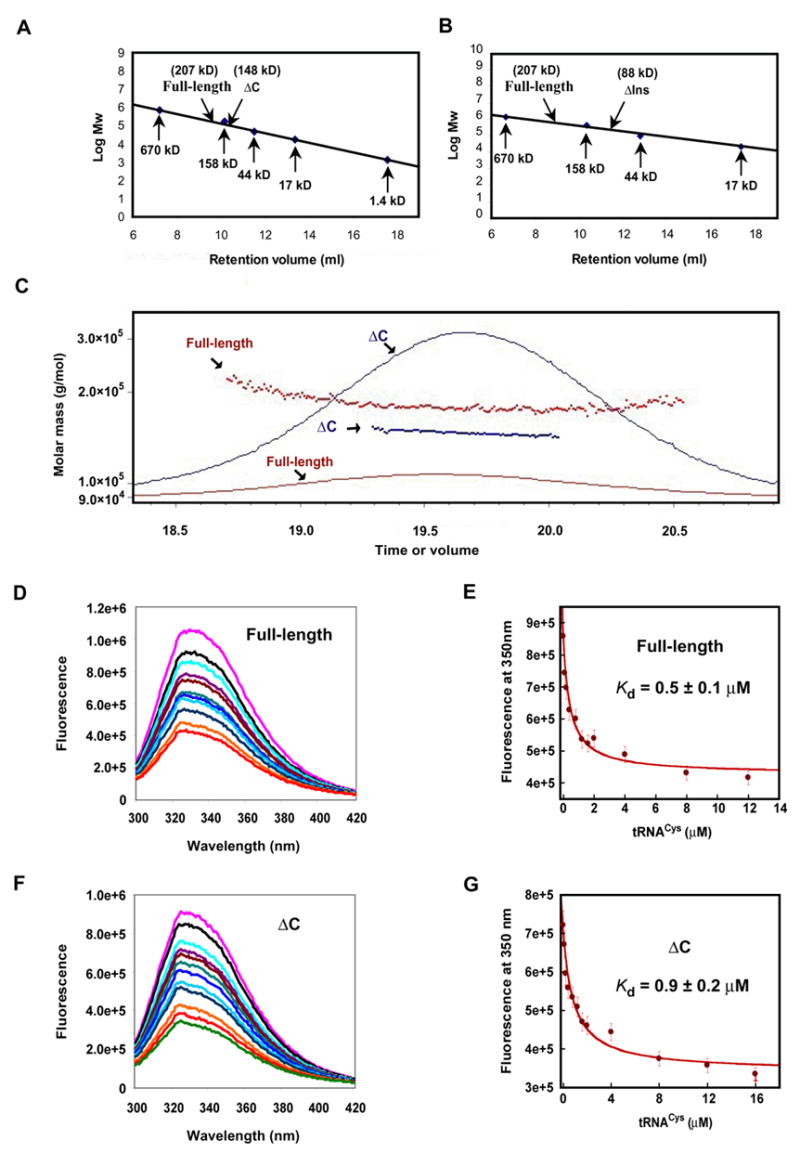
Analysis of human CysRS and the ΔC mutant. (A) Gel filtration of human CysRS (100 μg) and the ΔC mutant (100 μg) on Superose 12 calibrated by known molecular weight markers. (B) Gel filtration of the ΔIns variant that lacks the insertion peptide in the Rossmann-fold, showing an apparent molecular mass of 88 kDa, most consistent with the mass of a monomer. (C) Size exclusion profile of the wild-type and ΔC mutant of human CysRS. The solid lines indicate the trace from the refractive index detector, and the dots are weight-average molecular weights determined for each slice (red, wild-type enzyme; blue, truncation mutant). (D) Quenching of intrinsic tryptophan fluorescence of the full-length human CysRS through tRNA binding, showing emission scans in the presence of increasing amounts of tRNACys. The concentration of tRNA was varied from zero (purple line) to 12 μM (light green). The fluorescence intensity is given in an arbitrary unit. (E) Analysis of the full-length CysRS fluorescence versus tRNA concentration. At each tRNA concentration, the emission peak at 350 nm was quantified and fit to a hyperbolic binding equation for calculation of the Kd for tRNA. (F) Quenching of intrinsic tryptophan fluorescence of the ΔC mutant. (G) Fitting the ΔC fluorescence versus tRNA concentration to a hyperbolic binding equation to calculate the Kd for tRNA.
Other experiments showed that deletion of the insertion peptide that spans the two halves of the active-site Rossmann fold (Fig. 1A) caused the human CysRS enzyme to elute as a monomer from the Superose 12 column (Fig. 2B), suggesting that this insertion peptide may be responsible for the dimerization. Interestingly, the dimer interfaces of the only two dimeric class I tRNA synthetases, TyrRS and TrpRS, are also located within the active-site Rossmann fold 28; 29; 30. However, the crystal structure of the TyrRS-tRNATyr complex shows that tRNA binds across the two subunits, and that the enzyme binds in the major groove of the tRNA acceptor stem in a manner resembling tRNA binding in class II synthetases 28. Since there is extensive homology between human and E. coli CysRS enzymes in the conserved domains of the structure (Fig. 1B), it appears more likely that tRNA binding to human CysRS will follow the canonical class I mode of binding along the entire inside interface of the L-shaped tRNA 20. These considerations suggest that the human CysRS dimer may bind two tRNAs.
The C-terminal extension is important for stable binding of tRNACys
The contribution of the C-terminal extension to the binding affinity for tRNACys was examined by intrinsic tryptophan equilibrium fluorescence. The substrate was the T7 transcript of human tRNACys, which lacks the modifications found in the native tRNA. Kinetic analysis suggests that tRNA modifications are not crucial to aminoacylation in this system (see below). Titration by fluorescence measurements showed a progressive quenching of the enzyme intrinsic fluorescence through tRNA binding (Fig. 2D). The inner filter effect was corrected according to the method described 31. It was minimized by scanning the excitation spectrum and identifying a wavelength that was specific for CysRS (but not for BSA) to yield an emission spectrum. For the full-length enzyme, a hyperbolic fit of the fluorescence intensity versus tRNA concentration yielded Kd of 0.5 ± 0.1 μM (Fig. 2E), similar to values reported for other cognate synthetase-tRNA interactions 20; 32; 33. Titration of the ΔC mutant yielded Kd of 0.9 ± 0.2 μM (Fig. 2F,G), indicating that the C-terminal extension provides a 2-fold improvement in the tRNA binding affinity. However, as shown below, the contribution of the C-terminal extension to tRNA binding during the kinetics of aminoacylation is elevated significantly.
Importance of the C-terminal extension for tRNA aminoacylation
The contribution of the C-terminal extension to tRNA aminoacylation was first examined by steady-state analysis with respect to tRNACys (Table 1). A series of aminoacylation reactions at saturating ATP and near-saturating 35S-cysteine (50 μM) were conducted in the presence of varying concentrations of the unmodified T7 transcript of human tRNACys, and the steady-state rate of Cys-tRNACys formation was monitored by acid precipitation of the label on filter pads. For the full-length human CysRS enzyme, the Km of 0.9 ± 0.1 μM for tRNACys and kcat of 2.3 ± 0.1 s−1 were similar to the respective values obtained for E. coli CysRS with E. coli tRNACys (Km of 1.2 μM and kcat of 2.5 s−1; Zhang et al., 2003). Given the common function of CysRS in aminoacylation in the two organisms, it thus appears highly likely that modifications in the tRNA are not of crucial importance in the human cysteinylation system. Analysis of the ΔC mutant revealed a 5-fold increase in the Km for tRNACys (to 4.5 ± 0.7 μM) and a 11-fold decrease in kcat (to 0.2 ± 0.01 s−1), for an overall decrease of nearly 60-fold in kcat/Km. Thus, the C-terminal extension is important for efficient steady-state aminoacylation by human CysRS.
The steady-state parameters Km and kcat are complex terms that encompass all of the individual steps that occur in the course of the reaction (substrate association, conformational changes, aminoacylation chemistry, product release), such that the individual mechanistic steps are not resolvable. This inherent limitation of steady-state kinetics thus does not allow assignment of the catalytic defects implied by the altered steady-state parameters, to a particular step in the aminoacylation reaction. To address these limitations, transient-state kinetics was employed. In these experiments the enzyme was present as a stoichiometric reactant to allow the kinetics of individual steps to be determined directly.
We defined the single turnover rate for tRNA aminoacylation as kapp which is a composite term that includes both synthesis of cysteinyl adenylate and transfer of cysteine to tRNACys 34; 35. To determine kapp single turnover experiments using a molar excess of enzyme over tRNA were performed on a rapid chemical quench instrument, and the observed rate constants kapp were obtained by fitting the time courses to a single exponential equation. Control experiments established that 6.25 mM ATP and 0.5 mM cysteine were saturating for both the wild-type and mutant enzymes. Other control experiments showed that the reaction rate was independent of the manner in which the various substrates were premixed. Of interest is that, for both the full-length enzyme and ΔC mutant, measurement of the rate constant for the two-step aminoacylation (by adding CysRS to a mixture of all three substrates) and of the rate constant for the transfer step (by adding tRNA into a pre-mixed CysRS-ATP-cysteine complex) yielded the same kapp (data not shown). The identical rates suggest that the rate of the chemical transformation in the two-step aminoacylation is defined by the rate of aminoacyl transfer to tRNA in the second step. The same conclusion was reached in studies of E. coli CysRS 35.
To determine kchem for the full-length human CysRS, time courses over a range of tRNA concentrations (0.5–5.0 μM) were monitored, and the enzyme was maintained at a fixed 10-fold molar excess over tRNA (Fig. 3A). All time courses were well-fit to a first-order exponential function, indicating that the enzyme and tRNA were in rapid equilibrium in this concentration range. Fitting the dependence of the single turnover rate constant with enzyme concentration to a hyperbolic curve yielded a saturating kapp of 14.4 s−1 (Fig. 3B), very similar to the saturating kapp of 14.1 s−1 determined for the E. coli enzyme 35. Unexpectedly, the ΔC mutant showed a significantly faster saturating kapp as compared with the full-length enzyme (Figs. 3C, 3D). Notably, kapp increased as the tRNA concentration was increased from 0.5 μM to 6 μM. The use of higher concentrations of tRNA was not practical due to the requirement for a correspondingly 10-fold higher enzyme concentration. However, because partial saturation was apparent in fitting kapp vs the concentration of ΔC to a hyperbolic equation, it was possible to estimate the saturating kapp at approximately 120 s−1 (Fig. 3D), which is nearly 10-fold faster than the saturating kapp exhibited by the full-length CysRS. Therefore, removal of the C-terminal extension appears to have altered a rate-limiting step in the single-turnover aminoacylation kinetics by the full-length enzyme.
Fig. 3.
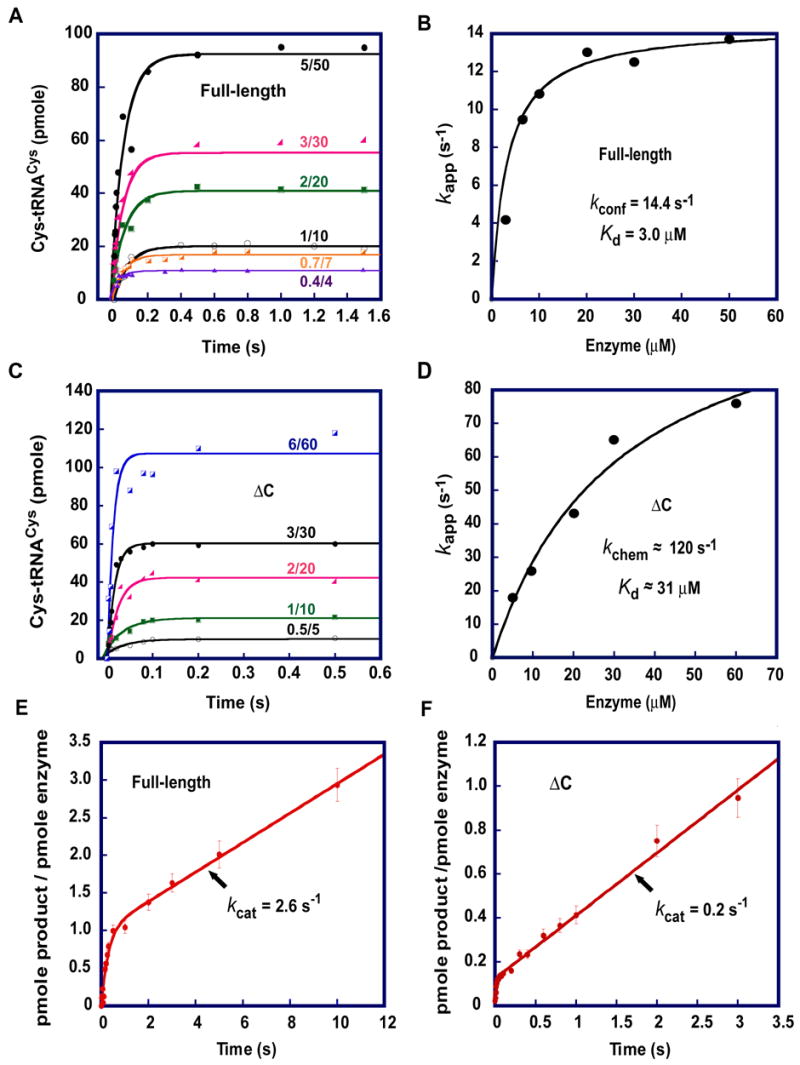
Transient-state kinetics of aminoacylation. (A) Time courses of single turnover aminoacylation of tRNACys by the full-length enzyme. The rate constant of kapp was derived by fitting the data to the equation y = y0 + A* (1−e−kapp*t), where Y0is the y intercept, A is a scaling constant, kapp is the apparent rate constant, and t is time in second. (B) Replot of kapp from (A) versus enzyme concentrations to derive the saturating rate constant kconf and the concentration of the enzyme that yielded the half kconf as Kd. (C) Time courses of single turnover aminoacylation of tRNACys by the ΔC enzyme. (D) Replot of kapp from (B) versus enzyme concentrations to derive the saturating rate constant kchem and the concentration of the enzyme that yielded the half kchem as Kd. (E, F) Time courses of pre-steady-state kinetics of aminoacylation by the full-length enzyme (0.5 μM) and ΔC mutant (0.05 μM), respectively. The rate constants kcat were derived by fitting the data to the equation y = y0 + A* (1−e− k1*t)+ k2*E0 * t by using KaleidaGraph, where is the apparent rate constant of the exponential phase, and is the rate constant of the steady-state phase. For panels (A) and (C) the ratio such as 5/50 indicates the concentrations of tRNA and enzyme used for a time course.
In interpreting these observations, it is important to recognize that the rapid mixing experiment returns a value for kapp that represents all of the reaction steps up to and including aminoacylation on the enzyme. Because kapp is determined by the rate of aminoacyl transfer to tRNA, this parameter in principle encompasses rates of tRNA association to the CysRS-Cys-ATP complex, induced-fit conformational arrangement of CysRS and tRNA, and the actual transfer step. Given the high enzyme and substrate concentrations used in these experiments, it is expected that the second-order association rates would be very fast (Uter et al., 2005), such that kapp reflects the chemical rate of the transfer (kchem) for both the full-length CysRS and the ΔC mutant. However, an alternative possibility is that the measured kapp in fact represents the rate of a slower conformational rearrangement step that would follow the initial substrate association and would be required for the productive juxtaposition of the reactive moieties of ATP, cysteine, and tRNACys in the active site of the enzyme. Because the C-terminal extension is outside of the active site, and yet the removal of the domain generates a nearly 10-fold improvement in kapp, it appears likely that the extension in the native enzyme is responsible for a slow conformational rearrangement required for formation of the catalytically productive CysRS:Cys:ATP:tRNA complex. For this reason, we defined the saturating kapp for the full-length enzyme as kconf to reflect the influence of enzyme conformational rearrangement on the overall chemistry of aminoacylation. For the ΔC mutant, because the slower rearrangement step is removed, the saturating kapp is likely to reflect the rate of chemistry (kchem).
The application of transient kinetics to the human CysRS reaction also allows examination of binding equilibria and kinetics. As an initial exploration of equilibrium binding by this approach, we examined the dependence of the single-turnover reaction rates on the concentrations of tRNACys, while maintaining a fixed molar excess of enzyme over tRNA. The titrations showed single-exponential kinetics at all concentrations tested, indicating that the CysRS-ATP-Cys complex was in rapid equilibrium with the tRNA substrate. Because the single turnover rate is driven by the enzyme concentration, but not by the tRNA concentration, the kinetic Kd is determined from a replot of kapp vs. [E] to a hyperbolic equation 35. This analysis yielded a kinetic Kd for tRNA of 3 μM for the full-length enzyme (Fig. 3B) and an estimated Kd for the ΔC mutant of ~31 μM (Fig. 3D). Thus, as in the case of the Kd of the binary enzyme-tRNA complex measured by fluorescence titration (Fig. 2D–G), the C-terminal extension strengthens the binding affinity in the enzyme transition state complex. However, compared to the 2-fold effect of the C-terminal extension on the Kd of the binary complex, the effect of the C-terminal extension on the kinetic Kd is 10-fold, suggesting that this domain plays a more important role in tRNA binding in the context of a catalytically active enzyme.
Both native CysRS and the ΔC mutant show values of saturating kapp that are faster than their respective steady-state turnover numbers (kcat), suggesting that in each case the enzyme might exhibit burst kinetics, where product release is rate limiting. In addition, both enzymes exhibit values of kinetic Kd greater than Km, consistent with a rate-limiting step subsequent to aminoacylation 36. The prediction of burst kinetics was confirmed by experiments performed under pre-steady-state conditions, in which cysteine and ATP were present at saturating concentrations while the enzyme:tRNA ratios and concentrations were adjusted such that it was possible to observe a single turnover followed by a subsequent steady-state rate. For the full-length enzyme, the time course of aminoacylation revealed a rapid burst of product synthesis, followed by a slower steady-state rate (Fig. 3E), indicating that a step following product formation on the enzyme and prior to rebinding of substrates in the subsequent turnover is rate-limiting in the steady state. Fitting the data to a burst equation yielded kcat = 2.6 s−1, similar to the value determined separately from the Michaelis-Menten steady-state plots. Similarly, the ΔC mutant also exhibited burst kinetics (Fig. 3F) and showed kcat = 0.2 s−1, again as found in the steady-state experiments. It should be noted that different amounts of enzyme were used for the full-length CysRS (0.5 μM) and ΔC mutant (0.05 μM) in order to clearly show the burst kinetics; thus, the amplitudes of the burst (which reflected the amount of the enzyme) were not identical. Nonetheless, the calculated amplitude of the full-length enzyme corresponded to 1 mole product/mole of enzyme. In contrast, the calculated amplitude of the ΔC enzyme corresponded to ~10% of the active fraction of the enzyme, which is determined by the aminoacyl-adenylate synthesis activity.
Taken together, the kinetic experiments show that while the C-terminal extension of human CysRS improves kcat in the steady state, it reduces the rate of kapp as determined by single turnover kinetics. These observations are consistent with a model in which the function of the C-terminal extension is to reduce the rate of an induced-fit conformational rearrangement that occurs after initial binding and before catalysis. Thus, the extension domain may play a role in the selection of cognate amino acid and/or tRNA, because the slower rearrangement might permit the enzyme to better use this step as a means of discrimination. The observation of burst kinetics suggests that product release remains rate-limiting for both the wild-type and truncated enzymes in the overall reaction.
The C-terminal extension selectively improves anticodon recognition
The C-terminal extension might in principle slow early steps along the catalytic pathway by making direct contacts with the tRNA, perhaps in the anticodon stem-loop or globular core portions of the tRNA molecule (Fig. 1B). Alternatively or additionally, the new domain might operate by facilitating interactions of the more distal N-terminal domains through protein-protein contacts. To examine these possibilities more closely, the abilities of the full-length human CysRS and ΔC mutant to aminoacylate an extensive set of mutants of tRNACys were examined. Mutations were created in the GCA anticodon, the G15-C48 base pair in the tertiary core, and the discriminator base U73 near the acceptor end. These nucleotides were chosen for mutagenesis because they are known recognition determinants of E. coli CysRS 20; 22. Although identity determinants in tRNAs are not always conserved across biological domains, the expected overall homology of the protein structures, and the common presence of the unusual U73 nucleotide and GCA anticodon, suggests that these nucleotides may be of importance to the human enzyme as well. Further, the ability of E. coli CysRS to recognize an unusual G15–G48 tertiary base pair in the core by an indirect readout mechanism made the 15–48 position of interest to examine in the case of human CysRS, even though human tRNACys possesses the standard G15-C48 pair (Fig. 4).
Fig. 4.
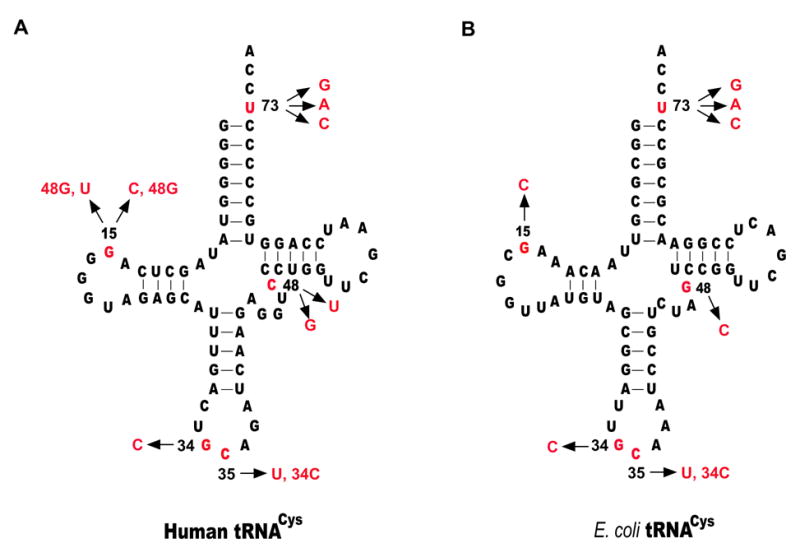
Sequence and cloverleaf structure of human and E. coli tRNACys. Nucleotides where mutations were made are shown in red, and substitutions were shown by arrows.
Ten tRNA mutants were examined by determination of the second-order specificity parameter kcat/Km, which was determined by measurement of the reaction velocities under steady-state conditions, at tRNA concentrations well below the Km (Table 2). The kcat/Km values for the full-length and ΔC enzymes obtained by this method were 1.35 × 106 M−1s−1 and 2.5 × 104 M−1s−1 respectively for the wild-type tRNA, each within two-fold of the value obtained from determination of the Michaelis-Menten kinetics (2.6 × 106 and 4.4 × 104; Table 1). To a first approximation, both the full-length and ΔC enzymes discriminated against mutations at U73 and in the anticodon by 4–6 orders of magnitude, but discriminated against mutations at G15-C48 in the core region by more modest effects. These data confirmed that the identity determinants for human CysRS recapitulate those for the E. coli enzyme. They also showed that the full-length enzyme discriminated better than did the ΔC mutant against tRNA mutations in all regions, and that the largest difference between the two enzymes was observed in the anticodon (Table 2, top half). For example, the comparison showed that the full-length enzyme discriminated nearly 80-fold more strongly than the ΔC mutant against a tRNA mutant containing the G34C substitution (compare FL/ΔC values in Table 2). This 80-fold improved specificity arose from the more efficient operation of the full-length enzyme toward the wild-type tRNA substrate, and from the poorer discrimination of the ΔC mutant against tRNA mutations. Similarly, the full-length enzyme was more discriminatory than the ΔC mutant against the G34C/C35U double mutant by nearly 120-fold. Again, the difference arose from more efficient aminoacylation by the full-length enzyme toward the wild-type tRNA and from the poorer discrimination of the mutant enzyme against mutant tRNAs.
Table II.
Kinetic Analysis of kcat/Km of tRNA aminoacylation.
| Full-length Human CysRS | Human CysRS ΔC mutant | E. coli CysRS | FL/ ΔC | FL/Ec | |||||
|---|---|---|---|---|---|---|---|---|---|
| Mutant Type | tRNA | kcat/Km (μM−1s−1) | Discrimination by x-fold | kcat/Km (μM−1s−1) | Discrimination by x-fold | kcat/Km (μM−1s−1) | Discrimination by x-fold | ||
| Hs (WT) | 1.35±0.12 | 1.0 | 0.025±0.03 | 1.0 | 1 | ||||
| Anticodon | Hs(G34C) | (6.2±0.2) ×10−5 | 2.2×104 | (8.5±0.3)×10−5 | 2.9×102 | 76 | |||
| mutants | Hs(G34C, C35U) | (1.6±0.3) ×10−5 | 8.4×104 | (3.6±0.6)×10−5 | 6.9×102 | 122 | |||
| U73 | Hs(U73A) | (2.0±0.1) ×10−5 | 6.8×104 | (2.2±0.1) ×10−6 | 1.1×104 | 6 | |||
| mutants | Hs(U73G) | (3.4±0.3)×10−6 | 4.0×105 | (1.1±0.1) ×10−6 | 2.3×104 | 17 | |||
| Hs(U73C) | (1.7±0.1)×10−5 | 7.9×104 | (2.1±0.1) ×10−6 | 1.2×104 | 7 | ||||
| Core | Hs(C48U) | 0.14±0.01 | 9.6 | 0.01±0.004 | 2.5 | 4 | |||
| mutants | Hs(C48G) | 0.052±0.003 | 26 | 0.025±0.001 | 1.0 | 26 | |||
| Hs(C15, G48) | 0.040±0.007 | 34 | (6.8±0.1)×10−3 | 3.7 | 9 | ||||
| Hs(U15, G48) | 0.047±0.002 | 29 | (6.1±0.2) ×10−3 | 4.1 | 7 | ||||
| Ec (WT) | 0.61 ± 0.03 | 1.0 | (8.6±0.7)×10−3 | 1.0 | 0.99 ± 0.06 | 1.0 | 1 | 1 | |
| Anticodon | Ec(G34C) | (1.9±0.1) ×10−5 | 3.2 ×104 | (6.3±0.7) ×10−5 | 1.4 ×102 | (5.6±0.3) ×10−4 | 1.8 ×103 | 229 | 18 |
| mutants | Ec(G34C, C35U) | (1.7±0.4) ×10−5 | 3.6 ×104 | (3.0±0.4) ×10−5 | 2.9 ×102 | (4.2±0.4)×10−4 | 2.4 ×103 | 124 | 15 |
| U73 | Ec(U73A) | (1.7±0.1) ×10−6 | 3.6 ×105 | (1.3±0.1) ×10−6 | 6.6 ×103 | (5.2±0.1) ×10−6 | 1.9 ×105 | 55 | 2 |
| mutants | Ec(U73G) | (1.0±0.4) ×10−6 | 6.1 ×105 | (1.0±0.1) ×10−6 | 8.6 ×103 | (6.8±0.1) ×10−6 | 1.5 ×105 | 71 | 4 |
| Ec(U73C) | (4.9±0.2) ×10−6 | 1.2 ×105 | (1.5±0.1) ×10−6 | 5.7 ×103 | (8.8±0.3) ×10−6 | 1.1 ×105 | 21 | 1 | |
| Core | Ec(G48C) | 0.043 ± 0.002 | 14 | (4.7±0.4)×10−4 | 18 | 0.01 ± 0.01 | 100 | 1 | 0.1 |
| mutants | Ec(G15C) | 0.048 ± 0.006 | 13 | (3.2±0.2)×10−3 | 2.7 | 0.02 ± 0.018 | 50 | 5 | 0.3 |
Hs: Homo sapiens. Ec: E. coli. FL: Full-length human CysRS. ΔC: C-terminal extension deletion mutant of human CysRS.
The effect of the C-terminal extension on the ability of human CysRS to discriminate against tRNA mutations at G15-C48 or at U73 was smaller than in the case of the anticodon. For example, the full-length enzyme discriminated by a factor of ~10 against the C48U core mutation, whereas the ΔC mutant discriminated by a factor of only 2.5 at this position - a difference of ~4-fold. Similarly, the full-length enzyme discriminated better than did the ΔC mutant against the C48G, G15C/C48G, and G15U/C48G mutants by 7 to 26-fold (Table 2, top half). At the U73 position, the full-length enzyme discriminated against mutations better than the ΔC mutant by 6–17 fold (Table 2, top half). Taken together, these data demonstrate that, within the context of a generally impaired capacity for tRNA recognition, the C-terminal extension has a role in enforcing discrimination throughout the entire tRNA, with a greater effect on mutations in the anticodon.
To complement these studies on human tRNACys, we also examined the ability of the human CysRS enzyme to discriminate among wild-type and mutated E. coli tRNACys substrates, and compared the ability of the human enzyme with that of the E. coli enzyme. This provides the possibility of a direct comparison between human and E. coli CysRSs in all three regions of interest in recognition of tRNACys, allowing further appraisal of the importance of the human C-terminal extension (Table 2, bottom half). This comparison was not made in the sequence context of human tRNACys (see above), because E. coli CysRS is not able to efficiently cross-acylate this tRNA 22. Kinetic analysis showed that, in the sequence context of E. coli tRNACys, the most discriminating enzyme against anticodon mutations was the full-length human CysRS. The specificity factor of the full-length human CysRS was 3.2 × 104 against the G34C mutation, while this parameter decreased to 1.8 × 103 for E. coli CysRS, and to 1.4 × 102 for the human ΔC mutant. This order of specificity among the three enzymes was also found against the G34C/C35U double mutations. In contrast, human and E. coli CysRS enzymes exhibited a similar specificity against mutations at U73 (~105-fold), which was higher than the specificity manifested by the ΔC mutant (~103-fold). Finally, as shown previously in the sequence context of E. coli tRNACys, while E. coli CysRS effectively discriminated against the G48C or G15C mutation in the tertiary core by a factor of 100 and 50, respectively 20; 22, human CysRS was more accommodating to these alterations, showing a reduced discrimination factor of only about 13–14-fold. In this case the ΔC mutant discriminated by factors similar to those of the full-length enzyme, again suggesting that the C-terminal extension has a smaller role in recognition of the tertiary core.
Probing tRNA recognition of tRNA by enzymatic footprinting
To determine whether the C-terminal extension domain of human CysRS influences how tRNA is bound by the enzyme, we employed an enzymatic footprinting approach. The sensitivity of the wild-type T7 transcript of human tRNACys to nuclease T1 cleavage was probed under native conditions in the presence of 10 mM MgCl2. Cleavage fragments were separated by denaturing PAGE, and positions of cleavage were determined from a sequencing ladder provided by alkaline hydrolysis. The G-specific T1 nuclease cleaved the unprotected tRNACys most prominently at G34 and G37, the two susceptible positions in the anticodon loop (Fig. 5A). Comparison of band intensities at specific positions relative to a reference non-specific reaction (at C40) revealed that the full-length enzyme and ΔC mutant conferred similar protection on nucleotides at G19, G30, G37, and G46 along the extended D-anticodon stem and loop (Fig. 5B). However, among the generally similar patterns of protection, a strong difference between the full-length and ΔC enzymes can be observed at G34, where analysis of the direct band intensities showed a 5–10-fold loss in protection by the ΔC mutant. In addition, two other differences of lesser magnitude were observed in the tertiary core: while the native enzyme protected G19 better, the truncation mutant provided more protection at G57. Together, these results reveal that the C-terminal extension of human CysRS modulates binding interactions at position G34 of the anticodon, and also affects interactions in the tRNA tertiary core.
Fig. 5.
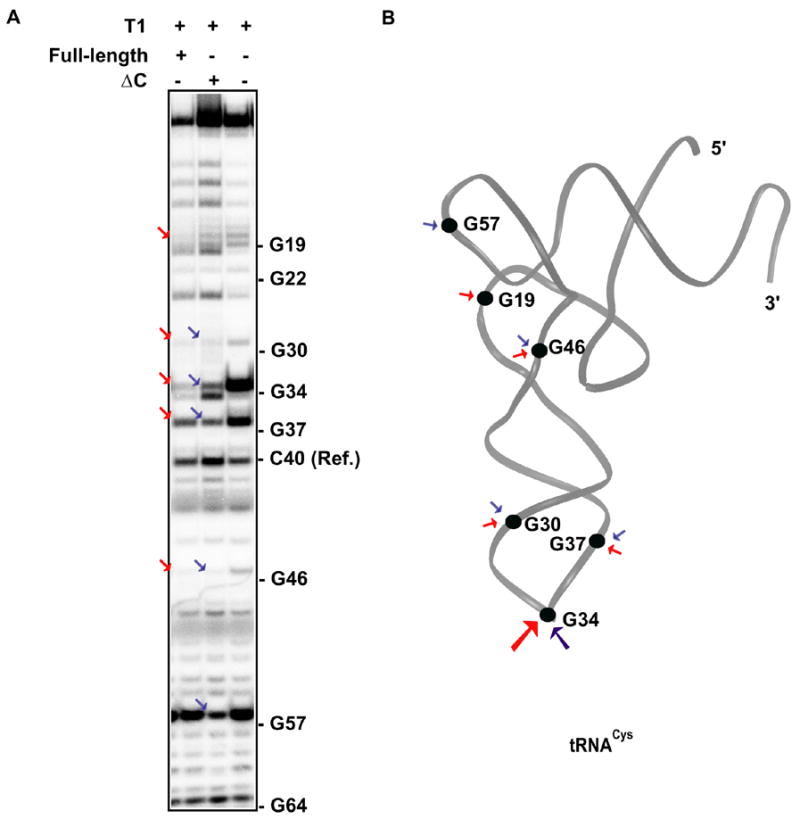
(A) The nuclease T1 cleavage of the human CysRS-tRNACys complex. The G residues that were sensitive to T1 cleavage in the absence of CysRS were marked on the side, whereas those that were protected by the full-length enzyme or ΔC mutant were indicated by red or blue arrows, respectively. (B) The G residues that were protected by the full-length enzyme are marked in the L-shaped tRNA structure taken from the crystal structure of the E. coli CysRS-tRNA complex 20, where red arrows indicate protection by the full-length and blue arrows indicate protection by the truncation mutant.
Discussion
A eukaryotic extension domain for improving anticodon recognition
While extension domains of eukaryotic synthetases within the multi-synthetase complex help to stabilize this large assembly, the function of extension domains in synthetases not associated with the complex has been largely obscure. Here we have employed a combination of single turnover, pre-steady-state, and steady-state kinetics to examine the function of the C-terminal extension of the non-complex human CysRS in aminoacylation. Our results show that the C-terminal extension plays an important role in the kinetic pathway of aminoacylation with effects on both kapp and kcat. The extension domain also strengthens the enzyme-tRNA binding affinity both in the binary complex and in the transition-state complex, although the effect of the domain on the latter is greater. However, the extension domain does not affect the rate-limiting step in steady-state kinetics, because both the wild-type and ΔC deletion enzymes exhibit burst kinetics, indicating that product release is rate-limiting. Mutational analysis reveals that the presence of the C-terminal extension enables the wild-type human CysRS to better discriminate against mutations, particularly in the anticodon region, than E. coli CysRS (Table 2, bottom half). Thus, it appears that the C-terminal extension uses its ability to modulate the rate of chemistry to endow the wild-type human CysRS with a greater capacity to examine the anticodon sequence. The enforced examination of the anticodon sequence conferred by the C-terminal extension is consistent with the proximity of the domain to the anticodon-binding site of the tRNA, as suggested both from the crystal structure of the E. coli homolog bound to tRNA, and from the results of the nuclease protection assay (Fig. 5).
The crystal structure of the E. coli CysRS-tRNACys complex shows that recognition of the anticodon is mediated by a major rearrangement of the anticodon-binding domain to provide direct hydrogen-bonds with base-specific functional groups of all three anticodon bases: D436 and R427 recognize G34; R439 and D451 recognize C35; and D451 recognizes A36 20. The most extensive contacts are made with G34, which is stabilized by an additional stacking interaction with the indole ring of W432. All of these amino acids emanate from the anticodon-binding domain and are conserved in human CysRS (D615, R603, R617, E630, and L611, respectively), suggesting that the direct readout mechanism for anticodon recognition is conserved from bacteria to humans. In the context of this conservation, since all of the direct interactions that are required for specificity are already made by conserved amino acids, it is reasonable to suppose that the improved specificity of human CysRS for anticodon is derived from an indirect role of the C-terminal extension. In this indirect role, the C-terminal extension may mediate the necessary conformational rearrangement of the preceding anticodon-binding domain of human CysRS to interact with G34 of the anticodon, as suggested by the footprinting experiments (Fig. 5). Future structural analysis of human CysRS will shed more light on how the C-terminal extension mediates the conformational rearrangement of the anticodon-binding domain.
The effects of the C-terminal extension in providing discrimination at the U73 and G15-C48 positions may arise from a tightening or stabilization of anticodon binding. This stabilization may in turn transmit a signal to the active site in the induced-fit process that likely follows tRNA binding. This domain-domain communication has recently been explicitly demonstrated for E. coli CysRS: physical separation of the anticodon-binding domain from the active-site domain 27, or separation of the anticodon stem-loop from the acceptor end in tRNACys 37, irreversibly severs the communication. Although the mechanism of communication is not well understood, structural analysis suggests that it includes both the ordering of the entire anticodon-binding domain and bending of the tRNA D-anticodon stem toward the enzyme 20.
The potential role of the C-terminal extension in the enzyme induced-fit process that follows tRNA binding is structurally plausible. In the E. coli CysRS-tRNACys complex, the C-terminus is spatially located between the helical bundle and anticodon-binding domains (Fig. 1B). The helical bundle domain makes contact with the anticodon stem and tertiary core, but leaves a substantial solvent accessibility around the core region 20. Possibly, the C-terminal extension domain may extend toward the tertiary core to complement the function of the helical bundle domain. Indeed, the C-terminal extension has an effect on discrimination of tRNA mutations in the core (Table 2), and T1 mapping has identified protection in the core enhanced by the extension (such as G19, Fig. 5). However, it appears unlikely on structural grounds that the new domain could extend far enough to make new contacts in the vicinity of U73. Thus, an induced-fit mechanism seems necessary to explain the effects on specificity at this position.
Quality control and adaptation to codon bias
The improved specificity for tRNA recognition promoted by the C-terminal extension is accompanied by a nearly 10-fold decrease in the single turnover rate constant kapp and by a 10-fold increase in the steady-state enzyme turnover kcat. This suggests the possibility of a kinetic quality control provided by the extension domain, whereby the quality of aminoacylation is largely controlled by kinetics, rather than by equilibrium binding. The term “quality control” is generally used to indicate an active rejection step. However, we extend its use here to include the involvement of a protein domain in slowing the rate of a required induced-fit conformational rearrangement, with the effect of improving specificity. Importantly, while the C-terminal extension domain contributes to kapp presumably by modulating a slow enzyme conformational rearrangement that is required for aminoacyl transfer, the domain does not alter the rate-limiting product release step. The appearance of burst kinetics for the wild-type and ΔC enzymes suggests that both are limited by product release. For the wild-type enzyme, the basis of the rate-limiting product release may be due to a higher enzyme affinity for the Cys-tRNACys product than for the tRNACys substrate, as is the case for the wild-type E. coli CysRS 35, which also exhibits burst kinetics. However, for the ΔC mutant, the nature of the rate-limiting product release may be more complicated. Possibly, the reduced kcat of the ΔC mutant compared to the wild-type might reflect a higher product release barrier that is associated with tighter affinity of the mutant to the Cys-tRNACys product than the wild-type. Alternatively, the reduced kcat of the ΔC mutant might reflect a defective ability to resume a functional active site after product release. Regardless of the nature of the C-terminal domain on kcat, it is clear that the domain impacts on both kapp and kcat, and that it may achieve the dual functions by slowing down the early steps of the aminoacylation pathway that involve tRNA-binding, induced-fit, and/or product formation on the enzyme, while at later stages it also accelerates release of aa-tRNA and facilitates enzyme turnovers.
While further studies are necessary to elucidate how the C-terminal extension of human CysRS alters the kinetic determinants of tRNA aminoacylation, the concept of a kinetic quality control appears crucial in other macromolecular systems as well. In particular, kinetic control is the key feature by which the ribosome enforces selectivity for aa-tRNA, as it is brought to the A-site in a ternary complex with the elongation factor EF-Tu and GTP. In that system, selectivity is dependent on the match between the anticodon of the aa-tRNA and codon of the mRNA on the ribosome. To ensure selectivity, the ribosome slows down the rate of GTP hydrolysis for near- and non-cognate aa-tRNA, relative to the rate of cognate aa-tRNA, in order to allow time for rejection of incorrect species 38. For incorrect aa-tRNAs, the ribosome also slows down the rates of accommodation to the A site and formation of peptide bond 38, providing additional levels of kinetic quality control after GTP hydrolysis.
Two features of the C-terminal extension of human CysRS deserve mention. First, the ability of this domain to improve kcat for aminoacylation is in contrast to the inhibitory effect of a similar C-terminal extension of human MetRS, which reduces kcat 11. In MetRS, the reduction in kcat has been interpreted as a means by which the C-terminal extension helps retain the aa-tRNA product for subsequent transfer to the eukaryotic EF-1α. Because MetRS is a member of the eukaryotic multi-synthetase complex that has direct interactions with EF-1α, its C-terminal extension may coordinate between product release and the EF-1α-assisted delivery of product to the ribosome. Second, the C-terminal extension of human CysRS contains lysine-rich clusters that are conserved among eukaryotic CysRS enzymes (Fig. 1A). This suggests the possibility that the C-terminal extension makes direct interactions with the negatively charged sugar-phosphate backbone of the tRNA. Such lysine-rich clusters also feature prominently in extension domains of many eukaryotic synthetases. For example, lysine-rich clusters in MetRS increase the affinity of the synthetase for the cognate tRNA 11, although whether these clusters are directed to a specific region of tRNA is unknown. In AspRS, AsnRS, and LysRS, lysine-rich clusters promote non-specific general tRNA binding 39. Thus, depending on sequence context, lysine-rich clusters may be exploited to enhance specific or non-specific interactions with tRNA.
The quality control provided by the C-terminal extension is an evolutionary development that appears to have been acquired by eukaryotic CysRSs to improve anticodon recognition over their bacterial counterparts. The improved specificity may simply reflect the challenges imposed by a larger and more complex tRNA pool in eukaryotes than in bacteria. Alternatively, the improved specificity may be an adaptive function in response to changes in codon usage from bacteria to humans. In all living organisms, the GCA sequence is the only anticodon for cysteine. If CysRS mis-reads the anticodon as CCA, it will synthesize Cys-tRNATrp, which will deliver cysteine to the tryptophan codon. Importantly, if CysRS mis-reads the anticodon as UCA, it will synthesize a cysteine opal suppressor, which will interfere with the ability of UGA to function as a stop codon, or will compete with tRNASel for the selenocysteine codon within coding regions. Interestingly, while UGA is modestly used in bacteria (29% among the three stop codons), it is the most heavily used stop codon in humans (47%) 40. The shift of codon bias from bacteria to humans thus imposes a stronger demand for G34 in the anticodon sequence. Given the data presented here, the C-terminal extension of human CysRS seems to fulfill this role by specifically strengthening enzyme recognition of G34.
The kinetic quality control of anticodon recognition is distinct from the previously characterized editing quality controls of tRNA synthetases, which monitor the selection of the amino acid substrate 41; 42. In the amino acid proofreading mechanism, for example, IleRS misactivates valine at a frequency (one in 180, 43) significantly higher than the error rate of protein synthesis (one in 3,000) 1. The reason that this inability to discriminate against valine does not compromise the fidelity of translation is that IleRS has an editing activity that removes valine from tRNAIle 44. Similar editing reactions exist for ValRS, LeuRS, ProRS, ThrRS, AlaRS, and PheRS 2; 45; 46. Recent studies have identified the editing domains in some of these synthetases 2; 47, as well as free-standing editing domains, which include those naturally present 48, those derived artificially from multi-domains 49, and the widespread D-Tyr-tRNATyr deacylases that catalyze removal of D-tyrosine from tRNATyr due to mis-acylation of TyrRS 50. While the quality control provided by these editing domains is manifested in an explicit hydrolytic activity, whereas that provided by the C-terminal extension of human CysRS functions by improving specificity in the synthetic reaction, both types of control operate to reduce errors in protein synthesis. The development of distinct mechanisms for quality control emphasize the high priority that the cellular machinery places on developing aaRSs to accurately match tRNAs with amino acids before these molecules enter the protein synthesis cycle.
Materials and Methods
Construction of plasmids and mutants
The expression plasmid for the full-length cDNA of human CysRS was constructed in the pET-24d vector 18. To construct the plasmid for the ΔC mutant, the full-length plasmid was restricted with AccI and XhoI, and the large fragment was isolated and re-ligated, so that the resulting plasmid encodes amino acids 1–642 with a His6-tag at the C-terminus. The gene for human tRNACys was cloned into the pTFMa vector behind the bacterial phage T7 RNA polymerase promoter 22. Mutations in tRNAs were generated by QuickChange mutagenesis (Stratagene, La Jolla, CA) and were confirmed by DNA sequence analysis.
Purification of proteins and tRNAs
The human and E. coli full-length CysRS enzymes and the human ΔC mutant, each bearing a C-terminal His6-tag, were over-expressed in the E. coli strain BL21(DE3). Culturing for the full-length enzymes was at 37 °C for 4 hours in LB in the presence of 100 μg/ml ampicillin, while the ΔC mutant was grown at room temperature for 9 hours, before induction with 0.4 mM IPTG (isopropyl-β-D-thiogalatoside). Purification of enzymes was achieved using the Talon resin (BD Clonetech), followed by Mono Q chromatography on an FPLC (Amersham Biosciences). The final products were homogeneous as assessed by SDS-PAGE. Enzyme concentrations were determined by the Bradford assay using BSA as a standard, and were further corrected by the active site burst assay 51. The tRNA gene was transcribed by T7 RNA polymerase from plasmids restricted with BstN1 (for E. coli tRNA) or Nsi I (for human tRNA). Transcripts of tRNAs were purified by a 12% PAGE/urea gel, excised, crushed and soaked in TE (10 mM Tris-HCl, pH 8.0, 1 mM EDTA). After ethanol precipitation, tRNAs were stored in TE and concentrations determined by absorption at 260 nm (1 OD260 unit equivalent to 40 μg/ml), and further corrected using plateau aminoacylation levels.
Gel filtration
Full-length CysRS and the ΔC mutant were analyzed by Superose 12 column chromatography on an Akta FPLC system (Amersham Biosciences), run at a flow rate of 0.75 ml/min at ambient temperature in 20 mM Tris-HCl (pH 7.5), 12 mM MgCl2, 5 mM β-mercaptoethanol. The condition of gel filtration of the variant lacking the insertion domain of the Rossmann-fold was slightly modified. The molecular mass markers (BioRad) for calibration consisted of: thyroglobulin (670 kDa), bovine γ-globulin (158 kDa), chicken ovalbumin (44 kDa), equine myoglobin (17 kDa), and vitamin B12 (1.4 kDa).
Size Exclusion Chromatography Light Scattering (SEC-LS)
The molecular weights of full-length human CysRS and the C mutant were determined at room temperature at a flow rate of 0.6 ml/min, in a buffer containing 20 mM Tris-HCl (pH 7.5), 12 mM MgCl2, 5 mM β-mercaptoethanol, 5% glycerol, and 150 mM NaCl. A sample containing 2 mg/ml of protein was filtered through a 0.22 uM Durapore membrane (Millipore) and applied to a Superdex 200 HR10/30 column coupled with an HPLC, a miniDawn laser light-scattering apparatus (Wyatt Technology Corp.), refractometer (Wyatt Technology Corp.), and UV detector. The weight average molecular weight of the elution peaks were calculated using ASTRA software, as described 52.
Determination of Kd by tryptophan fluorescence quenching
Equilibrium titrations were performed at room temperature with 0.1 μM full-length or ΔC human CysRS in 20 mM Tris-HCl (pH 6.8), 50 mM NaCl, 12 mM MgCl2, and 5 mM β-mercaptoethanol. Tryptophan fluorescence was excited at 295 nm and the emission was monitored from 310–400 nm. An emission wavelength of 350 nm was used to quantify binding after correction for dilution and for the inner filter effect 31. Control solutions of BSA or of tryptophan showed no fluorescence response to tRNA. The tRNA was titrated in the concentration range 0–12 μM for the full length CysRS and up to 20 μM for the ΔC mutant. The Kd values were determined by fitting the data to a hyperbolic decay equation using the KaleidaGraph software.
Steady-state and pre-steady-state kinetics
Steady-state aminoacylation was performed as described 24. The ATP-PPi exchange reaction was performed as described, using 1 mM γ-32P-ATP, 75 nM CysRS, and 4 to 128 μM cysteine 27. Reaction products were resolved on a PEI cellulose plate (Sigma) as described 53. Apparent parameters Km, kcat, and kcat/Km were derived by fitting the data to the Michaelis-Menten equation using SigmaPlot (SPSS), and were reported as the average of at least three independent assays. Transient kinetics experiments were performed at 37 °C on a rapid chemical quench-flow apparatus (RQF-3; KinTek) 34; 35; 54. Single-turnover measurements to determine kapp for aminoacylation were performed by mixing saturating concentrations of 35S-cysteine (0.5 mM) and ATP (6.25 mM) and indicated amounts of tRNA in one syringe, while CysRS in the second syringe. To maintain the single turnover condition, the concentrations of CysRS were in excess of tRNA so that the reaction could turn over only once. Burst kinetics was conducted with CysRS and tRNACys at a 1:10 molar ratio in the same buffer. Reaction aliquots of 15 μL were quenched in 2.5 M sodium acetate, pH 5.0, and mixed with 100 μl of a 5 × CAM solution (1.2 M iodoacetic acid and 0.5 M sodium acetate, pH 5.0), 40 μL of which was analyzed by acid precipitation of Cys-tRNACys on filter pads.
Determination of kinetic constants for tRNA mutants
The kcat/Km values were determined by measuring the initial rate of aminoacylation using concentrations of tRNA at least 10-fold below the Km. The Km was estimated for each mutant by preliminary titrations, and concentrations were then chosen to assure kcat/Km conditions in each case. The concentrations used were: 0.2–0.4 μM for mutants in the tRNA core region, 1–2 μM for mutants of the anticodon, and 10 μM for mutants with substitutions of U73.
T1 mapping
The human tRNACys transcript was 3′-end labeled using the CCA-adding enzyme and purified by a 12% PAGE/urea gel. The labeled tRNA (1 μM, 105 cpm) was incubated with CysRS (60 μM) in 20 mM Mes-KOH, pH 5.5, 1.2 mM MgCl2, 5 mM β-mercaptoethanol at room temperature for 20 min. Nuclease T1 (Roche, 0.7 units) was added to the CysRS-tRNA complex and incubated at room temperature for 20 min. Cleavage fragments of tRNA were resolved on a 10% PAGE/urea gel (20cm × 40 cm × 0.4 mm) run at 1850 volts for 2.5 hours, and visualized by phosphorimaging (Molecular Dynamics).
Acknowledgments
This work was supported by US National Institutes of Health grants GM56662 (to YMH) and GM63713 (to JJP). We thank Chun-Mei Zhang and Caryn Evilia for discussion.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Loftfield RB, Vanderjagt D. The frequency of errors in protein biosynthesis. Biochem J. 1972;128:1353–6. doi: 10.1042/bj1281353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ibba M, Francklyn C, Cusack S. In: The Aminoacyl-tRNA Synthetases. Ibba M, Francklyn C, Cusack S, editors. Landes Bioscience, Georgetown; Texas: 2005. [Google Scholar]
- 3.Rodnina MV, Gromadski KB, Kothe U, Wieden HJ. Recognition and selection of tRNA in translation. FEBS Lett. 2005;579:938–42. doi: 10.1016/j.febslet.2004.11.048. [DOI] [PubMed] [Google Scholar]
- 4.Fersht AR. Sieves in sequence. Science. 1998;280:541. doi: 10.1126/science.280.5363.541. [DOI] [PubMed] [Google Scholar]
- 5.Mirande M. Multi-aminoacyl-tRNA synthetase complexes. In: Ibba M, Francklyn C, Cusack S, editors. The aminoacyl-tRNA synthetases. Landes Bioscience; Georgetown, Texas: 2005. pp. 298–308. [Google Scholar]
- 6.Wolfe CL, Warrington JA, Treadwell L, Norcum MT. A three-dimensional working model of the multienzyme complex of aminoacyl-tRNA synthetases based on electron microscopic placements of tRNA and proteins. J Biol Chem. 2005;280:38870–8. doi: 10.1074/jbc.M502759200. [DOI] [PubMed] [Google Scholar]
- 7.Park SG, Ewalt KL, Kim S. Functional expansion of aminoacyl-tRNA synthetases and their interacting factors: new perspectives on housekeepers. Trends Biochem Sci. 2005;30:569–74. doi: 10.1016/j.tibs.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 8.Negrutskii BS, Stapulionis R, Deutscher MP. Supramolecular organization of the mammalian translation system. Proc Natl Acad Sci U S A. 1994;91:964–8. doi: 10.1073/pnas.91.3.964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Francin M, Kaminska M, Kerjan P, Mirande M. The N-terminal domain of mammalian Lysyl-tRNA synthetase is a functional tRNA-binding domain. J Biol Chem. 2002;277:1762–9. doi: 10.1074/jbc.M109759200. [DOI] [PubMed] [Google Scholar]
- 10.Cahuzac B, Berthonneau E, Birlirakis N, Guittet E, Mirande M. A recurrent RNA-binding domain is appended to eukaryotic aminoacyl-tRNA synthetases. Embo J. 2000;19:445–52. doi: 10.1093/emboj/19.3.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaminska M, Shalak V, Mirande M. The appended C-domain of human methionyl-tRNA synthetase has a tRNA- sequestering function. Biochemistry. 2001;40:14309–16. doi: 10.1021/bi015670b. [DOI] [PubMed] [Google Scholar]
- 12.Robinson JC, Kerjan P, Mirande M. Macromolecular assemblage of aminoacyl-tRNA synthetases: quantitative analysis of protein-protein interactions and mechanism of complex assembly. J Mol Biol. 2000;304:983–94. doi: 10.1006/jmbi.2000.4242. [DOI] [PubMed] [Google Scholar]
- 13.Rho SB, Kim MJ, Lee JS, Seol W, Motegi H, Kim S, Shiba K. Genetic dissection of protein-protein interactions in multi-tRNA synthetase complex. Proc Natl Acad Sci U S A. 1999;96:4488–93. doi: 10.1073/pnas.96.8.4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ling C, Yao YN, Zheng YG, Wei H, Wang L, Wu XF, Wang ED. The C-terminal Appended Domain of Human Cytosolic Leucyl-tRNA Synthetase Is Indispensable in Its Interaction with Arginyl-tRNA Synthetase in the Multi-tRNA Synthetase Complex. J Biol Chem. 2005;280:34755–63. doi: 10.1074/jbc.M413511200. [DOI] [PubMed] [Google Scholar]
- 15.Mirande M, Kellermann O, Waller JP. Macromolecular complexes from sheep and rabbit containing seven aminoacyl-tRNA synthetases. II. Structural characterization of the polypeptide components and immunological identification of the methionyl-tRNA synthetase subunit. J Biol Chem. 1982;257:11049–55. [PubMed] [Google Scholar]
- 16.Shiba K, Suzuki N, Shigesada K, Namba Y, Schimmel P, Noda T. Human cytoplasmic isoleucyl-tRNA synthetase: selective divergence of the anticodon-binding domain and acquisition of a new structural unit. Proc Natl Acad Sci U S A. 1994;91:7435–9. doi: 10.1073/pnas.91.16.7435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Raben N, Nichols R, Dohlman J, McPhie P, Sridhar V, Hyde C, Leff R, Plotz P. A motif in human histidyl-tRNA synthetase which is shared among several aminoacyl-tRNA synthetases is a coiled-coil that is essential for enzymatic activity and contains the major autoantigenic epitope. J Biol Chem. 1994;269:24277–83. [PubMed] [Google Scholar]
- 18.Davidson E, Caffarella J, Vitseva O, Hou YM, King MP. Isolation of two cDNAs encoding functional human cytoplasmic cysteinyl- tRNA synthetase. Biol Chem. 2001;382:399–406. doi: 10.1515/BC.2001.049. [DOI] [PubMed] [Google Scholar]
- 19.Newberry KJ, Hou YM, Perona JJ. Structural origins of amino acid selection without editing by cysteinyl- tRNA synthetase. Embo J. 2002;21:2778–2787. doi: 10.1093/emboj/21.11.2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hauenstein S, Zhang CM, Hou YM, Perona JJ. Shape-selective RNA recognition by cysteinyl-tRNA synthetase. Nat Struct Mol Biol. 2004;11:1134–41. doi: 10.1038/nsmb849. [DOI] [PubMed] [Google Scholar]
- 21.Komatsoulis GA, Abelson J. Recognition of tRNA(Cys) by Escherichia coli cysteinyl-tRNA synthetase [published erratum appears in Biochemistry 1993 Dec 7;32(48):13374] Biochemistry. 1993;32:7435–44. doi: 10.1021/bi00080a014. [DOI] [PubMed] [Google Scholar]
- 22.Lipman RS, Hou YM. Aminoacylation of tRNA in the evolution of an aminoacyl-tRNA synthetase. Proc Natl Acad Sci U S A. 1998;95:13495–500. doi: 10.1073/pnas.95.23.13495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hou YM, Motegi H, Lipman RS, Hamann CS, Shiba K. Conservation of a tRNA core for aminoacylation. Nucleic Acids Res. 1999;27:4743–50. doi: 10.1093/nar/27.24.4743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hou YM, Westhof E, Giege R. An unusual RNA tertiary interaction has a role for the specific aminoacylation of a transfer RNA. Proc Natl Acad Sci U S A. 1993;90:6776–80. doi: 10.1073/pnas.90.14.6776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Motorin Y, Le Caer JP, Waller JP. Cysteinyl-tRNA synthetase from Saccharomyces cerevisiae. Purification, characterization and assignment to the genomic sequence YNL247w. Biochimie. 1997;79:731–40. doi: 10.1016/s0300-9084(97)86931-3. [DOI] [PubMed] [Google Scholar]
- 26.Motorin Y, Waller JP. Purification and properties of cysteinyl-tRNA synthetase from rabbit liver. Biochimie. 1998;80:579–90. doi: 10.1016/s0300-9084(98)80010-2. [DOI] [PubMed] [Google Scholar]
- 27.Zhang CM, Hou YM. Domain-domain communication for tRNA aminoacylation: the importance of covalent connectivity. Biochemistry. 2005;44:7240–9. doi: 10.1021/bi050285y. [DOI] [PubMed] [Google Scholar]
- 28.Yaremchuk A, Kriklivyi I, Tukalo M, Cusack S. Class I tyrosyl-tRNA synthetase has a class II mode of cognate tRNA recognition. Embo J. 2002;21:3829–3840. doi: 10.1093/emboj/cdf373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Doublie S, Bricogne G, Gilmore C, Carter CW., Jr Tryptophanyl-tRNA synthetase crystal structure reveals an unexpected homology to tyrosyl-tRNA synthetase. Structure. 1995;3:17–31. doi: 10.1016/s0969-2126(01)00132-0. [DOI] [PubMed] [Google Scholar]
- 30.Yang XL, Otero FJ, Ewalt KL, Liu J, Swairjo MA, Kohrer C, Rajbhandary UL, Skene RJ, McRee DE, Schimmel P. Two conformations of a crystalline human tRNA synthetase-tRNA complex: implications for protein synthesis. Embo J. 2006;25:2919–29. doi: 10.1038/sj.emboj.7601154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lakowicz J. Principles of fluorescence spectroscopy. 2. Kluwer Academic/Plenum Publishers; New York: 1999. [Google Scholar]
- 32.Lipman RS, Chen J, Evilia C, Vitseva O, Hou YM. Association of an Aminoacyl-tRNA Synthetase with a Putative Metabolic Protein in Archaea. Biochemistry. 2003;42:7487–96. doi: 10.1021/bi0344533. [DOI] [PubMed] [Google Scholar]
- 33.Bovee ML, Yan W, Sproat BS, Francklyn CS. tRNA discrimination at the binding step by a class II aminoacyl-tRNA synthetase. Biochemistry. 1999;38:13725–35. doi: 10.1021/bi991182g. [DOI] [PubMed] [Google Scholar]
- 34.Uter NT, Perona JJ. Long-range intramolecular signaling in a tRNA synthetase complex revealed by pre-steady-state kinetics. Proc Natl Acad Sci U S A. 2004;101:14396–401. doi: 10.1073/pnas.0404017101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang CM, Perona JJ, Ryu K, Francklyn C, Hou YM. Distinct Kinetic Mechanisms of the Two Classes of Aminoacyl-tRNA Synthetases. J Mol Biol. 2006;361:300–11. doi: 10.1016/j.jmb.2006.06.015. [DOI] [PubMed] [Google Scholar]
- 36.Johnson KA. Transient state kinetic analysis of enzyme reaction pathways. The Enzymes. 1992;XX:1–61. [Google Scholar]
- 37.Hamann CS, Hou YM. Enzymatic aminoacylation of tRNA acceptor stem helices with cysteine is dependent on a single nucleotide. Biochemistry. 1995;34:6527–32. doi: 10.1021/bi00019a034. [DOI] [PubMed] [Google Scholar]
- 38.Rodnina MV, Wintermeyer W. Fidelity of aminoacyl-tRNA selection on the ribosome: kinetic and structural mechanisms. Annu Rev Biochem. 2001;70:415–35. doi: 10.1146/annurev.biochem.70.1.415. [DOI] [PubMed] [Google Scholar]
- 39.Frugier M, Moulinier L, Giege R. A domain in the N-terminal extension of class IIb eukaryotic aminoacyl- tRNA synthetases is important for tRNA binding. Embo J. 2000;19:2371–80. doi: 10.1093/emboj/19.10.2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nakamura Y, Gojobori T, Ikemura T. Codon usage tabulated from international DNA sequence databases: status for the year 2000. Nucleic Acids Res. 2000;28:292. doi: 10.1093/nar/28.1.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Freist W. Mechanisms of aminoacyl-tRNA synthetases: a critical consideration of recent results. Biochemistry. 1989;28:6787–95. doi: 10.1021/bi00443a001. [DOI] [PubMed] [Google Scholar]
- 42.Jakubowski H. Proofreading in vivo. Editing of homocysteine by aminoacyl-tRNA synthetases in Escherichia coli. J Biol Chem. 1995;270:17672–3. [PubMed] [Google Scholar]
- 43.Baldwin AN, Berg P. Transfer ribonucleic acid-induced hydrolysis of valyladenylate bound to isoleucyl ribonucleic acid synthetase. J Biol Chem. 1966;241:839–45. [PubMed] [Google Scholar]
- 44.Schmidt E, Schimmel P. Mutational isolation of a sieve for editing in a transfer RNA synthetase. Science. 1994;264:265–7. doi: 10.1126/science.8146659. [DOI] [PubMed] [Google Scholar]
- 45.Swairjo MA, Schimmel PR. Breaking sieve for steric exclusion of a noncognate amino acid from active site of a tRNA synthetase. Proc Natl Acad Sci U S A. 2005;102:988–93. doi: 10.1073/pnas.0409024102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roy H, Ling J, Alfonzo J, Ibba M. Loss of editing activity during the evolution of mitochondrial phenylalanyl-tRNA synthetase. J Biol Chem. 2005;280:38186–92. doi: 10.1074/jbc.M508281200. [DOI] [PubMed] [Google Scholar]
- 47.Dock-Bregeon AC, Rees B, Torres-Larios A, Bey G, Caillet J, Moras D. Achieving Error-Free Translation; The Mechanism of Proofreading of Threonyl-tRNA Synthetase at Atomic Resolution. Mol Cell. 2004;16:375–86. doi: 10.1016/j.molcel.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 48.Ahel I, Korencic D, Ibba M, Soll D. Trans-editing of mischarged tRNAs. Proc Natl Acad Sci U S A. 2003;100:15422–7. doi: 10.1073/pnas.2136934100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wong FC, Beuning PJ, Silvers C, Musier-Forsyth K. An isolated class II aminoacyl-tRNA synthetase insertion domain is functional in amino acid editing. J Biol Chem. 2003;278:52857–64. doi: 10.1074/jbc.M309627200. [DOI] [PubMed] [Google Scholar]
- 50.Rigden DJ. Archaea recruited D-Tyr-tRNATyr deacylase for editing in Thr-tRNA synthetase. Rna. 2004;10:1845–51. doi: 10.1261/rna.7115404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fersht AR, Ashford JS, Bruton CJ, Jakes R, Koch GL, Hartley BS. Active site titration and aminoacyl adenylate binding stoichiometry of aminoacyl-tRNA synthetases. Biochemistry. 1975;14:1–4. doi: 10.1021/bi00672a001. [DOI] [PubMed] [Google Scholar]
- 52.Folta-Stogniew E, Williams K. Determination of molecular masses of proteins in solution: Implementation of an HPLC size exclusion chromatography and laser light scattering service in a core laboratory. J Biomol Techniques. 1999;10:51–63. [PMC free article] [PubMed] [Google Scholar]
- 53.Uter NT, Gruic-Sovulj I, Perona JJ. Amino acid-dependent transfer RNA affinity in a class I aminoacyl-tRNA synthetase. J Biol Chem. 2005;280:23966–77. doi: 10.1074/jbc.M414259200. [DOI] [PubMed] [Google Scholar]
- 54.Guth E, Connolly SH, Bovee M, Francklyn CS. A substrate-assisted concerted mechanism for aminoacylation by a class II aminoacyl-tRNA synthetase. Biochemistry. 2005;44:3785–94. doi: 10.1021/bi047923h. [DOI] [PubMed] [Google Scholar]