

Abstract
Mutations in presenilin genes PS1 and PS2 account for ∼50% of early-onset familial Alzheimer’s disease (FAD). The PS1 and PS2 genes encode highly homologous transmembrane proteins related to theCaenorhabditis elegans sel-12 and spe-4 gene products. A hydrophilic loop region facing the cytoplasmic compartment is likely to be functionally important because at least 14 mutations in FAD patients have been identified in this region. We report here that the loop regions of PS1 and PS2 interact with nonmuscle filamin (actin-binding protein 280, ABP280) and a structurally related protein (filamin homolog 1, Fh1). Overexpression of PS1 appears to modify the distribution of ABP280 and Fh1 proteins in cultured cells. A monoclonal antibody recognizing ABP280 and Fh1 binds to blood vessels, astrocytes, neurofibrillary tangles, neuropil threads, and dystrophic neurites in the AD brain. Detection of ABP280/Fh1 proteins in these structures suggests that these presenilin-interacting proteins may be involved in the development of AD and that interactions between presenilins and ABP280/Fh1 may be functionally significant. The ABP280 gene is located on the human X chromosome, whereas the newly identified Fh1 gene maps to human chromosome 3. These results provide a new basis for understanding the function of presenilin proteins and further implicate cytoskeletal elements in AD pathogenesis.
Keywords: Alzheimer’s disease, presenilins, protein–protein interaction, actin-binding protein 280, filamin homolog 1, cytoskeletal elements
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized neuropathologically by large numbers of neurofibrillary tangles (NFT) and senile plaques in the hippocampus and cerebral cortex. In addition to advanced age, Down syndrome, and ApoE-ε4 genotype, the presence of a positive family history is a consistent risk factor for AD (Heyman et al., 1984; van Duijin et al., 1991; Corder et al., 1993; Chartier-Harlin et al., 1994). Genetic studies have identified a number of autosomal dominant mutations associated with early-onset familial AD (FAD) in three genes: amyloid precursor protein (APP) gene on chromosome 21 (for review, seeSelkoe, 1994; Lendon et al., 1997), presenilin 1 (PS1) on chromosome 14 (Sherrington et al., 1995), and presenilin 2 (PS2) on chromosome 1 (Levy-Lahad et al., 1995; Rogaev et al., 1995). Mutations in all three genes lead to changes in amyloid β-peptide (Aβ), suggesting that APP processing and Aβ deposition may play a central role in AD pathogenesis (Citron et al., 1992; Cai et al., 1993; Scheuner et al., 1996).
PS1 and PS2 are expressed in many tissues, including the brain (Levy-Lahad et al., 1995; Rogaev et al., 1995; Sherrington et al., 1995; Kovacs et al., 1996; Suzuki et al., 1996; Giannakopoulos et al., 1997; Takami et al., 1997). In mouse brain, PS1 mRNA and protein have been detected in both neuronal and glial cells (Lee et al., 1996). In human brains, PS1 immunoreactivity has been reported in cortical neurons (Elder et al., 1996; Giannakopoulos et al., 1997), in NFT (Murphy et al., 1996), and in neuritic plaques (Wisniewski et al., 1995).
PS1 and PS2 genes encode transmembrane (TM) proteins with sequence similarity to the Caenorhabditis elegans gene products Sel-12 and Spe-4 (Levitan and Greenwald, 1995; Sherrington et al., 1995). Sel-12 has been implicated in modulating the reception of intercellular signaling mediated by lin-12, a member of the Notch family of receptor molecules (Levitan and Greenwald, 1995). The normal function or functions of PS1 and PS2 are not clear yet, although PS2 has been proposed to be involved in apoptosis (Vito et al., 1996;Wolozin et al., 1996). Presenilin mutations are associated with increases in the levels of the longer forms of the amyloid β-peptide (Aβ1–42/43), demonstrating that presenilin genes may modulate APP processing (Scheuner et al., 1996).
Recent studies suggest that PS1 protein has six or eight TM domains with both the N- and C-terminal regions, as well as a large hydrophilic loop region, facing the cytosolic compartment (Doan et al., 1996;Lehmann et al., 1997). The possibility that this hydrophilic region plays an important role in PS1 function and AD development is suggested by the fact that at least 14 independent mutations, including one splice site mutation identified in FAD, are located in this loop region (Schellenberg, 1995; Cruts et al., 1996; Lendon et al., 1997). PS1 is processed by endoproteolysis between amino acid residues 290–300 within this hydrophilic loop region (Thinakaran et al., 1996; Podlisny et al., 1997), further supporting the hypothesis that this region is an important functional domain.
We report here that the actin-binding protein, ABP280 (also called nonmuscle filamin), and a protein related to filamin (filamin homolog 1, Fh1) can interact with both PS1 and PS2 proteins. The gene encoding Fh1 maps to human chromosome 3. In cells transiently transfected with PS1, overexpression of PS1 appears to modify the intracellular distribution of endogenous ABP280/Fh1 proteins as detected by indirect immunofluorescence microscopy. Immunohistochemical studies using a monoclonal antibody recognizing both ABP280 and Fh1 show that these proteins are expressed predominantly in blood vessels and astrocytes in the normal brain. In the AD brain, strong immunoreactivity was detected not only in astrocytes but also in NFT, neuropil threads, and dystrophic neurites within some senile plaques. Similar astrocytic immunostaining also was observed by using two different anti-PS1 antisera. Thus, our results demonstrate that presenilin proteins can interact both in vitro and in vivo with ABP280 and Fh1, cytoskeletal proteins of the actin-binding protein family, and that these cytoskeletal proteins are present in NFT. These observations provide new information on a possible function of the presenilin proteins in interaction with the cytoskeleton and their role in the pathogenesis of AD.
MATERIALS AND METHODS
Yeast two-hybrid cDNA library screening and protein–protein interaction assay. A human fetal brain cDNA library was constructed with the yeast plasmid pJG4–5, which expresses individual cDNAs as fusion proteins containing a transcription activation domain (Zervos et al., 1993). As detected by Western blotting with anti-LexA antibody, the PS1 loop region (amino acids 263–411) was expressed as a fusion protein with the DNA-binding protein LexA in yeast carrying the reporter genes leu2 andlacZ. The human brain cDNA library was screened as described (Wu and Maniatis, 1993; Zervos et al., 1993) in yeast expressing the PS1 bait as well as the reporter genes. Individual cDNAs were purified from the positive clones and retransformed into yeast to confirm that the cDNAs encode proteins that specifically interact with the PS1 bait but not other unrelated control proteins. The confirmed cDNAs were sequenced with an ABI automatic sequencer. To test interactions between PS1/2 and ABP280 or Fh1, we introduced the bait plasmids expressing either the wild-type PS1/2 loop region or the PS1 loop region containing individual FAD mutations as LexA fusion proteins into yeast expressing either ABP280 or Fh1 C-terminal regions as fusion proteins containing the transcription activation domain. Quantitative liquid β-galactosidase assays were performed on at least three independent colonies for each combination and compared with the background, as previously described (Wu and Maniatis, 1993). The background is defined as the amount of β-galactosidase activity detected in yeast cotransformed with corresponding prey plasmids and bait plasmid expressing the APP intracellular domain–LexA fusion protein, which has the same level of β-galactosidase activity as the yeast containing the LexA bait vector plasmid without any cDNA insert.
Chromosome mapping. A sequenced tagged site (STS) for the Fh1 gene was developed on the basis of the sequence of the Fh1 cDNA clones. PCR was performed by using DNA samples prepared from a panel of monochromosome somatic cell hybrids obtained from American Type Culture Collection (ATTC; Rockville, MD) to determine the chromosomal location of the Fh1 gene. The PCR primers used were CACCACAGGTATCCAGTC and CTGTCACCTTGGCCTTGAAG. PCR reactions were performed in a final reaction volume of 25 μl with 10 pmol of each primer, 200 μmdNTPs, 1.5 mm MgCl2, 1 μl of formamide, and 0.1 μl of Taq polymerase. The reaction was denatured at 94°C for 5 min, followed by 35 cycles of 94°C for 45 sec, 53°C for 30 sec, 72°C for 1 min, and a final extension at 72°C for 5 min. The PCR products from individual monochromosome somatic cell hybrids and control cell lines, as well as DNA molecular size markers, were separated on a 2% agarose gel and visualized under UV transillumination with ethidium bromide.
Coimmunoprecipitation. In vitro translation products of the ABP280 or Fh1 C-terminal region and of PS1 or PS2 loop region were labeled with [35S]methionine. Individual proteins or proteins after coincubation were immunoprecipitated with the monoclonal antibody NCL-FIL or monoclonal antibodies against the epitope tags present on the corresponding proteins. The precipitated products were separated on SDS-PAGE, and the dried gel was exposed to x-ray films.
Transfection and immunofluorescent microscopy. Cos-1 cells were grown on coverslips to ∼70% confluence and transiently transfected with a plasmid expressing full-length PS1, using lipofectin (Life Technologies, Gaithersburg, MD). At 36 hr after transfection, the cells were fixed with 4% paraformaldehyde for 20 min and permeabilized with 1% Triton X-100 in PBS for 10 min. After the cells were blocked with PBS/20% fetal bovine serum/0.05% Tween 20, the monoclonal antibody NCL-FIL (NovoCastra Laboratories, New Castle, UK) and polyclonal rabbit anti-PS1 antibodies recognizing the N terminus of PS1 [Ab14, Lee et al. (1996); an affinity-purified antibody raised against a PS1 synthetic peptide containing amino acid residues 30–46, Malin et al. (1997)] were added in blocking solution and incubated for 1.5 hr. Secondary antibodies (anti-mouse fluorescein conjugate or anti-rabbit Cy3 conjugate, Jackson ImmunoResearch Laboratories, West Grove, PA) were applied to the slides. After three washes, cells were mounted for viewing. Similar results were obtained by using polyclonal antibodies recognizing either the N terminus or the loop region of PS1.
Collection of tissue samples and diagnosis of AD. Seven AD brains and two age-matched control brains were obtained at autopsy; the detailed information about the human brain cases used is shown in Table1. The neuropathological diagnosis of AD relies on previously described methods (Berg et al., 1993; McKeel et al., 1993), using objective quantitative criteria as originally proposed by Khachaturian (1985). The criteria were modified on the basis of the average (10 mm2 microscopic fields) age-adjusted total argyrophilic (diffuse and neuritic) plaque score, which must be met or exceeded in at least a single standardized CERAD (Consortium to Establish a Registry for Alzheimer’s Disease) neocortical area (Mirra et al., 1991). This strategy prevents making a diagnosis of AD in cases that have only a focal cluster of cortical plaques.
Table 1.
Subject demographics
| Case | Age | Sex | PMI (hr) | DX |
|---|---|---|---|---|
| 1 | 78.2 | M | 12 | AD |
| 2 | 81.3 | F | 4 | AD |
| 3 | 89.2 | F | 12 | AD |
| 4 | 74.4 | M | 3 | AD |
| 5 | 91.3 | F | 6 | AD |
| 6 | 85.1 | F | 14 | AD |
| 7 | 75.3 | M | 7 | AD |
| 8 | 79.7 | F | 10 | Control |
| 9 | 87.0 | F | 15 | Control |
Shown in the table is information about the human brain cases that were examined, including age, sex, postmortem interval (PMI), and diagnosis (DX). All of the AD cases are of clinical dementia rating 3, and both control cases are of clinical dementia rating 0 (Morris, 1993).
Antibodies. The monoclonal antibody against human filamin was purchased from NovoCastra Laboratories. The polyclonal anti-human PS1 antisera were kindly provided by Dr. S. Gandy (Lee et al., 1996) and by Dr. J. M. Nerbonne (Malin et al., 1997). The final dilutions of corresponding antibodies used are described in the figure legends.
Immunohistochemistry. Cryostat and paraffin sections of midfrontal (CERAD standard area) cortex (6 μm each) that had been obtained at autopsy were used. The sections were fixed 24–48 hr in the refrigerator in either ethanol fixative (0.15N NaCl and 70% ethanol) or paraformaldehyde fixative (4% paraformaldehyde in 0.1 mneutral pH phosphate buffer). The paraffin was removed by heat and xylene, followed by 8 min microwave pH 6.0 citrate buffer antigen retrieval. Then sections were rinsed multiple times in buffer. The primary antibody NCL-FIL was applied in a moist chamber in the refrigerator overnight at working dilutions of 1:200 and 1:500. Subsequent steps used the Vector Elite ABC kit (Burlingame, CA), with DAB as substrate. Ethanol fixative gave more robust staining signals than paraformaldehyde fixative when the NCL-FIL monoclonal antibody was used. Sections were mounted in permanent mountant and photographed with a Leitz Dialux automated exposure 35 mm camera system. Photographic prints were reproduced from the original color negatives.
RESULTS
PS-1 interacts with nonmuscle filamin and a protein with sequence similarity to filamin, filamin homolog 1 (Fh1)
A human brain cDNA library was constructed with the yeast expression vector for yeast two-hybrid interaction cloning (Wu and Maniatis, 1993; Zervos et al., 1993). cDNAs in this library are expressed as proteins fused to a transcription activation domain. Human PS1 loop region (amino acids 263–411) was expressed as a fusion protein with the DNA-binding protein LexA in yeast carrying the reporter genes, as detected by Western blotting of the yeast extracts, using anti-LexA antibodies (data not shown). Although the PS1–LexA fusion protein by itself does not lead to activation of the reporter genes, interaction of the PS1 bait protein with a protein encoded by the cDNA in the library will bring together the DNA-binding domain and the transcription activation domain to form an active transcription complex, resulting in the activation of the reporter genes,leu2 and lacZ (Zervos et al., 1993).
Using the PS1 loop region as a bait, we screened ∼2 × 106 independent clones of the human brain cDNA library and identified a number of positive interaction clones. The individual cDNAs were isolated and retransformed into the yeast to confirm that these genes encode proteins that interact specifically with the PS1 loop region, but not with other unrelated proteins. DNA sequence analyses of these PS1 interacting clones revealed the presence of several distinct groups of cDNAs. These cDNAs currently are being characterized further and will be described elsewhere. Two of these cDNA clones encode the C-terminal 358 amino acid residues of human nonmuscle filamin (filamin 1, FLN1) (also named actin-binding protein 280, ABP280; Gorlin et al., 1990), and four cDNA clones encode the C-terminal 291 residues of a protein that is highly homologous to the nonmuscle filamin, which we have named Filamin homolog 1 (Fh1).
Sequence analysis shows that the Fh1 gene encodes a previously unidentified protein with significant sequence similarity to ABP280. The 291 amino acid region encoded by the Fh1 cDNA fragment obtained from the human brain cDNA yeast two-hybrid library shows 69% identity and 80% similarity to the C terminus of ABP280 (Fig.1). Northern analysis demonstrated that both ABP280 and Fh1 cDNA probes hybridized to two bands of ∼8 and 9 kb in several tissues, including heart, brain, placenta, lung, liver, kidney, and pancreas (data not shown).
Fig. 1.

Alignment of ABP280 and Fh1 peptide sequences in the C-terminal region. Two clones of ABP280 cDNA and four clones of Fh1 cDNA were identified from screening a human brain cDNA yeast two-hybrid library, using the PS1 loop region as bait. The ABP280 cDNAs encode the C-terminal 387 residues of the protein, whereas the Fh1 cDNAs encode the C-terminal 291 amino acids. The alignment of the C-terminal 291 residues of the two proteins as predicted from cDNA sequences is shown. The amino acid residues identical between the two proteins are inbold and underlined, with the similar residues underlined. Over this region of 291 residues, identity is 69% and similarity is 80% without the introduction of any gaps.
The full-length cDNA encoding ABP280 has been cloned, and sequence analysis predicts that ABP280 is a protein of 2647 amino acids, with the N-terminal 275 residues as the actin-binding domain, followed by 24 copies of a 96-residue repeat (Gorlin et al., 1990). ABP280 forms dimers with a leaf spring-like structure via intermolecular interaction between the C-terminal 96-residue repeat region of the protein (Gorlin et al., 1990). The ABP280 cDNA clones that we identified in the yeast two-hybrid screening encode part of the twenty first repeat and the last three 96-residue repeats.
Human Fh1 gene maps to chromosome 3
Two genes encoding ABP280 (FLN1) and a related protein (FLN2) have been identified previously. The FLN1 gene maps to human chromosome Xq28 and is expressed in many cell types, whereas the FLN2 gene maps to human chromosome 7q32-35 and is expressed predominantly in skeletal and cardiac muscle (Gorlin et al., 1993; Maetrini et al., 1993).
We developed a STS for the Fh1 gene and used PCR to determine the chromosomal location of this gene using a panel of monochromosome somatic cell hybrids obtained from ATCC. In total human genomic DNA, the STS generated two PCR fragments, one of the expected size (230 bp) and a second band of 800 bp. DNA sequence analysis of these bands confirmed that the 230 bp band represented the Fh1 genomic DNA fragment and that the 800 bp band was a nonspecific PCR product. The 230 bp band maps to chromosome 3 (Fig. 2). It should be noted that a putative locus for late-onset AD also has been mapped to chromosome 3 (Tanzi et al., 1996). Therefore, Fh1 represents a novel gene on chromosome 3 encoding a protein highly homologous to ABP280. This brings to three the number of filamin-like genes in the human genome.
Fig. 2.

Fh1 maps to human chromosome 3. A sequenced tagged site (STS) for the Fh1 gene was developed, and PCR was used to determine the Fh1 chromosomal location. The PCR products from individual monochromosome somatic cell hybrids and control cell lines, as well as DNA molecular size markers, were separated on a 2% agarose gel and visualized under UV transillumination with ethidium bromide. In total human DNA, the STS generated two PCR fragments, one of the expected size (230 bp) and a second band of 800 bp. Direct sequencing of the PCR products revealed that the 230 bp band was Fh1 and that the 800 bp band was a nonspecific PCR product. The 220 bp Fh1 band maps to chromosome 3.
ABP280 and Fh1 interact with loop regions of both PS1 and PS2 proteins
To test whether the PS1 loop region can interact with ABP280 or Fh1 in biochemical assays, we performed coimmunoprecipitation experiments. Because presenilin proteins are associated with membrane structures and ABP280/Fh1 are high molecular weight proteins associated with cytoskeletal elements, it is difficult to rule out the possibility that coimmunoprecipitation of the full-length proteins from solubilized cell extracts is an artifact caused by their association with membrane or cytoskeletal structures. Therefore, we decided to use in vitro-translated soluble protein fragments of presenilins and ABP/Fh1 to perform coimmunoprecipitation experiments. The cDNA fragments encoding the loop region of PS1 or encoding the C-terminal fragments of ABP280 or Fh1 identified from the yeast two-hybrid screening were inserted into a plasmid vector under the control of the T7 promoter, and the corresponding proteins were radiolabeled byin vitro translation in the presence of [35S]methionine. A monoclonal antibody against filamin (NCL-FIL, NovoCastra Laboratories) immunoprecipitated both ABP280 and Fh1 in vitro translation products (Fig.3, lanes 1 and4), but not PS1 protein (Fig. 3, lane 3), demonstrating that this monoclonal antibody recognized both ABP280 and Fh1 but did not cross-react with PS1. However, when ABP280 or Fh1in vitro translation products were incubated with in vitro-translated PS1 protein, PS1 was coimmunoprecipitated by this monoclonal antibody (Fig. 3, lanes 2 and 5, respectively).
Fig. 3.

ABP280 and Fh1 interact with PS1/2 in coimmunoprecipitation experiments. The individual proteins were labeled by in vitro translation in the presence of [35S]methionine. The monoclonal antibody NCL-FIL precipitated both ABP280 (labeled as ABP orA; lanes 1 and 6) and Fh1 (labeled as Fh1 or F;lanes 4 and 9) from in vitrotranslation products. The antibody did not cross-react with in vitro-translated PS1 or PS2 loop region (lanes 3and 8). After coincubation with either ABP280 or Fh1 proteins, PS1 (lanes 2 and 5) or PS2 (lanes 7 and 10) was coimmunoprecipitated by the antibody. The immunoprecipitated products were separated on SDS-PAGE. An autoradiograph of the gel is shown.
We also examined whether the PS2 loop region could interact with ABP280 or Fh1 in the coimmunoprecipitation assay. Although monoclonal antibody NCL-FIL did not immunoprecipitate in vitro-translated PS2 protein (Fig. 3, lane 8), PS2, like PS1, was coimmunoprecipitated by this antibody after being incubated with ABP280 or Fh1 (Fig. 3, lanes 7 and 10). Similar coimmunoprecipitation results were obtained by using monoclonal antibodies against epitope tags present in the in vitro-translated proteins (data not shown). Unrelated proteins that did not bind to ABP280/Fh1 in the yeast two-hybrid assay did not coimmunoprecipitate with ABP280/Fh1 in this assay, suggesting that coimmunoprecipitation of PS1/2 with ABP280/Fh1 was not attributable to nonspecific protein–protein interactions. These results demonstrate that both PS1 and PS2 loop regions are able to interact with ABP280 and Fh1 proteins in vitro.
Using the yeast two-hybrid assay, we have confirmed that Fh1 and ABP280 are able to interact with the PS2 loop region (Table2). Quantitative liquid β-galactosidase assays were performed with protein extracts prepared from yeast expressing the bait PS1 or PS2–LexA fusion proteins and the prey proteins Fh1 or ABP280 fused to the transcription activation domain. Because APP protein, another important player in the pathogenesis of AD, also contains a hydrophilic intracellular domain, we tested whether the APP intracellular domain could interact with Fh1 or ABP280. In yeast expressing the APP intracellular domain as a LexA fusion protein and the Fh1 or ABP280 prey proteins, only basal levels of β-galactosidase activity were detected, indicating that the APP intracellular domain does not interact with Fh1 or ABP280. When either PS1– or PS2–LexA fusion protein was expressed in yeast containing Fh1 or ABP280 prey proteins, significant activation of β-galactosidase activity was detected, indicating that both the PS1 and PS2 loop regions were able to interact with Fh1 or ABP280. These experiments confirm the results obtained from the coimmunoprecipitation experiments.
Table 2.
Protein–protein interactions detected by using the yeast two-hybrid assay
| Prey | Fh1 | ABP280 |
|---|---|---|
| Bait (LexA fusion) | ||
| APP | 1.0 | 1.0 |
| PS1 | 17.0 | 17.4 |
| PS2 | 15.5 | 32.6 |
| PS1E280A | 32.8 | 15.4 |
| PS1L286V | 23.3 | 19.0 |
| PS1P267S | 22.0 | 16.5 |
| PS1R269H | 41.2 | 15.9 |
| PS1dEx9 | 33.5 | 36.1 |
Quantitative liquid β-galactosidase assays were performed on at least three independent colonies for each combination. The numbers represent the fold activation above background. The background is defined as the amount of β-galactosidase activity detected in the yeast, with cotransformation of corresponding prey plasmids and the bait plasmid expressing APP intracellular domain–LexA fusion protein, which has the same level of β-galactosidase activity as the yeast containing the prey plasmid and the LexA bait vector plasmid without any cDNA insert.
We then investigated whether mutations found in the PS1 hydrophilic loop region in AD patients affected interactions with Fh1 or ABP280. Interestingly, all of the PS1 mutants examined, including P267S, R269H, E280A, L286V, and the exon 9 deletion (dEx9), showed similar or higher levels of β-galactosidase activation, indicating that these mutants retain the ability to interact with Fh1 or ABP280. It is possible that these PS1 mutants represent gain-of-function rather than loss-of function mutants. Whether these PS1 mutants indeed interact more strongly with Fh1 or ABP280 proteins in cells and the functional significance of such interactions will be examined further.
PS1 appears to modify intracellular distribution of Fh1 and ABP280 in transfected cells
To investigate whether PS1 interacts with ABP280 and Fh1 in cells, we examined the intracellular localization of these proteins in cells overexpressing PS1. After Cos-1 cells were transiently transfected with a plasmid expressing full-length PS1 protein, double immunofluorescence microscopy was performed by using the NCL-FIL monoclonal antibody to detect endogenously expressed ABP280 or Fh1 (Fig.4A) and polyclonal rabbit anti-PS1 antibodies (Ab14) to detect PS1 protein (Fig.4B). As observed in previous studies (Doan et al., 1996; Kovacs et al., 1996), in transfected cells PS1 is localized primarily in the endoplasmic reticulum, Golgi apparatus, or other intracellular membrane structures in the perinuclear region (Fig.4B). Low levels of PS1 immunostaining also were detected in nontransfected cells, which may represent immunoreactivity with endogenous PS1 protein (compare the four cells in the center with the surrounding cells in Fig. 4B), because no signal was detected by the same staining procedure, using the preimmune serum (data not shown). Although the NCL-FIL immunostaining was relatively uniform in the cytoplasm of Cos-1 cells not overexpressing PS-1 (see the cells in the periphery of Fig. 4A), a stronger NCL-FIL immunostaining signal was detected in the perinuclear region in cells overexpressing PS1 (Fig. 4A), suggesting that PS1 may be able to modulate the intracellular distribution of these actin-binding proteins. The PS1 immunoreactivity could be superimposed on the perinuclear signal from the NCL-FIL immunostaining. These results strongly suggest that PS1 can interact with ABP280 and Fh1 in cells.
Fig. 4.
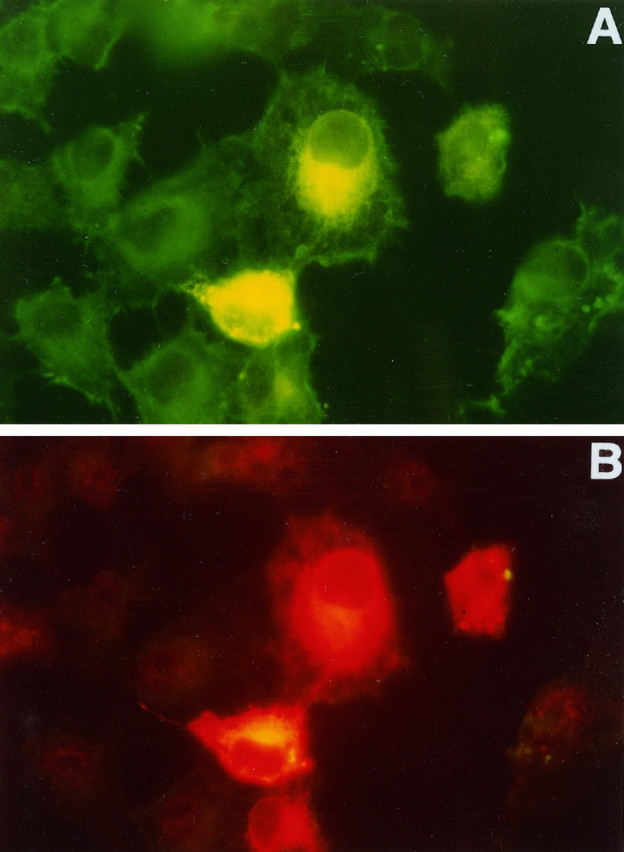
PS1 overexpression may modify intracellular distribution of ABP280/Fh1 proteins in transfected cells. After Cos-1 cells were transfected with a plasmid expressing the full-length PS1 protein, cells were fixed and double-stained, using polyclonal rabbit anti-PS1 antisera (B) and the monoclonal antibody NCL-FIL, which recognizes both ABP280 and Fh1 (A). Fluorescein-conjugated anti-mouse secondary antibody or Cy3-conjugated anti-rabbit secondary was used to detect ABP280/Fh1 or PS1 staining, respectively. ABP280/Fh1 staining is shown in A. The four cells in the center of the picture were expressing high levels of PS1, as detected by the anti-PS1 antibody shown in B. In cells not transfected by the PS1 plasmid, ABP280/Fh1 staining is more uniform throughout the cytoplasm, whereas the ABP280/Fh1 staining in cells overexpressing PS1 is perinuclear and similar in distribution to that of PS1 protein (compareA with B).
Immunodetection of PS1 and ABP280/Fh1 proteins in normal and AD brains
To investigate the possible role of ABP280 and Fh1 in AD pathogenesis, we examined seven AD brains and two age-matched control brains for expression of PS1 and ABP280/Fh1 proteins by immunohistochemical staining. On cryostat sections of the frontal cortex from both control and AD brains, robust immunostaining was detected in cells with fibrillary astrocyte morphology and in cell processes with either the monoclonal antibody NCL-FIL or two different polyclonal antisera raised against the N terminus of PS1 (Lee et al., 1996; Malin et al., 1997) (Fig. 5; data not shown). The number and the size of immunoreactive astrocytes appeared to be greater in the AD brains as compared with the control brains. The control brains showed an astrocyte distribution similar to that obtained with antibodies against glial fibrillary acidic protein, with most astrocytes in the subpial cortex and junction region between cortical gray and white matter, as well as in white matter. In AD brains, however, PS1-positive astrocytes are distributed throughout the cortical laminae. Weaker PS1 immunoreactivity was detected in the cytoplasm of some cells with neuronal morphology (Fig. 5B). No clear PS1 immunostaining signal was detected in senile plaques, NFT, neuropil threads, or blood vessels.
Fig. 5.

Expression of PS1 and ABP280/Fh1 proteins in control and AD brains. A–H show microscopic findings from nondemented controls and persons with advanced AD, using human brain sections that have been immunostained with polyclonal antisera to human PS-1 or monoclonal antibodies to human filamin (NCL-FIL) and hyperphosphorylated tau (PHF-1). All sections were immunostained with the Vector ABC Elite kit, with diaminobenzidine as a substrate, and were lightly counterstained with hematoxylin before being coverslipped.A, Frozen 6-μm-thick section of the frontal cortex from an 89-year-old woman with advanced AD has been fixed for 30 min in paraformaldehyde before being immunostained with NCL-FIL diluted 1:200. Numerous reactive fibrillary astrocytes are evident, as are immunopositive capillaries (250× magnification). B, A 6-μm-thick frozen section near that in A has been immunostained with polyclonal PS-1 antibody (1:10 dilution; Lee et al., 1996) to show reactive astrocytes in AD cortex. Theinset shows two other glial cells with astrocyte morphology. Lightly immunostained cells of uncertain nature are identified by arrows. The more densely labeled cell in the main panel has the morphology of a small neuron with labeled nonfibrillary cytoplasm (400× magnification). C, An entorhinal cortex paraffin section (6 μm) from the brain of a 78-year-old man with advanced AD has been fixed in ethanol saline before being immunostained with NCL-FIL at 1:200 dilution. The neurofibrillary tangles detected on this specimen are mostly extraneuronal tangles, which are not stained by NCL-FIL. The filamin antibody labels capillaries (arrow) and arterioles and reveals reactive astrocytic gliosis (400× magnification).D, Cerebral cortex paraffin section from paraformaldehyde-fixed brain of a 79-year-old nondemented woman shows intense transmural immunostaining of both meningeal muscular arteries and parenchymal arteries, as well as capillaries, with filamin antibody diluted at 1:200. Veins and venules (not shown) also were stained by the filamin antibody. Comparable patterns of filamin immunoreactivity were seen in control and AD brains (100× magnification).E, The same specimen as in A andB shows large reactive filamin-positive astrocytes surrounding and within an ill-defined circular immunopositive area with a cortical senile plaque profile (NCL-FIL at 1:200 dilution; 400× magnification). F, The same brain as inA, B, and E showing filamin staining pattern of hippocampal CA1 fields in advanced AD. Several intraneuronal globoid tangles (arrows) are immunopositive, as is a neurite contributing to a senile plaque (arrowhead). Adjacent extraneuronal ghost tangles (asterisk) are not labeled (NCL-FIL at 1:500 dilution; 400× magnification). G, Frontal cortex paraffin section of ethanol saline-fixed brain stained with NCL-FIL at 1:500. The specimen is the same brain illustrated in A, B, E, andF. Four triangles, numerous neuropil threads, and the bulbous dystrophic neurites within two senile plaques (insets) are immunostained (400× magnification). H, A section near that shown in G has been immunostained with a monoclonal antibody against tau (PHF-1; Greenberg and Davies, 1990) to show a similar pattern to the filamin antibody-labeling pattern inG. PHF-1 labeled more tangles and neuropil threads than did NCL-FIL in all AD brains (PHF-1 at 1:100 after microwave-citrate and formic acid antigen retrieval procedures; 400× magnification).
On cryostat sections strong NCL-FIL immunoreactivity was detected in astrocytes and fine cellular processes in the neuropil of the AD brains (Fig. 5A). Similar but less intense immunoreactivity also was seen in the control brains (data not shown). The phenotypic appearance and distribution of these NCL-FIL-positive astrocytes overlapped with those of PS1-labeled astrocytes. NCL-FIL-positive astrocytosis also was detected in paraffin sections of AD brains that had been fixed in ethanol (Fig. 5C). In addition, robust NCL-FIL immunostaining was detected in blood vessels of all caliber, including capillaries and some meningeal vessels (Fig.5C,D). This immunostaining pattern in meningeal vessels resembled that observed with Aβ peptide antibody 10D5 (Athena Neurosciences, San Francisco, CA) (data not shown). Most interestingly, in the AD brains unequivocal and strong NCL-FIL immunostaining was detected in the majority of cortical and hippocampal CA1 intraneuronal NFTs and neuropil threads, as well as dystrophic neurites within some senile plaques, demonstrating that ABP280/Fh1 proteins were present in these structures (Fig. 5E–G). This NCL-FIL immunostaining pattern in the AD brain is similar to that obtained by using a monoclonal antibody against hyperphosphorylated tau protein, PHF-1 (Greenberg and Davies, 1990) (compare Fig. 5G,H), except that PHF-1 labels more tangles and neuropil threads in all of the AD brains we examined. This suggests that only a subset of NFT is labeled by the filamin antibody.
DISCUSSION
Numerous studies indicate that mutations in the presenilin genes play an important role in the development of early-onset FAD. PS1 and PS2 are members of a highly conserved family of TM proteins with sequence similarity to the C. elegans Sel-12 and Spe-4 gene products. Recent studies suggest that PS1/2 proteins have six or eight TM domains with the N and C termini, as well as the largest hydrophilic loop region, facing the cytoplasmic compartment. Identification of a large number of mutations in the hydrophilic loop region suggests that this region is critical for presenilin function. We therefore set out to identify proteins that interact with the loop region of the presenilin proteins. Among the proteins identified that interact with both PS1 and PS2 are actin-binding protein ABP280 and a closely related protein, Fh1. The protein–protein interactions between the ABP280/Fh1 and PS1/2 are specific, because there were no detectable interactions observed between ABP280/Fh1 and LexA fusion proteins containing the intracellular domain of APP (Table 2) or other unrelated bait proteins rich in hydrophilic amino acid residues (data not shown). The interactions between ABP280/Fh1 and PS1/2 proteins not only were detected in the yeast two-hybrid assay but also were confirmed in coimmunoprecipitation experiments. Interaction between PS1 and ABP280/Fh1 is supported further by the observation that overexpression of PS1 in Cos-1 cells by transient transfection leads to a change in the intracellular distribution of the endogenous ABP280/Fh1 proteins such that they colocalize with PS1. These results implicate a potential role of presenilin proteins in modulating the cytoskeleton in cells. Consistent with our results, it has been reported that the C-terminal fragments of both PS1 and PS2 are associated with cytoskeleton (Kim et al., 1997).
Our results demonstrating interactions between presenilins and cytoskeletal-associated proteins ABP280/Fh1 provide insights into the possible functions of the presenilin proteins. ABP280 protein has been found to promote branching of actin filaments (Hartwig et al., 1980;Niederman et al., 1983; Hartwig and Shevlin, 1986) and is proposed to modulate cell shape, polarity, and motility via changes in actin filament organization (Stossel, 1993; Matsudaira, 1994; Drubin and Nelson, 1996; Mitchison and Cramer, 1996). In eukaryotic cells the cytoskeleton is composed mainly of microtubules, microfilaments, and intermediate filaments. Biochemical and structural studies indicate that these polymeric systems are interacting with each other in cells (Pollard et al., 1984; Green et al., 1987). Actin-binding proteins such as ABP280 have been proposed to interconnect the microfilament and intermediate filament cytoskeletal systems (Brown and Binder, 1992). Recently, the actin cytoskeleton has been implicated in regulating the activity of the cystic fibrosis TM conductance regulator (Prat et al., 1995). It is conceivable that, by interacting with these cytoskeletal proteins, presenilins may modulate the activities of these and other related proteins in cells.
Both ABP280 and Fh1 are expressed in human brain tissue, as detected by Northern blotting and immunostaining. Our results of immunohistochemical staining of human brain sections indicate that both PS1 and ABP280/Fh1 proteins colocalize to astrocytes and delicate cellular processes in both gray matter and white matter of the cerebral cortex and hippocampal formation. Although we could not detect strong PS1 immunostaining in somatodendritic regions of neurons by using two different polyclonal antisera against PS1 protein on either paraffin or cryostat sections, strong PS1 immunoreactivity with both PS1 antisera was detected in fibrous astrocytes, consistent with a previous report that both mRNA and protein of PS1 are present in astrocytes (Lee et al., 1996). In the AD brains there are more PS1-positive astrocytes distributed throughout the cortical laminae, as compared with the control brains. Immunostaining of astrocytes and fine cellular processes by the monoclonal antibody recognizing ABP280/Fh1 is similar to that detected by PS1 antibodies. Many studies suggest that astrocytes play an important role in the amyloid β deposition and neurodegeneration observed in AD (Potter, 1992; Nieto-Sampedro and Mora, 1994; Pike et al., 1995). Our study demonstrating that both PS1 and its interacting proteins ABP280/Fh1 are expressed in astrocytes raises the possibility that presenilin protein in astrocytes plays an important role in AD pathogenesis, providing additional support for the involvement of astrocytes in development of AD.
Robust ABP280/Fh1 immunoreactivity also was seen in blood vessels, NFTs, neuropil threads, and some senile plaque neurites. The specificity of the monoclonal antibody NCL-FIL is demonstrated by the observation that this antibody recognizes one protein band of 250 kDa, which may represent both ABP280 and Fh1, and that it did not cross-react with other proteins in the cell, including presenilin proteins, other cytoskeletal proteins, or unrelated proteins (NovoCastra Laboratories data) (Fig. 3; data not shown). Our PS1 antibodies did not show NFT immunoreactivity, although PS1 staining in NFTs has been reported (Murphy et al., 1996). This discrepancy may reflect differences in the specificity of different PS1 antisera. NFTs are one of the major neuropathological characteristics of the AD brain. The accumulation of NFTs is highly correlated with the loss of pyramidal cells and with dementia. Ultrastructural and biochemical studies of NFTs have revealed that the major constituent of the paired helical filaments is polymerized and hyperphosphorylated tau (Wischik et al., 1995). Molecular and immunohistochemical experiments demonstrated that NFTs also contain a number of cytoskeletal proteins, including neurofilament proteins, vimentin, actin, ubiquitin, and MAP2 (Wischik et al., 1995). Because actin has been detected in NFTs, it is not surprising that actin-binding proteins are also present in NFTs. Our result that PS1 and PS2 interact directly with components of NFTs, ABP280/Fh1 proteins, raises the possibility that the presenilins may play an important role in the formation of NFTs. These observations also suggest that protein–protein interactions between presenilins and ABP280/Fh1 may be functionally significant. It is noted that only a subset of NFTs was detected by the filamin monoclonal antibody. In particular, in the AD brains we have examined, only intraneuronal NFTs, but not extraneuronal NFTs, were labeled by the filamin antibody. The exact mechanism of this apparent epitope selectivity is at present uncertain and awaits further investigation by comparing results from dual labeling and differential antigen retrieval and by using competing antigens.
Human ABP280 has been mapped to the X chromosome (Xq28) (Gorlin et al., 1993; Maetrini et al., 1993). The new filamin-related gene Fh1, which was identified in our study, maps to human chromosome 3. It is interesting to note that a putative locus for late onset FAD recently has been mapped to chromosome 3, although detailed information is not available yet (Tanzi et al., 1996). The protein–protein interactions between Fh1 and presenilin proteins and the presence of Fh1 in NFT in the AD brain suggest that Fh1 is a candidate FAD gene. We are presently testing whether Fh1 could be the putative AD locus on chromosome 3.
Footnotes
This work is supported by grants from the Alzheimer’s Disease Research Center at Washington University School of Medicine, from the Missouri Alzheimer’s Disease and Related Disorder Program, the Alzheimer’s Association, and the American Health Assistance Foundation to J.Y.W.; by National Institutes of Health Grant AG-05861 from the National Institute of Aging to Washington University School of Medicine (WUSM) ADRC (L. Berg, M.D.); and by Grants from National Institutes of Health (AG00634 and AG05681), the Western Southern Foundation, and the Metropolitan Life Foundation to A.G. The anti-PS1 and PHF1 antibodies were generously provided by Drs. S. Gandy, J. Nerbonne, J. Gitlin, and P. Davies. We thank Deborah Carter and Raymond Taylor for expert assistance in preparation of tissue sections and immunohistochemical staining procedures. We acknowledge the contributions of the WUSM ADRC Clinical Core (John Morris, M.D., director) and the Neuropathology/Tissue Resource Core (Daniel McKeel, M.D., director), and components of WUSM ADRC for providing diagnoses, patient assessment, and tissue preparation. We also thank Drs. A. L. Schwartz and Y. Rao for critical reading of this manuscript and members of the WUSM Neuropathology Division (R. E. Schmidt, M.D., Ph.D.) for providing expert neuropathological diagnoses.
Correspondence should be addressed to Dr. Wu at the above address.
REFERENCES
- 1.Berg L, McKeel DW, Jr, Miller JP, Baty J. Neuropathologic indexes of Alzheimer’s disease in demented and nondemented people aged 80 years and older. Arch Neurol. 1993;50:349–358. doi: 10.1001/archneur.1993.00540040011008. [DOI] [PubMed] [Google Scholar]
- 2.Brown KD, Binder LI. Identification of the intermediate filament-associated protein gyronemin as filamin, implication for a novel mechanism of cytoskeletal interaction. J Cell Sci. 1992;102:19–30. doi: 10.1242/jcs.102.1.19. [DOI] [PubMed] [Google Scholar]
- 3.Cai X-D, Golde TE, Younkin SG. Release of excess amyloid β protein from a mutant amyloid β protein precursor. Science. 1993;259:514–516. doi: 10.1126/science.8424174. [DOI] [PubMed] [Google Scholar]
- 4.Chartier-Harlin M-C, Parfitt M, Legrain S, Perez-Tur J, Brousseau T, Evans A, Berr C, Vidal O, Roques P, Gourlet V, Delacourte A, Rossor M, Amouyel P. Apolipoprotein E ε4 allele as a major risk factor for sporadic early and late-onset forms of Alzheimer’s disease: analysis of the 19q13.2 chromosomal region. Hum Mol Genet. 1994;3:569–574. doi: 10.1093/hmg/3.4.569. [DOI] [PubMed] [Google Scholar]
- 5.Citron M, Oltersdorf T, Haass C, McConlogue L, Hung AY, Seubert P, Vigo-Pelfry C, Lieberburg I, Selkoe DJ. Mutation of the β-amyloid precursor protein in familial Alzheimer’s disease increases β-protein production. Nature. 1992;360:672–674. doi: 10.1038/360672a0. [DOI] [PubMed] [Google Scholar]
- 6.Corder E, Saunders A, Strittmatter W, Schmechel D, Gaskell P, Small G, Roses A, Haines J, Pericak-Vance M. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 7.Cruts M, Hendriks L, Van Broeckhoven C. The presenilin genes: a new gene family involved in Alzheimer’s disease pathology. Hum Mol Genet. 1996;5:1449–1455. doi: 10.1093/hmg/5.supplement_1.1449. [DOI] [PubMed] [Google Scholar]
- 8.Doan A, Thinakaran G, Borchelt DR, Slunt HH, Rotovitsky T, Podlisny M, Selkoe D, Seeger M, Gandy SE, Price DL, Sisodia SS. Protein topology of presenilin 1. Neuron. 1996;17:1023–1030. doi: 10.1016/s0896-6273(00)80232-9. [DOI] [PubMed] [Google Scholar]
- 9.Drubin DG, Nelson WJ. Origin of cell polarity. Cell. 1996;84:335–344. doi: 10.1016/s0092-8674(00)81278-7. [DOI] [PubMed] [Google Scholar]
- 10.Elder GA, Tesapsidis J, Carter J, Shioi J, Bouras C, Li HC, Johnston JM, Efthimiopoilos S, Friedrich VL, Robakis NK. Identification and neuron-specific expression of the S182/presenilin 1 protein in human and rodent brains. J Neurosci Res. 1996;45:308–320. doi: 10.1002/(SICI)1097-4547(19960801)45:3<308::AID-JNR13>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 11.Giannakopoulos P, Bouras C, Kovari E. Presenilin immunoreactive neurons are preserved in late-onset Alzheimer’s disease. Am J Pathol. 1997;150:429–436. [PMC free article] [PubMed] [Google Scholar]
- 12.Gorlin JB, Yamin R, Egan S, Stewart M, Stossel TP, Kwiatkowski DJ, Hartwig JH. Human endothelial actin-binding protein (ABP280, non-muscle filamin): a molecular leaf spring. J Cell Biol. 1990;111:1098–1105. doi: 10.1083/jcb.111.3.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gorlin JB, Henske E, Warren ST, Kunst CB, D’urso M, Palmieri G, Hartwig JH, Bruns G, Kwiatkowski DJ. ABP280 filamin gene maps telomeric to the color vision locus and centrimeric to G6PD in Xq28. Genomics. 1993;17:496–498. doi: 10.1006/geno.1993.1354. [DOI] [PubMed] [Google Scholar]
- 14.Green KJ, Geiger B, Jones JCR, Talian JC, Goldman RD. The relationship between intermediate filaments and microfilaments before and during the formation of desmosomes and adherence-type junctions in mouse epidermal keratinocytes. J Cell Biol. 1987;104:1389–1402. doi: 10.1083/jcb.104.5.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Greenberg SG, Davies P. A preparation of Alzheimer paired filaments that displays distinct tau proteins by polyacrylamide gel electrophoresis. Proc Natl Acad Sci USA. 1990;87:5827–5831. doi: 10.1073/pnas.87.15.5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hartwig J, Shevlin P. The architecture of actin filaments and the ultrastructural location of actin-binding protein in the periphery of lung macrophage. J Cell Biol. 1986;103:1007–1020. doi: 10.1083/jcb.103.3.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hartwig J, Tyler J, Stossel T. Actin-binding protein promotes the bipolar and perpendicular branching of actin filaments. J Cell Biol. 1980;87:841–848. doi: 10.1083/jcb.87.3.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heyman A, Wilkinson WE, Stafford JA, Helms MJ, Sigmon AH, Weinberg T. Alzheimer’s disease: a study of epidemiological aspects. Ann Neurol. 1984;15:335–341. doi: 10.1002/ana.410150406. [DOI] [PubMed] [Google Scholar]
- 19.Khachaturian ZS. The diagnosis of Alzheimer’s disease. Arch Neurol. 1985;42:1097–1105. doi: 10.1001/archneur.1985.04060100083029. [DOI] [PubMed] [Google Scholar]
- 20.Kim TW, Pettingell WH, Hallmark OG, Moir RD, Wasco W, Tanzi RE. Endoproteolytic cleavage and proteasomal degradation of presenilin 2 in transfected cells. J Biol Chem. 1997;272:11006–11010. doi: 10.1074/jbc.272.17.11006. [DOI] [PubMed] [Google Scholar]
- 21.Kovacs DM, Fausett HJ, Page KJ, Kim T, Moir RD, Merriam DE, Hollister RD, Hallmark OG, Mancinim R, Felsenstein KM, Hyman BT, Tanzi RE, Wasco WL. Alzheimer associated presenilins 1 and 2: neuronal expression in brain and localization to intracellular membranes in mammalian cells. Nat Med. 1996;2:224–229. doi: 10.1038/nm0296-224. [DOI] [PubMed] [Google Scholar]
- 22.Lee MK, Slunt HH, Martin LJ, Thinakaran G, Kim G, Gandy SE, Seeger M, Koo E, Price DL, Sisodia SS. Expression of presenilin 1 and 2 in human and murine tissues. J Neurosci. 1996;16:7513–7525. doi: 10.1523/JNEUROSCI.16-23-07513.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lehmann S, Chiesa R, Harris D. Evidence for a six-transmembrane domain structure of presenilin 1. J Biol Chem. 1997;272:12047–12051. doi: 10.1074/jbc.272.18.12047. [DOI] [PubMed] [Google Scholar]
- 24.Lendon CL, Ashall F, Goate AM. Exploring the etiology of Alzheimer’s disease using molecular genetics. JAMA. 1997;277:825–831. [PubMed] [Google Scholar]
- 25.Levitan D, Greenwald I. Facilitation of lin-12-mediated signaling by sel-12, a Caenorhabditis elegans S182 Alzheimer’s disease gene. Nature. 1995;377:351–355. doi: 10.1038/377351a0. [DOI] [PubMed] [Google Scholar]
- 26.Levy-Lahad E, Wasco W, Poorkaj P, Romano D, Oshima J, Pettingell W, Yu C, Jondro P, Schmidt S, Wang K, Crowley A, Fu Y-H, Guenette S, Galas D, Nemens E, Wijsman E, Bird T, Schellenberg G, Tanzi R. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science. 1995;269:973–977. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- 27.Maetrini E, Patrosso C, Rivella S, Rocchi M, Repetto M, Villa A, Frattini A, Zoppe M, Vezzoni P, Toniolo D. Mapping of two genes encoding isoforms of ABP280, a dystrophin like protein, to Xq28 and to chromosome 7. Hum Mol Genet. 1993;2:761–766. doi: 10.1093/hmg/2.6.761. [DOI] [PubMed] [Google Scholar]
- 28.Malin SA, Guo WXA, Jafari G, Goate AM, Nerbonne JM (1998) Presenilins upregulate functional K+ channel currents in mammalian cells. Neurobiol Dis, in press. [DOI] [PubMed]
- 29.Matsudaira P. Actin crosslinking proteins at the leading edge. Semin Cell Biol. 1994;5:165–174. doi: 10.1006/scel.1994.1021. [DOI] [PubMed] [Google Scholar]
- 30.McKeel DW, Jr, Ball MJ, Price JL, Smith DS, Miller JP, Berg L, Morris JC. Interlaboratory histopathologic assessment of Alzheimer neuropathology: different methodologies yield comparable diagnostic results. Alzheimer Dis Assoc Disord. 1993;7:136–151. doi: 10.1097/00002093-199307030-00003. [DOI] [PubMed] [Google Scholar]
- 31.Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 32.Mitchison TJ, Cramer LP. Actin-based cell motility and cell locomotion. Cell. 1996;84:371–379. doi: 10.1016/s0092-8674(00)81281-7. [DOI] [PubMed] [Google Scholar]
- 33.Morris JC. The clinical dementia rating (CDR): current version and scoring rules. Neurology. 1993;43:2412–2414. doi: 10.1212/wnl.43.11.2412-a. [DOI] [PubMed] [Google Scholar]
- 34.Murphy GM, Forno LS, Ellis WG, Nochlin D, Levy-Lahad E, Poorkaj P, Bird TD, Jiang Z, Cordell B. Antibodies to presenilin proteins detect neurofibrillary tangles in Alzheimer’s disease. Am J Pathol. 1996;149:1839–1846. [PMC free article] [PubMed] [Google Scholar]
- 35.Niederman R, Amrein P, Hartwig J. The three dimensional structure of actin filaments in solution and an actin gel made with actin-binding protein. J Cell Biol. 1983;96:1400–1413. doi: 10.1083/jcb.96.5.1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nieto-Sampedro M, Mora F. Active microglia, sick astroglia, and Alzheimer type dementias. NeuroReport. 1994;5:375–380. doi: 10.1097/00001756-199401120-00001. [DOI] [PubMed] [Google Scholar]
- 37.Pike CJ, Cummings BJ, Cotman CW. Early association of reactive astrocytes with senile plaques in Alzheimer’s disease. Exp Neurol. 1995;132:172–179. doi: 10.1016/0014-4886(95)90022-5. [DOI] [PubMed] [Google Scholar]
- 38.Podlisny M, Citron M, Amarante P, Sherrington R, Xia WM, Zhang JM, Diel T, Levesque G, Fraser P, Haass C, Koo EM, Seubert P, St. George-Hyslop P, Teplow DB, Selkoe DJ. Presenilin proteins undergo heterogenous endoproteolysis between Thr291 and Ala299 and occur as stable N- and C-terminal fragments in normal and Alzheimer brain tissue. Neurobiol Dis. 1997;3:325–337. doi: 10.1006/nbdi.1997.0129. [DOI] [PubMed] [Google Scholar]
- 39.Pollard TD, Selden SC, Maupin P. Interaction of actin filaments with microtubules. J Cell Biol. 1984;99:33s–37s. doi: 10.1083/jcb.99.1.33s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Potter H. The involvement of astrocytes and an acute phase response in the amyloid deposition of Alzheimer’s disease. Prog Brain Res. 1992;94:447–458. doi: 10.1016/s0079-6123(08)61772-0. [DOI] [PubMed] [Google Scholar]
- 41.Prat AG, Xiao Y-F, Ausiello DA, Cantiello HF. cAMP-independent regulation of CFTR by the actin cytoskeleton. Am J Physiol. 1995;268:1552–1561. doi: 10.1152/ajpcell.1995.268.6.C1552. [DOI] [PubMed] [Google Scholar]
- 42.Rogaev EI, Sherrington R, Rogaeva EA, Levesque G, Ikeda M, Liang Y, Chi H, Lin C, Holman K, Tsuda T. Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature. 1995;376:775–778. doi: 10.1038/376775a0. [DOI] [PubMed] [Google Scholar]
- 43.Schellenberg GD. Genetic dissection of Alzheimer’s disease, a heterogeneous disorder. Proc Natl Acad Sci USA. 1995;92:8552–8559. doi: 10.1073/pnas.92.19.8552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, Larson E, Levy-Lahad E, Viitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D, Younkin S. Secreted amyloid-β-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by PS1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat Med. 1996;2:864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 45.Selkoe DJ. Cell biology of the amyloid β-protein precursor and the mechanism of Alzheimer’s disease. Annu Rev Cell Biol. 1994;10:373–403. doi: 10.1146/annurev.cb.10.110194.002105. [DOI] [PubMed] [Google Scholar]
- 46.Sherrington R, Rogaev E, Liang Y, Rogaeva E, Levesque G, Ikeda M, Chi H, Lin C, Holman K, Tsuda T, Rainero I, Pinessi L, Nee L, Chumakov I, Pollen D, Brookes A, Sanseau P, Polinsky R, Wasco W, da Silva H, Haines J, Pericak-Vance M, Tanzi R, Roses A, Fraser P, Rommens J, St. George-Hyslop P. Cloning of a gene bearing mis-sense mutations in early-onset familial Alzheimer’s disease. Nature. 1995;375:754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- 47.Stossel TP. On the crawling of animal cells. Science. 1993;260:1086–1094. doi: 10.1126/science.8493552. [DOI] [PubMed] [Google Scholar]
- 48.Suzuki T, Nishiyama K, Murayama S. Regional and cellular presenilin 1 gene expression in human and rat tissues. Biochem Biophys Res Commun. 1996;219:708–713. doi: 10.1006/bbrc.1996.0299. [DOI] [PubMed] [Google Scholar]
- 49.Takami K, Terai K, Matsuo A, Walker DG, McGeer PL. Expression of presenilin 1 and 2 mRNAs in rat and Alzheimer’s disease brains. Brain Res. 1997;748:122–130. doi: 10.1016/s0006-8993(96)01274-7. [DOI] [PubMed] [Google Scholar]
- 50.Tanzi RE, Kovacs DM, Kim TW, Moir RD, Guenette SY, Wasco W. The gene defects responsible for familial Alzheimer’s disease. Neurobiol Dis. 1996;3:159–168. doi: 10.1006/nbdi.1996.0016. [DOI] [PubMed] [Google Scholar]
- 51.Thinakaran G, Borchelt DR, Lee MK, Slunt HH, Spitzer L, Kim G, Rotovitsky T. Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron. 1996;17:181–190. doi: 10.1016/s0896-6273(00)80291-3. [DOI] [PubMed] [Google Scholar]
- 52.van Duijin CM, Stijnen T, Hofman A (1991) Risk factors for Alzheimer’s disease: overview of the EURODEM collaborative re-analysis of case-control studies. Int J Epidemiol 20[Suppl 2]:S4–S12. [DOI] [PubMed]
- 53.Vito P, Lacana E, D’Adamio L. Interfering with apoptosis: Ca binding protein ALG-2 and Alzheimer’s disease gene ALG-3. Science. 1996;271:521–525. doi: 10.1126/science.271.5248.521. [DOI] [PubMed] [Google Scholar]
- 54.Wischik CM, Lai R, Harrington CR, Mukaetova-Ladinska E, Xuereb F, Mena R, Edwards P, Roth M. Structure, biochemistry, and molecular pathogenesis of paired helical filaments in Alzheimer’s disease. In: Goate AM, Ashall F, editors. Pathobiology of Alzheimer’s disease. Academic; New York: 1995. pp. 9–40. [Google Scholar]
- 55.Wisniewski T, Palha G, Ghiso J, Frangione B. S182 protein in Alzheimer’s disease neuritic plaques. Lancet. 1995;346:8986. doi: 10.1016/s0140-6736(95)92379-9. [DOI] [PubMed] [Google Scholar]
- 56.Wolozin B, Iwasaki K, Vito P, Ganjei JK, Lacana E, Sunderland T, Zhao B, Kusaik JW, Wasco W, D’Adamio L. Participation of presenilin 2 in apoptosis-enhanced basal activity conferred by an Alzheimer mutation. Science. 1996;274:1710–1713. doi: 10.1126/science.274.5293.1710. [DOI] [PubMed] [Google Scholar]
- 57.Wu JY, Maniatis T. Specific interactions between proteins implicated in splice site selection and regulated alternative splicing. Cell. 1993;75:1061–1070. doi: 10.1016/0092-8674(93)90316-i. [DOI] [PubMed] [Google Scholar]
- 58.Zervos AS, Gyuris J, Brent R. Mix1, a protein that specifically interacts with Max to bind Myc-Max recognition sites. Cell. 1993;72:223–232. doi: 10.1016/0092-8674(93)90662-a. [DOI] [PubMed] [Google Scholar]