

Abstract
Background and purpose:
Racemic (R,S) AM1241 is a cannabinoid receptor 2 (CB2)-selective aminoalkylindole with antinociceptive efficacy in animal pain models. The purpose of our studies was to provide a characterization of R,S-AM1241 and its resolved enantiomers in vitro and in vivo.
Experimental approach:
Competition binding assays were performed using membranes from cell lines expressing recombinant human, rat, and mouse CB2 receptors. Inhibition of cAMP was assayed using intact CB2-expressing cells. A mouse model of visceral pain (para-phenylquinone, PPQ) and a rat model of acute inflammatory pain (carrageenan) were employed to characterize the compounds in vivo.
Key results:
In cAMP inhibition assays, R,S-AM1241 was found to be an agonist at human CB2, but an inverse agonist at rat and mouse CB2 receptors. R-AM1241 bound with more than 40-fold higher affinity than S-AM1241, to all three CB2 receptors and displayed a functional profile similar to that of the racemate. In contrast, S-AM1241 was an agonist at all three CB2 receptors. In pain models, S-AM1241 was more efficacious than either R-AM1241 or the racemate. Antagonist blockade demonstrated that the in vivo effects of S-AM1241 were mediated by CB2 receptors.
Conclusions and implications:
These findings constitute the first in vitro functional assessment of R,S-AM1241 at rodent CB2 receptors and the first characterization of the AM1241 enantiomers in recombinant cell systems and in vivo. The greater antinociceptive efficacy of S-AM1241, the functional CB2 agonist enantiomer of AM1241, is consistent with previous observations that CB2 agonists are effective in relief of pain.
Keywords: AM1241, cannabinoid, CB2, enantiomer, protean agonism
Introduction
First cloned from a macrophage cell line from human spleen, the CB2 cannabinoid receptor, a G-protein-coupled receptor (GPCR) that signals through Gi, is one of at least two cell surface receptors capable of transducing the signals of endocannabinoid ligands (Munro et al., 1993). Another Gi-coupled GPCR, the CB1 receptor is highly expressed in the central nervous system (CNS) (Howlett et al., 2004), and preliminary evidence suggests that additional endocannabinoid receptors may exist (Fride et al., 2003; Baker et al., 2006). While CB2 is expressed mainly in tissues of the immune system (Howlett et al., 2004), recent reports provide evidence of expression in the CNS (Cabral and Marciano-Cabral, 2005; Van Sickle et al., 2005; Beltramo et al., 2006) and inducible expression in peripheral sensory neurons (Wotherspoon et al., 2005). DNA sequence analysis of rodent orthologues of CB2 (Shire et al., 1996; Griffin et al., 2000; Brown et al., 2002) reveals mouse and rat CB2 to be, respectively, 79 and 81% identical to human CB2 in predicted primary amino-acid composition and 93% identical to each other.
Agonists of CB2 are thought to possess therapeutic promise in a number of diseases, including cancer (Flygare et al., 2005; Herrera et al., 2005), osteoporosis (Ofek et al., 2006), atherosclerosis (Steffens et al., 2005) and amyotropic lateral sclerosis (Kim et al., 2006; Shoemaker et al., 2007). However, the therapeutic potential of agonists of the CB2 receptor has been most strongly demonstrated in animal models of inflammatory and neuropathic pain. Much of this evidence has been generated using the racemic mixture of the synthetic ligand AM1241 (Makriyannis, 2002). The in vitro selectivity of R,S-AM1241 for CB2 vs CB1 receptors has been demonstrated to be approximately 80-fold in binding studies, employing natively expressing tissues (Ibrahim et al., 2003) and recombinant cell systems (Yao et al., 2006). In pain efficacy studies, the action of R,S-AM1241 at CB2 receptors has been demonstrated either pharmacologically using CB2-selective antagonists, such as AM630 or SR144528, or genetically, using animals lacking the CB2 receptor (CB2−/−). Similarly, efficacy through CB1 receptor activation has been ruled out through the use of either CB1-selective antagonist compounds (AM251 or SR141716A) or CB1−/− animals (Malan et al., 2001; Ibrahim et al., 2003, 2005, 2006; Nackley et al., 2003, 2004; Quartilho et al., 2003; Hohmann et al., 2004; LaBuda et al., 2005; Beltramo et al., 2006).
In the present report, we provide an extensive in vitro pharmacological characterization of R,S-AM1241, measuring binding affinity and functional inhibition of forskolin stimulated cyclic adenosine monophosphate (cAMP) accumulation in CHO-K1 cell lines overexpressing human, rat or mouse CB2. We reveal not only species-specific effects of R,S-AM1241, but in extending this analysis to the separated enantiomers of R,S-AM1241, we also demonstrate stereoisomer-specific pharmacology for this synthetic cannabinoid ligand both in vitro and in vivo.
Methods
Cloning and cell culture
CHO-K1 cells expressing hCB1 and hCB2 receptors (Euroscreen, Gosselies, Belgium) were cultured in Hams F12 medium containing 10% foetal bovine serum (FBS), penicillin (10 IU ml−1)/streptomycin (10 μg ml−1) and 400 μg ml−1 G418. Mouse and rat CB2 receptor open reading frame sequences were PCR amplified from commercially prepared spleen cDNA (BD Biosciences, San Jose, CA, USA) using oligonucleotide primers spanning the start and stop sites designed from published sequences (GenBank accession numbers X86405 (mouse) and AF176350 (rat)). Restriction sites (5′ HindIII and 3′ EcoRI) were included in the sequence of the PCR primers to facilitate cloning into pcDNA3.1(+) (Invitrogen, Carlsbad, CA, USA). Transfection of CHO-K1 cells was with Lipofectamine Plus (Invitrogen) according to the manufacturer's instructions. Initial selection of transfectants was with 800 μg ml−1 G418. Cell lines stably expressing mCB2 and rCB2 receptors were cultured in Dulbecco's modified Eagle's medium containing 10% FBS, penicillin (10 IU ml−1)/streptomycin (10 μg ml−1), non-essential amino acids and 500 μg ml−1 G418. All tissue culture reagents were from Invitrogen.
Chiral separation of R,S-AM1241
The enantiomers of R,S-AM1241 were separated by chiral HPLC on a 2 × 25 cm Chiralcel OD column (elution solvent: 20% isopropanol/0.1% diethylamine in hexane, 22 ml min−1). S-AM1241 eluted at 12.2 min, and R-AM1241 eluted at 17.26 min. Optical rotations were obtained with a Jasco P-1020 polarimeter with a 5 cm cell. S-AM1241: [α]25D=−46°(c 1.0, dimethyl sulphoxide (DMSO)); R-AM1241 [α]25D=+40° (c 1.0, DMSO).
The absolute stereochemistry of the enantiomers was determined by vibrational circular dichroism (VCD). The VCD spectra were measured with the VCD instrument, ChiralIR (BioTools Inc., Wauconda, IL, USA). Each sample was dissolved in CDCl3 (10 mM) and placed in a BaF2 cell with a 0.1 mm pathlength. The VCD spectrum of each sample and solvent was measured for 4 h with a 4 cm−1 resolution and the photo elastic modulators optimized at 1400 cm−1. The VCD baseline was obtained by subtracting the VCD of one enantiomer from that of the other, then dividing by two. The infrared (IR) baseline was obtained by subtracting the IR spectrum of CDCl3 from that of the sample.
The (S)-conformers of the molecule and a truncated molecule were built with Hyperchem 7 (Hypercube Inc., Gainesville, FL, USA). (Truncation accelerates the geometry optimizations and the VCD calculations). The conformational search was performed with the semi-empirical PM3 method and resulted in 15 conformers for the whole molecule and 18 conformers for the truncated molecule. Six conformers of the truncated molecule have matches among the conformers of the whole molecule. The geometry optimization and VCD spectra of the six (S)-conformers were calculated with Gaussian 03 (Frisch et al., 2003) at density functional theory (DFT) level with the b3lyp/6–31G(d) basis set. The average and the Boltzmann sum of the VCD and IR spectra of the six conformers were calculated and compared with the measured spectra. S-AM1241 was confirmed as the S-enantiomer, and R-AM1241 was confirmed as the R-enantiomer.
Membrane preparation
Confluent 245 cm2 dishes of cells were washed twice with cold phosphate-buffered saline (PBS). Cells were scraped in 10 ml cold buffer (20 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulphonic acid (HEPES) pH 7.5, 10 mM ethylenediaminetetraacetic acid (EDTA)), homogenized in a Dounce homogenizer and pelleted at 32 000 g. Cell pellets were resuspended in storage buffer (20 mM HEPES, pH 7.5, 100 mM NaCl, 1 mM MgCl), homogenized again, aliquoted and frozen at −80°C. Protein concentrations were determined using Bio-Rad Protein Assay reagents as per the manufacturer's instructions (Biorad, Hercules, CA, USA).
Radioligand binding
Binding assays were conducted using 30 μg (hCB2), 50 μg (rodent CB2) or 12 μg (hCB1) membrane protein per tube and 1–3 nM [3H]-CP55,940 (Perkin Elmer, Boston, MA, USA) as the radioligand; compounds were diluted to 10 × concentrations in 4% DMSO/H2O, and all reagents were combined in the assay buffer (50 mM Tris, pH 7.5, 2.5% bovine serum albumin, 2.5 mM EDTA). The assay was incubated at 30°C for 60 min and filtered on Whatman GFB filter mats treated with 0.15% polyethyleneimine using a Brandel 96-channel harvester (Brandel, Gaithersburg, MD, USA). Radioactivity was determined by liquid scintillation counting.
cAMP inhibition assays
Cells cultured in T-175 flasks were harvested by washing twice with PBS, followed by addition of 5 ml cell dissociation solution (Mediatech, Herndon, VA, USA). After 3–5 min incubation at room temperature, the dissociated cells were removed, mixed with 10 ml Krebs assay buffer (118 mM NaCl, 5 mM KCl, 1.2 mM KH2PO4, 25 mM NaHCO3, 11.1 mM glucose, 1.2 mM MgSO4, 2.4 mM CaCl2) and pelleted. Cell pellets were resuspended in Krebs and counted. Cannabinoid ligands were serially diluted in Krebs containing 1 μM forskolin. Per well of a 96-well plate (Corning 3912), the ligand/forskolin mixture was combined with 1.5 × 104 cells and incubated at 37°C for 30 min. cAMP determinations were performed using the HitHunter cAMP XS Assay according to the manufacturer's protocol (DiscoveRx, Fremont, CA, USA). Chemiluminescence was counted using a Wallac Victor V after a 3 h incubation. For the Pertussis toxin study, cells were incubated in the presence of 100 ng ml−1 Pertussis toxin for 4 h before forskolin stimulation.
In vivo studies
All animal procedures were approved by an institutional animal care and use committee and were conducted in accordance with the International Association for the Study of Pain guidelines on the use of animals in experimental research (Zimmermann, 1983).
Acute analgesia (tail flick and hot plate)
Acute analgesia was investigated using the tail-flick (D'Amour and Smith, 1941) and hot-plate assays (Woolfe and MacDonald, 1944). For the tail-flick assay, male Sprague–Dawley rats (n=10 per group) were placed on the apparatus (Ugo Basile, Varese, Italy), and an infrared beam was focused 5 cm from the tip of the tail. The latency to tail flick was measured to the nearest 0.1 s with a cutoff of 20 s. For the hot-plate assay, male Sprague–Dawley rats (n=10 per group) were placed on a metal plate (Ugo Basile, Varese, Italy) maintained at 52°C. The latency to nocifensive response, defined as hindpaw lift, flutter, licking or escape behaviour, was measured to the nearest 0.1 s with a cutoff of 30 s. Approximately, 1 h after determination of baseline latency, animals received a single intraperitoneal (i.p.) dose of vehicle (0.5% methylcellulose and 2% Tween, 2 ml kg−1) or 1, 3 or 10 mg kg−1 R,S-AM1241, R-AM1241 or S-AM1241. Dosing of the positive control (10 mg kg−1 morphine) was by subcutaneous (s.c.) injection. Tail-flick and hot-plate latencies were determined 30 and 90 min after drug administration.
Acute visceral pain (PPQ)
The ability of compounds to attenuate painful abdominal stretching (also referred to as writhing) was assessed in male CD-1 mice following i.p. injection of 2 mg kg−1 para-phenylquinone (PPQ) (dissolved in 4% ethanol). Delivery of R,S-AM1241, R-AM1241 or S-AM1241 (s.c.) was as a suspension in vehicle (1% DMSO, 0.5% methylcellulose and 2% Tween) 30 min before PPQ injection. Following PPQ administration, mice were placed individually in a Plexiglas observation cage, and stretching movements were recorded for two periods of 1 min each, at 5 and 10 min post-injection. Percent blockade was calculated according to the following equation:
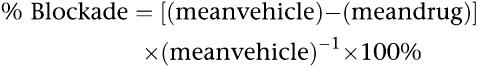 |
Acute inflammatory pain (carrageenan)
Latency of paw withdrawal from a thermal stimulus (Hargreaves et al., 1988) was assessed in male Sprague–Dawley rats in response to focusing a radiant heat source (ITTC, Woodland Hills, CA, USA) on the plantar surface of the left hindpaw. Intraplantar injection of 2% carrageenan (50 μl volume) into the left hindpaw took place under anaesthesia (3% isoflurane/O2), 24 h after baseline withdrawal latency was measured. Following a 30 min habituation period on a heated glass surface (32°C), withdrawal latency was measured to the nearest 0.1 s, with a cutoff of 20 s to avoid tissue damage. Delivery of R,S-AM1241, R-AM1241 or S-AM1241 (i.p., 2.5 h after carrageenan administration) was as a solution in a vehicle of 0.5% methylcellulose and 2% Tween. Three withdrawal latency measurements were taken for each rat 30 min post-drug administration. Paw volume was measured with a plethysmometer (Ugo Basile, Varese, Italy) before and 3.5 h after carrageenan injection. Percent reversal was calculated according to the following equation:
For the antagonist experiments, two consecutive i.p. injections were administered 2.5 h post-carrageenan. The first injection was either vehicle (0.5% methylcellulose and 2% Tween) or 10 mg kg−1 S-AM1241 in vehicle; the second injection was either vehicle (1% DMSO, 0.5% methylcellulose and 2% Tween) or 1 mg kg−1 AM630 (Tocris Bioscience, Ellisville, MO, USA) in vehicle. A positive control group (indomethacin, Sigma, St Louis, MO, 10 mg kg−1, per os) was included.
Statistical analysis of data
From the radioligand binding experiments, Ki values were determined using GraphPad Prism (GraphPad Software, San Diego, CA, USA). From the cAMP inhibition experiments, EC50 values were determined using GraphPad Prism (GraphPad Software, San Diego, CA, USA). For all in vivo pain studies, raw data were analysed by one-way ANOVA using a customized SAS-Excel application (SAS Institute, Cary, NC, USA). Significant (P<0.05) main effects were analysed further post hoc, using least significant difference analysis.
Results
R,S-AM1241 binds to CB2 receptors
The human, rat and mouse CB2 receptors were expressed stably in CHO-K1 cells. Radioligand saturation binding analysis using [3H]-CP55,940 indicated that the levels of expression were comparable (7.8, 9.5 and 23.1 pmol mg−1, respectively). In binding studies, the control compound WIN55,212-2 displaced [3H]-CP55,940 from human, rat and mouse receptors with Ki values of 2.8±0.6, 129±36 and 209±34 nM, respectively (data not shown). This increased affinity for the human receptor was not reflected by the functional studies, in which WIN55,212-2 was nearly equipotent at all three receptors (see below). R,S-AM1241 displaced [3H]-CP55,940 from all three CB2 receptors with near-equal affinity (Ki values 20–30 nM) (Table 1, Figure 1). To investigate the pharmacology of R,S-AM1241 further, we resolved its enantiomers. R-AM1241 had similar affinities at all three species of CB2 receptors, although these affinities were approximately twofold greater for R-AM1241 than the racemate, as reflected by Ki values. S-AM1241 had a much lower affinity, with Ki values ranging from 600 to 900 nM. The Ki value of R-AM1241 for the hCB1 receptor was approximately 5 μM, while the corresponding values for racemic AM1241 and S-AM1241 exceeded 10 μM (Table 1).
Table 1.
Affinity of R,S-AM1241 and its enantiomers, R-AM1241 and S-AM1241, for the human, rat and mouse CB2 receptor and the human CB1 receptor
| Species of receptor | R,S-AM1241 Ki (nM) | R-AM1241 Ki (nM) | S-AM1241 Ki (nM) |
|---|---|---|---|
| hCB2 | 28.7±2.01 | 15.1±4.18 | 658±44.2 |
| rCB2 | 26.7±0.44 | 12.0±1.27 | 893±58.5 |
| mCB2 | 23.8±4.36 | 13.2±0.76 | 577±58.4 |
| hCB1 | >10 × 103 | 5.0 × 103±300 | >10 × 103 |
Membranes prepared from CHO-K1 cells stably expressing human, rat or mouse CB2 or human CB1 receptors were incubated in the presence of drug and 1–3 nM [3H]-CP55,940 for 1 h. The membranes were filtered onto Whatman GFB paper and radioactivity determined. Ki values were calculated using the Cheng–Prusoff equation. Data are expressed as mean±s.e.m. for three independent experiments.
Figure 1.

Radioligand displacement curves for R,S-AM1241, R-AM1241 and S-AM1241 at the human (a), rat (b) and mouse (c) CB2 receptor. Membranes prepared from CHO-K1 cells expressing the human, rat or mouse CB2 receptor were incubated in the presence of drug and 1–3 nM [3H]-CP55,940 for 1 h. Membranes were filtered onto Whatman GFB paper and radioactivity determined. Ki values were calculated using the Cheng–Prusoff equation. Data are expressed as mean±s.e.m. for three independent experiments.
CB2 receptor agonists decrease cAMP levels
For all CB2 functional assays, 1 μM forskolin was used to stimulate cAMP production. The effects of the non-selective cannabinoid agonist WIN55,212-2 on forskolin stimulated cAMP accumulation are shown in Figure 2a. A robust response was seen in cells with the human receptors, with a maximal inhibition of approximately 80%. However, stimulation of the rat and mouse CB2 receptor resulted in a smaller inhibition of cAMP formation (approximately 40%), despite the high level of expression in the murine cell line. The inverse agonist SR144528 (Rinaldi-Carmona et al., 1998), which increased forskolin-stimulated cAMP by 50–100% in cells expressing any of the three CB2 receptors (Figure 2b, Table 2), provided evidence for constitutive activity of the CB2 receptors, with the mouse CB2 receptor displaying the greatest amount.
Figure 2.

Effects of the non-selective cannabinoid agonist WIN55,212-2 (a) and the CB2-selective antagonist/inverse agonist SR144528 (b) on cAMP levels in cells expressing the human, rat or mouse CB2 receptor. Cells were stimulated in the presence of 1 μM forskolin for 30 min. Data are expressed as mean±s.e.m. from a representative dose–response curve.
Table 2.
Effects of a non-selective cannabinoid agonist (WIN55,212-2) and a CB2-selective antagonist/inverse agonist on cAMP levels in cells expressing the human, rat or mouse CB2 receptor
| Species of receptor | WIN 55,212-2 EC50 (nM) | SR144528 EC50 (nM) |
|---|---|---|
| hCB2 | 103±22.0 | 1720±879 |
| rCB2 | 234±29.4 | 1050±467 |
| mCB2 | 134±25.0 | 1240±758 |
Abbreviation: cAMP, cyclic adenosine monophosphate.
Cells were stimulated in the presence of 1 μM forskolin for 30 min. cAMP levels were determined using HitHunter. Data are expressed as mean±s.e.m. for three independent experiments.
R,S-AM1241 and its enantiomers display species-dependent in vitro pharmacology
At the human CB2 receptor, R,S-AM1241 demonstrated partial agonist activity with a decrease of forskolin-stimulated cAMP by a maximum of 60% with an EC50 of 28 nM; in comparison, WIN55,212-2 produced a maximal inhibition of approximately 80%. Surprisingly, an opposite effect was observed when either rodent CB2 receptor was stimulated. At these receptors, R,S-AM1241 acted as an inverse agonist, increasing forskolin-stimulated cAMP levels by 30–70% (Figure 3a). Interestingly, stereoisomer-specific pharmacology was observed at the rodent receptors. As seen with the racemate, R-AM1241 was an agonist at the human receptor and an inverse agonist at each of the rodent receptors. Similar to SR144528, R-AM1241 increased the levels of cAMP to a greater extent in the mouse cell line than the rat (Figure 3b). S-AM1241 was a potent (131 nM) agonist at the human receptor, but in contrast to the R-enantiomer, was also an agonist at the rodent receptors, albeit with lower potency than at the human receptor (Figure 3c, Table 3). The CB2-specificity of the effects of R,S-AM1241 and its enantiomers was demonstrated by the absence of effects on forskolin-stimulated cAMP in parental CHO-K1 cells (data not shown). The effects of all three ligands in all three CB2-expressing cells were sensitive to Pertussis toxin (data not shown), indicating that the observed inverse agonist effects of R,S-AM1241 and R-AM1241 were the result of Gi-coupled signalling and not the result of rodent CB2 receptors signalling through an alternative G-protein in response to these ligands.
Figure 3.

Effects of R,S-AM1241 (a) and its enantiomers, R-AM1241 (b) and S-AM1241 (c) on cAMP accumulation in CHO-K1 cells expressing the human, rat or mouse CB2 receptor. Cells were stimulated in the presence of 1 μM forskolin for 30 min. Data are expressed as mean±s.e.m. for three independent experiments. cAMP, cyclic adenosine monophosphate.
Table 3.
Effects of R,S-AM1241 and its enantiomers, R-AM1241 and S-AM1241, on cAMP levels in cells expressing the human, rat or mouse CB2 receptor
| Species of receptor | R,S-AM1241 EC50 (nM) | R-AM1241 EC50 (nM) | S-AM1241 EC50 (nM) |
|---|---|---|---|
| hCB2 | 190±184 (agonist) | 118±112 (agonist) | 131±46.9 (agonist) |
| rCB2 | 216±71.9 (inverse agonist) | 315±180 (inverse agonist) | 785±564 (agonist) |
| mCB2 | 463±199 (inverse agonist) | 341±94.4 (inverse agonist) | 2000±475 (agonist) |
Abbreviation: cAMP, cyclic adenosine monophosphate.
Cells were stimulated in the presence of 1 μM forskolin for 30 min, and cAMP levels were determined using the HitHunter assay. In parentheses, the agonist or inverse agonist designation is given. Data are expressed as mean±s.e.m. for three independent experiments.
R,S-AM1241 and its enantiomers are not analgesic
R,S-AM1241 and its separated enantiomers were tested for acute nociception in rats using the tail-flick and hot-plate assays (data not shown). I.p. administration of each of R,S-AM1241, R-AM1241 and S-AM1241 did not affect hot-plate or tail-flick latency at 30 or 90 min following administration of doses up to 10 mg kg−1. In contrast, morphine (10 mg kg−1, s.c.), a positive control in these assays, produced a significant increase in both the tail-flick and hot-plate latencies at both 30 and 90 min post-administration (data not shown).
S-AM1241 blocks visceral pain and thermal hyperalgesia associated with chemical irritants
R,S-AM1241 and its enantiomers, R-AM1241 and S-AM1241, were evaluated in a dose–response study in the PPQ model of acute visceral pain. R,S-AM1241 did not produce a statistically significant blockade of PPQ induced stretching at the doses tested. At the 10 mg kg−1 dose, R-AM1241 produced a small (17%) reversal, 30 min post-PPQ injection, while S-AM1241 produced a relatively greater (35%) reversal of stretching (Figure 4a).
Figure 4.

Effects of R,S-AM1241 and its enantiomers, R-AM1241 and S-AM1241, on visceral pain and thermal hyperalgesia associated with chemical irritants. (a) Male CD-1 mice (25–30 g; n=10 per group) were pretreated with vehicle or compound (s.c.) 30 min before PPQ administration and tested 10 min post-administration. Data (mean±s.e.m.) are expressed as percent blockade relative to vehicle-treated mice. (b) Male SD rats (220–250 g; n=8 per group) received an intraplantar injection of 50 μl of saline or 2% carrageenan into the hindpaw, followed 2.5 h later by i.p. administration of vehicle, R,S-AM1241, R-AM1241 or S-AM1241. Data (mean±s.e.m.) are expressed as percent reversal relative to vehicle-treated rats. (c) Male SD rats (220–250 g; n=10 per group) received an intraplantar injection of 50 μl of saline or 2% carrageenan into the hindpaw, followed 2.5 h later by i.p. administration of either vehicle or 10 mg kg−1 S-AM1241 and either vehicle or 1 mg kg−1 AM630. Data (mean±s.e.m.) are expressed as paw withdrawal latencies; pre-carrageenan baseline data not shown. I.p., intraperitoneal; SD rat, Sprague–Dawley rat.
In the rat carrageenan model of inflammatory pain, R,S-AM1241 produced a reversal of carrageenan-induced thermal hyperalgesia, but only at the two highest doses tested. R-AM1241 did not reverse thermal hyperalgesia at any dose tested. In contrast, S-AM1241 was more efficacious than the racemate, producing a reversal of thermal hyperalgesia at all doses (Figure 4b). Neither the racemate nor either of the enantiomers produced a significant change in carrageenan-induced paw oedema at any of the doses tested (data not shown).
The CB2-selective antagonist AM630 was used to confirm the CB2 specificity of the S-AM1241 anti-hyperalgesic effects in the carrageenan model (Figure 4c). S-AM1241 at a 10 mg kg−1 dose produced a complete reversal of carrageenan-induced thermal hyperalgesia, similar to that produced by the positive control, treatment with indomethacin. This anti-hyperalgesic effect of S-AM1241 was blocked by the antagonist, AM630 at 1 mg kg−1. The paw withdrawal latency resulting from co-administration of S-AM1241 and AM630 was not different from that resulting from administration of AM630 alone.
Discussion and conclusions
In this paper, we describe the in vitro and in vivo pharmacology of R,S-AM1241 and its resolved enantiomers, as summarized in Table 4. The affinity of R,S-AM1241 for the murine CB2 receptor (28 nM) was lower than a previous report of 2 nM in mouse spleen membranes (Nackley et al., 2003). This discrepancy may reflect differences in the G-protein coupling of the CB2 receptors between native and heterologous expression systems, wherein any differences in stoichiometry of the receptor, G-proteins and other signalling molecules may be expected to affect agonist affinity. We were unable to distinguish between high- and low-affinity states, consistent with the report of a single Ki in mouse spleen (Nackley et al., 2003).
Table 4.
Qualitative summary of in vitro and in vivo results for R,S-AM2141 and its enantiomers, R-AM2141 and S-AM1241
| In vitro activity |
In vivo efficacy |
||||
|---|---|---|---|---|---|
| hCB2 | rCB2 | mCB2 | Carrageenan | PPQ | |
| R,S-AM1241 | Agonist | Inverse agonist | Inverse agonist | ++ | − |
| R-AM1241 | Agonist | Inverse agonist | Inverse agonist | − | + |
| S-AM1241 | Agonist | Agonist | Agonist | +++ | ++ |
Abbreviation: PPQ, para-phenylquinone.
In vitro pharmacological characterizations are as measured in Table 3. Categories of in vivo efficacy: −, no statistically significant reversal of the chemically induced pain state; +, statistically significant reversal of the pain state; ++, statistically significant >30% reversal of the pain state; +++, statistically significant >60% reversal of the pain state.
Consistent with the coupling of CB2 receptors to the inhibitory G-protein α-subunit Gi, stimulation of the receptor resulted in decreased cAMP levels following activation of adenylyl cyclases by forskolin. In agreement with previous data (Pertwee, 1997), the agonist WIN55,212-2 decreased cAMP formation by 80% in hCB2-expressing cells. The reason for the more modest 40–50% decrease seen in both rodent CB2 cell lines is not clear, but may be due to differences in coupling of the receptor to the G-protein complex. An increase in cAMP levels above those stimulated by forskolin was observed in response to the CB2 antagonist SR144528, as would be expected based on this compound's characterization as an inverse agonist (Rhee and Kim, 2002). Inverse agonism is an operative term used to describe inhibition of basal coupling or constitutive activity of the ligand-unbound receptor. As shown by its higher maximal response to either SR144528 or R-AM1241, the cells with the mCB2 receptors would appear to have a higher level of constitutive activity than those with the human or rat receptors, perhaps corresponding to a more effective coupling of this receptor to the cellular signal transduction machinery.
R,S-AM1241 inhibited cAMP production stimulated by treatment of the h CB2-expressing cell line with 1 μM forskolin, consistent with this racemate acting as an agonist of hCB2 receptors. The forskolin concentration used in our studies was lower than those used in a similar study (Yao et al., 2006), wherein it was reported that the function of R,S-AM1241 (partial agonist or neutral antagonist) in cyclase assays was sensitive to the concentration of forskolin (8 or 37 μM, respectively) used to stimulate hCB2-expressing cells. In our characterization of the rodent receptors, R,S-AM1241 demonstrated inverse agonist properties at the same concentration of forskolin (1 μM) that was associated with agonist activity at the hCB2 receptors. S-AM1241 was seen to be an agonist at human, mouse and rat CB2 receptors, whereas R-AM1241 was observed to be an agonist at the human receptor and an inverse agonist in the cells with the rodent receptors. The functional properties of the racemate are dominated by those of the R-enantiomer, reflecting its more than 40-fold higher CB2 affinity compared with the S-enantiomer.
In an analysis of racemic AM1241 in hCB2 receptor assays (Yao et al., 2006), functional activity varied depending on the end point that was measured (antagonist of calcium mobilization; agonist of extracellular signal-regulated kinase activation; and antagonist or partial agonist of cAMP inhibition, depending on the level of stimulation). The authors proposed the diverse functional effects of R,S-AM1241 as a case of protean agonism (Yao et al., 2006), a phenomenon wherein the state of constitutive receptor activity can determine the functional effect of a ligand-receptor interaction (Kenakin, 2001). Under the protean agonist hypothesis, two receptor states, a ligand-bound and a constitutively active, ligand-unbound form, compete for G-proteins. If the efficacy of the constitutively active receptor is higher than that of the ligand-bound receptor, then the protean agonist, by inducing a less active receptor conformation, will appear as an inverse agonist. In the absence of constitutive activity, the same ligand will act as a partial agonist. Differing levels of receptor activation in different cell-based assay systems can thus suffice to produce varying functional outcomes. It is tempting, therefore, to speculate that the inverse agonist activity of R-AM1241 at the rodent CB2 receptors, in contrast to its agonist activity at the human receptor, results from different levels of CB2 constitutive activity between our rodent and human receptor expression systems, giving rise to a case of protean agonism. However, the observation that the human receptor displays higher basal activity than the rat receptor is at odds with this hypothesis and suggests that other, as yet undefined, mechanisms may be involved.
The in vivo efficacy of R,S-AM1241 and its enantiomers was assessed in rodent models of acute, inflammatory and visceral pain. Neither R,S-AM1241 nor either of its enantiomers showed evidence of acute nociception in either the tail-flick or hot-plate assay. This is the first report of the effects of the AM1241 enantiomers in an assay of acute nociceptive pain. Our results, although in contrast with an earlier report demonstrating analgesic effects of racemic AM1241 (Malan et al., 2001), are consistent with studies demonstrating that other CB2 agonists are not analgesic in vivo (Valenzano et al., 2005; Whiteside et al., 2005).
S-AM1241 was efficacious in the mouse PPQ model, as was R-AM1241. However, the latter compound had only a modest antinociceptive effect, and the racemate had no statistically significant effect in this model. The lone previous report of in vivo efficacy of a resolved stereoisomer of AM1241 was an investigation of (+)-AM1241 (the R-enantiomer) in a mouse pain model that used intraplantar formalin injection (Beltramo et al., 2006). In light of our characterization of the resolved enantiomers, particularly the antinociceptive effects of S-AM1241, it would be of interest to compare the efficacy of both enantiomers in the formalin-induced pain model.
In the rat carrageenan model of inflammatory pain, S-AM1241, an agonist at rCB2 receptors, was more efficacious than the racemate against thermal hyperalgesia, whereas R-AM1241, an inverse agonist, lacked statistically significant efficacy. The antihyperalgesic effect of S-AM1241 was blocked by the CB2 antagonist AM630, demonstrating that the activity of S-AM1241 was mediated by CB2 receptors. Additional off-target effects of S-AM1241 cannot be ruled out, but the magnitude of the AM630-induced blockade should be interpreted as evidence that any non-CB2 components of this effect would be minor in comparison to the CB2 component. Our results in the carrageenan model are consistent not only with previous reports of antinociceptive efficacy following administration of racemic AM1241, but also with reports of efficacy achieved with other CB2 agonists in models of inflammatory pain (Clayton et al., 2002; Elmes et al., 2005; Valenzano et al., 2005).
Whereas the in vivo efficacy of S-AM1241 in rodent pain models is consistent with the in vitro functional characterization of this enantiomer as a rodent CB2 agonist, the in vivo efficacy of R,S-AM1241 and R-AM1241 in the same rodent pain models appears to be inconsistent with their in vitro characterization as inverse agonists. In the absence of constitutive CB2 receptor activity in vivo, the prediction following from the protean agonist hypothesis is that R-AM1241 would behave as a partial agonist. In that case, the efficacy of R-AM1241 in the mouse formalin (Beltramo et al., 2006) and PPQ models and the efficacy of the racemate in multiple pain models would be consistent with the in vitro characterization of these compounds. However, constitutive activation of receptors is an elusive property to measure in vivo. In one case in which this property has been deduced for CB2 receptors, the in vivo efficacy of CB2-selective inverse agonists in the inhibition of leucocyte trafficking (Lunn et al., 2006) provides evidence of the existence of constitutive CB2 receptor activity in rodents. This condition, if it holds in our rodent pain models, would argue against any expectation of partial agonist properties of R-AM1241 in vivo.
It is noteworthy that our study is not the first reported example of a discrepancy between the in vitro characterization of cannabinoid ligands and their in vivo effects. Formalin-induced hyperalgesia in mice was shown to be exacerbated by each of two fatty acid-derived compounds whose in vitro properties indicate them to be CB1 partial agonists (Cascio et al., 2006), an observation that is not consistent with the expectation of CB1 receptor agonism being antihyperalgesic. Expectations about the effects of cannabinoid receptor inverse agonist compounds are further confused by reports of anti-inflammatory effects of CB2 inverse agonists (Iwamura et al., 2001; Ueda et al., 2005). Without direct in vivo measurements of the basal state of CB2 receptor activation, in particular, in cell types known to mediate the responses to exogenous CB2 ligands, the behavioural studies we report herein can at the best be viewed as a characterization of R,S-AM1241 and its enantiomers, and not as a direct test of the protean agonist hypothesis.
In summary, we have reported for the first time an in vitro functional characterization of R,S-AM1241 in rodent CB2 heterologous expression systems. In addition, we have provided the first in vitro and in vivo pharmacological assessment of this compound's resolved enantiomers. Despite the observation that S-AM1241, the enantiomer that displayed rodent CB2 receptor agonist properties, was more efficacious than either R-AM1241 or the racemate in rodent pain models, a full understanding of the relevance of the species-dependent and stereoisomer-dependent pharmacology we present herein will require further characterization.
Acknowledgments
We thank Drs Alexandros Makriyannis, Adam M Gilbert and Kathryn E Rogers for beneficial discussion of this work.
Abbreviations
- PPQ
para-phenylquinone
- SNL
spinal nerve ligation
Conflict of interest
The authors state no conflict of interest.
References
- Baker D, Pryce G, Davies WL, Hiley CR. In silico patent searching reveals a new cannabinoid receptor. Trends Pharmacol Sci. 2006;27:1–4. doi: 10.1016/j.tips.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Beltramo M, Bernardini N, Bertorelli R, Campanella M, Nicolussi E, Fredduzzi S, et al. CB2 receptor-mediated antihyperalgesia: possible direct involvement of neural mechanisms. Eur J Neurosci. 2006;26:1530–1538. doi: 10.1111/j.1460-9568.2006.04684.x. [DOI] [PubMed] [Google Scholar]
- Brown SM, Wager-Miller J, Mackie K. Cloning and molecular characterization of the rat CB2 cannabinoid receptor. Biochim Biophys Acta. 2002;1576:255–264. doi: 10.1016/s0167-4781(02)00341-x. [DOI] [PubMed] [Google Scholar]
- Cabral GA, Marciano-Cabral F. Cannabinoid receptors in microglia of the central nervous system: immune functional relevance. J Leukoc Biol. 2005;78:1192–1197. doi: 10.1189/jlb.0405216. [DOI] [PubMed] [Google Scholar]
- Cascio MG, Bisogno T, Palazzo E, Thomas A, van der Stelt M, Brizzi A, et al. In vitro and in vivo pharmacology of synthetic olivetol- or resorcinol-derived cannabinoid receptor ligands. Br J Pharmacol. 2006;149:431–440. doi: 10.1038/sj.bjp.0706888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton N, Marshall FH, Bountra C, O'Shaughnessy CT. CB1 and CB2 cannabinoid receptors are implicated in inflammatory pain. Pain. 2002;96:253–260. doi: 10.1016/S0304-3959(01)00454-7. [DOI] [PubMed] [Google Scholar]
- D'Amour FE, Smith D. A method for determining loss of pain sensation. J Pharmacol Exp Ther. 1941;72:74–79. [Google Scholar]
- Elmes SJ, Winyard LA, Medhurst SJ, Clayton NM, Wilson AW, Kendall DA, et al. Activation of CB1 and CB2 receptors attenuates the induction and maintenance of inflammatory pain in the rat. Pain. 2005;118:327–335. doi: 10.1016/j.pain.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Flygare J, Gustafsson K, Kimby E, Christensson B, Sander B. Cannabinoid receptor ligands mediate growth inhibition and cell death in mantle cell lymphoma. FEBS Letters. 2005;579:6885–6889. doi: 10.1016/j.febslet.2005.11.020. [DOI] [PubMed] [Google Scholar]
- Fride E, Foox A, Rosenberg E, Faigenboom M, Cohen V, Barda L, et al. Milk intake and survival in newborn cannabinoid CB1 receptor knockout mice: evidence for a “CB3” receptor. Eur J Pharmacol. 2003;461:27–34. doi: 10.1016/s0014-2999(03)01295-0. [DOI] [PubMed] [Google Scholar]
- Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, et al. Gaussian 2003Pittsburgh, PA: Gaussian, Inc; Revision B.03. [Google Scholar]
- Griffin G, Tao Q, Abood ME. Cloning and pharmacological characterization of the rat CB(2) cannabinoid receptor. J Pharmacol Exp Ther. 2000;292:886–894. [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Herrera B, Carracedo A, Diez-Zaera M, Guzman M, Velasco G. p38 MAPK is involved in CB2 receptor-induced apoptosis of human leukaemia cells. FEBS Lett. 2005;579:5084–5088. doi: 10.1016/j.febslet.2005.08.021. [DOI] [PubMed] [Google Scholar]
- Hohmann AG, Farthing JN, Zvonok AM, Makriyannis A. Selective activation of cannabinoid CB2 receptors suppresses hyperalgesia evoked by intradermal capsaicin. J Pharmacol Exp Ther. 2004;308:446–453. doi: 10.1124/jpet.103.060079. [DOI] [PubMed] [Google Scholar]
- Howlett AC, Breivogel CS, Childers SR, Deadwyler SA, Hampson RE, Porrino LJ. Cannabinoid physiology and pharmacology: 30 years of progress. Neuropharmacology. 2004;47:345–358. doi: 10.1016/j.neuropharm.2004.07.030. [DOI] [PubMed] [Google Scholar]
- Ibrahim M, Porreca F, Lai J, Albrecht P, Rice F, Khodorova A, et al. CB2 cannabinoid activation produces antinociception by stimulating peripheral release of endogenous opioids. Proc Natl Acad Sci USA. 2005;102:3093–3098. doi: 10.1073/pnas.0409888102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim M, Rude M, Stagg N, Mata H, Lai J, Vanderah T, et al. CB2 cannabinoid receptor mediation of antinociception. Pain. 2006;122:36–42. doi: 10.1016/j.pain.2005.12.018. [DOI] [PubMed] [Google Scholar]
- Ibrahim MM, Deng H, Zvonok A, Cockayne DA, Kwan J, Mata HP, et al. Activation of CB2 cannabinoid receptors by AM1241 inhibits experimental neuropathic pain: pain inhibition by receptors not present in the CNS. Proc Natl Acad Sci USA. 2003;100:10529–10533. doi: 10.1073/pnas.1834309100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamura H, Suzuki H, Ueda Y, Kaya T, Inaba T. In vitro and in vivo pharmacological characterization of JTE-907, a novel selective ligand for cannabinoid CB2 receptor. J Pharmacol Exp Ther. 2001;296:420–425. [PubMed] [Google Scholar]
- Kenakin T. Inverse, protean, and ligand-selective agonism: matters of receptor conformation. FASEB J. 2001;15:598–611. doi: 10.1096/fj.00-0438rev. [DOI] [PubMed] [Google Scholar]
- Kim K, Moore DH, Makriyannis A, Abood ME. AM1241, a cannabinoid CB2 receptor selective compound, delays disease progression in a mouse model of amyotrophic lateral sclerosis. Eur J Pharmacol. 2006;542:100–105. doi: 10.1016/j.ejphar.2006.05.025. [DOI] [PubMed] [Google Scholar]
- LaBuda CJ, Koblish M, Little PJ. Cannabinoid CB2 receptor agonist activity in the hindpaw incision model of postoperative pain. Eur J Pharmacol. 2005;527:172–174. doi: 10.1016/j.ejphar.2005.10.020. [DOI] [PubMed] [Google Scholar]
- Lunn CA, Fine JS, Rojas-Triana A, Jackson JV, Fan X, Kung TT, et al. A novel cannabinoid peripheral cannabinoid receptor-selective inverse agonist blocks leukocyte recruitment in vivo. J Pharmacol Exp Ther. 2006;316:780–788. doi: 10.1124/jpet.105.093500. [DOI] [PubMed] [Google Scholar]
- Makriyannis A.University of Connecticut, Receptor selective cannabimimetic aminoalkylindoles 2002. WO 02060447 A1
- Malan TP, Ibrahim MM, Deng H, Liu Q, Mata HP, Vanderah T, et al. CB2 cannabinoid receptor-mediated peripheral antinociception. Pain. 2001;93:239–245. doi: 10.1016/S0304-3959(01)00321-9. [DOI] [PubMed] [Google Scholar]
- Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- Nackley A, Zvonok A, Makriyannis A, Hohmann A. Activation of cannabinoid CB2 receptors suppresses C-fiber responses and windup in spinal wide dynamic range neurons in the absense and presence of inflammation. J Neurophysiol. 2004;92:3562–3574. doi: 10.1152/jn.00886.2003. [DOI] [PubMed] [Google Scholar]
- Nackley AG, Makriyannis A, Hohmann AG. Selective activation of cannabinoid CB(2) receptors suppresses spinal fos protein expression and pain behavior in a rat model of inflammation. Neuroscience. 2003;119:747–757. doi: 10.1016/s0306-4522(03)00126-x. [DOI] [PubMed] [Google Scholar]
- Ofek O, Karsak M, Leclerc N, Fogel M, Frenkel B, Wright K, et al. Peripheral cannabinoid receptor, CB2, regulates bone mass. Proc Natl Acad Sci USA. 2006;103:696–701. doi: 10.1073/pnas.0504187103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG. Pharmacology of cannabinoid CB1 and CB2 receptors. Pharmacol Ther. 1997;74:129–180. doi: 10.1016/s0163-7258(97)82001-3. [DOI] [PubMed] [Google Scholar]
- Quartilho A, Mata HP, Ibrahim MM, Vanderah TW, Porreca F, Makriyannis A, et al. Inhibition of inflammatory hyperalgesia by activation of peripheral CB2 cannabinoid receptors. Anesthesiology. 2003;99:955–960. doi: 10.1097/00000542-200310000-00031. [DOI] [PubMed] [Google Scholar]
- Rhee MH, Kim SK. SR144528 as inverse agonist of CB2 cannabinoid receptor. J Vet Sci. 2002;3:179–184. [PubMed] [Google Scholar]
- Rinaldi-Carmona M, Barth F, Millan J, Derocq JM, Casellas P, Congy C, et al. SR 144528, the first potent and selective antagonist of the CB2 cannabinoid receptor. J Pharmacol Exp Ther. 1998;284:644–650. [PubMed] [Google Scholar]
- Shire D, Calandra B, Rinaldi-Carmona M, Oustric D, Pessegue B, Bonnin-Cabanne O, et al. Molecular cloning, expression and function of the murine CB2 peripheral cannabinoid receptor. Biochim Biophys Acta. 1996;1307:132–136. doi: 10.1016/0167-4781(96)00047-4. [DOI] [PubMed] [Google Scholar]
- Shoemaker JL, Seely KA, Reed RL, Crow JP, Prather PL. The CB2 cannabinoid agonist AM-1241 prolongs survival in a transgenic mouse model of amyotrophic lateral sclerosis when initiated at symptom onset. J Neurochem. 2007;101:87–98. doi: 10.1111/j.1471-4159.2006.04346.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffens S, Veillard NR, Arnaud C, Pelli G, Burger F, Staub C, et al. Low dose oral cannabinoid therapy reduces progression of atherosclerosis in mice. Nature. 2005;434:782–786. doi: 10.1038/nature03389. [DOI] [PubMed] [Google Scholar]
- Ueda Y, Miyagawa N, Matsui T, Kaya T, Iwamura H. Involvement of cannabinoid CB(2) receptor-mediated response and efficacy of cannabinoid CB(2) receptor inverse agonist, JTE-907, in cutaneous inflammation in mice. Eur J Pharmacol. 2005;520:164–171. doi: 10.1016/j.ejphar.2005.08.013. [DOI] [PubMed] [Google Scholar]
- Valenzano KJ, Tafesse L, Lee G, Harrison JE, Boulet JM, Gottshall SL, et al. Pharmacological and pharmacokinetic characterization of the cannabinoid receptor 2 agonist, GW405833, utilizing rodent models of acute and chronic pain, anxiety, ataxia and catelepsy. Neuropharmacology. 2005;48:658–672. doi: 10.1016/j.neuropharm.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Van Sickle MD, Duncan M, Kingsley PJ, Mouihate A, Urbani P, Mackie K, et al. Indentification and functional characterization of brainstem cannabinoid CB2 receptors. Science. 2005;310:329–332. doi: 10.1126/science.1115740. [DOI] [PubMed] [Google Scholar]
- Whiteside GT, Gottshall SL, Boulet JM, Chaffer SM, Harrison JE, Pearson MS, et al. A role for cannabinoid receptors, but not endogenous opioids, in the antinociceptive activity of the CB2-selective agonist, GW405833. Eur J Pharmacol. 2005;528:65–72. doi: 10.1016/j.ejphar.2005.10.043. [DOI] [PubMed] [Google Scholar]
- Woolfe G, MacDonald AD. The evaluation of the analgesic action of pethidine. J Pharmacol Exp Ther. 1944;80:300–307. [Google Scholar]
- Wotherspoon G, Fox A, McIntyre P, Colley S, Bevan S, Winter J. Peripheral nerve injury induces cannabinoid receptor 2 protein expression in rat sensory neurons. Neuroscience. 2005;135:235–245. doi: 10.1016/j.neuroscience.2005.06.009. [DOI] [PubMed] [Google Scholar]
- Yao BB, Mukherjee S, Fan Y, Garrison TR, Daza AV, Grayson GK, et al. In vitro pharmacological characterization of AM1241: a protean agonist at the cannabinoid CB(2) receptor. Br J Pharmacol. 2006;149:145–154. doi: 10.1038/sj.bjp.0706838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain. 1983;16:109–110. doi: 10.1016/0304-3959(83)90201-4. [DOI] [PubMed] [Google Scholar]