

SUMMARY
Little is known about practice patterns regarding the diagnosis and management of idiopathic pulmonary fibrosis (IPF). This study attempts to define the practice patterns of academic pulmonologists caring for patients with idiopathic pulmonary fibrosis. Academic pulmonologists in the United States were surveyed electronically. Completed surveys were received from 272 respondents (representing approximately 10% of academic pulmonologists). The majority agreed that high-resolution computed tomography scanning can establish the diagnosis of idiopathic pulmonary fibrosis, and that surgical lung biopsy is indicated when the diagnosis remains unclear. Bronchoscopy is little utilized. Most respondents treat patients with medications, but there is no consensus regarding treatment regimen. These results suggest there is general consensus regarding the approach to diagnosis, but that there is no consensus about medical management in IPF.
INTRODUCTION
Idiopathic pulmonary fibrosis is the most common form of idiopathic interstitial pneumonia, with an estimated prevalence of up to 42 per 100,000 people. [1] There are no proven therapies for IPF and median survival is around 3 years from the time of diagnosis. [2-5] Because of its rarity, severity and lack of response to therapy, IPF is confusing and frustrating to practicing pulmonologists.
The American Thoracic Society (ATS), European Respiratory Society (ERS) and the American College of Chest Physicians (ACCP) produced a joint statement in 2000 outlining the current definition of IPF and recommending an approach to its diagnosis and management. [2] This statement defined IPF as a specific form of chronic fibrosing interstitial pneumonia with the histologic appearance of usual interstitial pneumonia (UIP) on surgical (thoracoscopic or open) biopsy. In the absence of surgical lung biopsy, criteria for a presumptive clinical diagnosis of IPF were developed that centered around a typical appearance on high-resolution computed tomography (HRCT) scan of the chest. [2] These clinical criteria have since been validated, and although they have poor sensitivity, they are highly specific. [6-8] While careful to state that no therapy has been proven beneficial in the treatment of IPF, the ATS/ERS/ACCP statement recommended considering combination therapy with corticosteroid and cytotoxic (azathioprine or cyclophosphamide) drugs.
Little is known about current practice patterns regarding the diagnosis and management of IPF. In addition, it is unknown whether these practice patterns are consistent with the recent consensus recommendations. Academic physicians are leaders in the continuing education of the pulmonary community through publication of research and review articles, presentations at conferences, and day-to-day interaction with community providers and physicians in training. Knowledge of academic physicians' approach to caring for patients with IPF would, therefore, directly inform the current standard of care for diagnosis and management of this condition. This study was designed to define the practice patterns of academic pulmonologists caring for patients with IPF and assess the degree to which these patterns are consistent with consensus recommendations.
METHODS
Survey Population
A search for Accreditation Council for Graduate Medical Education-accredited training programs in Pulmonary and Critical Care Medicine in the United States was performed through the website (www.acgme.org/acWebsite/home/home.asp). Each program was contacted to obtain email addresses of their pulmonary and critical care faculty for use in administering an anonymous, voluntary electronic survey. Individual email addresses were requested for direct distribution of the survey. In cases where individual email addresses were not available, a central administrative contact was identified for internal distribution of the survey at that institution.
This protocol was approved by the Committee on Human Research at the University of California San Francisco and the Institutional Review Board at Vanderbilt University.
Survey Administration
The survey was electronically administered from July 1 to August 31, 2006 using a professional online survey website (www.surveymonkey.com). An introductory email describing the survey and providing a link for interested respondents was sent to all faculty or faculty administrative contacts. This was followed by two reminder emails that were sent only to those who had yet to respond. Respondents were allowed to complete a partial survey. Only one response per respondent was allowed. All responses were anonymous, they could not be linked to any respondent information not provided in the survey.
Data Analysis
Individual survey responses were compiled with additional variables (e.g. combination treatment regimens) created based on further analysis of the primary survey data. For determination of region of practice, the United States Census Bureau regions were adopted. Summary statistics were calculated and displayed as percentages. Comparisons across demographic and geographic variables were performed using chi-squared and Fisher's exact procedures where appropriate. A p value of < 0.05 was considered statistically significant. All data analyses were performed using SAS v9.2 (SAS Institute, Cary, NC.)
RESULTS
Survey Respondents
One hundred and thirty training programs were identified. Eight hundred and thirty four individual emails were obtained from 39 programs in 22 states and the District of Columbia. Three additional institutions provided administrative email contacts and internally distributed the survey to their faculty. Assuming an equal number of faculty at each institution listed responded to the survey, this pool represents 32% of all academic pulmonologists. Responses were received from 272 respondents (33% survey response rate), or an estimated 10.5% (33% of 32%) of all academic pulmonologists in the United States. Information on individual program response rates was not available due as survey responses were anonymous. Respondents were asked to provide some general demographic information which is summarized in Table 1. There was at least one response from all 22 states and the District of Columbia. Twenty two (8%) of responses were incomplete.
Table 1.
Respondent Characteristics
| Characteristic | Number of Respondents (percent) |
|---|---|
| Years in practice | |
| < 5 years | 55 (22) |
| 5-15 years | 92 (36) |
| > 15 years | 105 (42) |
| Number of idiopathic pulmonary fibrosis patients seen annually | |
| None | 9 (3) |
| 1-5 | 93 (37) |
| 5-10 | 80 (32) |
| 11-20 | 38 (15) |
| > 20 | 32 (13) |
| Primary academic position | |
| Clinician educator | 105 (42) |
| Researcher (clinical or basic science) | 128 (51) |
| Administrative | 19 (7) |
| Geographic region of practice | |
| Midwest | 46 (19) |
| Northeast | 54 (21) |
| South | 78 (31) |
| West | 74 (29) |
Diagnosis and Monitoring
The overwhelming majority of respondents reported that a HRCT scan should be obtained in all patients suspected of having IPF (90%), and that a surgical lung biopsy should be obtained if the HRCT is atypical (76%). Seventy one percent reported that a definitive HRCT scan could establish the diagnosis of IPF in the proper clinical setting. A minority of respondents reported that a surgical biopsy should be obtained in all patients with IPF (12%). Most respondents did not feel bronchoscopy was important to the diagnosis of IPF (82%).
Most respondents (85%) reported that analysis of serial changes in both the forced vital capacity (FVC) and diffusing capacity for carbon monoxide (DLCO) were used to judge treatment response. Other commonly reported outcome measures included breathlessness and dyspnea (74%), 6 minute walk test distance (59%), exercise oxygen saturation (55%) and HRCT appearance (50%).
Screening for Comorbidities
Thirty-five percent of respondents routinely screen for sleep apnea, generally by history and physical examination. Slightly more respondents (55%) routinely screen for gastroesophageal reflux disease, again generally by history. A minority report screening with pH probe (15%) and barium swallow (7%). The vast majority of respondents screen for exertional hypoxemia (87%), generally (59%) with formal 6 minute walk testing, and for autoimmune disease (92%), generally with history (87%), antinuclear antibody (95%), rheumatoid factor (90%) and scl-70 antibody (72%).
Acute Exacerbation of IPF
Nearly all respondents (97%) reported observing acute exacerbation of IPF, defined as acute worsening of respiratory status with no clear etiology. (Figure 1A) Nearly all respondents (87%) required a combination of subjective worsening of breathlessness and at least one objective measure to identify an episode as an acute exacerbation of IPF. Objective measures commonly required in diagnosing an acute exacerbation included: worsening oxygenation/gas exchange (92%); absence of infection (86%); absence of cardiac events (83%); and absence of pulmonary embolism (82%). A new opacity identified on the chest CT scan was required by 49% of respondents.
Figure 1. Figure 1A. Frequency of Acute Exacerbations. Figure 1B. Outcome of Acute Exacerbations.
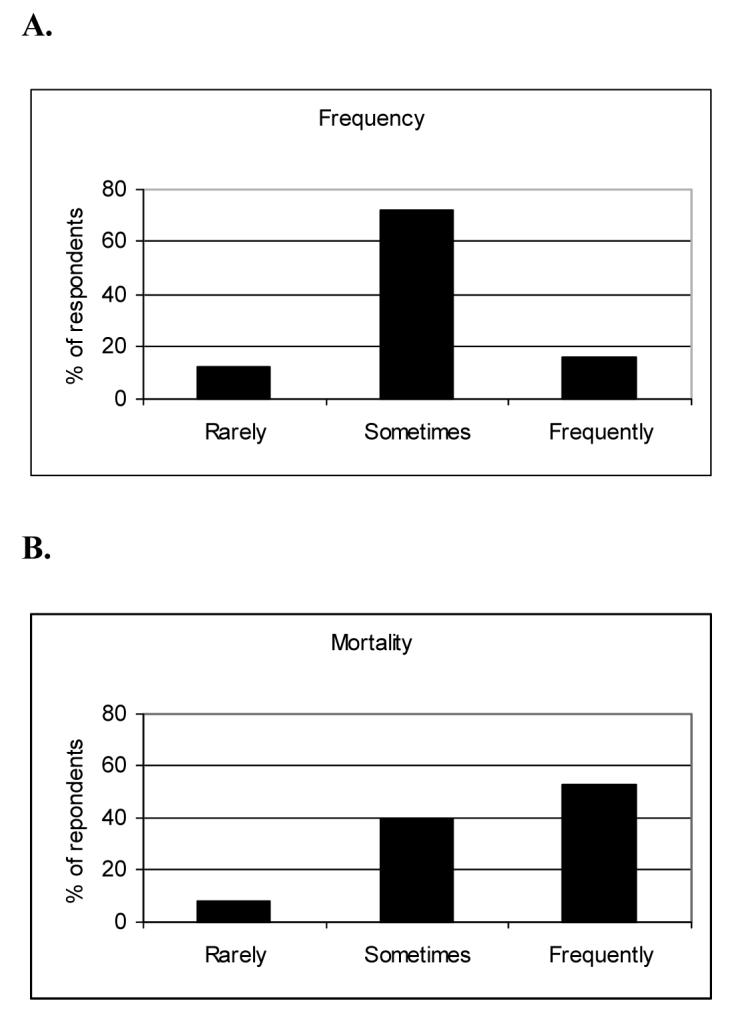
Figure 1A. Eighty-eight percent of respondents felt acute exacerbations occur “sometimes” or “frequently”.
Figure 1B. Ninety-two percent of respondents felt that acute exacerbations were “sometimes” or “frequently” fatal.
Fifty-one percent of respondents reported treating acute exacerbations with corticosteroids alone; another 30% report treating with combination corticosteroids and cytotoxic therapy (azathioprine or cyclophosphamide). Eight percent of respondents reported using anticoagulation for the treatment of acute exacerbations, usually as a single agent. Most respondents reported that such events are highly morbid. (Figure 1B)
Pharmacologic Therapy
The majority of respondents reported (52%) “frequently” or “always” treating patients with IPF at the time of diagnosis. Another 35% reported treating patients with IPF “sometimes”. Twelve percent reported that they “rarely” or “never” treat IPF with medications. Most first-line treatment regimens (74%) contained prednisone with or without the addition of immunosuppressive agents and N-acetylcysteine. (Figure 2) Twenty-three percent reported using combination corticosteroid and cytotoxic therapy (azathioprine or cyclophosphamide). Salvage therapy (i.e., therapy for patients unresponsive to initial treatment) was much less likely to include prednisone (18%) and more likely to include interferon gamma 1b (17%) or no therapy (20%.) (Figure 2) Respondents seeing fewer than 5 patients with IPF a year were more likely to use prednisone alone as a first choice regimen (p = 0.01) and appeared less likely to report no therapy as an initial approach (p = 0.08) (Table 2).
Figure 2. Pharmacologic therapy in IPF.

Initial (black bars) and salvage (grey bars) treatment regiments reported by survey respondents (n = 260).
Abbreviations: P&C = prednisone plus cytotoxic therapy; P = prednisone only; NAC = N-acetylcysteine only; PA&NAC = prednisone, azathioprine and N-acetylcysteine; IFN = interferon gamma 1b; None = no therapy.
Table 2.
Initial Treatment Regimen by Various Subgroups
| By Geographic Region | |||||
|---|---|---|---|---|---|
| West (n=74) |
Midwest (n=46) |
South (n=78) |
Northeas t (n=54) |
p value | |
| Prednisone only (%) | 23 | 15 | 14 | 19 | 0.51 |
| Prednisone plus cytotoxic agent (%) |
27 | 17 | 23 | 22 | 0.68 |
| Prednisone, azathioprine, N- acetylcysteine (%) |
15 | 13 | 19 | 7 | 0.29 |
| Any prednisone regimen * | 62 | 37 | 50 | 43 | 0.03 |
| No therapy (%) | 4 | 13 | 17 | 4 | 0.02 |
| By Years in Practice | ||||
|---|---|---|---|---|
| < 5 (n=55) |
5-15 (n=92) |
>15 (n=105) |
p value | |
| Prednisone only (%) | 15 | 15 | 22 | 0.36 |
| Prednisone plus cytotoxic agent (%) |
20 | 25 | 23 | 0.78 |
| Prednisone, azathioprine, N- acetylcysteine (%) |
16 | 12 | 15 | 0.71 |
| Any prednisone regimen * | 49 | 47 | 52 | 0.73 |
| No therapy (%) | 16 | 8 | 8 | 0.15 |
| By Number of Idiopathic Pulmonary Fibrosis Patients Seen Annually | |||||
|---|---|---|---|---|---|
| 0-5 (n=102) |
5-10 (n=80) |
11-20 (n=38) |
>20 (n=32) |
p value | |
| Prednisone only (%) | 25 | 19 | 13 | 0 | 0.01 |
| Prednisone plus cytotoxic agent (%) |
28 | 18 | 29 | 13 | 0.12 |
| Prednisone, azathioprine, N- acetylcysteine (%) |
5 | 24 | 13 | 22 | 0.002 |
| Any prednisone regimen * | 52 | 55 | 47 | 31 | 0.14 |
| No therapy (%) | 7 | 6 | 18 | 16 | 0.08 |
Any prednisone regimen combines three choices (prednisone only; prednisone, azathioprine; prednisone, azathioprine, N-acetylcysteine)
Non-pharmacological Therapy
Lung transplantation was reported as a treatment option for IPF by 54% of respondents. There was significant variation in the reported use of transplantation, with higher reported use in the Midwest (p=0.01), in respondents in practice for less than 5 years (p=0.01) and in respondents seeing over 20 patients with IPF annually (p=0.0007.) (Table 3)
Table 3.
Transplantation by Various Subgroups
| By Geographic Region | |||||
|---|---|---|---|---|---|
| West (n=74) |
Midwest (n=46) |
South (n=78) |
Northeast (n=54) |
p value | |
| Transplantation (%) | 41 | 70 | 60 | 50 | 0.01 |
| By Years in Practice | ||||
|---|---|---|---|---|
| < 5 years (n=55) |
5-15 years (n=92) |
>15 years (n=105) |
p value | |
| Transplantation (%) | 65 | 60 | 43 | 0.01 |
| By Number of Idiopathic Pulmonary Fibrosis Patients Seen Annually | |||||
|---|---|---|---|---|---|
| 0-5 (n=102) |
5-10 (n=80) |
11-20 (n=38) |
>20 (n=32) |
p value | |
| Transplantation (%) | 41 | 59 | 55 | 81 | 0.0007 |
Two thirds (68%) of respondents reported that they routinely refer patients with IPF for pulmonary rehabilitation. Of those that do not refer for pulmonary rehabilitation, the majority (76%) respond that they do not do so because pulmonary rehabilitation has unproven benefit in IPF. One third (32%) of respondents reported presumptively treating patients with IPF for gastroesophageal reflux disease.
DISCUSSION
This study represents the first large-scale assessment of clinical practice patterns of academic physicians caring for patients with IPF. Importantly, this is the first survey performed since the publication of the ATS/ERS/ACCP statement on IPF [2]. The findings of this study show the following: (1) There is general consensus regarding the approach to diagnosing and monitoring IPF, and this approach is largely consistent with the consensus recommendations. The exception is the use of bronchoscopy, which is recommended but is rarely performed. (2) There is no consensus about the medical management of patients with IPF, with a minority of respondents following consensus treatment recommendations. (3) There is uniform recognition that acute exacerbations of IPF occur and that they are common and highly morbid events. (4) Lung transplantation is considered as a treatment option for half of patients with IPF.
Previous Surveys of Practice Patterns in IPF
In the last 20 years, three surveys of practice patterns in IPF have been published in the English language literature. In 1989, Smith and colleagues surveyed 109 graduates of the University of California San Diego pulmonary fellowship training program and received 25 analyzable responses (23% response rate). [9] A minority reported routine use of open biopsy (30%) while a majority reported the use of transbronchial biopsy (65%) for the diagnosis of IPF. Initial treatment was generally with prednisone alone (72%) or prednisone plus cytotoxic agent (8%). Salvage therapy was most commonly prednisone plus cytotoxic agent (52%). Disease monitoring was by pulmonary function tests (80%), exercise studies (64%) chest x-ray (40%), gallium scan (36%) and symptoms (32%). Lung transplant was little used (4%), likely because at that time it was not widely available.
In 1993, Johnson and colleagues published a retrospective chart review of 200 IPF patients seen in the United Kingdom. [10] They found very few cases were diagnosed by surgical biopsy (8%), with a larger minority diagnosed by transbronchial biopsy (33%). Treatment was most commonly with prednisone alone (55%) and uncommonly with prednisone plus a cytotoxic agent (11%). Thirty four percent received no therapy. A follow up study by Johnson and colleagues in 1997 utilized a prospective registry of 558 patients entered by 150 providers over 2 years. [11] Their findings confirmed that surgical biopsy was rare (12%), that prednisone alone remained the most common therapy (34%), and that 48% of patients received no therapy.
Our survey expands upon these previous efforts in several important ways. First, it involves a larger sample of pulmonologists than any previous study. Secondly, it provides insight into the current standard of care being taught to practicing pulmonologists and physicians in training. Thirdly, it is broader in scope than previous surveys, investigating diagnosis, pharmacologic and non-pharmacologic management, natural history, disease monitoring and screening for comorbidities. Fourthly, it is the only study to incorporate the use of HRCT scanning. Lastly, it is the only study conducted after the ATS/ERS/ACCP statement revised the definition of IPF and recommended an approach to diagnosis and management and can therefore comment on the degree to which expert opinion is being adopted by the larger academic community.
Management Patterns in Patients with IPF
These results suggest that, unlike diagnosis, the management of IPF remains far from standardized; there is no consensus approach among academic pulmonologists. There are likely several factors involved. First, recent data on the impact of corticosteroid and cytotoxic drugs has been published that suggest a lack of efficacy and confirm the common occurrence of side effects. [12-14] Second, published studies of agents such as interferon gamma 1b, pirfenidone, and N-acetylcysteine have been interpreted as showing promise. [15-21] Third, the increasingly accepted pathophysiologic paradigm of abnormal wound healing in which inflammation plays a limited role argues against the traditional approach to therapy (corticosteroids and cytotoxic agents) proposed by the consensus statement.[22] In the absence of compelling data or rationale, pulmonologists are left to make treatment decisions based on intuition and anecdote, and this understandably leads to a variety of approaches.
Study Limitations and Future Directions
There are important limitations to our survey. With any survey, a critical issue is how well respondents represent the target population. Overall, this survey received responses from approximately 10% of academic pulmonologists in the United States. Sampling bias (i.e. bias introduced due to differences in the intended target population and the cohort surveyed) is always present, and effort must be made to reduce its impact. Randomly choosing potential respondents from the target population is ideal, but not always practical. In this survey, sampling was not random; emails were sent to potential respondents based on whether or not their training program provided contact information. However, a large percentage of the general population of interest was surveyed (32%) reducing the impact of sampling bias. A second important issue is the survey response rate. Ideally, 100% of those sampled would respond, but again, this is often impractical, particularly with large, geographically diverse cohorts such as this one. The lower the response rate, the more potential for responder bias (i.e. those who responded are somehow different from those who did not). Our response rate of 33% is typical of physician surveys published in the medical literature. [23] Finally, there is always the potential for survey questions to be misinterpreted.
It is hoped that the observations in this survey serve as a benchmark for understanding how academic physicians approach the diagnosis and management of IPF. Further, these results may provide guidance to thought leaders involved in physician education and practice guideline development regarding continued educational needs.
Acknowledgement
The authors wish to acknowledge the efforts of the 272 participating pulmonologists, and thank them for taking time from their busy careers to contribute to medical research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement
None of the authors has a conflict of interest to declare in relation to this work.
Contributor Information
Harold R. Collard, Department of Medicine, San Francisco General Hospital, University of California San Francisco, 1001 Potrero Avenue, 5K1, San Francisco, CA, 94110.
James E. Loyd, Department of Medicine, Vanderbilt University Medical Center, 1161 21st Avenue, Rm T1219, Nashville, TN, 37232..
Talmadge E. King, Jr., Department of Medicine, San Francisco General Hospital, University of California San Francisco, 1001 Potrero Avenue, 5K1, San Francisco, CA 94110..
Lisa H. Lancaster, Department of Medicine, Vanderbilt University Medical Center, 1301 22nd Avenue South, Rm B817, Nashville, TN, 37232..
REFERENCES
- 1.Raghu G, Weycker D, Edelsberg J, Bradford WZ, Oster G. Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2006 Oct 1;174(7):810–6. doi: 10.1164/rccm.200602-163OC. [DOI] [PubMed] [Google Scholar]
- 2.American Thoracic Society Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS) Am J Respir Crit Care Med. 2000;161:646–64. doi: 10.1164/ajrccm.161.2.ats3-00. [DOI] [PubMed] [Google Scholar]
- 3.Bjoraker JA, Ryu JH, Edwin MK, et al. Prognostic significance of histopathologic subsets in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1998;157:199–203. doi: 10.1164/ajrccm.157.1.9704130. [DOI] [PubMed] [Google Scholar]
- 4.Flaherty KR, Toews GB, Travis WD, et al. Clinical significance of histological classification of idiopathic interstitial pneumonia. Eur Respir J. 2002;19(2):275–83. doi: 10.1183/09031936.02.00182002. [DOI] [PubMed] [Google Scholar]
- 5.Nicholson AG, Colby TV, Dubois RM, Hansell DM, Wells AU. The Prognostic Significance of the Histologic Pattern of Interstitial Pneumonia in Patients Presenting with the Clinical Entity of Cryptogenic Fibrosing Alveolitis. Am J Respir Crit Care Med. 2000;162:2213–7. doi: 10.1164/ajrccm.162.6.2003049. [DOI] [PubMed] [Google Scholar]
- 6.Flaherty KR, Thwaite EL, Kazerooni EA, et al. Radiological versus histological diagnosis in UIP and NSIP: survival implications. Thorax. 2003;58(2):143–8. doi: 10.1136/thorax.58.2.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hunninghake G, Zimmerman MB, Schwartz DA, et al. Utility of lung biopsy for the diagnosis of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2001;164:193–6. doi: 10.1164/ajrccm.164.2.2101090. [DOI] [PubMed] [Google Scholar]
- 8.Raghu G, Mageto YN, Lockhart D, et al. The accuracy of the clinical diagnosis of new-onset idiopathic pulmonary fibrosis and other interstitial lung disease: A prospective study. Chest. 1999;116(5):1168–74. doi: 10.1378/chest.116.5.1168. [DOI] [PubMed] [Google Scholar]
- 9.Smith CM, Moser KM. Management for interstitial lung disease. State of the art. Chest. 1989 Mar;95(3):676–8. doi: 10.1378/chest.95.3.676. [DOI] [PubMed] [Google Scholar]
- 10.Johnston ID, Gomm SA, Kalra S, et al. The management of cryptogenic fibrosing alveolitis in three regions of the United Kingdom [see comments] Eur Respir J. 1993;6(6):891–3. [PubMed] [Google Scholar]
- 11.Johnston ID, Prescott RJ, Chalmers JC, Rudd RM. British Thoracic Society study of cryptogenic fibrosing alveolitis: current presentation and initial management. Fibrosing Alveolitis Subcommittee of the Research Committee of the British Thoracic Society. Thorax. 1997 Jan;52(1):38–44. doi: 10.1136/thx.52.1.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Collard HR, Ryu JH, Douglas WW, et al. Combined corticosteroid and cyclophosphamide therapy does not alter survival in idiopathic pulmonary fibrosis. Chest. 2004 Jun;125(6):2169–74. doi: 10.1378/chest.125.6.2169. [DOI] [PubMed] [Google Scholar]
- 13.Douglas WW, Ryu JH, Schroeder DR. Idiopathic pulmonary fibrosis: Impact of oxygen and colchicine, prednisone, or no therapy on survival. American Journal of Respiratory and Critical Care Medicine. 2000;161(4 Pt 1):1172–8. doi: 10.1164/ajrccm.161.4.9907002. [DOI] [PubMed] [Google Scholar]
- 14.Flaherty KR, Toews GB, Lynch JP, III, et al. Steroids in Idiopathic Pulmonary Fibrosis: A Prospective Assessment of Adverse Reactions, Response to Therapy, and Survival. Am J Med. 2001;110:278–82. doi: 10.1016/s0002-9343(00)00711-7. [DOI] [PubMed] [Google Scholar]
- 15.Bajwa EK, Ayas NT, Schulzer M, et al. Interferon-gamma1b therapy in idiopathic pulmonary fibrosis: a metaanalysis. Chest. 2005 Jul;128(1):203–6. doi: 10.1378/chest.128.1.203. [DOI] [PubMed] [Google Scholar]
- 16.King TE, Jr, Safrin S, Starko KM, et al. Analyses of Efficacy End Points in a Controlled Trial of Interferon-{gamma}1b for Idiopathic Pulmonary Fibrosis. Chest. 2005 January 1;127(1):171–7. doi: 10.1378/chest.127.1.171. 2005. [DOI] [PubMed] [Google Scholar]
- 17.Raghu G, Brown KK, Bradford WZ, et al. A Placebo-Controlled Trial of Interferon Gamma-1b in Patients with Idiopathic Pulmonary Fibrosis. N Engl J Med. 2004 January 8;350(2):125–33. doi: 10.1056/NEJMoa030511. 2004. [DOI] [PubMed] [Google Scholar]
- 18.Strieter RM, Starko KM, Enelow RI, Noth I, Valentine VG. Effects of interferon-gamma 1b on biomarker expression in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2004 Jul 15;170(2):133–40. doi: 10.1164/rccm.200312-1670OC. [DOI] [PubMed] [Google Scholar]
- 19.Azuma A, Nukiwa T, Tsuboi E, et al. Double-blind, Placebo-controlled Trial of Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med. 2005 May 1;171(9):1040–7. doi: 10.1164/rccm.200404-571OC. 2005. [DOI] [PubMed] [Google Scholar]
- 20.Azuma A, Tsuboi E, Abe S, et al. A Placebo Control And Double Blind Phase II Clinical Study Of Pirfenidone In Patients With Idiopathic Pulmonary Fibrosis In Japan. Am J Respir Crit Care Med. 2002;165:A729. doi: 10.1164/rccm.200404-571OC. [DOI] [PubMed] [Google Scholar]
- 21.Demedts M, Behr J, Buhl R, et al. High-dose acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med. 2005 Nov 24;353(21):2229–42. doi: 10.1056/NEJMoa042976. [DOI] [PubMed] [Google Scholar]
- 22.Selman M, King TE, Jr., Pardo A. Idiopathic Pulmonary Fibrosis: Prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med. 2001;134:136–51. doi: 10.7326/0003-4819-134-2-200101160-00015. [DOI] [PubMed] [Google Scholar]
- 23.Asch DA, Jedrziewski MK, Christakis NA. Response rates to mail surveys published in medical journals. J Clin Epidemiol. 1997 Oct;50(10):1129–36. doi: 10.1016/s0895-4356(97)00126-1. [DOI] [PubMed] [Google Scholar]