

Abstract
AKXD recombinant inbred (RI) strains develop a variety of leukemias and lymphomas due to somatically acquired insertions of retroviral DNA into the genome of hematopoetic cells that can mutate cellular proto-oncogenes and tumor suppressor genes. We generated a new set of tumors from nine AKXD RI strains selected for their propensity to develop B-cell tumors, the most common type of human hematopoietic cancers. We employed a PCR technique called viral insertion site amplification (VISA) to rapidly isolate genomic sequence at the site of provirus insertion. Here we describe 550 VISA sequence tags (VSTs) that identify 74 common insertion sites (CISs), of which 21 have not been identified previously. Several suspected proto-oncogenes and tumor suppressor genes lie near CISs, providing supportive evidence for their roles in cancer. Furthermore, numerous previously uncharacterized genes lie near CISs, providing a pool of candidate disease genes for future research. Pathway analysis of candidate genes identified several signaling pathways as common and powerful routes to blood cancer, including Notch, E-protein, NFκB, and Ras signaling. Misregulation of several Notch signaling genes was confirmed by quantitative RT-PCR. Our data suggest that analyses of insertional mutagenesis on a single genetic background are biased toward the identification of cooperating mutations. This tumor collection represents the most comprehensive study of the genetics of B-cell leukemia and lymphoma development in mice. We have deposited the VST sequences, CISs in a genome viewer, histopathology, and molecular tumor typing data in a public web database called VISION (Viral Insertion Sites Identifying Oncogenes), which is located at http://www.mouse-genome.bcm.tmc.edu/vision.
Introduction
The oncogenic transformation of a cell requires several mutations conferring abnormal properties that may include independence from the requirement for growth stimulatory signals, loss of sensitivity to growth inhibitory signals, the acquisition of unlimited proliferative capacity, suppression of apoptotic signals, and the successful evasion of immune responses (Dunn et al. 2002; Hanahan and Weinberg 2000). A common feature of human leukemias and lymphomas is nonrandom, somatically acquired chromosomal translocations and inversions that are often diagnostic criteria of specific subtypes of malignancies. It has been estimated that 65% of all cases of human acute leukemias involve chromosomal abnormalities (Look 1997). Oncogenic translocations frequently act by upregulation of proto-oncogenes or inactivation of tumor suppressor genes (TSGs).
In the mouse, loci involved in the onset and progression of hematologic malignancies have been identified primarily by the isolation of common proviral insertion sites (CISs). Slow-transforming retroviruses cause a subset of reinfected cells to become oncogenic by altering the expression of genes near their insertion sites in a manner reminiscent of translocations in human malignancy. Aberrant gene expression can be caused by placing the affected locus under the direct control of viral long terminal repeat (LTR) promoters, long-range enhancer activity of the LTRs, disruption of the affected locus by proviral insertion within coding sequences, or local alterations of chromatin. The rare cases in which an insertion affects the expression of a proto-oncogene or TSG can result in the neoplastic transformation and clonal expansion of the affected cell. Because proviral insertions are near the loci whose expression they alter, these insertions can be used as a molecular tag to isolate and identify affected loci. Identification of these loci can be important for understanding the pathogenesis of human neoplasms because genes proximal to viral insertion sites in mouse tumors are often deregulated in human blood cancers, highlighting the applicability of slow-transforming retroviral mutagenesis as a model for human disease (Erkeland et al. 2006). Systematic surveys of CISs found in well-established mouse models of lymphoid and myeloid malignancies have implicated a wide range of genes involved in a diversity of cellular functions as potential oncogenes and TSGs. As with chromosomal translocations in human leukemia and lymphomas, many genes implicated in tumor incidence are known to be key regulators of normal hematopoiesis or have hematopoietic lineage-specific expression patterns (Suzuki et al. 2002). In addition, insertion sites that are commonly found in a particular class of neoplasia may identify loci whose altered expression is a common event in the development of that cancer subtype (reviewed in Jonkers and Berns 1996). The AKXD mouse strains chosen for the present study were previously found to develop a high incidence of B-cell lymphomas allowing us to focus our efforts on understanding the molecular basis underlying the pathogenesis of the most common human hematopoietic neoplasms (http://www.seer.cancer.gov).
The AKXD RI strains were generated from crosses of the T-cell lymphoma-prone AKR/J strain of mice with the DBA/2J strain of mice to generate 21 highly lymphomatous strains that vary in tumor incidence and tumor subtype. High rates of lymphoma correlated with the presence of transposable MuLV, indicating that MuLV is the primary mutagen in this model. Seven strains develop primarily B-cell neoplasms, whereas six develop T-cell, seven mixed B- and T-cell, and one primarily myeloid neoplasia (Gilbert et al. 1993). Many other mouse strains used for studying the molecular genetics of tumor development develop primarily myeloid disease (Eriksson et al. 1999; Jonkers et al. 1997; Liao et al. 1997; Moskow et al. 1995; Nakamura et al. 1996; Shaughnessy et al. 1999; Szymanska et al. 1999; Valk et al. 1997; van Lohuizen and Berns 1990; for review see Voisin et al. 2006), with the exception of the NFS.V+ strain, which also develops predominantly B-cell lineage neoplasms (Hartley et al. 2000). Each of these strains has proven to be very useful for identifying potential oncogenes and TSGs involved in the development of lymphoid and myeloid malignancies.
In this article we describe a new tumor subset from AKXD mice and perform a complete genomic analysis of the insertion sites. The use of our novel viral insertion site amplification (VISA) technique permits the rapid sequence-based identification of loci that potentially play a role in the development of these tumors (Hansen et al. 2000). The VISA approach is based on a technique called restriction-site PCR (Sarkar et al. 1993), which allows priming from a single site flanking the provirus with no prior knowledge of the genomic sequence and does not require a subcloning step, reducing false-positive detection. Here we have isolated 550 VISA sequence tags (VSTs) from screening tumors that developed in 204 mice from nine AKXD strains. These tags identify 74 CISs, of which 21 have not previously been identified, and represent new candidate proto-oncogenes and TSGs for lymphoid tumors.
Materials and methods
AKXD mice
Animals from nine AKXD RI strains of mice that develop either predominantly B-cell or mixed T/B-cell lymphomas were obtained from The Jackson Laboratory (Bar Harbor, ME), aged to 18 months, and monitored for signs of illness. Moribund animals or normal animals at the age of 18 months were autopsied and evaluated. Both normal and tumor tissues from a single lymphomatous animal was snap-frozen in liquid nitrogen.
Classification of tumors
A portion of each tumor was fixed in 10% formalin and embedded in paraffin. These fixed tumors were analyzed for histopathology as previously described (Morse et al. 2001) at the Laboratory of Immunopathology at the National Institute of Allergy and Infectious Diseases at the National Institutes of Health (Bethesda, MD).
Genomic DNA was extracted from both normal and tumor tissues as described (Wu et al. 1995) followed by a single phenol/chloroform extraction and ethanol precipitation (Sambrook et al. 1989). Restriction enzyme-digested genomic DNAs from tumors were electrophoresed and transblotted (Sambrook et al. 1989). Membranes were prehybridized and hybridized as described (Church and Gilbert 1984). Probes for IgH, IgK, JB1, and JB2 were described previously (Kronenberg et al. 1985; Mucenski et al. 1986). Tumors were classified as B, T, or mixed lineage based on BCR (IgH and IgK) or TCR (JB1 or JB2) gene rearrangements. In cases where histopathology and genomic rearrangements were contradictory, histopathology data took precedence in classifying the tumor, but all information is deposited in the database.
VST identification
The VISA procedure was performed as described previously (Hansen et al. 2000). Briefly, the amplification of mouse genomic DNA flanking proviral insertion elements was performed in a two-step PCR. The first round was performed using a degenerate, restriction-site-specific primer containing the M13 sequence and a nested primer (SP1) specific to the ecotropic MuLV LTR. The second round was performed using 1 μl of first-round PCR product as template, a nested LTR-specific primer, and the M13F primer. The products of second-round PCR were analyzed by gel electrophoresis and the tumor-specific VISA products were excised (reducing endogenous virus detection), purified, and sequenced directly. DNA isolated from brain tissue, which is rarely infiltrated by tumor cells, was used as a control to identify all nontarget amplification products such as endogenous proviral insertion sites, internal proviral sequences, or single primer amplification products generated by the restriction-site-anchored primer. Purified DNAs were amplified using the ABI Prism BigDye Terminator Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) and electrophoresed on an ABI Model 377 DNA sequencer (Applied Biosystems). Sequences were analyzed using Sequencher software (Gene Codes Corp., Ann Arbor, MI). Sequences were edited to remove ambiguous bases and examined for sequence overlap. As a control for specificity, each sequence from the second-round PCR was analyzed to confirm the presence of the target restriction enzyme (RE) recognition site and viral LTR sequence. When a single insertion mutation was amplified multiple times using different RE primers from a single tumor, only the longest sequence was designated as a VISA sequence tag (VST) and was used for further analysis. Repeat-masked VST sequences were compared using BLASTn (ftp://ftp.ncbi.nih.gov/blast/executables/) against the mouse genome assembly (Ensembl release 40 from NCBI assembly m36), which was downloaded from the Ensembl genome annotation website (ftp://ftp.ensembl.org/pub/current_mus_musculus/data/fasta/dna/). The BLAST results were visualized by an in-house-developed program (VST Visualization) to confirm that the highest-ranking alignment was the only highly ranked alignment and the match was not a repeat sequence. All valid hits from the BLAST results were deposited in our MS-SQL Server databases. Based on the information from the BLAST results, VSTs were mapped to the mouse genome (http://www.ensembl.org/Multi/martview), and flanking genes for each of the VSTs were identified. All Ensembl annotated genes within 200 kb of each VST location are displayed in the VISION database (http://www.mouse-genome.bcm.tmc.edu/vision).
Calculation of false positives
The proportion of cancers caused by genetic background versus virus insertion can be differentiated using basic probability:
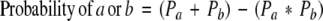 |
Cancer rate in the absence of transposable virus is 12.5% (Mucenski et al. 1986); overall AKXD cancer rate (with transposable virus) is 43% (this article):
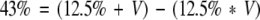 |
where V is the cancer rate due to viral insertion and is 39%. The proportion of cancer that can be attributed to viral insertion in this study is
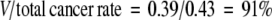 |
Since viral insertion is causative in 91% of cancers and the viral insertion number in cancers is low, one would expect a low rate of false positives. The average number of proviral insertions required to cause cancer in this model is unknown but it must be at least 1 (since viral insertion is the primary mutagen) and less than 5.5 (average viral insertions per tumor). This allows us to calculate a range of true positives that we should expect to recover:
204 tumors * 91% requiring viral insertion * 1–5.5 insertions required to induce cancer = 186–1021 insertions contributing to cancer * 49% detection rate of VISA = 91–500 true positive VSTs recovered
Calculation of degree of saturation
We have used a truncated Poisson distribution to determine the number of undiscovered alleles from the frequency of alleles with more than one mutation (f), using the equation:
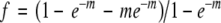 |
(Johnsen 2000) where m is mean number of insertions per locus, f is multiple-hit common insertion loci/total loci (from above):
f = 46/186 = 0.2473
f = 46/1021 = 0.0451
% undiscovered loci = e−m = 58–91%
saturation = 1−e−m = 9–42%
Real-time quantitative RT-PCR (QPCR)
Total RNA was isolated from frozen tumor tissue samples with RNA-stat (Tel-test Inc., Friendswood, TX). cDNA was prepared from 2 μg of RNA with the Superscript II Reverse Transcription Kit (Invitrogen, Carlsbad, CA) using random hexamers. QPCR primers were designed using Primer Express (Applied Biosystems) with amplicons spanning exon junctions in most cases. Optimal concentrations were determined for each primer pair by running all pairwise combinations of 50-, 300-, and 900-nM primers on wild-type mouse blood, spleen, or embryo cDNA. Real-time PCR was performed in the ABI 7000 TaqMan. Applied Biosystems SYBR Green master mix was used with 1 μl of cDNA/well in 96-well optical plates. PCR cycling conditions were 1 × (95°C for 10 min) and 40 × (95°C for 15 sec; 60°C for 1 min), with a dissociation curve performed following amplification.
Results
Screening for cancer loci
A total of 469 male and female mice from nine different AKXD RI strains that develop primarily B-cell or mixed B/T-cell leukemia or lymphoma were aged for 18 months. Although tumor incidence varied by strain, 43% (204/469) of aged mice developed lymphomas (Table 1). Tumors were collected, and histopathology was performed to determine cell type. Somatic recombination of immunoglobulin (IgH, IgK) and T-cell receptor β-chain genes (JB1 and JB2) was analyzed by Southern blot to further refine lymphoid cell typing. The combined histologic and molecular criteria used to determine cell type demonstrate the high frequency of B-cell lineage neoplasms (48%; Table 2) (Morse et al. 2001). The molecular cell type, stage, and nature of each tumor can be found at the VISION database.
Table 1.
AKXD mouse, tumor, and VST information by strain
| Statistics | Total | AKXD strain | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| AK-2 | AK-3 | AK-9 | AK-10 | AK-13 | AK-14 | AK-16 | AK-18 | AK-27 | ||
| Animals aged | 469 | 32 | 96 | 32 | 29 | 58 | 14 | 71 | 63 | 74 |
| Lymphomatous animals | 204 | 7 | 57 | 18 | 4 | 18 | 4 | 13 | 42 | 41 |
| % Tumor incidence | 43 | 22 | 59 | 56 | 14 | 31 | 29 | 18 | 67 | 55 |
| VST distribution | ||||||||||
| in high-confidence CISs | 140 | 6 | 18 | 13 | 1 | 21 | 2 | 6 | 30 | 43 |
| in single-hit CISs | 25 | 0 | 5 | 1 | 0 | 5 | 0 | 3 | 4 | 7 |
| single hits | 237 | 13 | 53 | 18 | 7 | 28 | 6 | 25 | 40 | 47 |
| with no BLAST hits | 28 | 2 | 8 | 1 | 0 | 5 | 0 | 0 | 3 | 9 |
| repeat sequence hits | 120 | 2 | 30 | 19 | 1 | 21 | 2 | 6 | 10 | 29 |
| endogenous site hits1 | 66 | 5 | 21 | 11 | 0 | 13 | 2 | 5 | 2 | 7 |
| total identified | 616 | 28 | 135 | 63 | 9 | 93 | 12 | 45 | 89 | 142 |
Number of animals aged, tumor incidence, and the number of VSTs varied by strain. VST data are broken down by results of genomic alignments and clustering analysis. VSTs that gave no significant BLAST result or mapped to repeat sequences were excluded from further study. Significant BLAST results were analyzed for clustering and multiple hit CISs were constructed. Several multiple-hit CISs were excluded as likely endogenous insertion sites. The remaining nonclustering single hits were compared to other viral mutation cancer screens to identify single-hit CISs (single hits in this screen that map to the location of previously identified CIS)
Table 2.
Distribution of tumors collected from AKXD mice
| Tumor type | % |
|---|---|
| B cell | 27 |
| Pre-B cell | 21 |
| Progenitor | 15 |
| T cell | 13 |
| Pre-T cell | 1 |
| Mixed B/T cell | 13 |
| Myeloid | 4 |
| Undetermined | 6 |
Almost half of the tumors (48%) type as B cell or pre-B cell. Molecular cell type was determined by histopathology and Southern blot of IgH, IgK, Jβ1, and Jβ2 genes to detect somatic recombination in lymphoid tumors
Tumors were screened by the VISA technique to identify the location of somatic retroviral insertion sites (Hansen et al. 2000). To avoid redundancy, multiple sequence tags identified by different restriction enzyme primers from a single animal that overlap were edited into a single VISA sequence tag (VST) representing the longest unique flanking sequence (Fig. 1A). The fact that our technique amplified from the 5′ end of the provirus alone ensured that nonoverlapping tags from a single tumor were not acquired. From over 3000 sequence tags, a total of 550 nonendogenous VSTs were identified in tumors that occurred in 204 animals.
Fig. 1.
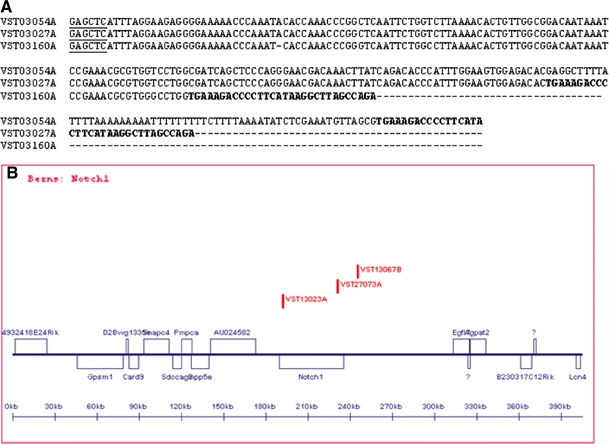
A VISA screening yields the exact base pair of insertions. The VSTs that define Evi1 are shown aligned by CLUSTAL W to illustrate VST clustering analysis. VST sequences are generated from amplification from a restriction enzyme site (underlined) anchored genomic primer and a virus specific primer. The start of proviral sequence (shown in bold) marks the exact base of viral insertion. B Clustered VSTs are displayed graphically in the VISION database with all annotated candidate genes within 200 kb of any insertion. A screen capture of the Notch1 locus is shown here for illustration
VST sequences were subjected to BLAST against the Ensembl mouse genome sequence (NCBI m36). After exclusion of low-scoring matches and repetitive sequence matches, 468 VSTs yielded single significant matches to the genome. These insertions were analyzed for genomic clustering to identify CISs (Fig. 1B). CISs representing endogenous virus loci were identified and excluded based on locations of known endogenous virus, all hits occurring at the same base, and/or VST matches to viral sequence. Of the 468 BLAST matches, 66 map to the locations of 16 endogenous proviruses or virus-like LTR sequences; thus, 402 VSTs identify potential tumor-specific tags.
Identification of CISs from these 402 VSTs was based primarily on proximity of the insertions (average ∼25 kb) but in some cases it also included Southern blotting to confirm that rearrangements were tumor specific, a characterization of aberrant expression of genes at the locus, and/or concurrence with previously identified loci. When VSTs surrounded a single gene but were spread over longer distances, we designated the site as a single CIS (e.g., Evi16; Table 3). We identified 49 CISs that were ranked with high confidence because they have multiple hits in our AKXD tumors (Table 3). When a CIS unique to this screen was defined by multiple insertions, we designated the CIS a lymphoid viral insertion site (Lvis). Loci previously identified in other screens are named in accordance with the published nomenclature. We identified 25 VSTs in a second class of loci consisting of single insertions that identify CISs previously identified in screens of other tumors (Table 4). The 74 CISs are distributed across the entire mouse genome and lie in or near known proto-oncogenes and TSGs, in the vicinity of candidate TSGs and proto-oncogenes, and near a set of predicted genes.
Table 3.
Common insertion sites (CISs)
| VST hitsa | Sb | Chr | Designationc | Candidate genesd | Tumor typee |
|---|---|---|---|---|---|
| 2, 2s | 2 | 1 | Evi32 | Prdm14, Ncoa2, ENSMUSG00000062226, Slco5a1 | T/B(2) |
| 4, 1s, 1r | 1 | Lvis10 | Wdfy1*, Mrpl44, serpine2, Ap1s3 | B, pT, pB | |
| 2, 2r | 1 | Sag | Sag, 5830472F04Rik, Usp40, Atg16l1 | pB | |
| 2 | 1 | Lvis11 | Arl7, Glrp1 | B, T | |
| 2 | 1 | Lvis12 | Jarid1b/Plu1*, Rabif, Klhl12, Adipor1 | T, pro | |
| 3, 4s, 2m | 2 | Notch1 | Notch1*, AU024582, Inpp5e, Egfl7 | T(2), T/B | |
| 1 | 1 | 2 | Lvis5 | Alx4* | B |
| 2 | 2 | Lvis13 | Pofut1*, Asxl1*, Kif3b, 8430427H17Rik | pro,T/B | |
| 3 | 3 | Evi1 | Evi1* | pro(2), B/M | |
| 3, 3s, 1h | 3 | Evi53 | 1700021C14Rik, Cct3, Rhbg, 0610031J06Rik | pB(3) | |
| 5, 5s | 3 | Evi54 | Notch2, NP_001028951.1*, Adam30, Sec22b | pB(2), pro, B | |
| 2, 1s | 3 | Nras | Nras*, CSDE1_MOUSE, 5730470L24Rik, Ampd1 | pro, pB | |
| 2, 1r | 3 | Lvis14 | Mcoln2*, Mcoln3, Edg7, Wdr63 | B(2) | |
| 2, 2r | 4 | Fgr | Fgr*, Adhc1, Q3U0Y7, ENSMUSG00000073749 | T/B(2) | |
| 4, 7s,15m, 3h | 5 | Gfi1 | Gfi1*, Glmn, AW060207, Evi5 | B(2), pB, T | |
| 2, 1r | 6 | Lvis15 | Tcf3*, Retsat*, Rbed1, Tgoln1 | B, pro | |
| 7 | 6 | Lvis16 | Rassf8, Tuba7, Bhlhb3, Sspn | B(3), T(2), pB, pro | |
| 3 | 7 | Lvis17 | Ceacam1*, Erf, Cic, BC024561 | pB, B/M, M | |
| 5, 4s, 3m | 7 | Evi84 (Lvis2) | Rras2, Spon1, Copb1, Psma1 | B(2), T(2), pro | |
| 2, 2s, 1h | 7 | Ccnd1 | Ccnd1, Oraov1, Fgf15, Fgf4 | pB, T | |
| 2, 2s, 4r | 8 | Rasa3 | Rasa3*, 1700029H14Rik, Gas6, ENSMUSG00000057052 | pro(2) | |
| 2, 1r | 8 | Lvis18 | Cdyl2, Dynlrb2, ENSMUSG00000074073 | pB | |
| 2 | 8 | Lvis4 (Ris2) | Cbfa2t3h (ETO-2), Ris2, Aprt, Galns | pB | |
| 2, 1h | 9 | Lvis19 | Btg4, Snf1lk2, Layn, Pou2af1 | B, pro | |
| 2, 1h | 9 | Sh3bgrl | Sh3bgrl2, Elovl4, Ttk | pB(2) | |
| 2, 2s, 1m | 9 | Evi100 | Ccr4, Cnot10, Q8C888_MOUSE, Glb1 | pB, B | |
| 1, 1h | 1 | 11 | Lvis7 | 4933435A13Rik, Rel, Pex13, Papolg | pB |
| 2, 3s | 0 | 11 | Evi108 | Smg6*, Hic1, Ovca2, Srr | pro, pB |
| 2, 7r | 11 | Ccr7 | Ccr7*, Smarce1, 6330509G02Rik, Tns4 | pB, B | |
| 2, 2r | 11 | Lvis20 | Map3k14*, 1700023F06Rik, Arhgap27, Hexim1, Hexim2 | B(2) | |
| 3, 1s | 12 | Nmyc1 | n-Myc1(Mycn), ENSMUSG00000066629 | T(2), B | |
| 2, 2r | 12 | Pnma1 | Pnma1, ENSMUSG00000042962, ENSMUSG00000072946, ENSMUSG00000042507 | B, T | |
| 3 | 12 | Lvis21 | Chga, Itpk1*, Golga5, Moap1 | pro, pB, T/B | |
| 2, 1s, 1r | 12 | Lvis22 | Bcl11b*, ENSMUSG00000072842, Setd3, Ccnk | pro, T | |
| 3 | 12 | Lvis23 | Igh6, Tmem121, Gm900, 4930427A07Rik | pro | |
| 12, 55s, 8h | 13 | Evi16 (Lvis3, 9) | Sox4, XP_914375.1 | pro(3), pB(2), B(2), T(2), T/B, M | |
| 2, 2r | 13 | Auh | Auh*, ENSMUSG00000065771, Nfil3, Syk | pB, T/B | |
| 3, 4s | 15 | Myc | Pvt1, Myc, ENSMUSG00000056590 | pro, pB, B | |
| 1, 1r | 4 | 15 | Lvis24 | Tnfrsf13c(Baffr) | pB |
| 2 | 17 | Lvis25 | XM_001005577.1, XM_001001995.1, XR_004526.1 | T(2) | |
| 2 | 17 | Ccnd3 (Lvis8) | Ccnd3, Bysl, Trfp, Usp49 | pB, B | |
| 6, 5s | 2 | 18 | Evi3 | Evi3 | pB(5), pro |
| 2, 1s, 1r | 18 | Lvis26 | Cxxc5*, 2610307O08Rik*, XP_913389.2, 1700066B19Rik | T, B | |
| 2, 1r | 18 | Lvis27 | Malt1, Alpk2, ENSMUSG00000057009, ENSMUSG00000065402 | pro, M | |
| 2, 2s | 18 | Evi138 | Nfatc1, Atp9b, Ctdp1 | pro, pB | |
| 3 | 19 | Lvis28 | Ighmbp2, Saps3, Gal, Mtl5 | T/B | |
| 11, 23s, 1h | 15 | 19 | Lvis1 | Hex, Exoc6, Eg5, Ide | B(4), pB(2), T/B(2), pro, M, pT |
| 2, 1r | 19 | Lvis29 | Wnt8b, Scd1-4, Chuk (Ikkα) | pro, pB | |
| 3, 1r | X | Lvis30 | Tlr7, Tlr8, ENSMUSG00000072409, Tmsb4x | pB, pro, B |
VST sequences were mapped to the mouse genome by BLAST and cosegregating hits are listed as common sites
a# hits column lists the number of VSTs that map to the locus from this screen (no label) and the number of hits at this locus published in Suzuki et al. (s), Mikkers et al. (m), Shin et al. (h), or other hits from the RTCGD (r)
bSouthern blotting of tumor DNAs revealed insertions not detected by VISA
cPreviously described loci are named consistently with the literature. Novel loci are named Lvis #
dUp to four candidate genes proximal to insertions are listed for each locus. Predicted transcripts and ESTs are listed as Ensembl, Riken, UniProtKB/TrEMBL, or Refseq designations. An asterisk (*) following the transcript name indicates that an insertion lies within the transcript. Best candidates (closest gene or known function) are listed first
eTumor cell type is presented as tumor type letter code followed by the number of tumors of that type, if more than one, in parenthesis. T = T cell; B = B cell; T/B = mixed T and B cell; M = myelod; E = erythroid; pB = pre B; pT = pre T; pro = progenitor. A. High confidence hits were chosen based on VST clustering, concurrence with established loci, Southern blotting, and expression information. B. Several single hits in this screen coincided with common sites from other screens
Table 4.
CISs with one tag in our database
| VST hits | Chr | Designation | Candidate genes | Tumor type |
|---|---|---|---|---|
| 1, 1m | 1 | Cis1 | ENSMUSG00000067125, ENSMUSG00000026238, 1700019O17Rik, Pde6d | B |
| 1, 3s, 17m | 2 | Bmi1 | Bmi1*, Commd3, BC061194, XP_895476.1 | pB |
| 1, 7s, 3m, 1h | 2 | Evi18 1 | XP_984911.1, Spred1, ENSMUSG00000065794, Rasgrp1 | pro |
| 1, 7s, 3m, 1h | 2 | Evi18 1 | Rasgrp1, 2610510H03Rik, ENSMUSG00000065845, XR_002866.1 | T/B |
| 1, 3s | 2 | Evi47 1 | Hck*, Pofut1, Asxl1, Plagl2 | pB |
| 1, 2s | 2 | Evi50 | Gnas*, ENSMUSG00000060124, ENSMUSG00000067549, ENSMUSG00000065410 | B |
| 1, 6s, 2h | 4 | Evi59 | Bach2, ENSMUSG00000073980, Gja10, ENSMUSG00000073981 | T |
| 1, 2s | 4 | Evi162 | Q6R5H0_MOUSE, Klf4 | B |
| 1, 2s | 4 | Evi62 | Cnr2, Fuca1, Pnrc2, Fusip1 | |
| 1, 1h | 4 | Evi11 | Rpl11, Id3, E2f2 | pro |
| 1, 4s | 5 | Evi29 | Card11*, Gna12, 6530401C20Rik | T |
| 1, 3s | 6 | Evi80 | Lrmp*, Casc1, 4930469P12Rik, Kras | B |
| 1, 6s | 7 | Evi24 | Gmfg, Lrfn1, Samd4b, Paf1 | pro |
| 1, 2s | 8 | Evi88 | Tacc1, Plekha2, XP_988283.1, Tm2d2 | T/B |
| 1, 2s | 8 | Evi96 | Foxf1a, Mthfsd, Foxc2, NM_008024.1 | B |
| 1, 1m, 1h | 10 | Txnrd1 | 1700028I16Rik*, Nfyb, Txnrd1, Hcfc2 | pB |
| 1, 1m | 11 | Evi106 1 | 1110064P04Rik*, ENSMUSG00000073112, ENSMUSG00000065591, 1700093K21Rik | pB |
| 1, 1m | 11 | Evi1061 | Rel, 4933435A13Rik, Papolg, Pex13 | pB |
| 1, 2s, 1m | 11 | Evi159 | Rnf43*, Supt4h1, ENSMUSG00000065420, Bzrap1 | pB |
| 1, 1m | 11 | Cis11 | Stat5b*, Stat5a., D11Lgp1e, Hcrt | pB |
| 1, 1m | 13 | Cis13 | Dusp22, Uqcrfs1, Irf4, Exoc2 | B |
| 1, 9m | 17 | Pim1 | Fgd2, Mtch1, Pi16, Pim1 | B |
| 1, 2m | 19 | Dkmi29 | Cd5, A430093F15Rik, Vps37c, Cd6 | pro |
| 1, 2m | X | Pim2 | Hdac6, Gata1, Eras, Pcsk1n | pro |
| 1, 1m | X | Nki37 | Suhw3, Slc25a14, Rab33a, Gpr119 | pro |
Twenty-four single-hit VSTs identified established CISs. Table information same as Table 3; see footnotes. 1. For Evi18 and Evi106, VSTs fell on either side of the established locus (within 200 kb) but VSTs were >200 kb apart and are listed as separate CISs
The Viral Insertion Sites Identifying Oncogenes (VISION) Database
We have constructed a public internet-based graphical database that provides specific information on VSTs, including genomic locations, VST sequence, tumor typing and histopathology, insertion-proximal genes, and pathway analysis. Users can use the graphic view to browse CISs by chromosome, viewing distance and orientation of genes with links to additional gene information from Ensembl, and Biocarta pathway information. Users can access primary VST sequence data, histopathology results, and molecular tumor-typing information by clicking on the red vertical bars representing individual VSTs in the graphic view (Fig. 1B), or by clicking on the VST name in the list view. The list view allows sorting of VSTs by chromosome, tumor type, accession number, or VST name, as well as links to individual VST records. The database can be accessed at http://www.mouse-genome.bcm.tmc.edu/vision. VST sequences are also available in GenBank and the RTCGD (http://www.rtcgd.abcc.ncifcrf.gov/) to make these data as widely available as possible.
Estimation of false-positive rate and degree of saturation
MuLV insertions can occur in any chromosome in a sequence-independent manner, but MuLV exhibits a slight preference for insertions near transcriptional start sites and CpG islands (Lewinski et al. 2006; reviewed in Wu et al. 2003). In vitro studies indicate that chromatin conformation also influences target site selection (Muller and Varmus 1994; Pryciak and Varmus 1992; Pryciak et al. 1992). The identification of false positives is possible through the sequencing of clonal mutations that have no association with cancer initiation or progression. Compared to positively selected oncogenic mutations, these uninformative insertions are less likely to occur in the same locus in independent tumors. False-positive CIS identification rates have been estimated by several different methods. Random insertion based on genome size estimates the rate of false-positive detection of cancer loci defined by two proviral insertions in a 26-kb window as one common site in 250 VSTs distributed randomly in the genome (Mikkers et al. 2002). Monte Carlo simulations estimate 17% of CISs are false positive (16.3 simulations/95 detected) using 1200 random insertions (Suzuki et al. 2002; supplemental). Experiments using MuLV in immortalized human cervical cell lines estimate as high as two thirds of two-hit CISs are false positive (Wu et al. 2006). With such a wide variation in estimates, we found it necessary to address this point.
Using experimental data, we first calculated the detection efficiency of VISA by comparing VSTs recovered by VISA to screening by Southern blot hybridization for clonal proviral insertions, a more thorough but lower-throughput method of insertion detection. We recovered an average of 2.7 VSTs per tumor by the VISA method (550 nonendogenous VSTs/204 tumors). Previous analyses showed that 1–10 somatically acquired proviruses can be detected by Southern blotting in these tumors (data not shown). Screening by Southern blot at eight loci yielded several insertions not detected by VISA, showing that our VISA tagging identified approximately 49% (24/49) of the insertion events at these sites. This allows us to estimate an average of 5.5 somatic proviral insertions per tumor (2.7 VSTs/49% detection rate). Using the 550 nonendogenous VSTs (including hits in repeat sequence and hits with no BLAST match), we calculate 1122 potential VSTs (550/49%). By isolating cancer rates due to viral insertion, we calculate that there are between 186 and 1020 true-positive VSTs in the pool of 1122 total (based on 1–5.5 proviral insertions required for tumor formation; see Materials and methods section), so 17–91% of the 550 collected VSTs identify true-positive cancer-associated VSTs. The lower end of this range represents an average of 1 viral insertion/tumor required to cause cancer and the upper end represents an average of 5.5 insertions/tumor required to cause cancer. These 550 nonendogenous VSTs were narrowed to 165 VSTs at 74 loci by clustering analysis, representing a dramatic minimization of false positives.
Calculations of mutagenic saturation provide a rough estimate of the completeness of a data set. Modeling insertion frequency with a Poisson distribution estimates that 9–42% of the readily identifiable loci have been identified by at least a single insertion (see Materials and methods section). This is a low degree of saturation and fits with the low degree of overlap with other screens, low multiplicity of insertion at CISs, and a high degree of unique data from this study. This calculation of saturation does not account for variability in insertion efficiency at different loci and may not accurately predict the number of oncogenes in hematopoietic cells. However, the calculation does provide an estimate of the number of sites likely to be identified by retroviral insertion in the AKXD strains used in this screen and what proportion have been identified by our VST screen.
Expression of genes flanking proviral insertion sites
In many instances there are a number of genes in the vicinity of a specific CIS with one, and possibly more, being affected by the proviral integration event. Retroviral insertions are known to alter gene expression at distances of over 50 kb (Hansen and Justice 1999b; Jonkers and Berns 1996), so the analysis of gene expression in tumors allows for the validation of candidate genes and a determination of whether insertions upregulate or downregulate expression of the flanking genes. We selected 16 candidate genes with one or more proximal insertions at nine different loci to test expression by real-time quantitative PCR (QPCR). The loci were selected based on the presence of interesting candidate genes. Several genes were selected based on their association with the Notch signaling pathway (Notch1, Notch2, Pofut1, Psen1, Numb, Lfng, Dtx2, ADAM precursor, and ADAM 3, 9, 19, 32) or because they map in close proximity to Notch genes (Egfl7). The remaining genes were selected because they were sites of very high numbers of insertions (Sox4, rRas2, and Copb1). Nineteen tumors with insertions and two control nontumor AKXD tissues were assayed for expression of all 16 genes. Three of four CISs showed significant upregulation of one or more proximal genes and two of five single insertions showed upregulation. Significant upregulation of gene expression generally correlated with the presence of insertions proximal to the upregulated gene, particularly at CISs (Fig. 2). In some cases we found genes to be upregulated without proximal insertions. This could be due to failure to detect a proximal insertion or mutation of another gene in the pathway that led to upregulation.
Fig. 2.
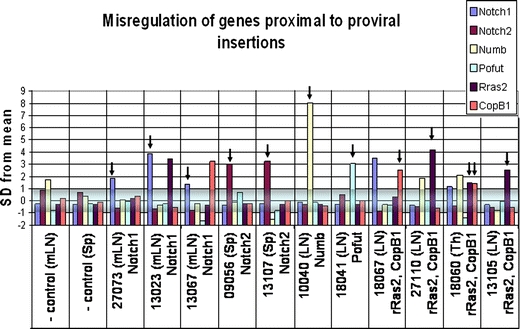
Misregulation of genes proximal to insertion loci. A set of 16 genes at 9 loci (4 CISs, 5 single hits) with proximal insertions was tested by real-time QPCR for misregulation in 19 tumors bearing those insertions and 2 control tissues. Tumors are labeled according to the organ of origin (mLN = mesenteric lymph node; LN = axillary, cervical, and/or branchial lymph nodes). Only genes that displayed significant misregulation are displayed here. Three CISs assayed (Notch1, Notch2, Ras/CopB1) showed misregulation of at least one proximal gene, two of the five single hits showed significant change of one gene. Real-time PCR threshold cycle (Ct) readings were normalized for cDNA content comparison to the average Ct of 16 genes assayed for each tumor using the formula: ΔCt = (Ctsample) - (CTavg. for tumor). To normalize scale across several genes, fold change relative to average ΔCt for each gene across all tumors (except tumors with insertions near the gene) was calculated using the equation: 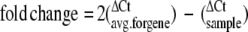 . To determine significant changes in gene expression, the average and standard deviation of normalized values were calculated for each gene across all tumors. Values on the table are expressed as the number of standard deviations from the mean of fold changes for each gene. The shaded box designates −1 to +1 standard deviations. Values outside this box are considered significant. Black arrows denote significant misregulation of genes in tumors harboring insertions near the misregulated genes. rRas2 and Copb1 are at the same locus and most insertions fall between the two genes
. To determine significant changes in gene expression, the average and standard deviation of normalized values were calculated for each gene across all tumors. Values on the table are expressed as the number of standard deviations from the mean of fold changes for each gene. The shaded box designates −1 to +1 standard deviations. Values outside this box are considered significant. Black arrows denote significant misregulation of genes in tumors harboring insertions near the misregulated genes. rRas2 and Copb1 are at the same locus and most insertions fall between the two genes
Discussion
Using VISA, we identified the genomic sequences flanking somatically acquired proviral insertions in tumors from nine AKXD RI strains that develop a high incidence of B-cell or mixed B/T-cell leukemia and lymphoma. The VISA technique has an advantage over PCR techniques that require a subcloning step because PCR products are sequenced directly, reducing the likelihood of sequencing contaminating products. Furthermore, because a rare PCR product is not likely to be subcloned and sequenced, such a technique focuses on proviral insertions that are monoclonal in the tumor population and, therefore, are likely cause tumor formation. Analysis of the cellular sequences allowed us to identify 74 loci that identify one or more candidate proto-oncogenes or TSGs.
Assessing data quality was a priority, with reports of potentially high rates of false-positive CISs (de Ridder et al. 2006; Wu et al. 2003). In AKXD mice, viral integration is the primary mutagen contributing to cancer formation. Base cancer rates from AKXD-20 mice that lack a transposable AKV virus were compared to rates from AKXD mice containing active virus in this study, allowing us to calculate that 91% of cancers in these AKXD mice are causally associated with at least one viral insertion. Of our 550 nonendogenous VSTs, we calculate 17–91% identify true-positive CISs. This range is dependent on the average number of true-positive insertions required to cause cancer, with 17% (true-positive percentage) of 550 VSTs representing a situation where only one viral insertion per cell on average was required to cause cancer. To reduce the false-positive rate, we used clustering of insertions to identify cancer-associated loci. If insertions are identified at the same locus in independent tumors, they are more likely to be a result of positive selection and representative of oncogenic mutations. Clustering analysis refines the pool of 550 VSTs to 165 VSTs, reducing the false-positive rate. These 165 VSTs represent 74 frequent insertion loci selected for oncogenicity. To reconcile these observations with the high observed false-positive rate in immortalized human cervical cell lines (Wu et al. 2006), we have only to point out that as cancer-associated insertions are positively selected, insertions that are not associated with cancer are negatively selected, reducing the background false-positive rate. Because every sample analyzed here was cancerous, we have enriched for insertions causing cancer, reducing the number of “random” insertions and the false-positive error rate. Furthermore, the degree of saturation of our VST detection indicates that we have likely not identified each of the CISs causative of cancer in this lymphoid tumor bank.
A comparison to similar screens in other mice (Mikkers et al. 2002; Shin et al. 2004; Suzuki et al. 2002) indicates that the CISs identified in our study are likely to be influenced by differences in genetic background. We compare our data to the screen described by Suzuki et al. (2002) because it was of similar scale, included AKXD RI strains and lymphoid neoplasms, and neither screen was based on a known sensitizing mutation. The Suzuki screen identified 493 tags at 152 CISs, whereas we identified 140 tags at 46 CISs. As a measure of saturation, allele frequencies for CISs (number of insertions/number of loci) in our VST screen were comparable to the Suzuki screen: 3.04 (140/46) (this article) vs. 3.24 (493/152) (Suzuki et al. 2002). One would expect to find a much greater allele frequency/saturation with more CIS tags when screening the same number of possible cancer loci. The discrepancies between the two screens for allele frequency and number of sites identified can be partially explained by the number of different strains used for each screen and strain genetic background. Target site preferences of provirus have been shown to vary among mouse strains and among AKV subtypes (Martin-Hernandez et al. 2006). Our screen used a subset of AKXD strains that are susceptible to B-cell neoplasms and the Suzuki screen used more AKXD strains, fewer aged mice per strain, and NFS.V+ and BXH2 RI mice. It is likely that strain variations, particularly predisposing mutations in individual AKXD strains, are influencing the pool of alleles recovered in common. Therefore, the calculated saturation is measuring primarily the mutation of loci that cooperate with existing cancer-predisposing mutations in these strains, rather than all possible cancer loci.
Additional comparisons between the two screens showed that only 15 of the 46 multihit CISs were identified as CISs in the previous screen. Low overlap between the screens suggests that many oncogenes and TSGs identified at these CISs will be unique to this AKXD tumor subset. Except for hits at the Lvis1 and Sox4 loci, the allele frequency was nearly the same for the remaining 14 shared loci with 3.15 (41/13) insertions/locus in our screen vs. 3.46 (45/13) in the Suzuki screen, indicating similar levels of saturation. This also demonstrates that Suzuki et al. (2002) were sampling from a larger pool of mutations because they found very similar mutation frequencies for most targeted genes that were common between the screens, yet they mapped more than twice as many sequence tags. The implication of this finding is that these screens are identifying cancer loci that cooperate with predisposing cancer loci carried in these genetic backgrounds at a high rate while identifying all blood cancer loci at a lower rate. This also raises the possibility that strain-specific germ line insertions may be acting as sensitizing mutations, influencing the frequency with which specific loci are identified as well as the occurrence of specific tumor types.
Another measure of the quality of the data is the ability to replicate previous work and to identify validated oncogenes. As expected, many of the insertional mutations identified CISs that were known from previous screens, including Evi1, 3, 11, 16, 32, 38, 53, 54, 84; Lvis1-9; Myc, N-Myc, Ccnd1, Ccnd3, Ris2, Nras, Notch, and Gfi1 (Hansen and Justice 1999a; Hansen et al. 2000; Li et al. 1999; Mikkers et al. 2002; Mucenski et al. 1988a; Shin et al. 2004; Suzuki et al. 2002). This validates the VISA method for identification of CISs; however, the most interesting data are the 21 loci not previously identified as CISs. These sites may be specific to a lymphoid subset of tumors, and, in particular, many may be unique to the B-cell lineage. Several of these novel sites have been validated as novel cancer-causing genes by gene expression in tumors or by oncogenicity after bone marrow transductions, or in transgenic mice, including Hex (George et al. 2003), Evi3 (Hentges et al. 2005), Tnfrsr13c (Hentges et al. 2002), Evi1 (Boyd et al., 2006), Sox4 (Boyd et al. 2006), and Eg5 (Castillo et al., unpublished). Of note, the Lvis24 (Tnfrsf13c) site was first detected as only a single hit in our VST database, but was pursued because it was commonly overexpressed in human lymphoid tumors in the Cancer Genome Anatomy Project (CGAP) database, showing that some unclustered single insertions are true positives.
The Lvis1 site is the most frequently rearranged locus in this tumor subset, with retroviral insertions detected in 26 independent tumors. Subsequent tests of gene expression showed that that although the insertions occurred 50 kb distant of the hematopoietic transcription factor Hex, they upregulated the expression of Hex as well as the kinesin-related spindle protein Eg5. Subsequently, both genes have shown oncogenicity in bone marrow transductions (George et al. 2003) or transgenic mice (Castillo et al., unpublished), indicating that retroviral insertions may influence the expression of two or more oncogenes at a single site and affect genes more than 50 kb distant. Such an observation led us to develop the VISION database such that all genes in the region of the proviral insertions are displayed, rather than indicating only the closest gene to the retrovirus. Similarly, all genes in the region of the proviral insertion site are listed in Table 1 because any or all of them are candidate proto-oncogenes.
Histologic analysis reveals that this tumor collection is composed of primarily lymphoid tumors; therefore, we would expect to identify known or suspected proto-oncogenes and TSGs implicated in B-cell lymphomagenesis and/or normal B-cell differentiation. Indeed, several genes of this type are identified, including Bcl11b (Cismasiu et al. 2006), Plu-1 (Lu et al. 1999), Atp1b1 (Chen et al. 2006; Le Page et al. 2006), Blzf1/Jem1(Duprez et al. 1997), Ccr4 (Yoshie et al. 2002), Ccr7 (Albert 2006), Ceacam I and II (Obrink 1997), and Traf1 and Malt1 (Nakagawa et al. 2005). Many of these have been implicated in lymphoma development by virtue of aberrant expression in tumors or as targets of translocation in human cancers. Finding these genes not only provides support for the validity of our methods but it also provides further evidence that these genes are a cause rather than a consequence of cancer. In some cases several genes that are known to function within an established or proposed signaling pathway were identified in our screen. Insertions identifying genes that act within oncogenic pathways support a functional interaction and delineate pathways involved in cancer.
The first such pathway is the Notch signaling pathway. Several members of the Notch pathway are identified as candidates, including Notch1, Notch2, Dtx2, Lfng, Psen1, Numb, Pofut1, Tcf3/E2A, and ADAM3, 5, 9, 18, and 32. We find insertions not only near Notch1 (a regulator of E2A and EBF) (Smith et al. 2005) and E2A, but also in Evi3, which modulates EBF activity (Hentges et al. 2005). Insertions in E2A/Tcf3 lie within the gene, which may indicate a loss of function followed by loss of heterozygosity. This is consistent with the ability of Notch1 to repress E2A activity and the observed upregulation of Notch1 in tumors. Cbfa2t3h (ETO-2) is 75% identical to ETO, which is a frequent target of translocations (with AML1) in humans. The resulting AML-ETO fusion protein, like Notch, inhibits E protein activity (Plevin et al. 2006). Independent of AML-ETO fusions, ETO itself is able to inhibit E-protein-dependent transactivation activity (Zhang et al. 2004). Likewise, ETO-2 binds SCL (a T-ALL oncoprotein) in a complex that inhibits E-protein activity. SCL also binds GFI1B (Schuh et al. 2005), the functional equivalent to Gfi1 in blood cells (Fiolka et al. 2006). Gfi1 is a frequent target of insertions in this screen and is the homolog of the Drosophila gene senseless, a modulator of Notch signaling (Nolo et al. 2000). Not only do we observe frequent insertion near Notch signaling genes, but these genes are misexpressed in the cancers. QPCR was used to assess expression levels of several candidate genes, including several Notch signaling genes, where Notch1, Notch2, Numb, and Pofut all showed significant upregulation (Fig. 2). Associations between these genes are illustrated in Fig. 3. Mutation of many genes that affect Notch signaling and E protein signaling identifies these pathways as common and powerful routes to cancer in blood cells.
Fig. 3.
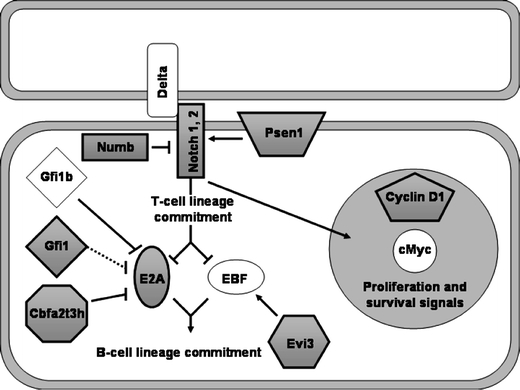
The Notch pathway is a frequent target of insertion. Members of the Notch signaling pathway were identified repeatedly as candidate oncogenes. Shaded boxes indicate genes with proximal insertions. Arrows represent direct or indirect effects that occur in one or more hematopoietic cell types. These associations are not necessarily occurring contemporaneously or in the same cell type
NFκB signaling is frequently involved in proliferation and survival signals of many blood cell lineages. NFκB signaling genes Ikka/Chuk, Malt1, and Traf1 were identified by multiple insertions in this screen. MALT1, IKKB, and IKKA/CHUK form a complex that is able to activate NFκB signaling in T and B cells (O’Mahony et al. 2000; Wegener et al. 2006; for review see Thome 2004). TRAF1 is able to activate NFκB during B-cell activation via CD40 but may have many roles in lymphoid development (Xie et al. 2006; for review see Lee and Lee 2002). These data indicate that aberrant activation of NFκB contributes to lymphoid cancer and that other members of this pathway are potential oncogenes.
Evi1 was originally identified through viral mutagenesis as a myeloid oncogene (Mucenski et al. 1988b). A screen for targets of EVI1 transcription factor activity yielded several candidate genes, including Map3k14 (Yatsula et al. 2005). Our screen identified three insertions near Evi1 and two insertions near Map3k14, suggesting a functional pathway for Evi1 and Map3k14 in cancer.
A pair of related genes, Hck and Fgr, were identified as candidate oncogenes by this screen. Notably, both of these Src family kinases are used for β-integrin outside-in signaling (Giagulli et al. 2006). In acute myeloid leukemia blasts, the expression of FGR and HCK is associated with early commitment and differentiation of myeloid lineages (Willman et al. 1991). These molecules are also critical for pre-B ALL induction by BCR/ABL (Hu et al. 2004). Furthermore, comparative genomic hybridization showed amplification of FGR in endometrial cancer (O’Toole et al. 2006). The somatic mutations found in the Fgr and Hck loci provide additional evidence linking downstream signaling of these two genes to cancer. The preceding pathways are all normally involved in proliferation or differentiation of lymphoid lineages, lending further validity to their identification as oncogenes or TSGs in a primarily lymphoid cancer collection.
The screen of the lymphoid tumors described here provides a new collection of 21 loci that are associated with leukemia and lymphoma. The majority of tumors recovered were of B-cell origin, indicating that the loci are identifying genes involved in normal B-cell development and, when expressed aberrantly, in cancer. Indeed, many of the genes identified are B-cell-specific genes with known functions in development and maturation. Insertions near several members of multiple pathways identify networks of signaling active in the development and progression of blood cancers. Notably, members of the Notch signaling pathway were frequently identified and, for some, deregulated expression was verified by QPCR. The identification of signaling networks active in cancer will allow us to understand cancer as a misregulation of cell processes rather than a collection of mutations leading to a phenotype. This is particularly useful for the evaluation and prioritization of drug targets and the development of effective treatments for disease.
Acknowledgments
The authors thank Shamsha Damani for assistance with QPCR and Toni Jago for assistance in aging mice and obtaining tumors. Dr. Richard Behringer is thanked for his helpful comments on the manuscript. This work was supported by a grant from the National Cancer Institute (RO1 CA63229 to MJJ) and in part by the Intramural Research Program of the NIH, National Institute of Allergy and Infectious Diseases (HCM).
Footnotes
Keith C. Weiser and Bin Liu are authors that contributed equally to this work.
References
- Albert Z (2006) Chemokines and cancer. Int J Cancer 119:2026–2029 [DOI] [PubMed]
- Boyd KE, Xiao Y-Y, Fan K, Poholek A, Copeland NG, et al. (2006) Sox4 cooperates with Evi1 in AKXD-23 myeloid tumors via transactivation of proviral LTR. Blood 107:733–741 [DOI] [PMC free article] [PubMed]
- Chen Q, Watson JT, Marengo SR, Decker KS, Coleman I, et al. (2006) Gene expression in the LNCaP human prostate cancer progression model: Progression associated expression in vitro corresponds to expression changes associated with prostate cancer progression in vivo. Cancer Lett 244:27–288 [DOI] [PubMed]
- Church GM, Gilbert W (1984) Genomic sequencing. Proc Natl Acad Sci USA 81:1991–1995 [DOI] [PMC free article] [PubMed]
- Cismasiu VB, Ghanta S, Duque J, Albu DI, Chen H-M, et al. (2006) BCL11B participates in the activation of IL2 gene expression in CD4+ T lymphocytes. Blood 108:2695–2702 [DOI] [PMC free article] [PubMed]
- de Ridder J, Uren A, Kool J, Reinders M, Wessels L (2006) Detecting statistically significant common insertion sites in retroviral insertional mutagenesis screens. PLoS Comput Biol 2:1530–1542 [DOI] [PMC free article] [PubMed]
- Dunn GP, Bruce AT, Ikeda H, Oid LJ, Schreiber RD (2002) Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol 3:991–998 [DOI] [PubMed]
- Duprez E, Tong J, Derre J, Chen S, Berger R, et al. (1997) JEM-1, a novel gene encoding a leucine-zipper nuclear factor upregulated during retinoid-induced maturation of NB4 promyelocytic leukaemia. Oncogene 14:1563–1570 [DOI] [PubMed]
- Eriksson B, Johansson A-S, Roos G, Levan G, Holmberg D (1999) Establishment and characterization of a mouse strain (TLL) that spontaneously develops T-cell lymphomas/leukemia. Exp Hematol 27:682–688 [DOI] [PubMed]
- Erkeland SJ, Verhaak RGW, Valk PJM, Delwel R, Lowenberg B, et al. (2006) Significance of murine retroviral mutagenesis for identification of disease genes in human acute myeloid leukemia. Cancer Res 66:622–626 [DOI] [PubMed]
- Fiolka K, Hertzano R, Vassen L, Zeng H, Hermesh O, et al. (2006) Gfi1 and Gfi1b act equivalently in haematopoiesis, but have distinct, non-overlapping functions in inner ear development. EMBO Rep 7:326–333 [DOI] [PMC free article] [PubMed]
- George A, Morse HC III, Justice MJ (2003) The homeobox gene Hex induces T cell-derived lymphomas when overexpressed in hematopoietic precursor cells. Oncogene 22:6764–6773 [DOI] [PubMed]
- Giagulli C, Ottoboni L, Caveggion E, Rossi B, Lowell C, et al. (2006) The Src family kinases Hck and Fgr are dispensable for inside-out, chemoattractant-induced signaling regulating beta2 integrin affinity and valency in neutrophils, but are required for beta2 integrin-mediated outside-in signaling involved in sustained adhesion. J Immunol 177:604–611 [DOI] [PubMed]
- Gilbert DJ, Neumann PE, Taylor BA, Jenkins NA, Copeland NG (1993) Susceptibility of AKXD recombinant inbred mouse strains to lymphomas. J Virol 67:2083–2090 [DOI] [PMC free article] [PubMed]
- Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100:57–70 [DOI] [PubMed]
- Hansen G, Justice M (1999a) A mouse chromosome 19 genetic map including the Lvis1 viral insertion site. Genomics 18:6531–6539 [DOI] [PubMed]
- Hansen GM, Justice MJ (1999b) Activation of Hex and Eg5 by retroviral insertion may contribute to mouse B-cell leukemia. Oncogene 18:6531–6539 [DOI] [PubMed]
- Hansen GM, Skapura D, Justice MJ (2000) Genetic profile of insertion mutations in mouse leukemias and lymphomas. Genome Res 10:237–243 [DOI] [PMC free article] [PubMed]
- Hartley J, Chattopadhyay S, Lander M, Taddesse-Heath L, Naghashfar Z, et al. (2000) Accelerated appearance of multiple B cell lymphoma types in NFS/N mice congenic for ecotropic murine leukemia viruses. Lab Invest 80:159–169 [DOI] [PubMed]
- Hentges KE, Yarlagadda S, Justice MJ (2002) Tnrfr13c (BAFF receptor) is misexpressed in tumors with murine leukemia virus insertions at Lvis22. Genomics 80:204–212 [DOI] [PubMed]
- Hentges KE, Weiser KC, Schountz T, Woodward LS, Morse HC III, et al. (2005) Evi3, a zinc-finger protein related to EBFAZ, regulated EBF activity in B-cell leukemia. Oncogene 24:1220–1230 [DOI] [PubMed]
- Hu Y, Liu Y, Pelletier S, Buchdunger E, Warmuth M, et al. (2004) Requirement of Src kinases Lyn, Hck and Fgr for BCR-ABL1-induced B-lymphoblastic leukemia but not chronic myeloid leukemia. Nat Genet 36:453–461 [DOI] [PubMed]
- Johnsen RC, Jones SJ, Rose AM (2000) Mutational accessibility of essential genes on chromosome I(left) in Caenorhabditis elegans. Mol Gen Genet 263:239–252 [DOI] [PubMed]
- Jonkers J, Berns A (1996) Retroviral insertional mutagenesis as a strategy to identify cancer genes. Biochim Biophys Acta 1287:29–57 [DOI] [PubMed]
- Jonkers J, Korswagen H, Acton D, Breuer M, Berns A (1997) Activation of a novel proto-oncogene, Frat1, contributes to progression of mouse T-cell lymphomas. EMBO J 16:441–450 [DOI] [PMC free article] [PubMed]
- Kronenberg M, Goverman J, Haars R, Malissen M, Kraig E, et al. (1985) Rearrangement and transcription of the beta-chain genes of the T-cell antigen receptor in different types of murine lymphocytes. Nature 313:647–653 [DOI] [PubMed]
- Le Page C, Ouellet V, Madore J, Ren F, Hudson T, et al. (2006) Gene expression profiling of primary cultures of ovarian epithelial cells identifies novel molecular classifiers of ovarian cancer. Br J Cancer 94:436–445 [DOI] [PMC free article] [PubMed]
- Lee NK, Lee SY (2002) Modulation of life and death by the tumor necrosis factor receptor-associated factors (TRAFs). J Biochem Mol Biol 35:61–66 [DOI] [PubMed]
- Lewinski M, Yamashita M, Emerman M, Ciuffi A, Marshall H, et al. (2006) Retroviral DNA integration: viral and cellular determinants of target-site selection. PLoS Pathol 2:e60 [DOI] [PMC free article] [PubMed]
- Li J, Shen H, Himmel KL, Dupuy AJ, Largaespada DA, et al. (1999) Leukaemia disease genes: large-scale cloning and pathway predictions. Nat Genet 23:348–353 [DOI] [PubMed]
- Liao X, Du Y, Morse HC III, Jenkins N, Copeland N (1997) Proviral integrations at the Evi5 locus disrupt a novel 90 kDa protein with homology to the Tre2 oncogene and cell-cycle regulatory proteins. Oncogene 14:1023–1029 [DOI] [PubMed]
- Look AT (1997) Oncogenic transcription factors in the human acute leukemias. Science 278:1059–1064 [DOI] [PubMed]
- Lu PJ, Sundquist K, Baeckstrom D, Poulsom R, Hanby A, et al. (1999) A novel gene (PLU-1) containing highly conserved putative DNA/chromatin binding motifs is specifically up-regulated in breast cancer. J Biol Chem 274:15633–15645 [DOI] [PubMed]
- Martin-Hernandez J, Sorensen AB, Pedersen FS (2006) Non-identical patterns of proviral insertions around host transcription units in lymphomas induced by different strains of murine leukemia virus. Virology 353:193–199 [DOI] [PubMed]
- Mikkers H, Allen J, Knipscheer P, Romeyn L, Hart A, et al. (2002) High-throughput retroviral tagging to identify components of specific signaling pathways in cancer. Nat Genet 32:153–159 [DOI] [PubMed]
- Morse HC III, Qi C-F, Chattopadhyay SK, Hori M, Taddesse-Heath L, et al. (2001) Combined histologic and molecular features reveal previously unappreciated subsets of lymphoma in AKXD recombinant inbred mice. Leukemia Res 25:719–733 [DOI] [PubMed]
- Moskow J, Bullrich F, Huebner K, Daar I, Buchberg A (1995) Meis1, a PBX1-related homeobox gene involved in myeloid leukemia in BXH-2 mice. Mol Cell Biol 15:5434–5443 [DOI] [PMC free article] [PubMed]
- Mucenski ML, Taylor BA, Jenkins NA, Copeland NG (1986) AKXD recombinant inbred strains: models for studying the molecular genetic basis of murine lymphomas. Mol Cell Biol 6:4236–4243 [DOI] [PMC free article] [PubMed]
- Mucenski ML, Bedigian HG, Shull MM, Copeland NG, Jenkins NA (1988a) Comparative molecular genetic analysis of lymphomas from six inbred mouse strains. J Virol 62:839–846 [DOI] [PMC free article] [PubMed]
- Mucenski ML, Taylor BA, Ihle JN, Hartley JW, Morse HC III, et al. (1988b) Identification of a common ecotropic viral integration site, Evi-1, in the DNA of AKXD murine myeloid tumors. Mol Cell Biol 8:301–308 [DOI] [PMC free article] [PubMed]
- Muller H, Varmus H (1994) DNA bending creates favored sites for retroviral integration: an explanation for preferred insertion sites in nucleosomes. EMBO J 13:4704–4714 [DOI] [PMC free article] [PubMed]
- Nakagawa M, Hosokawa Y, Yonezumi M, Izumiyama K, Suzuki R, et al. (2005) MALT1 contains nuclear export signals and regulates cytoplasmic localization of BCL10. Blood 106:4210–4216 [DOI] [PubMed]
- Nakamura T, Largaespada D, Shaughnessy JJ, Jenkins N, Copeland N (1996) Cooperative activation of Hoxa and Pbx1-related genes in murine myeloid leukaemias. Nat Genet 12:149–153 [DOI] [PubMed]
- Nolo R, Abbott LA, Bellen HJ (2000) Senseless, a Zn finger transcription factor, is necessary and sufficient for sensory organ development in Drosophila. Cell 102:349–362 [DOI] [PubMed]
- O’Mahony A, Lin X, Geleziunas R, Greene WC (2000) Activation of the heterodimeric Ikappa B kinase alpha (IKKalpha )-IKKbeta complex is directional: IKKalpha regulates IKKbeta under both basal and stimulated conditions. Mol Cell Biol 20:1170–1178 [DOI] [PMC free article] [PubMed]
- O’Toole SA, Dunn E, Sheppard BL, Klocker H, Bektic J, et al. (2006) Genome-wide analysis of deoxyribonucleic acid in endometrial cancer using comparative genomic hybridization microarrays. Int J Gynecol Cancer 16:834–842 [DOI] [PubMed]
- Obrink B (1997) CEA adhesion molecules: multifunctional proteins with signal-regulatory properties. Curr Opin Cell Biol 9:616–626 [DOI] [PMC free article] [PubMed]
- Plevin MJ, Zhang J, Guo C, Roeder RG, Ikura M (2006) The acute myeloid leukemia fusion protein AML1-ETO targets E proteins via a paired amphipathic helix-like TBP-associated factor homology domain. Proc Natl Acad Sci USA 103:10242–10247 [DOI] [PMC free article] [PubMed]
- Pryciak PM, Varmus HE (1992) Nucleosomes, DNA-binding proteins, and DNA sequence modulate retroviral integration target site selection. Cell 69:769–780 [DOI] [PubMed]
- Pryciak PM, Muller H, Varmus HE (1992) Simian virus 40 minichromosomes as targets for retroviral integration in vivo. Proc Natl Acad Sci USA 89:9237–9241 [DOI] [PMC free article] [PubMed]
- Sambrook J, Fritsch E, Maniatis T (1989) Molecular Cloning: a laboratory manual (Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press)
- Sarkar G, Turner R, Bolander M (1993) Restriction-site PCR: a direct method of unknown sequence retrieval adjacent to a known locus by using universal primers. PCR Methods Appl 2:318–322 [DOI] [PubMed]
- Schuh AH, Tipping AJ, Clark AJ, Hamlett I, Guyot B, et al. (2005) ETO-2 associates with SCL in erythroid cells and megakaryocytes and provides repressor functions in erythropoiesis. Mol Cell Biol 25:10235–10250 [DOI] [PMC free article] [PubMed]
- Shaughnessy JDJ, Largaespada DA, Tian E, Fletcher CF, Cho BC, et al. (1999) Mrvi1, a common MRV integration site in BXH2 myeloid leukemias, encodes a protein with homology to a lymphoid-restricted membrane protein Jaw1. Oncogene 18:2069–2084 [DOI] [PubMed]
- Shin MS, Fredrickson TN, Hartley JW, Suzuki T, Agaki K, et al. (2004) High-throughput retroviral tagging for identification of genes involved in initiation and progression of mouse splenic marginal zone lymphomas. Cancer Res 64:4419–4427 [DOI] [PubMed]
- Smith EMK, Akerblad P, Kadesch T, Axelson H, Sigvardsson M (2005) Inhibition of EBF function by active Notch signaling reveals a novel regulatory pathway in early B-cell development. Blood 106:1995–2001 [DOI] [PubMed]
- Suzuki T, Shen H, Akagi K, Morse HC III, Malley JD, et al. (2002) New genes involved in cancer identified by retroviral tagging. Nat Genet 32:166–174 [DOI] [PubMed]
- Szymanska H, Sitarz M, Krysiak E, Piskorowska J, Czarnomska A, et al. (1999) Genetics of susceptibility to radiation-induced lymphomas, leukemias and lung tumors studied in recombinant congenic strains. Int J Cancer 83:674–678 [DOI] [PubMed]
- Thome M (2004) CARMA1, BCL-10 and MALT1 in lymphocyte development and activation. Nat Rev Immunol 4:348–359 [DOI] [PubMed]
- Valk PJ, Hol S, Vankan Y, Ihle JN, Askew D, et al. (1997) The genes encoding the peripheral cannabinoid receptor and alpha-L-fucosidase are located near a newly identified common virus integration site, Evi11. J Virol 71:6796–6804 [DOI] [PMC free article] [PubMed]
- van Lohuizen M, Berns A (1990) Tumorigenesis by slow-transforming retroviruses–an update. Biochim Biophys Acta 1032:213–235 [DOI] [PubMed]
- Voisin V, Barat C, Hoang T, Rassart E (2006) Novel insights into the pathogenesis of the Graffi murine leukemia retrovirus. J Virol 80:4026–4037 [DOI] [PMC free article] [PubMed]
- Wegener E, Oeckinghaus A, Papadopoulou N, Lavitas L, Schmidt-Supprian M, et al. (2006) Essential role for I[kappa]B kinase [beta] in remodeling Carma1-Bcl10-Malt1 complexes upon T cell activation. Mol Cell 23:13–23 [DOI] [PubMed]
- Willman CL, Stewart CC, Longacre TL, Head DR, Habbersett R, et al. (1991) Expression of the c-fgr and hck protein-tyrosine kinases in acute myeloid leukemic blasts is associated with early commitment and differentiation events in the monocytic and granulocytic lineages. Blood 77:726–734 [PubMed]
- Wu X, Li Y, Crise B, Burgess SM (2003) Transcription start regions in the human genome are favored targets for MLV integration. Science 300:1749–1751 [DOI] [PubMed]
- Xie P, Hostager BS, Munroe ME, Moore CR, Bishop GA (2006) Cooperation between TNF receptor-associated factors 1 and 2 in CD40 signaling. J Immunol 176:5388–5400 [DOI] [PubMed]
- Yatsula B, Lin S, Read AJ, Poholek A, Yates K, et al. (2005) Identification of binding sites of EVI1 in mammalian cells. J Biol Chem 280:30712–30722 [DOI] [PubMed]
- Yoshie O, Fujisawa R, Nakayama T, Harasawa H, Tago H, et al. (2002) Frequent expression of CCR4 in adult T-cell leukemia and human T-cell leukemia virus type 1-transformed T cells. Blood 99:1505–1511 [DOI] [PubMed]
- Zhang J, Kalkum M, Yamamura S, Chait BT, Roeder RG (2004) E protein silencing by the leukemogenic AML1-ETO fusion protein. Science 305:1286–1289 [DOI] [PubMed]