

Abstract
Melanization, an insect immune response, requires a set of hemolymph proteins including pathogen recognition proteins that initiate the response, a cascade of mostly unknown serine proteinases, and phenoloxidase. Until now, only initial and final proteinases in the pathways have been conclusively identified. Four such proteinases have been purified from the larval hemolymph of Manduca sexta: hemolymph proteinase 14 (HP14), which autoactivates in the presence of microbial surface components, and three prophenoloxidase-activating proteinases (PAP1-3). In this study, we have used two complementary approaches to identify a serine proteinase that activates proPAP3. Partial purification from hemolymph of an activator of proPAP3 resulted in an active fraction with two abundant polypeptides of ∼32 and ∼37 kDa. Labeling of these polypeptides with a serine proteinase inhibitor, diisopropyl fluorophosphate, indicated that they were active serine proteinases. N-terminal sequencing revealed that both were cleaved forms of the previously identified hemolymph serine proteinase, HP21. Surprisingly, cleavage of proHP21 had occurred not at the predicted activation site but more N-terminal to it. In vitro reactions carried out with purified HP14 (which activates proHP21), proHP21, proPAP3, and site-directed mutant forms of the latter two proteinases confirmed that HP21 activates proPAP3 by limited proteolysis. Like the HP21 products purified from hemolymph, HP21 that was activated by HP14 in the in vitro reactions was not cleaved at its predicted activation site.
Melanization of invading microbes is one mechanism that protects insects against infection. In hemolymph (insect blood), pathogen recognition is followed by the activation of one or more cascades of mostly unknown serine proteinases (1). The terminal proteinases cleave prophenoloxidase (proPO),2 and active PO oxidizes the catechols involved in melanin formation (2). Until now, only proteinases in the first and final positions of the melanization pathways have been conclusively identified. Just one initiating proteinase has been discovered: Manduca sexta hemolymph proteinase 14 (HP14), which autoactivates in the presence of microbial surface components (3, 4). In contrast, several proPO activativing proteinases (PAPs, also named PPAF and PPAE) have been identified through biochemical studies of M. sexta, Holotrichia diomphalia, and Bombyx mori (5-9). In addition, approximately a dozen proteinases with unknown functions in the melanization process have been identified through genetic screens using Drosophila melanogaster and Anopheles gambiae (10-15).
HP14 is a complex, multidomain serine proteinase that associates with peptidoglycan or β-1,3-glucan found on the surface of bacteria and fungi, respectively (3, 4). At least in some cases, the association is mediated by a pathogen recognition protein; for example, HP14 combines with a β-1,3-glucan recognition protein (βGRP2) to bind to β-1,3-glucan (4). Upon binding to these microbial polysaccharides, HP14 undergoes autoactivation, presumably via an initial conformational change that enables one molecule of HP14 to cleave a second molecule at its activation site (GTEL*VLGG). Key residues in the putative substrate binding pocket of HP14 (Gly585, Ala616, and Thr630) are consistent with cleavage after a bulky, hydrophobic residue such as leucine (3). The cleaved form of HP14 can initiate activation of proPO in hemolymph (3, 4).
All known proPO-activating proteinases have a C-terminal catalytic domain and one or two N-terminal clip domains (5-9). The function of the clip domain is unknown, but it is assumed to be regulatory (22). Active B. mori PPAE is competent to activate proPO by cleavage after an activation site arginine (9). In contrast, H. diomphalia PPAF-I and the M. sexta PAPs require the presence of one or more cofactors to fully activate proPO (16-18). The H. diomphalia cofactor (PPAF-II) and the M. sexta cofactors (SPH1 and SPH2) are serine proteinase homologs (proteins with high sequence similarity to serine proteinases but lacking the active site serine) that need to be cleaved to be functional. The proteinase activator of PPAF-II has been identified (19), while the activator of the M. sexta SPHs is still unknown. H. diomphalia proPO is activated in two steps: PPAF-I cleaves proPO to generate a 76-kDa form, then PPAF-II (activated by PPAF-III) cleaves the 76-kDa form to generate an active, 60-kDa form of PO (16, 17, 19). M. sexta proPO is activated by a single cleavage event that requires an active PAP and the presence of active SPHs (20, 21). The significance of the differences between these three mechanisms of activation is unknown.
As part of our long term goal of understanding immune related proteinase cascades, our objective for this study was to identify an activator of proPAP3 in M. sexta plasma. ProPAP3 is a constitutively expressed and secreted proteinase zymogen with two clip domains (7, 23). It is activated when it is cleaved after the P1 lysine within its activation site (VGNK*IIGG) (7); however, the proteinase that carries out this activation step has been unknown. PAP3 hydrolyzes IEAR-p-nitroanilide (IEAR-pNA) and other artificial substrates with a P1 arginine. We took two complementary approaches to identify an activator of proPAP3. First, we assayed fractions from chromatographic separations of hemolymph proteins or their ability to activate proPAP3. By this method, we identified hemolymph proteinase 21 (HP21) as a candidate activator of proPAP3. HP21 is a previously discovered serine proteinase with an N-terminal clip domain (24). Our second approach was to perform in vitro reactions with purified proteins and thereby establish directly that HP21 is capable of activating proPAP3. This work was made possible by the finding, reported separately, that HP14 can activate proHP21.3
EXPERIMENTAL PROCEDURES
Partial Purification of an Activator of proPAP3 from M. sexta Hemolymph
Hemolymph was collected from bar stage prepupae into saturated ammonium sulfate at 4 °C as described previously (6). Ammonium sulfate was adjusted to 50% saturation, and samples were frozen at -80 °C until 290 ml of hemolymph had been collected. All subsequent purification steps were done at 4 °C. Proteins that were precipitated at 50% saturation of ammonium sulfate were collected by centrifugation, and the pellet (containing 5.6 g of protein) was dissolved in 140 ml of 10 mm potassium phosphate, 0.5 m NaCl, pH 6.8, 0.001% phenylthiourea, 10 mm diethyldithiocarbamic acid. This fraction did not activate proPAP3; therefore, curdlan (an insoluble β-1,3-glucan) was added to elicit activity. (The ability of curdlan to elicit activation of proPAP3 in prepupal hemolymph was reported previously (7). ProPAP3 activation was detected using amidase and immunoblot assays described below.) The fraction was incubated with 0.36 g of curdlan for 10 min on ice with gentle shaking and the curdlan was removed by centrifugation. Further ammonium sulfate fractionation was done, and the pelleted 10 -30% saturated ammonium sulfate fraction (620 mg) was dissolved in 119 ml of 20 mm Tris-HCl, 0.5 m NaCl, pH 7.4. Proteins were applied to a concanavalin A-Sepharose column (2.5 cm i.d. × 4 cm), and bound proteins (104 mg) were eluted with 0.5 m methyl-α-d-mannopyranoside and dialyzed twice against 3.5 liters of 20 mm Tris-HCl, 100 mm NaCl, pH 8. This fraction was applied to a Q-Sepharose column (1.5 cm i.d. × 7.5 cm), and proteins were eluted with 100 -500 mm NaCl in 20 mm Tris-HCl, pH 8. Approximately 40% of the protein but about two-thirds of the activity was in the fraction that did not bind to Q-Sepharose in 20 mm Tris-HCl, 100 mm NaCl, pH 8. This fraction (42 mg) was concentrated 20-fold using MacroSep 10 kDa MWCO units. Forty percent of the concentrated protein fraction was dialyzed against 2 liters of 20 mm potassium phosphate, 10 mm NaCl, pH 6.8. This fraction was applied to a CM Sepharose column (1 cm i.d. × 5.6 cm), and proteins were eluted with 10 -700 mm NaCl in 20 mm potassium phosphate, pH 6.8. Sixty-five percent of the protein and all of the activity was in the flow-through fraction. An equal volume of 100 mm NaCl was added to the unbound protein fraction and loaded on a prepacked ceramic hydroxyapatite type II (Bio-Rad) column (1 ml). Proteins were eluted with 10 - 400 mm potassium phosphate, 50 mm NaCl, pH 6.8. Approximately 15% of the protein and 80% of the activity were present in the unbound fractions, which were dialyzed twice against 4 liters of 20 mm Tris-HCl, pH 8.5. Proteins were loaded on a Q-Sepharose column (1 cm i.d. × 4.5 cm), the column was washed with 20 mm Tris-HCl, pH 8.5, and proteins were eluted with a linear gradient of 0 - 400 mm NaCl in 20 mm Tris-HCl, pH 8.5. Activity was detected in the flow-through fraction and in fractions that eluted at 20-120 mm NaCl. The unbound protein fraction was chosen for further biochemical characterization, because its activity was relatively high and it contained just three polypeptides detectable by silver staining. This fraction was concentrated using MacroSep and NanoSep 10-kDa MWCO units, and the buffer was exchanged to 20 mm Tris-HCl, 100 mm NaCl, pH 8, using the NanoSep units. This active fraction contained 0.1 mg of protein at 0.5 mg/ml.
Amidase Assays
IEAR-pNA is an artificial substrate of proPAP3 (7) and so can be used to detect PAP3 activity. Assays were done by mixing column fractions or in vitro reactions with 50 μm IEAR-pNA in reaction buffer (typically 0.1 m Tris-HCl, 0.1 m NaCl, 5 mm CaCl2, pH 8.0) and recording the absorbance at 405 nm every 30 s for 20 min. The reaction rate was calculated from the slope of the initial, linear portion of each curve generated. One unit of activity was defined as a change in A405 of 0.001 per min.
Immunoblot Assay of proPAP3 Activation
Aliquots of column fractions were mixed with 70 ng of recombinant proPAP3 and incubated at room temperature for 10 -30 min. The reaction volume was typically 15 μl. Reactions were stopped by the addition of SDS-PAGE sample buffer. Samples were analyzed by reducing SDS-PAGE, transfer to nitrocellulose, and detection with antiserum to proPAP3 (7). Activity was assessed by the presence of specific PAP3 cleavage products (the ∼40-kDa catalytic domain and the ∼20-kDa dual clip domain).
The effect of proteinase inhibitors on proPAP3 activation was determined by incubating 400 ng of active fraction with inhibitor for 20 min at room temperature and then adding 70 ng of proPAP3 and assaying for activity by immunoblot analysis as described above. Inhibitors tested included 1 mm and 10 mm diisopropyl fluorophosphate (DFP), 1 mm phenylmethylsulfonyl fluoride (PMSF), and 1 mm (4-amidino-phenyl)-methanesulfonyl fluoride (APMSF), which were diluted from concentrated stocks immediately before use with 0.1 m MOPS, pH 6.9.
Diisopropyl Fluorophosphate Labeling of Serine Proteinases
22 μg of the hemolymph fraction containing a partially purified activator of proPAP3 (described above) was incubated with 0.01 mm [3H]diisopropyl fluorophosphate ([3H]DFP) for 30 min at 37 °C. Labeled proteins were analyzed by reducing SDS-PAGE and detected by fluorography (26). Masses were estimated by comparison with 14C-labeled standards. X-ray film exposure to the treated, dried gel was for 5 weeks at -70 °C.
N-terminal Protein Sequencing
Proteins in 7.5 μg of the active fraction were resolved by SDS-PAGE and transferred to a polyvinylidene difluoride membrane. Polypeptides were visualized by Coomassie Blue staining, and two major bands (∼32 kDa and ∼37 kDa) were excised for further analysis. N-terminal sequencing was performed on an Applied Biosciences 494 cLc sequencer by the W. M. Keck Facility at Yale University.
Purification of HP14 from M. sexta Hemolymph
ProHP14 was purified from immune induced larval hemolymph as described previously (4). Briefly, purification included ammonium sulfate fractionation and several chromatography steps (hydroxyapatite, Sephacryl S100, concanavalin-A Sepharose, and dextran sulfate-Sepharose). During initial purification trials, we discovered that proHP14 became activated upon binding to Macro-Prep High Q beads (Bio-Rad); therefore, we were able to purify active HP14 by binding purified proHP14 to a High Q column in 20 mm bis-Tris-HCl, 10 mm NaCl, pH 6.8 and eluting with a linear salt gradient (10-1000 mm NaCl). From 55 ml of hemolymph, we purified 60 μg of active HP14. HP14 activity was verified by three methods: ability of the purified HP14 to initiate activation of proPO and to complex with recombinant serpin-1I in vitro,4 and the ability to activate proHP21 (see “Results”).
Expression and Purification of Recombinant Proteins
Five proteins were expressed using a baculovirus expression system (Bac-toBac, Invitrogen). A cDNA containing the full coding region of proHP21 was cloned into pFH6 (pFastBac1 modified to encode a C-terminal hexahistidine tag) (27), and a cDNA containing the full coding region of proPAP3 was cloned into pFastBac1. Mutations were introduced into these cDNAs with the QuikChange Multi Site-directed Mutagenesis kit (Strat-agene). The proHP21[Ser→ Ala] mutation was made by changing the codon for the active site serine (Ser344) from AGC to GCC, and the proHP21[Leu→ Lys] mutation by changing the codon for the P1 leucine (Leu151) from CTT to AAG. The proPAP3[Ser→ Ala] mutation was generated by changing the codon for the active site serine (Ser358) from TCT to GCT. DNA sequencing confirmed that the mutant cDNA sequences were correct. Recombinant baculoviruses were generated for each of the five cDNAs. Plaque assays were used to determine titers of amplified virus stocks. For expression, typically, 1 liter of Sf9 cells (2 × 106 cells/ml serum-free medium [Sf-900 II SFM]) was infected with baculovirus at a multiplicity of infection of 5, and cells were incubated at 27 °C with shaking for 48 h. Cells were removed by two centrifugation steps (500 × g for 10 min).
Purification of proHP21 and its two mutant variants began with binding proteins in the culture medium to concanavalin-A-Sepharose followed by eluting with 0.5 m methyl-α-d-mannopyranoside in 20 mm Tris-HCl, 0.5 m NaCl, pH 7.4 (4 °C). Eluted proteins were dialzyed against 20 mm Tris-HCl, pH 8.0 (4 °C) and loaded on a High Q column (Bio-Rad). Proteins were eluted with a linear gradient of NaCl in 20 mm Tris-HCl (20 - 1000 mm NaCl). Fractions containing the proHP21 variant were pooled and then separated by Sephacryl S100HR chromatography using 20 mm Tris-HCl, 150 mm NaCl, pH 8 at 1.85 ml/min. In the next step, nickel affinity chromatography took advantage of the C-terminal His tag on these recombinant proteinases. Proteins were bound to nickle-NTA agarose beads (Qiagen) in 20 mm Tris-HCl, 150 mm NaCl, pH 8.0 and eluted with 50 mm Tris-HCl, 150 mm NaCl, 250 mm imidazole, pH 8. The final purification step was a second fractionation with a High Q column, this time using a shallow elution gradient (60 ml, 20 - 400 mm NaCl). At each step, fractions containing proHP21 were detected by immunoblot analysis. The purified proHP21 variants were stored at -80 °C.
The first step for purifying proPAP3 and proPAP3[Ser→ Ala] was binding and eluting from concanavalin A-Sepharose. Eluted proteins were dialyzed against 20 mm Tris-HCl, pH 8.7 (4 °C) followed by purification by Q-Sepharose chromatography. Proteins were eluted from the Q column with a 300 ml linear gradient from 0 -50 mm NaCl. These two steps were adequate for purifying proPAP3, but an additional step was used for proPAP3[Ser→ Ala]. Q-Sepharose fractions containing proPAP3[Ser→ Ala] were pooled, concentrated with Centriplus 10-kDa MWCO units, dialyzed against 10 mm potassium phosphate, pH 6.8, and fractionated by ceramic hydroxyapatite II (Bio-Rad) chromatography. Fractions containing pure proPAP3[Ser→ Ala] were pooled, and the sample buffer was exchanged by dialysis to 20 mm Tris-HCl, 40 mm NaCl, pH 8.5 (4 °C). The purified proPAP3 variants were concentrated and stored at -80 °C in small aliquots. For all proteins, purity was assessed by SDS-PAGE followed by a sensitive silver staining method (SilverXpress kit, Invitrogen).
In Vitro Proteinase Reactions
The proteinase stocks used for in vitro reactions were HP14 (40 ng/μl in 20 mm Bis-Tris-HCl, ∼175 mm NaCl, pH 6.8), proHP21 variant (14 ng/μl in 20 mm Tris-HCl, ∼100 mm NaCl, pH 8), proPAP3 (130 ng/μl in 20 mm Tris-HCl, ∼40 mm NaCl, pH 8.5 [4 °C]), and proPAP3[Ser→ Ala] (45 ng/μl in 20mm Tris-HCl, 40 mm NaCl, pH 8.5 [4 °C]). Sodium chloride concentrations of the first three buffers listed are estimations based on the predicted salt concentrations of the ion exchange column fractions containing the purified enzymes. Proteinases were mixed in various combinations. Amounts added were 80 ng of HP14, 70 ng of proHP21 variant, and 90 ng of proPAP3 variant, or an equal volume of the appropriate buffer was substituted. The reaction volume was 30 μl, of which 13 μl was the buffer 20 mm Tris-HCl, 20 mm NaCl, 0.002% Tween-20, pH 7.5. Samples were incubated at 37 °C for 1 h.
RESULTS
We partially purified an activator of proPAP3 from Manduca prepupal hemolymph. Details of the purification are described under “Experimental Procedures.” The active fraction contained 0.1 mg of protein and included six polypeptides detectable by Coomassie staining (Fig. 1A). Incubation of 250 ng of active fraction with 70 ng of proPAP3 for 10 min resulted in complete cleavage of proPAP3 into its ∼40-kDa catalytic domain and ∼20-kDa dual clip domain (Fig. 1B). Activity of the cleaved form of PAP3 was verified by an amidase assay that used IEAR-pNA as the substrate (Fig. 1C). DFP, an irreversible and nonspecific serine proteinase inhibitor, was used to determine which of the proteins in the active fraction might be active serine proteinases. The active fraction was incubated with [3H]DFP for 30 min, and labeled proteins were identified by SDS-PAGE followed by fluorography. A long (5 week) exposure was required to detect labeled polypeptides. Of these weakly labeled proteins, two (of ∼32 kDa and ∼37 kDa) were most prominent (Fig. 2A). Partial inhibition of the fraction’s activity by 10 mm DFP suggested that one of these two proteins may be an activator of proPAP3 (Fig. 2B).
FIGURE 1. An activator of proPAP3 from Manduca hemolymph.

A, active fraction (3.75 μg), isolated as described under “Experimental Procedures,” was analyzed by reducing SDS-PAGE followed by Coomassie staining. Arrows indicate two abundant polypeptides that correspond to polypeptides similarly marked in Figs. 2 and 3. B, immunoblot assay using PAP3 antiserum demonstrated that 250 ng of active fraction cleaved 70 ng of proPAP3 in 10 min. The resulting polypeptides (separated by reducing SDS-PAGE) were the expected masses of the PAP3 catalytic and clip domains. C, amidase assay demonstrated that the cleaved PAP3 visible by immunoblot analysis had IEARase activity. The 125-ng active fraction was incubated with 70 ng of proPAP3 and 50 μm IEAR-pNA for 20 min, and the change in absorbance at 405 nm was recorded. The error bar signifies mean ± S.D. Activity was zero when proPAP3 or the active fraction alone was incubated with IEAR-pNA.
FIGURE 2. A serine proteinase inhibitor labels polypeptides in the active fraction and partially inhibits activity.

A, polypeptides in the active fraction were labeled with [3H]DFP. 22.5 μg of active fraction was incubated with 0.01 mm [3H]DFP for 30 min at 37 °C. Labeled proteins were analyzed by reducing SDS-PAGE and detected by fluorography. Arrows point to the same bands as those in Fig. 1A. (Note that the molecular mass standards are different than those used for all other experiments, and the apparent masses were slightly different than those of the standards shown in the other figures.) B, effects of serine proteinase inhibitors on proPAP3 activation were determined by immunoblot analysis. For each reaction, 400 ng of the active fraction was incubated with inhibitor for 20 min at room temperature, 70 ng of proPAP3 was added and incubated for 10 min at room temperature, and the reactions were analyzed by reducing SDS-PAGE followed by immunoblotting using PAP3 antiserum.
N-terminal sequences of the two polypeptides matched residues in the previously identified Manduca serine proteinase, proHP21 (Fig. 3A). The ∼32-kDa band had a strong primary sequence that exactly matched 10 of 11 residues in proHP21. One of the 11 residues (presumably an arginine) could not be identified. The ∼37-kDa band was a mixture, but 9 of the identified residues matched those of proHP21. A cysteine within this region of proHP21 was not detected. The predicted masses of the truncated HP21 polypeptides (29.6 kDa and 34.3 kDa) are close to those estimated by SDS-PAGE (32 and 37 kDa). As expected, the two polypeptides reacted with antiserum against proHP21 (Fig. 3B). Column fractions from some of the purification steps were assayed for the presence of HP21. Each fraction that contained the ∼32-kDa and/or ∼37-kDa form of HP21 had activator of proPAP3 activity; however, some fractions were active but did not contain detectable HP21.4 Taken together, these results suggested that HP21 is an activator of proPAP3 and that at least one other activator is present in hemolymph.
FIGURE 3. Cleaved forms of HP21 are present in the active fraction.

A, N-terminal sequences of two major polypeptides in the active fraction (indicated by arrows in Fig. 1A) matched with sequences in HP21 (underlined). Also highlighted is the predicted signal peptide (italics), predicted activation site (asterisk), P1 leucine (shaded), and three residues in the putative substrate binding pocket that are predicted to be important for substrate specificity (bold). B, immunoblot analysis using antiserum against HP21 confirmed that the two major bands in the active fraction were forms of HP21.
A surprising discovery was that the truncated forms of HP21 were cleaved not at the predicted activation site but more N-terminal to it (Fig. 3A). This finding suggested that atypical forms of HP21 (that have been cleaved at sites other than the predicted cleavage site) may have some proteinase activity. In addition, these results might explain the relatively weak DFP labeling and low DFP sensitivity of the active fraction if the atypical forms of HP21 are less than fully active.
Data from two other studies strengthen the model of HP21 as a proPAP3 activator. We have recently demonstrated that HP21 forms a stable complex with serpin-4, a serine proteinase inhibitor that does not inhibit PAP3 or other PAPs but does inhibit proPO activation in hemolymph (28). This finding suggests that HP21 acts upstream of the PAPs in at least one proPO activation pathway. More direct evidence is the discovery that HP21, when cleaved at its predicted activation site, activates Manduca proPAP2, a proteinase related to proPAP3.3
To confirm that HP21 is an activator of proPAP3, we carried out a set of in vitro experiments using purified proteinases. Five proteinases were expressed as recombinant zymogens in an insect cell culture system (Fig. 4). Two forms of proPAP3 were made: wild-type and an active site mutant (serine to alanine). ProPAP3[Ser→ Ala] was useful for testing proPAP3 as a substrate for HP21 without the potential complication of PAP3 proteolytic activity (for example, the potential for PAP3 autoactivation). Three variants of proHP21 were produced: wild-type, an active site mutant (serine to alanine), and a P1 activation site site mutant (leucine to lysine). ProHP21[Ser→ Ala] was used to test whether HP21 activity was required for proPAP3 activation, and proHP21[Leu→ Lys] proved to be a useful variant because of its unexpected constitutive activity (see below). To assay for HP21 activity, we needed a means of activating proHP21. Fortunately, it was recently discovered that Manduca HP14 can activate proHP21.3 For this study, proHP14 was purified from larval hemolymph and then autoactivated through contact with Macro-Prep High Q chromatography beads.
FIGURE 4. SDS-PAGE analysis of purified proteinases.
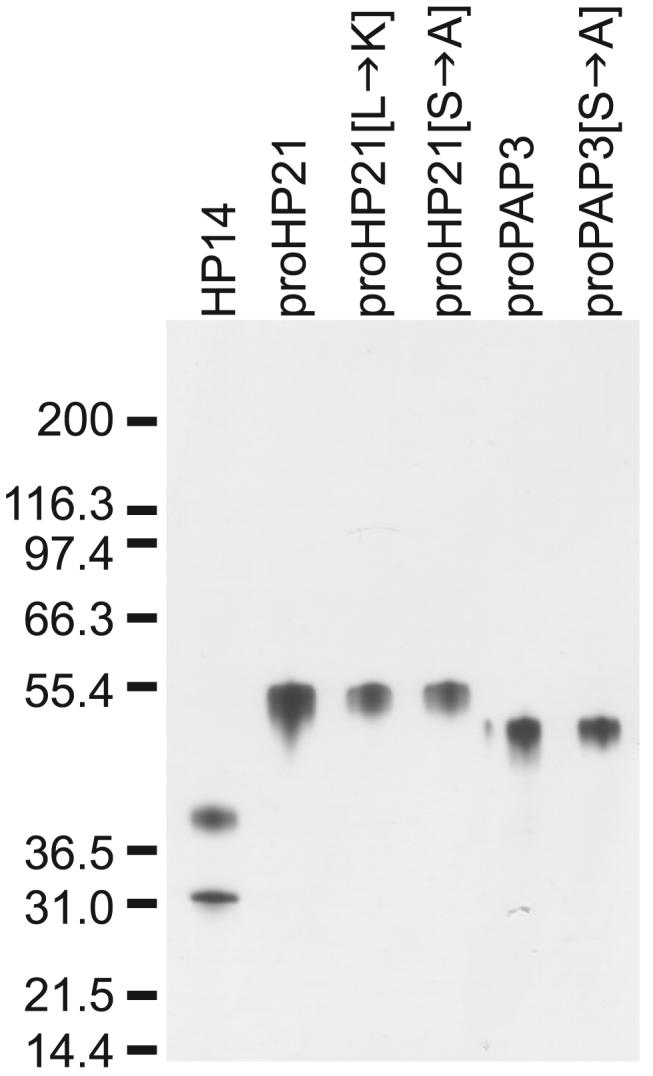
20 -50 ng of each protein was analyzed by reducing SDS-PAGE followed by highly sensitive silver staining. Proteins were judged to be highly purified. Note that active HP14 separates under reducing conditions into a ∼40-kDa regulatory domain and a ∼30-kDa catalytic domain.
The first approach we used to test whether HP21 can activate proPAP3 was in vitro reactions followed by immunoblot analysis of the resulting polypeptides (Fig. 5). We found that neither HP14 nor proHP21 alone cleaved a detectable amount of proPAP3; in contrast, 90 ng proPAP3 incubated with a mixture of HP14 and proHP21 was completely cleaved within 1 h. This result strongly suggested that HP21 can cut proPAP3. Substituting proPAP3 with proPAP3[Ser→ Ala] made no difference in proPAP3 cleavage, thus, no autoactivation of PAP3 was detected. Substituting proHP21 with proHP21[Ser→ Ala] eliminated cleavage of proPAP3. This result verified the requirement for active HP21 during proPAP3 activation. The effect of proHP21[Leu→ Lys] was unexpected. Even in the absence of HP14, proHP21[Leu→ Lys] cut some proPAP3. This result was interesting in light of our previous data suggesting that HP21 can be active even when it has not been cleaved at its activation site. The addition of HP14 to proHP21 plus proPAP3 resulted in activation of proHP21 and complete cleavage of proPAP3; in contrast, addition of HP14 to proHP21[Leu→ Lys] plus proPAP3 resulted in only slight activation of proHP21 and partial cleavage of proPAP3. This result demonstrates that the P1 residue of proHP21 is important for the activation of proHP21 by HP14.
FIGURE 5. HP21 cleaves proPAP3.

Purified proteinases were mixed in various combinations (shown beneath the graph), incubated for 60 min at 37 °C, and analysed by immunoblotting. The primary antibody used for each experiment is indicated above the blots. The reactions included 80 ng of HP14, 70 ng of proHP21 variant, and 90 ng of proPAP3 variant, or an equal volume of the appropriate buffer was substituted. The reaction volume was 30 μl, of which 13 μl was the buffer 20 mm Tris-HCl, 20 mm NaCl, 0.002% Tween-20, pH 7.5. The PAP3 polyclonal antiserum detects the clip domain better than the catalytic domain; therefore, the catalytic domain band in lanes 5, 6, 12, and 13 is difficult to see. The last three lanes in the figure show reactions in which more proPAP3[Ser→ Ala] was used as the substrate to improve the visibility of the PAP3 catalytic domain generated in the last two reactions.
Surprisingly, we did not detect any cleavage of proHP21 by HP14 (Fig. 5). Wang and Jiang3 detected a ∼30-kDa HP21 catalytic domain resulting from the incubation of proHP21 with active HP14, but we did not detect a polypeptide of this mass in any reaction during this study. Altering buffer components, concentrations of proteinases, method of activating HP14, or reaction time did not make a difference.4 (Note that a ∼40-kDa HP21 band was observed in reactions that contained active PAP3. This band was not seen in reactions in which proPAP3 was substituted with proPAP3[Ser→ Ala]; therefore, PAP3 must be able to cleave proHP21 at a site N-terminal to the P1 residue.) Although we do not have a definitive explanation for this difference in outcome (see “Discussion”), this investigation demonstrates that HP21 can be active without being cleaved.
The second method used to test whether HP21 is an activator of proPAP3 was to test the reactions described above for amidase activity using the artificial PAP3 substrate IEAR-pNA (Fig. 6). Proteinases were mixed in various combinations, incubated for 1 h at 37 °C and added to 50 μm IEAR-pNA for assay of PAP3 activity. In agreement with the immunoblot analyses, we observed the highest amidase activity when proPAP3 was incubated with a mixture of HP14 and proHP21. This more sensitive assay of PAP3 activation detected a low level of amidase activity in reactions of HP14 with proPAP3 and of proHP21 with proPAP3. Activation of proPAP3 by HP14 may be due to nonspecific cleavage. We have observed that HP14 degrades other proteins under certain circumstances.4 Negligible activation of proPAP3 by proHP21 may be due to a low level of constitutive activity of proHP21. As expected from the immunoblot assays, some proPAP3 was activated by the constitutively active proHP21[Leu→Lys] and slightly more (p = 0.04, unpaired t test) was activated when both HP14 and proHP21[Leu→Lys] were present. As expected, proHP21[Ser→Ala] and proPAP3 [Ser→Ala] showed no evidence of activity.
FIGURE 6. HP21 activates proPAP3.

Purified proteinases were mixed in the combinations shown below the graph (as described in the legend for Fig. 5) and incubated for 60 min at 37 °C. Reactions were then added to 50 μm IEAR-pNA, and activity was detected as increased absorbance at 405 nm. Error bars signify mean ± S.D.
DISCUSSION
By using a combination of two experimental approaches, we discovered that HP21 is an activator of proPAP3. We recognized HP21 as a candidate activator when we identified truncated forms of it in a hemolymph fraction that could activate proPAP3. We verified this putative function of HP21 by performing in vitro reactions using purified proteinases. In these reactions, proHP21 was activated by HP14, and activated HP21 cleaved proPAP3 to generate the active, two-chain form of PAP3. HP21 contains features consistent with that of a PAP activator. It is constitutively expressed and secreted as a zymogen into the hemolymph, and its expression is up-regulated in response to an immune challenge (24). Like most serine proteinases implicated in insect immune responses, it contains an N-terminal clip domain. Critically, key residues in its catalytic domain predict that substrates of HP21 will have an arginine or lysine in the P1 position. HP21 has been linked to the melanization process because of its ability to complex with serpin-4, a serine proteinase inhibitor that can block proPO activation in hemolymph (28). Our results are consistent with the finding that HP21 can activate proPAP2, a proteinase with considerable amino acid similarity to proPAP3.3
Our current model of one immune-inducible melanization pathway in M. sexta is shown in Fig. 7. In this model, HP14 autoactivates by directly binding peptidoglycan on the surface of Gram-positive bacteria or by binding to β-1,3-glucan on the surface of fungi via βGRP. HP14 activates proHP21 either by proteolytic cleavage or by eliciting a conformational change. HP21 cleaves proPAP2 and proPAP3, and the PAPs cleave proPO in the presence of cleaved SPH1 and SPH2 to generate active PO. The activator of M. sexta SPHs is not known. An activator of an SPH has been identified in H. diomphalia (19), but no clear ortholog of this proteinase (PPAF-III) has been identified in M. sexta. Negative regulation is accomplished by at least four serpins: serpin-4 inhibits HP21 (28), serpin-1J and serpin-3 inhibit PAP25 and PAP3 (7, 29), and serpin-6 inhibits PAP3 (30, 31). Several lines of evidence suggest that this is not the only melanization pathway in M. sexta. 1) ProPAP1 is not activated by HP21, thus, another proteinase is likely to be the activator of proPAP1.3 2) Complexes of HP21 with serpin-4 were detected in plasma incubated with Gram-positive but not Gram-negative bacteria; therefore, HP21 may not be involved in activation of proPO in response to Gram-negative bacteria infections (28). 3) We identified hemolymph fractions that could activate proPAP3 but that did not contain detectable HP21; therefore, at least one other activator of proPAP3 is probably present in hemolymph. Genetic analyses suggest at least two melanization pathways in D. melanogaster, one that activates proPO in response to bacteria and fungi and one that responds just to fungi (15). Likewise, RNA interference studies indicate that more than one melanization pathway is induced in response to infection in A. gambiae (11, 14).
FIGURE 7.

Model of a prophenoloxidase activation pathway in M. sexta hemolymph.
Our discovery that HP21 activates proPAP3 is significant because identifying proteinase components of the melanization process has been quite difficult. Biochemical strategies have been hindered by the difficulty of purifying active serine proteinases from hemolymph. The few successes include purification of HP14, several activators of proPO and an activator of an SPH (3-9, 19). A genetics approach to this problem was facilitated by the recent development of RNA interference as a means to silence candidate genes. By this method, several serine proteinases that function during melanization have been identified from D. melanogaster and A. gambiae (10-15). The disadvantage of the RNAi approach is that the specific location within a pathway remains unknown for the proteinases identified by this technique. The strategy used in this study, combining biochemistry with a candidate enzyme approach, has enabled us to identify a serine proteinase with a known activator (HP14) and a known substrate (proPAP3).
A surprising result of this study was that we observed activation but not cleavage of proHP21 by HP14. This outcome was unexpected for two reasons: serine proteinases usually are activated by limited proteolysis (32), and another study did detect cleavage of proHP21 by HP14.3 Nevertheless, all of our data that address this question are consistent with the existence of an active but uncleaved form of HP21. For example, the truncated forms of HP21 present in the active hemolymph fraction were cleaved in a location N-terminal to the predicted activation site. These truncated forms could be labeled with DFP, a serine proteinase inhibitor with very low affinity for zymogen forms of proteinases (33). In addition, the P1 activation site mutant form of proHP21, which was not cleaved, had constitutive catalytic activity. Taken together, our data indicate that HP21 can be active in an uncleaved form.
In contrast, Wang and Jiang3 demonstrated cleavage of proHP21 by HP14. A possible explanation for this difference is the requirement for an unknown cofactor for cleavage of HP21 by HP14, which was not present in the purified protein samples used in this study. Because of differences in purification methods, the proHP21 preparation that is cleaved by HP143 contains more impurities than the proHP21 preparation that is not cleaved (this study). A protein that acts as a cofactor for HP14 in vivo may be present in the former sample. Whether HP21 is cleaved at its predicted activation site in vivo is not clear. We have observed cleavage of proHP21 in hemolymph after the addition of elicitors of the melanization response, but the major cleavage product typically has a mass larger than that predicted for the catalytic or N-terminal domains,4 as we observed in this study (Fig. 3). However, one study did demonstrate that incubation of plasma with Gram-positive bacteria can generate a ∼30-kDa form of HP21 (28). It is possible that both mechanisms of activation of HP21 occur in vivo. Resolution of this problem requires further study.
Serine proteinases that are activated by conformational change not associated with cleavage are uncommon, but several are well characterized. Most are autoactivating enzymes positioned at the top of a proteinase cascade. For these proteinases, autoactivation occurs via a conformational change that is independent of peptide bond cleavage, and these activation processes occur within multi-protein complexes in the blood. For example, during fibrinolysis, in the presence of fibrin, the uncleaved form and the two-chain form of tissue-type plasminogen activator have similar activities toward plasminogen (a natural substrate) (34). Two other well known examples of this type of serine proteinase are mannose-binding lectin-associated serine proteinase-2 and horseshoe crab clotting factor C (35-36). Complement factor D, although cleaved at a putative zymogen activation site, is not active until it binds to its natural protein substrate C3bB, which induces a realignment of the catalytic triad to a functional conformation (37). Factor D and HP21 have in common the properties of being relatively insensitive to inhibition by DFP (requiring concentrations of 10 -20 mm) and that they do not efficiently hydrolyze small peptide substrates4 (25, 37-39).3 We propose that proHP21 is activated by a conformational change that occurs when proHP21 binds to HP14 and proPAP3. Support for the formation of such a complex in vivo is our recent finding that a curdlan-bound fraction of hemolymph contains these three proteinases as well as a curdlan binding protein, βGRP, and the target of the proteinase cascade, proPO.6
Acknowledgments
We thank the W. M. Keck Facility at Yale University for N-terminal sequencing and Nancy Williams for analyzing sequences.
Footnotes
This work was supported by National Institutes of Health Grant GM41247. This is contribution 07-126-J from the Kansas Agricultural Experiment Station. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
- proPO
- prophenoloxidase
- HP14
- hemolymph serine proteinase 14
- PAP
- prophenoloxidase-activating proteinase
- PPAF
- prophenoloxidase-activating factor
- PPAE
- prophenoloxidase-activating enzyme
- βGRP2
- β-1,3-glucan recognition protein
- SPH
- serine proteinase homolog
- IEAR-pNA
- IEAR-p-nitroanilide
- PTU
- phenylthiourea
- DETC
- diethylthiolcarbamate
- DFP
- diisopropyl fluorophosphate
- APMSF
- (4-ami-do-phenyl)-methane-sulfonyl fluoride
- MOPS
- 3-(N-morpholino) propane-sulfonic acid
Y. Wang and H. Jiang, submitted manuscript.
M. J. Gorman and M. R. Kanost, unpublished observations.
Y. Wang and H. Jiang, unpublished observations.
E. J. Ragan, M. J. Gorman and M. R. Kanost, unpublished observations.
REFERENCES
- 1.Kanost MR, Jiang H, Yu XQ. Immunol. Rev. 2004;198:97–105. doi: 10.1111/j.0105-2896.2004.0121.x. [DOI] [PubMed] [Google Scholar]
- 2.Cerenius L, Soderhall K. Immunol. Rev. 2004;198:116–126. doi: 10.1111/j.0105-2896.2004.00116.x. [DOI] [PubMed] [Google Scholar]
- 3.Ji C, Wang Y, Guo X, Hartson S, Jiang H. J. Biol. Chem. 2004;279:34101–34106. doi: 10.1074/jbc.M404584200. [DOI] [PubMed] [Google Scholar]
- 4.Wang Y, Jiang H. J. Biol. Chem. 2006;281:9271–9278. doi: 10.1074/jbc.M513797200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jiang H, Wang Y, Kanost MR. Proc. Natl. Acad. Sci. U. S. A. 1998;95:12220–12225. doi: 10.1073/pnas.95.21.12220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang H, Wang Y, Yu XQ, Kanost MR. J. Biol. Chem. 2003;278:3552–3561. doi: 10.1074/jbc.M205743200. [DOI] [PubMed] [Google Scholar]
- 7.Jiang H, Wang Y, Yu XQ, Zhu Y, Kanost M. Insect. Biochem. Mol. Biol. 2003;33:1049–1060. doi: 10.1016/s0965-1748(03)00123-1. [DOI] [PubMed] [Google Scholar]
- 8.Lee SY, Cho MY, Hyun JH, Lee KM, Homma KI, Natori S, Kawabata SI, Iwanaga S, Lee BL. Eur. J. Biochem. 1998;257:615–621. doi: 10.1046/j.1432-1327.1998.2570615.x. [DOI] [PubMed] [Google Scholar]
- 9.Satoh D, Horri A, Ochiai M, Ashida M. J. Biol. Chem. 1999;274:7441–7453. doi: 10.1074/jbc.274.11.7441. [DOI] [PubMed] [Google Scholar]
- 10.Castillejo-Lopez C, Hacker U. Biochem. Biophys. Res. Commun. 2005;338:1075–1082. doi: 10.1016/j.bbrc.2005.10.042. [DOI] [PubMed] [Google Scholar]
- 11.Volz J, Osta MA, Kafatos FC, Muller HM. J. Biol. Chem. 2005;280:40161–40168. doi: 10.1074/jbc.M506191200. [DOI] [PubMed] [Google Scholar]
- 12.Leclerc V, Pelte N, El Chamy L, Martinelli C, Ligoxygakis P, Hoffman JA, Reichart JM. EMBO Rep. 2006;7:231–235. doi: 10.1038/sj.embor.7400592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paskewitz SM, Andreeva O, Shi L. Insect Biochem. Mol. Biol. 2006;36:701–711. doi: 10.1016/j.ibmb.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 14.Voltz J, Muller H-M, Zdanowicz A, Kafatos FC, Osta MA. Cell. Microbiol. 2006;8:1392–1405. doi: 10.1111/j.1462-5822.2006.00718.x. [DOI] [PubMed] [Google Scholar]
- 15.Tang H, Kambris Z, Lemaitre B, Hashimoto C. J. Biol. Chem. 2006;281:28097–28104. doi: 10.1074/jbc.M601642200. [DOI] [PubMed] [Google Scholar]
- 16.Lee SY, Kwon TH, Hyun JH, Choi JS, Kawabata S-I, Iwanaga S, Lee BK. Eur. J. Biochem. 1998;254:50–57. doi: 10.1046/j.1432-1327.1998.2540050.x. [DOI] [PubMed] [Google Scholar]
- 17.Kwon TH, Kim MS, Choi HW, Joo CH, Cho MY, Lee BL. Eur. J. Biochem. 2000;267:6188–6196. doi: 10.1046/j.1432-1327.2000.01695.x. [DOI] [PubMed] [Google Scholar]
- 18.Yu X-Q, Jiang H, Wang Y, Kanost MR. Insect Biochem. Mol. Biol. 2003;33:197–208. doi: 10.1016/s0965-1748(02)00191-1. [DOI] [PubMed] [Google Scholar]
- 19.Kim MS, Baek MJ, Lee MH, Park JW, Lee SY, Soderhall K, Lee BL. J. Biol. Chem. 2002;277:39999–40004. doi: 10.1074/jbc.M205508200. [DOI] [PubMed] [Google Scholar]
- 20.Wang Y, Jiang H. Insect Biochem. Mol. Biol. 2004;34:731–742. doi: 10.1016/j.ibmb.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 21.Gupta S, Wang Y, Jiang H. Insect Biochem. Mol. Biol. 2005;35:241–248. doi: 10.1016/j.ibmb.2004.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Piao S, Song Y-L, Kim JH, Park SY, Park JW, Lee BL, Oh B-H, Ha N-C. EMBO J. 2005;24:4404–4414. doi: 10.1038/sj.emboj.7600891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zou Z, Jiang H. Insect Mol. Biol. 2005;14:433–442. doi: 10.1111/j.1365-2583.2005.00574.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang H, Wang Y, Gu Y, Guo X, Zou Z, Scholz F, Trenczek TE, Kanost MR. Insect Biochem. Mol. Biol. 2005;35:931–943. doi: 10.1016/j.ibmb.2005.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baker BC, Campbell CJ, Grinham CJ, Turcatti G. Biochem. J. 1991;279:775–779. doi: 10.1042/bj2790775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Skinner MK, Griswold MD. Biochem. J. 1983;209:281–284. doi: 10.1042/bj2090281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ji C, Wang Y, Ross J, Jiang H. Protein Expression Purif. 2003;29:235–243. doi: 10.1016/s1046-5928(03)00020-2. [DOI] [PubMed] [Google Scholar]
- 28.Tong Y, Jiang H, Kanost MR. J. Biol. Chem. 2005;280:14932–14942. doi: 10.1074/jbc.M500532200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu Y, Wang Y, Gorman MJ, Jiang H, Kanost MR. J. Biol. Chem. 2003;278:46556–46564. doi: 10.1074/jbc.M309682200. [DOI] [PubMed] [Google Scholar]
- 30.Wang Y, Jiang H. Insect Biochem. Mol. Biol. 2004;34:387–395. doi: 10.1016/j.ibmb.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 31.Zou Z, Jiang H. J. Biol. Chem. 2005;280:14341–14348. doi: 10.1074/jbc.M500570200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khan AR, James MNG. Protein Sci. 1998;7:815–836. doi: 10.1002/pro.5560070401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morgan PH, Robinson NC, Walsh KA, Neurath N. Proc. Natl. Acad. Sci. U. S. A. 1972;69:3312–3316. doi: 10.1073/pnas.69.11.3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stubbs MT, Renatus M, Bode W. Biol. Chem. 1998;379:95–103. doi: 10.1515/bchm.1998.379.2.95. [DOI] [PubMed] [Google Scholar]
- 35.Gal P, Harmat V, Kocsis A, Bian T, Barna L, Ambrus G, Vegh B, Balczer J, Sim RB, Naray-Szabo G, Zavodszky P. J. Biol. Chem. 2005;280:33435–33444. doi: 10.1074/jbc.M506051200. [DOI] [PubMed] [Google Scholar]
- 36.Nakamura T, Morita T, Iwanaga S. Eur. J. Biochem. 1986;154:511–521. doi: 10.1111/j.1432-1033.1986.tb09427.x. [DOI] [PubMed] [Google Scholar]
- 37.Volanakis JE, Narayana SVL. Protein Sci. 1996;5:553–564. doi: 10.1002/pro.5560050401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jing H, Macon KJ, Moore D, DeLucas LJ, Volanakis JE, Narayana SVL. EMBO J. 1999;18:804–814. doi: 10.1093/emboj/18.4.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lesavre PH, Muller-Eberhard HJ. J. Exp. Med. 1978;148:1498–1509. doi: 10.1084/jem.148.6.1498. [DOI] [PMC free article] [PubMed] [Google Scholar]