

Abstract
Brain-derived neurotrophic factor (BDNF) signaling through its receptor, trkB, is essential for the proper development and function of the nervous system. Here we identify a novel regulatory element designated TCaRE3 (TRKB Ca2+ Response Element 3) required for CREB-dependent TRKB transcription in neurons. TCaRE3-inactivating mutations abolished both Ca2+- and cAMP-stimulated TRKB expression, despite the presence of upstream CREs. TCaRE3 mutations also reduced basal expression by at least 80%. Electrophoretic mobility shift assays revealed the presence of a neuronal nuclear factor able to bind TCaRE3 in a sequence-specific manner and we have identified krüppel-like factor 7 (KLF7) as a candidate TCaRE3 transcription factor. Importantly, despite limited overall sequence homology between the promoter regions of the human and mouse TRKB genes, TCaRE3 exhibits 100% sequence identity. Mutation analysis of the human TRKB promoter region demonstrated that the role of TCaRE3 is also conserved, suggesting that the functional interaction between CREB bound to the CREs and KLF7 bound to TCaRE3 is essential for the proper regulation of TRKB in neurons.
The neurotrophin, BDNF, signals through its receptor trkB to mediate neuron survival and differentiation, neurite outgrowth, axon guidance, and activity-dependent synaptic plasticity (for reviews, see Huang and Reichardt, 2001; 2003). Studies of mice with forebrain-targeted deletions in the genes coding for BDNF or trkB displayed defects in neuron survival, long term potentiation, and learning and memory (Korte et al., 1996; Patterson et al., 1996; Xu et al., 2000). Abnormal BDNF/trkB signaling has been linked to neurological and psychiatric disorders such as Alzheimer’s and Parkinson’s diseases (Ferrer et al., 1999; Allen et al., 1999; Murer et al., 2001; Holsinger et al., 2000; Fumagalli et al., 2006a,b), as well as depression and schizophrenia (Nestler et al., 2002; Angelucci et al., 2005; Hashimoto et al., 2004;).
The expression of BDNF in cultured cortical neurons is upregulated by depolarization (Ghosh et al., 1994) due to the activation of Ca2+-dependent regulatory promoter elements in BDNF, the gene encoding BDNF (Tao et al., 1998; Shieh et al., 1998). Similarly, we demonstrated that depolarization-induced Ca2+ signaling specifically stimulated expression of catalytically-active, full-length trkB without affecting the inactive, truncated (T1) isoform (Kingsbury et al., 2003). The increased full-length trkB was readily auto-phosphorylated in response to BDNF, indicating that the newly synthesized receptors were at the plasma membrane and functionally active. Ca2+-dependent up-regulation of full-length trkB was accompanied by preferential stimulation of the downstream TRKB promoter, P2 (Kingsbury et al., 2003). Thus, Ca2+-dependent activation of TRKB P2 can increase neuronal responsiveness to BDNF by preferentially upregulating expression of the active isoform trkB. This coordinated regulation of both ligand (BDNF) and receptor (trkB) expression by Ca2+-dependent activation of gene transcription may lead to synergistic enhancement of the strength of BDNF trkB signaling in response to neuronal activity (Nagappan and Lu, 2005).
Stimulation of BDNF and TRKB transcription by Ca2+ depends on a Ca2+/cAMP response element binding protein (CREB) family member acting via a CRE in BDNF promoter III (Tao et al., 1998; Shieh et al., 1998) or a pair of tandem CREs in TRKB P2 (Kingsbury et al., 2003). Ca2+-responsive promoters, including BDNF PIII (Chen et al., 2003), frequently possess additional regulatory elements that modulate the transcriptional responses of the CRE (e.g. Robertson, 1995; West et al., 2001). It is this functional interaction between multiple promoter elements that allows ubiquitous second messengers such as Ca2+ and cAMP to be translated into gene- and cell-specific transcriptional responses. We therefore investigated the possibility that additional regulatory elements within the TRKB promoter shape the response of TRKB P2 to Ca2+ signaling. Here we report that a novel DNA element, designated TCaRE3, is required for TRKB P2 to respond to Ca2+ signals, and identify krüppel-like factor 7 (KLF7) as a candidate transcription factor for TCaRE3. We also show that the sequence and function of TCaRE3 are conserved in the human TRKB promoter.
RESULTS
Elements downstream of the P2 transcription initiation site modulate Ca2+-dependent TRKB P2 expression
Deletion analysis of TRKB P2 revealed the presence of additional regulatory sites important for Ca2+-dependent P2 expression located downstream from the P2 transcription initiation site. For the TRKB promoter, the translation initiation site has been numbered +1, placing the P2 transcription initiation site at −448 (Barettino et al., 1999). Beginning with the −944/+2 TRKB-luciferase construct, which we previously showed responds to depolarization-induced Ca2+ influx with a 2-fold increase in luciferase activity (Kingsbury et al., 2003), we generated a series of P2 TRKB-luciferase reporter constructs by progressive 3’ truncation (Figure 1). Each reporter was transiently transfected into mouse cortical neurons and assayed for Ca2+ responsiveness following depolarization with elevated KCl (50 mM). Neurons transfected with −944/+2, −944/−77 or −944/−162 TRKB-luciferase constructs all responded with a 2-fold stimulation following depolarization, suggesting that the sequences downstream from bp −162 do not participate in the Ca2+ responsiveness of TRKB P2. Constructs −944/−241, −944/−325 and −944/−339 exhibited a 3-fold increase in reporter activity in response to depolarization. This increased stimulation suggests that there is a Ca2+-dependent inhibitory element located between −162 and −241; we have not characterized this element further. As previously reported for −944/+2, removal of extracellular Ca2+ with EGTA blocked all depolarization-induced expression of these promoter constructs (data not shown). Further truncation to generate −944/−363, −944/−389 and −944/−444 resulted in constructs that were no longer stimulated in response to KCl-induced Ca2+ signaling. Thus, removal of the sequences between −339 and −363 eliminated Ca2+-stimulated P2 expression. Since all of these constructs contain the tandem CREs located between −922 and −904, these findings indicate that both the CREs and an additional site located downstream from the transcription start site (−448) are required for Ca2+-stimulated P2 expression. In addition to the loss of Ca2+-stimulated expression, the −944/−363, −944/−389 and −944/−444 constructs also exhibited a 92–96% reduction in basal TRKB P2-luciferase (* in Fig. 1), as compared to the original −944/+2 TRKB-luciferase construct (see below). In contrast, −944/−325 and −944/−339 showed wild type basal activity.
Figure 1. Identification of a domain required for Ca2+-dependent TRKB P2 expression.
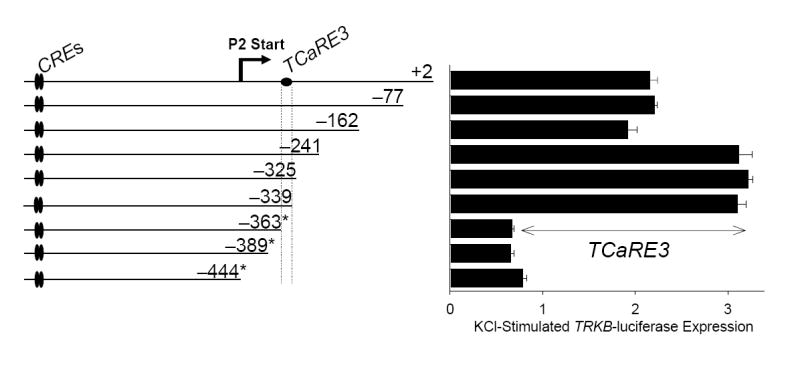
TRKB P2-luciferase reporter genes containing progressive 3’ truncations (left) were transiently transfected into cortical neurons. Two days later, cultures were treated with 50 mM KCl for 5 hours prior to harvesting for luciferase assays (right). Individual luciferase measurements were normalized to co-transfected TK-Renilla values to correct for sample-to-sample variation in transfection efficiency. For each construct, Renilla-normalized luciferase measurements from depolarized cells are presented relative to those of control, unstimulated cells. Bars show mean ± sem (N=4). Deletion of bp −162 to −241 increased the magnitude of Ca2+ stimulation, suggesting the presence of an inhibitory element within that sequence. Deletion of bp −339 to −363 abolished Ca2+-stimulation of P2 luciferase activity, defining the domain of TCaRE3 (TRKB Ca2+-response element 3). * denotes constructs exhibiting reduced basal activity.
In light of the multiple elements involved in Ca2+ stimulated TRKB P2 expression, we have adopted a nomenclature similar to that used for Ca2+ response elements (CaREs) in rat BDNF promoter III (West et al., 2001) referring to TRKB promoter elements as TCaREs (TRKB Ca2+ Response Elements). Using this nomenclature, the new element located between −363 and −339 is designated TCaRE3 (TRKB Ca2+ response element #3) and the pair of tandem CREs between −926 and −904 is now designated TCaRE2.
TCaRE3 is a novel element enabling Ca2+-dependent gene expression
The sequence of the region from −363 to −339 required for TCaRE3 function is shown in Figure 2. Using a scanning mutagenesis approach, a series of −944/−325 TRKB-luciferase constructs harboring the indicated point mutations was generated in order to more precisely delineate the location of TCaRE3. The basepair substitutions introduced in M1 were the same mutations introduced into −944/+2 to generate deletion construct −944/−363. In contrast to the −944/−363 deletion mutation, M1 had no effect on Ca2+-dependent P2 expression, indicating that truncation to −363 failed to respond due to the removal of downstream sequences (Fig. 2, M1), rather than introduction of mutations into a critical site. Mutations that were found to disrupt Ca2+ responsiveness of TRKB P2 mapped the critical sequence of TCaRE3 to 16 basepairs (Fig. 2, bold). Analysis of this sequence using TESS (Transcription Element Search Software, Univ. of Pennsylvania, Philadelphia, PA; URL: http://www.cbil.upenn.edu/tess), failed to identify a known Ca2+ response element, suggesting it contains a novel promoter element required for TRKB P2 Ca2+-stimulated expression.
Figure 2. Mutational analysis of TCaRE3.
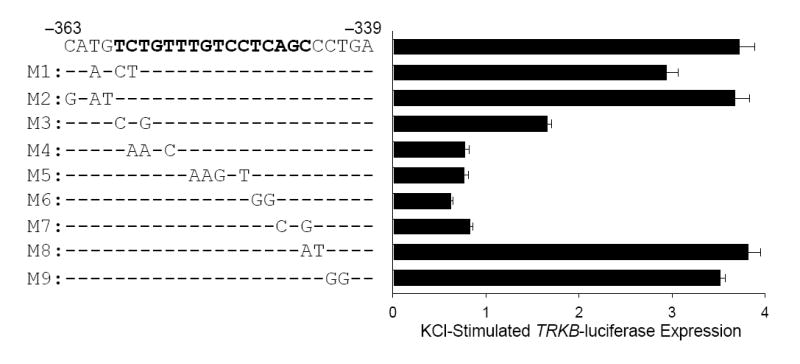
The −944/−325 TRKB P2-luciferase construct was subjected to scanning mutagenesis from −399 to −363 to map the nucleotides essential for TCaRE3 function. Cortical neurons were transiently transfected with corresponding P2-luciferase reporter constructs (left). Two days after transfection, cultures were stimulated with 50 mM KCl and harvested for luciferase assays (right). Bars show the averages of quadruplicate determinations ± sem. This mutation analysis revealed that the region from −344 to −359 (bold) is required for Ca2+-stimulated expression of TRKB P2.
Mutation of TCaRE3 also reduced basal TRKB expression. Figure 3 (left) shows the effect of mutation M4 (c.f., Fig. 2) on TRKB P2 luciferase activity. Note that, in addition to blocking Ca2+-stimulated expression, introduction of M4 into the −944/−325 P2-luciferase construct decreased activity to less than 5% of wild type expression in unstimulated samples. All of the other TCaRE3 mutations that abolished Ca2+-dependent P2 stimulation (M3–M7) also reduced basal TRKB expression by 80–95% (not shown). Mutations of the CREs, located within TCaRE2, also reduced basal TRKB transcription (see below).
Figure 3. Mutation of TCaRE3, but not the neighboring TBP site disrupts Ca2+-dependent P2 expression.
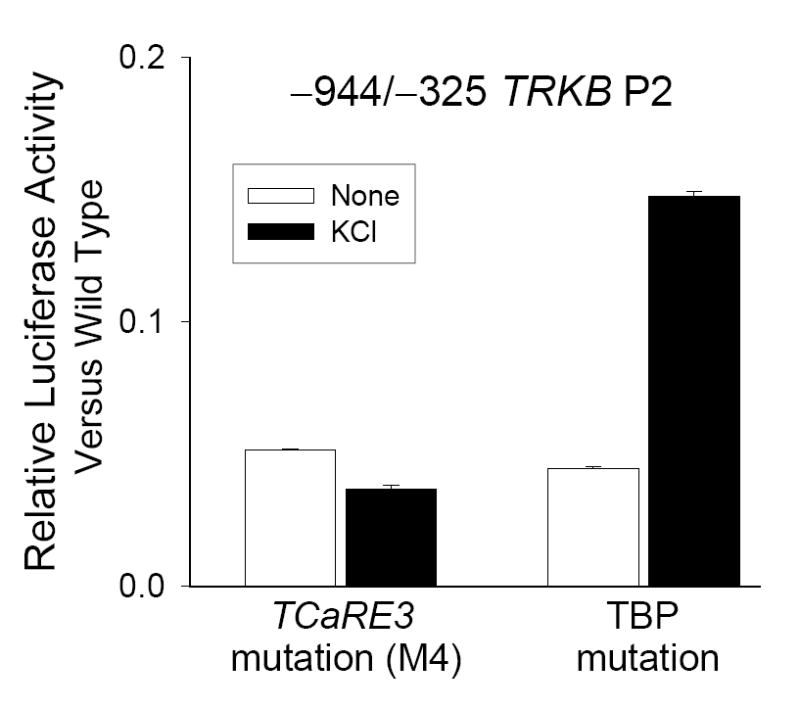
−944/−325 TRKB P2-luciferase constructs were transiently transfected into cortical neurons and assayed for Ca2+-stimulated expression following depolarization for 5 hours. Bars show the averages of quadruplicate determinations ± sem. Samples were normalized to basal expression of wild type −944/−325 construct assayed in parallel. Mutation of either TCaRE3 (left) (M4, c.f., Fig. 2) or the TBP site (right) (GTCTATTAG→GTCTAAGCT) resulted in a similar decrease in basal P2 expression, but only the TCaRE3 mutation abolished Ca2+- stimulated TRKB P2 expression. Bars show mean ± sem (N=4).
Analysis of the sequences flanking TCaRE3 using TESS revealed a potential TBP binding site (GTCTATTAG), located between bp −379 and −371 (c.f., Fig. 8). Since TBP functions to enable transcription of numerous promoters (Lee and Young, 1998), we examined the possibility that the TBP site is also involved in TRKB P2 expression. Mutations (GTCTAAGCT) were introduced into the −944/−325 P2-luciferase construct to disrupt the candidate TBP site. As observed for mutations in TCaRE3, mutation of the TBP site resulted in a 94% reduction in basal TRKB P2-luciferase expression (Fig. 3, right); in contrast to TCaRE3 mutations, however, the construct containing the TBP mutations still exhibited a 3.1-fold stimulation in response to elevated KCl, similar to the 3.2-fold increase observed in the −944/−325 wild type construct (Fig 1). Thus, although the TBP site is required for basal P2 transcription, it is not essential for the ability of Ca2+ to stimulate P2 activity.
Figure 8. Multiple promoter elements regulate TRKB P2 transcription.
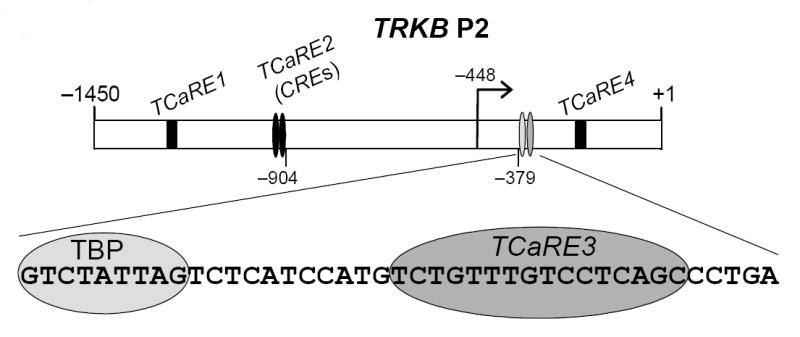
We have identified four promoter elements that are involved in regulating the responsiveness of TRKB P2 to Ca2+ signaling. We refer to these elements as TCaREs (TRKB Ca2+-response elements). TCaRE2 and TCaRE3 are required for Ca2+-dependent P2 expression. TCaRE2 consists of a pair of tandem CREs (Kingsbury et al., 2003). TCaRE3 is a novel element required for CRE-dependent expression (Figs. 1, 2, 4, 5). TCaRE1 enhances Ca2+-dependent TRKB P2 expression (Kingsbury, et al., 2003). TCaRE4 is a suppressor site located downstream from TCaRE3 (c.f., Fig. 1), which has not be further characterized. The TBP site is required for basal but not Ca2+-dependent expression in mouse TRKB P2 (c.f., Fig 3).
TCaRE3 is also required for cAMP responsiveness of TRKB P2
We (Kingsbury et al., 2003) and others (Deogracias et al., 2004) have shown that P2 TRKB-luciferase reporters can also be stimulated cAMP signaling induced by forskolin. The requirement for TCaRE3 in mediating P2 induction following forskolin treatment was tested using the TRKB-luciferase constructs generated to map TCaRE3. TRKB reporters lacking a functional TCaRE3 failed to respond to cAMP signaling (Fig. 4), indicating that TCaRE3 is required for both Ca2+ and cAMP stimulation of TRKB P2. Thus, CREB cannot stimulate TRKB P2 via the upstream tandem CREs in the absence of a functional TCaRE3.
Figure 4. TCaRE3 mediates cAMP-dependent P2 expression.
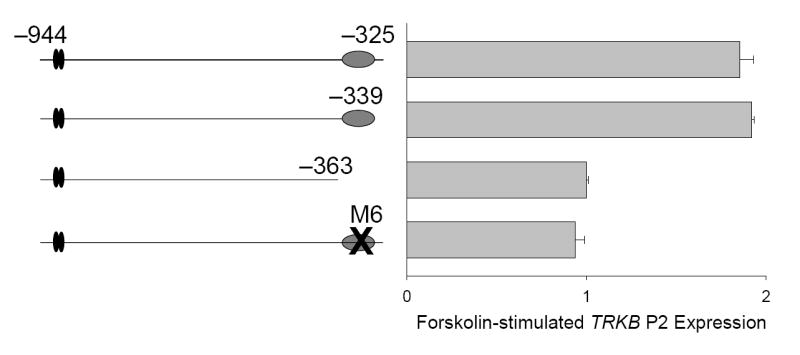
Cortical neurons were transiently transfected with −944/−325, −944/−339 or −944/−363 TRKB P2 luciferase constructs or −944/−325 with TCaRE3 mutation M6 (c.f., Fig. 2). Two days after transfection, cultures were stimulated for 5 hours with 40 μM forskolin. Bars show mean ± sem (N=4).
TCaRE3 functions as a DNA element regulating TRKB P2 transcription
Previous studies (Barettino et al., 1999) demonstrated that P2 generates a single transcript, which does not undergo splicing in the 5’ UTR. Since TCaRE3 is located in this 5’ UTR sequence of the mRNA, our results could be explained if TCaRE3 acted to promote Ca2+-dependent RNA stability or translation, rather than to mediate Ca2+-stimulated transcription. To distinguish between such transcriptional and post-transcriptional effects, the ability of TCaRE3 to mediate Ca2+-stimulated P2 TRKB-luciferase activity was analyzed in the presence of the transcription inhibitor actinomycin D (actD). Cortical neurons transiently transfected with −944/+2, −944/−325 or −944/−444 were depolarized with 50 mM KCl in either the presence or absence of actD (Fig. 5A). Ca2+-dependent stimulation of P2 was blocked by actD in all constructs containing TCaRE3 including the −944/−325 TRKB P2-luciferase construct, which lacks the downstream Ca2+-responsive inhibitory element between −241 and −162 (Fig. 1). This demonstrates that a post-transcriptional role for TCaRE3 was not being masked by the presence of this additional inhibitory element and that TCaRE3 acts by regulating transcription. For each of the three constructs, there was a similar, minimal effect of actD on basal P2-luciferase activity over the 5 hour time course of the experiment, suggesting that the absence of TCaRE3 did not increase RNA turnover.
Figure 5. TCaRE3 functions as a DNA element.
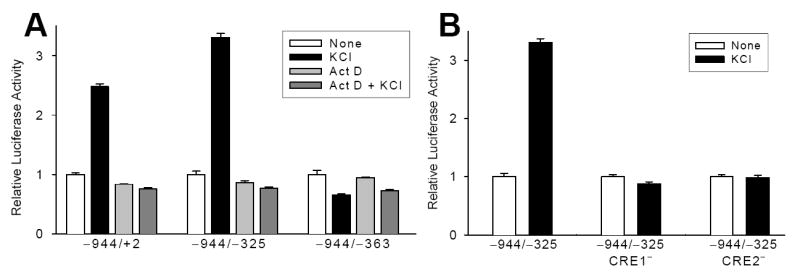
A. Cortical neurons were transfected with −944/+2,−944/−325 (inhibitory element, TCaRE4, deleted) or −944/−363 (TCaRE3-deleted) constructs. The ability of 50 mM KCl to stimulate P2 expression was tested in the presence or absence of actD (40 μM). Despite the presence of TCaRE3 sequence in RNA transcripts, neither −944/+2 nor −944/−325 exhibited any stimulation of luciferase activity following 5 hours KCl treatment. B. Mutation of either of the upstream CREs in TCaRE2 abolished depolarization-stimulated P2 expression even though TCaRE3 was still present. Thus, the presence of TCaRE3 in the RNA is not sufficient to mediate Ca2+-dependent stimulation of TRKB. With both CRE mutations, unstimulated TRKB transcription was reduced to an extent similar to that observed with TCaRE3 mutations (c.f., Fig. 3). In this figure, basal luciferase activity for each CRE mutation has been normalized to 1.0 to highlight the absence of Ca2+ dependence with the CRE mutations. Bars show mean ± sem (N=4).
In order to provide further evidence that TCaRE3 functions as a DNA element, we tested the effect of CRE mutations on the ability of Ca2+ to stimulate the −944/−325 TRKB luciferase construct. We previously showed that Ca2+-dependent transcription of TRKB P2 −944/+2 was abolished by introduction point mutations in either CRE in TCaRE2 (Kingsbury et al., 2003). Mutation of either CRE abolished Ca2+-dependent stimulation of TRKB expression, despite the continued presence of TCaRE3 (Fig. 5B). Both TCaRE2 mutations also reduced basal TRKB transcription by 70–80%, similar to what was observed with TCaRE3 mutations (c.f., Fig. 3). The absence of any residual Ca2+-dependent increase further supports the conclusion that TCaRE3 functions at the level of DNA transcription, rather than RNA stability or translation, since any effect of TCaRE3 at the level of TRKB RNA should still be functional, independently of the transcriptional blockade due to mutation of the CREs.
Neurons contain an endogenous transcription factor for TCaRE3
If TCaRE3 functions as a promoter element, DNA sequences containing TCaRE3 should interact with endogenous transcription factors. Nuclear extracts were prepared from cortical neurons that were either left untreated, or depolarized with high KCl for 30 minutes. Nuclear extracts were then subjected to electrophoretic mobility shift assays (EMSAs) with P-32 labeled oligonucleotide sequences containing either wildtype or mutant TCaRE3. The wild type sequence used is indicated in Figure 2 (bold). In the presence of nuclear extract, the wild type TCaRE3-containing probe underwent a mobility shift, resulting in a slower migrating band (arrow, Fig. 6A, lanes 1–2). The shift was observed in extracts generated from both control and depolarized neurons, suggesting that depolarization-induced Ca2+ signaling is not essential for the interaction of TCaRE3 with its binding factor. Mutations that disrupt TCaRE3 function in TRKB-luciferase reporter assays (Fig. 2) inhibited the mobility shift (Fig. 6A, lanes 3–4). Thus, a factor in neuronal nuclei interacts with the TCaRE3 sequence in a sequence-dependent manner.
Figure 6. TCaRE3 interacts with KLF7.
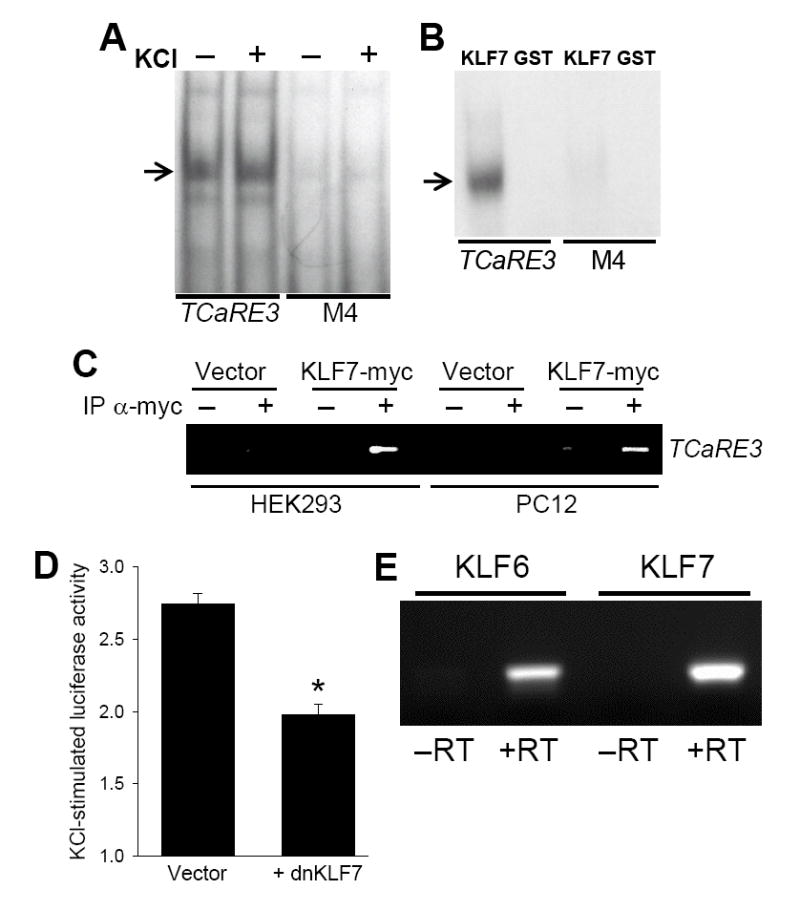
A. Neurons contain a TCaRE3 binding factor. Nuclear extracts from cortical neurons were subjected to electrophoretic mobility shift assays using either wild type or mutant TCaRE3 sequences as probe. A gel shift (arrow) was observed with wild type probe but not with probes with TCaRE3 mutation M4 (c.f., Fig. 2). Depolarization of the neurons prior to extraction did not affect the gel shift. B. Recombinant KLF7 interacts with TCaRE3. GST or GST-KLF7c was purified from E. coli. Binding to TCaRE3 was investigated assayed using EMSAs as described in A. KLF7 caused a shift of wild-type TCaRE3 probe, but not a probe containing an inactivating mutation (M4). No shift was observed with GST alone. C. KLF7 binds chromosomal TRKB promoter. HEK293 cells (lanes 1–4) and PC12 cells (lanes 5–8) were transiently transfected with either empty vector or KLF7-myc. Three days after transfection, cells were crosslinked and subjected to ChIP with anti-myc to precipitate DNA sequences associated with KLF7-myc; ChIP with IgG served as control. Immunoprecipitation of TCaRE3 containing DNA fragments was detected by PCR. D. Dominant-negative KLF7 inhibits depolarization-induced TRKB P2 transcription. Cortical neurons were co-transfected with −944/−325 and either a dominant-negative KLF7 construct or an empty (CMV) vector. Two days after transfection, cells were stimulated by addition of 50 mM KCl for 5 hours prior to harvesting for luciferase assays. Bars show mean ± sem (N=4). Similar results were obtained in two additional experiments. *, significantly different from control, p<.05. E. Mouse cortical neurons express both KLF6 and KLF7 mRNA. PCR using primers specific for KLF6 or KLF7 revealed single band at the predicted sizes. No bands were observed when reverse transcriptase (−RT) was omitted demonstrating that the products were not due to contaminating genomic DNA.
KLF7 binds TCaRE3 in vitro
In order to identify TCaRE3-interacting factors, we performed a yeast one-hybrid (Y1H) screen against a human fetal brain library using a construct containing three tandem copies of TCaRE3 as bait. We identified krüppel-like factor 7 (KLF7) as a factor interacting with TCaRE3 in Y1H screens. Cultured mouse cortical neurons contain KLF7 mRNA as determined by PCR (Fig. 6E; see below). The in vitro interaction of KLF7 with TCaRE3 was investigated using recombinant KLF7. Purified GST-KLF7c, which consists of the DNA binding domain of KLF7 fused to GST (Lei et al., 2001), was examined for its ability to bind TCaRE3 in electrophoretic mobility shift assays (Fig 6B). In the presence of GST-KLF7, the wild type TCaRE3 probe exhibited reduced mobility. GST alone had no effect on probe mobility. Introduction of mutations shown to inactivate TCaRE3 inhibited the appearance of this slower migrating band, indicating that the interaction with GST-KLF7c required intact TCaRE3.
KLF7 can bind to the chromosomal TRKB promoter
The ability of KLF7 to interact with the endogenous TRKB promoter was assayed using chromatin immunoprecipitation (ChIP). Since specific antibodies to KLF7 are not available, HEK293T or PC12 cells were transiently transfected with either KLF7-myc expression vector or empty control vector (c.f., Lei et al., 2005). Three days after transfection, cells were subjected to formaldehyde cross-linking and the nuclear chromatin was harvested and processed for ChIP. Anti-myc antibody was used to immunoprecipitate the KLF7. Following immunoprecipitation, PCR amplification of the region surrounding TCaRE3 was conducted to test for the presence of TRKB P2. In cells transfected with KLF7-myc, anti-myc antibody, but not IgG, immunoprecipitated TCaRE3-containing TRKB chromosomal DNA (Fig. 6C). In contrast, anti-myc-immunoprecipitates from cells transfected with empty vector were not enriched for TRKB P2 promoter DNA. Thus, KLF7 can interact with the chromosomal TRKB promoter.
Dominant-negative KLF7 inhibits Ca2+-dependent TRKB P2 expression
In order to examine the role of KLF7 in regulating TCaRE3, we generated a dominant-negative KLF7 allele (dn-KLF7) by deleting the activation domain of KLF7. The effect of dn-KLF7 on Ca2+-stimulated TRKB P2 expression was analyzed by co-transfecting the dn-KLF7 expression vector with the −944/−325 TRKB-luciferase vector into cortical neurons. Ca2+-stimulated expression of P2 was inhibited by the presence of dn-KLF7 (Fig. 6D). In contrast, co-transfection with empty vector had no effect on TRKB P2 transcription. We did not observe a significant decrease in basal P2-luciferase expression in the presence of dn-KLF7.
KLF7 and KLF6 mRNAs are expressed in cultured mouse cortical neurons
We used RT-PCR to confirm that KLF7 message is expressed in cultured mouse cortical neurons and to test for the presence of the closely-related KLF6. KLF7 and KLF6 are not only highly homologous in their DNA binding domain, but they also share a nearly identical stretch of 47 amino acids sequence at their amino terminus, required for transcriptional activation (Matsumoto et al., 1998). RNA was subjected to reverse-transcription followed by PCR using either KLF7 or KLF6-specific oligonucleotide primers. Both KLF7 and KLF6 transcripts were readily detected (Fig. 6E). The PCR products were absent from samples not treated with reverse transcriptase, demonstrating that the amplification of the two KLF cDNAs was due to the presence of RNA transcripts and not contaminating genomic DNA.
TCaRE3 is a conserved promoter element
Comparison of the genomic sequence of TRKB P2 (−944 to +2) with 1500 bp of the human TRKB (hTRKB; NTRK2) promoter region revealed only five blocks of high sequence conservation from mouse to human. As illustrated in Fig 7A, TCaRE2 (CTGCGTCAgcccTCACGTCA) is located within a block of 60 bp that is identical between mouse and human TRKB. Several additional domains of 46, 99, 37, and 28 bp with 77–93% conservation are also present in the hTRKB promoter region. Within one of these, the sequence we have identified as TCaRE3 is 100% conserved (Fig. 7A). In the mouse and the human promoters, TCaRE2 and TCaRE3 are separated by 545 and 634 bases, respectively. To test the ability of the hTRKB promoter to respond Ca2+, 1100 bp of the human promoter encompassing TCaRE2 and TCaRE3 were cloned into pGL3Basic to generate an hTRKB-luciferase reporter gene. Transient transfection of hTRKB-luciferase into mouse cortical neurons demonstrated that hTRKB could also be stimulated by either depolarization-induced Ca2+ signaling or for-skolin-induced cAMP signaling (Fig 7B). Introduction of the TCaRE3 mutation, M4, inhibited both Ca2+-stimulated and cAMP-stimulated hTRKB-luciferase expression. Interestingly, mutation of TCaRE3 also reduced basal hTRKB-dependent luciferase activity by approximately 80%. Thus, both the sequence and function of TCaRE3 in TRKB expression are conserved in the human gene.
Figure 7. TCaRE3 in the human TRKB promoter.
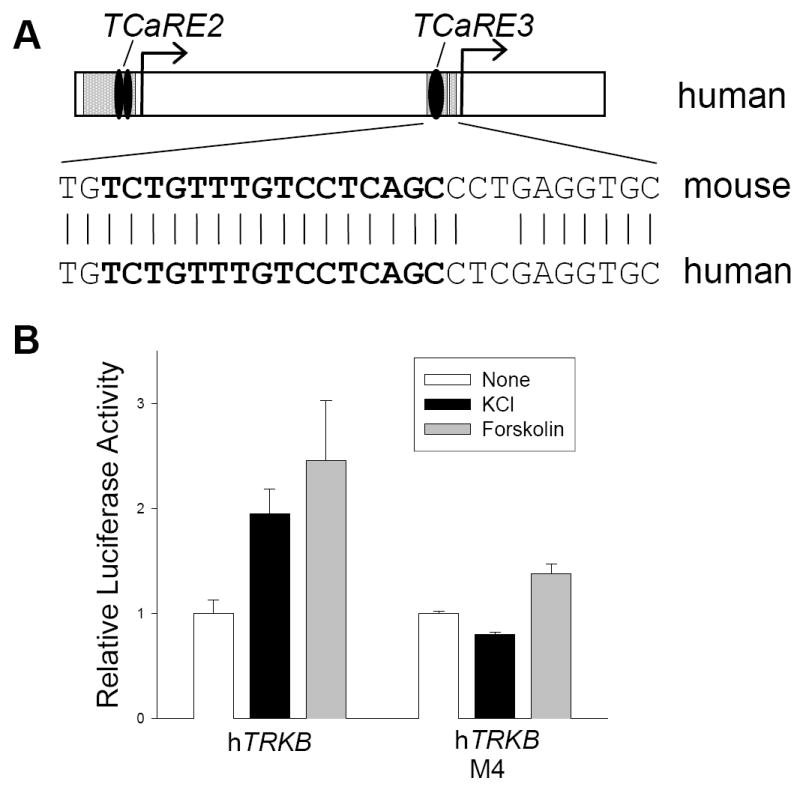
A. Sequence comparison between mouse TRKB P2 (−944/+2) and the human TRKB promoter region revealed varying degrees of sequence homology. Upper panel: The sequences of TCaRE2 and TCaRE3 (black ovals) are identical in the mouse and human promoters and fall within 100- and 35-bp regions of sequence identity (gray). Lower panel: The nucleotide sequence bracketing TCaRE3 is shown for both mouse and human TRKB; identity is indicated by the vertical lines. The TCaRE3 sequence is shown in bold. B. Mouse cortical neurons were transiently trans-fected with human TRKB-luciferase reporter genes containing either wild type or mutated (M4; Fig. 1B) TCaRE3. One day later, cultures were treated with either 50 mM KCl or 50 μM forskolin for 5 hours prior to harvesting for luciferase assays. Bars show mean ± sem (N=4). Similar results were obtained in two additional experiments.
Requirement for TCaRE3 in Ca2+-stimulated TRKB P2 expression is cell type-specific
The requirement for TCaRE3 to enable Ca2+-dependent TRKB P2 expression was tested in several neuronal and non-neuronal cell types. Cells were transiently transfected with either wild type or mutated (M6 c.f., Fig. 2) −944/−325 luciferase reporter constructs and assayed for the ability of depolarization-induced Ca2+ signaling to stimulate reporter activity. Our results revealed that both the responsiveness of TRKB P2 to Ca2+ and the requirement for TCaRE3 to mediate Ca2+-dependent expression are cell-type dependent. The results of these analyses are summarized in Table 1. Depolarization of primary E18 rat cortical neurons and PC12 cells stimulated P2 to the same extent as in primary E16 mouse neurons and this stimulation was abolished by mutation of TCaRE3. Treatment of human embryonic kidney (HEK293) cells with 2 μM ionomycin to elevate [Ca2+], stimulated both wild-type and mutant (M6; c.f., Fig. 2) TRKB P2 expression. In contrast, neither mouse cortical astrocytes nor the rat insulin secreting cell line, RIN-1046, exhibited any stimulation of TRKB P2 in response to ionomycin or KCl, respectively. Interestingly, however, mutation of TCaRE3 reduced basal TRKB P2-luciferase expression in all cell types (Table 1).
Table 1. Cell-type specific regulation of TRKB P2-luciferase.
TRKB P2-luciferase constructs harboring either wild type TCaRE3 sequence or mutated M6 (c.f., Fig. 2) sequence were transiently transfected into cells and assayed for expression following 5 hours stimulation with Ca2+ signaling triggered by either depolarization (neurons, RIN 1046 and PC12 cells) or ionomycin treatment (astrocytes and HEK 293 cells).
| Cell Type | Ca2+-dependent Expression
(+Ca2+/Untreated) |
Basal Expression
(M6/WT) |
|
|---|---|---|---|
| TCaRE3 WT | TCaRE3 M6 | ||
| Cortical neurons (mouse) | 3.0 | 0.5 | 0 .15 |
| Cortical neurons (rat) | 2.5 | 0.7 | 0 .12 |
| Astrocytes (mouse) | 1.0 | 1.0 | 0 .13 |
| PC12 (rat) | 2.0 | 1.0 | 0 .04 |
| RIN 1046 (rat) | 0.5 | 0.5 | 0 .13 |
| HEK 293 (human) | 2.6 | 2.2 | 0 .13 |
DISCUSSION
CREB is a ubiquitous regulator of gene expression, with thousands of mammalian genes containing one or more CREs in their promoters (Zhang et al., 2005). The response of a particular CRE-containing promoter to a Ca2+ or cAMP signal is a reflection of the functional interactions among promoter elements as determined by both developmental and cell-type specific expression of the corresponding transcriptional modulators (Mayr and Montminy, 2001). How the activation of CREB by specific intracellular signals or in different cell types translates to distinct gene activation patterns remains largely unknown and will require the systematic analysis of CREB-dependent activation of many genes in order to identify common themes (Conkright et al., 2003). Although the promoters of a number of CREB-dependent genes, including cFos and BDNF, have been shown to contain additional regulatory elements that impose specificity on CREB-mediated responses, our understanding of the TRKB promoter elements that function to shape the transcriptional response to Ca2+ and cAMP has been limited to the role of the tandem CREs (TCaRE2) (Kingsbury et al., 2003; Deogracias et al., 2004).
Here we report the identification of a novel DNA element, designated TCaRE3, that is required for the CREB-dependent stimulation of TRKB P2 transcription by Ca2+ and cAMP. Mutations of TCaRE3 abolished depolarization-induced P2 reporter gene expression despite the continued presence of the pair of tandem CREs in TCaRE2 (Figs. 1, 2), demonstrating that the tandem CREs (TCaRE2) are not sufficient to promote CREB-dependent activation of TRKB P2 by either Ca2+ (Fig. 1, 2) or cAMP (Fig. 4). Using both EMSA (Fig. 6B) and ChIP (Fig. 6C), we further show that KLF7, which is expressed in the neurons (Fig. 6E), can interact with TCaRE3 and that a dominant-negative allele of KLF7 inhibits Ca2+-dependent TRKB P2 expression (6D), suggesting that KLF7 functionally interacts with CREB to promote TRKB P2 transcription. Although this is the first report implicating KLF7 in Ca2+-dependent transcription, KLF4 has previously been demonstrated to interact with CREB to regulate bradykinin B2 receptor expression during nephron differentiation (Saifudeen et al., 2005), suggesting that there may be functional interactions between CREB and multiple members of the KLF family during development.
KLF7, the mouse homolog of UKLF, belongs to the krüppel-like factor family of transcription factors (Turner and Crossley, 1999; Bieker 2001). KLF7 has been proposed to play a role in neurogenesis due to its early expression in postmitotic neurons during differentiation of the cerebral cortex (Laub et al., 2001). KLF7 has also been shown to regulate the expression of TRKA, the gene coding for the NGF receptor, trkA (Lei et al., 2001; Lei et al., 2005), although this regulation is mediated by a different regulatory element (Ikaros2) in the TRKA promoter. There is no sequence homology between TCaRE3 and Ikaros2, moreover, mutation of a putative Ikaros2 site located in TRKB P2 had no effect on P2 reporter gene expression in our assays (data not shown). Thus, despite differences in mechanism, these results reveal that KLF7 may play a general role in coordinately regulating the expression of different classes of trk receptors in the nervous system.
KLF7 knockout mice have recently been generated and reported to exhibit reduced trkA expression in dorsal root ganglion neurons (Lei et al., 2005); trkB expression levels in brain were not reported. It will be interesting to determine if trkB is altered in the KLF7 knockout brain and whether changes in trkB expression contribute to the reduced cortical thickness and decreased dendritic arborization observed by Laub et al. (2005). Future investigation of the role of KLF7 in regulating trkB expression in vivo will need to take into account the fact that TRKB transcription can be initiated from two distinct promoters (Barettino et al., 1999; Kingsbury et al., 2003) and that the presence of KLF6 may compensate for the loss of KLF7.
A summary of our current understanding of the promoter elements involved in shaping the Ca2+ responsiveness of TRKB P2 is shown in Figure 8. Since TCaRE2 and TCaRE3 are separated by more than 500 bp, the functional interaction we observed indicates that DNA looping would be necessary to promote a physical interaction between their corresponding transcription factors. In addition to TCaRE2 (tandem CREs) and TCaRE3, TRKB P2 contains two other novel elements that can either enhance (TCaRE1, Kingsbury et al., 2003) or repress (TCaRE4; c.f., Fig. 1A) the amplitude of the promoter response, but are not absolutely required for Ca2+- and cAMP-dependent transcription.
Mutation of either TCaRE3 or the TBP site dramatically suppressed basal TRKB P2 expression (Fig. 3). TCaRE2 mutations also suppressed basal transcription. This finding is not unique; a role for CREs in basal transcriptional activity was previously reported (Quinn et al., 1995). One possible explanation for these findings is that CREB, acting at TCaRE2, KLF7, acting at TCaRE3, and TBP all contribute to the formation of the basal TRKB P2 transcription initiation complex via Ca2+-independent mechanisms, while CREB-KLF7 interactions, but not TBP, are required for Ca2+-dependent transcription. An alternative explanation is that resting Ca2+ levels in unstimulated neurons may be sufficient to partially activate TRKB expression. In either case, the requirement for TCaRE3 for basal TRKB transcription may explain triiodo-thyronine (T3)-dependent inhibition of TRKB reported in rat neuroblastoma cells (Pombo et al., 2000). TCaRE3 encompasses one of four potential T3 half sites (TGTCCT) required for T3-dependent inhibition TRKB; activation of T3 receptors may inhibit TRKB expression by displacing KLF7.
The requirement for interactions among promoter elements provides opportunities for the integration of multiple intracellular signaling pathways and allows diversity in cellular responses to identical stimuli (Maniatis et al., 1987). The importance of the functional interaction between the CREs, located within TCaRE2, and TCaRE3 is highlighted by the fact that, despite only limited domains of homology between the mouse and human TRKB promoters, the sequences of TCaRE2 and TCaRE3 are identical in the two species (Fig. 7A). Mutation of TCaRE3 in the human promoter abolished Ca2+-stimulated hTRKB transcription and caused similar reductions in basal activity (Fig. 7B), demonstrating that the role of TCaRE3 in TRKB expression is conserved in the human gene. This observation suggests that the interaction of CREB with a TCaRE3 transcription factor such as KLF7 (or KLF6) is essential for the proper regulation of TRKB expression in neurons. Cell-specific regulation of TCaRE3, as could occur via expression of distinct KLF family members, may underlie the range of TRKB P2 responses reported in Table 1, which include the failure of Ca2+ to activate TRKB P2 in astrocytes and the activation of P2 by Ca2+ signaling in HEK cells even in the absence of TCaRE3.
EXPERIMENTAL METHODS
Cell culture and Transfection
Cortical neuron cultures were isolated from embryonic day 16 (E16) mouse embryos and plated on laminin-coated dishes at a density of 5-6 × 105/well in 24-well plates for luciferase assays or 1.5 × 107 cells/100-mm dish for nuclear extract preparation. Neuron cultures were maintained in Neurobasal™ medium supplemented with 2% B27 (Invitrogen, Carlsbad, CA), 1 mM glutamine, and penicillin/streptomycin and incubated in 5% CO2 at 37°C. Neurons were transiently transfected 3–5 days after plating using a Ca2+-phosphate protocol (Xia et al, 1996). Typical transfection rates with this protocol range from 5-10% of the neurons.
Luciferase Assays
Luciferase assays were conducted as previously described (Kingsbury, et al., 2003). Individual luciferase measurements were normalized to co-transfected TK-Renilla values to correct for sample-to-sample variation in transfection efficiency. One day after transfection with reporter constructs, cells were stimulated for 5 hours by the addition of 50 mM KCl, 40 μM forskolin or 2 μM ionomycin (Sigma, St. Louis, MO). When indicated, 40 μM actD (Sigma) was added 30 min prior to stimulation.
Plasmids
The TRKB P2 −944/+2 luciferase reporter was previously described (Kingsbury, et al., 2003). Constructs −944/−77 and −944/−325 were generated by digesting −944/+2 with HindIII/StuI or HindIII/EV, respectively, treating with Klenow DNA polymerase to generate blunt ends and then re-ligating. All other truncations were produced by introduction of a HindIII site using QuickChange site-directed mutagenesis (Stratagene, La Jolla, CA) followed by HindIII digestion and re-ligation. The sequences of sense strand primers used were: −162: GGATCCTGAGCAGGTTG;−241: GGACATAGTCCTATAC; −339: GTCCTCAGCCCTGAAGCTTGCACCGTATCGATAT; 389: TCCCAGTTGGTGAAGCTTGGGGTCTATTAGTC; 444: GGATCCAGAAGAATGTCTGAGTG Scanning mutagenesis of −944/−325 was also conducted using Quickchange site-directed mutagenesis. The corresponding sense oligonucleotide sequences were: M1: CTATTAGTCTCATCCAAGCTTGTTTGTCCTCAGC; M2: TCTATTAGTCTCATCGAATTCTGTTTGTCCTCAGC; M3: ATTAGTCTCATCCATGCCGGTTTGTC-CTCA-GCCC; M4: ATTAGTCTCATCCATGTAAGCTTGTCCTCAGCCCTG; M5: TCATCCATGTCTGTTAAGCTTCAGCCCTGAGGTGCG; M6: ATCCATGTCTGTTTGTCCGGAGCCCTGAGGTGCG; M7: ATGTCTGTTTGTCCTCCGGCCTGAGGTGCGCAC; M8: GTCTGTTTGTCCTCAGATCTGAGGTGCGCACC; M9: TGTTTGTCCTCAGCCGGGAGGTGCGCACCGAT; TBP Mutation: GTGTGTGGGGTCTAAGCTTCTCATCCATGTCTG. A dominant-negative KLF7 allele was generated by amplifying the DNA-binding domain using the primers AAGCTTCTAGACCCCTTGCT and CTCGAGTTAGATATGTCTCTT and cloning into pcDNA3.1 EcoR1 to XhoI.
Electrophoretic Mobility Shift Assays (EMSAs)
Nuclear cortical neuron extracts were generated and EMSA conducted as previously described (Andrews and Faller, 1991). GST or GST-KLF7c were purified from E. coli. Single stranded oliogonucleotides (−339 to −361 with and without mutations) were radiolabeled with P-32 using T4 kinase (Invitrogen) prior to annealing. Binding reactions were incubated at room temperature for 20 min prior to separation on a 6% TBE gel and autoradiography.
Yeast One-Hybrid (Y1H) Assay
Yeast one-hybrid assay was conducted using Matchmaker One-hybrid system from Clontech according to manufacturer’s instructions. Three tandem copies of TCaRE3 were synthesized by Integrated DNA Technologies, Inc. (Coralville, IA): GGATCCGAATTCGTTTGTCCTCAGCCCTGAGGTGCGGTTTGTCCTCAGCCCTGAGGTGCGGTTTGTCCTCAGCCCTGAGGTG CGTCTAGATCT and cloned into target vectors using EcoRI and XbaI. Yeast were screened for His+ colonies following transformation with human fetal brain library (Clontech).
KLF7 and KLF6 PCR
KLF7 and KLF6 mRNAs were detected by reverse transcriptase-PCR using the following primer pairs: KLF7: AAGAGGGTCCATCGCTGTCAGTTT and ACCTGTGTGTTTCCTGTAGTGCCT; KLF6: TTTCTGCTCGGACTCCTGATCGTT and AAGATAGCGTTCCAACTCCAGGCA. When reverse transcriptase was omitted from the reaction (−RT), no PCR product was observed demonstrating that the assay was not detecting genomic DNA.
Chromatin Immunoprecipitation (ChIP)
The interaction between epitope-tagged KLF7 and endogenous TCaRE3 was examined as described by Lei et al. (2005) for the KLF7-Ikaros2 interaction. HEK-293T or PC12 cells were transfected with either KLF7-myc or empty vector using Lipofectamine 2000 three days prior to harvesting for ChIP assays. Cells were cross-linked with formaldehyde and chromatin extracted from isolated nuclei was digested with mung bean nuclease to an average size of approximately 300 base pairs. Immunoprecipitations were conducted using a kit from Upstate Biotechnology (Lake Placid, NY), with Protein G-agarose (Upstate) substituted for Protein A-agarose. Anti-myc (9E10) was purchased from Roche Applied Science (Indianapolis, IN). Reverse crosslinking was conducted overnight at 65°C. DNA was extracted with phenol-chloroform, ethanol-precipitated and re-suspended in water for PCR analysis. Oligonucleotide primers for PCR of the TCaRE3 domain were: GCTCAGTC-TTACGCGTGTCTGTTT and CGGCATACGAACCGCTTTACA.
Acknowledgments
We thank L.L. Bambrick, S.G. Dorsey, D.D. Ginty and P.D. Murray for helpful discussions. We thank C.D. Roby and S. Connolly for technical support. Funding provided by grants NIH R01NS40492, NIH R01NS048095 and DOD W81XWH-04-1-0176 (BKK) and by NIH training grant T32 NS07375 (TJK). TJK is a BIRCWH Scholar supported by NICHD/ORWH/NIDDK (NICHD K12 HD43489).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allen SJ, Wilcock GK, Dawbarn D. Profound and selective loss of catalytic TrkB immunoreactivity in Alzheimer’s disease. Biochem Biophys Res Comm. 1999;264:648–651. doi: 10.1006/bbrc.1999.1561. [DOI] [PubMed] [Google Scholar]
- Andrews NC, Faller DV. A rapid micropreparation technique for extraction of DNA-binding proteins from limiting numbers of mammalian cells. Nucleic Acids Res. 1991;19:2499. doi: 10.1093/nar/19.9.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelucci F, Brene S, Mathe AA. BDNF in schizophrenia, depression and corresponding animal models. Mol Psychiatry. 2005;10:345–52. doi: 10.1038/sj.mp.4001637. [DOI] [PubMed] [Google Scholar]
- Barettino D, Pombo PMG, Espliguero G, Rodriguez-Pena A. The mouse neurotrophin receptor trkB gene is transcribed from two different promoters. Biochim Biophys Acta. 1999;1446:24–34. doi: 10.1016/s0167-4781(99)00056-1. [DOI] [PubMed] [Google Scholar]
- Bieker JJ. Kruppel-like factors: three fingers in many pies. J Biol Chem. 2001;276:34355–34358. doi: 10.1074/jbc.R100043200. [DOI] [PubMed] [Google Scholar]
- Chen WG, West AE, Tao X, Corfas G, Szentirmay MN, Sawadogo M, Vinson C, Greenberg ME. Upstream stimulatory factors are mediators of Ca2+-responsive transcription in neurons. J Neurosci. 2003;23:2572–2581. doi: 10.1523/JNEUROSCI.23-07-02572.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conkright MD, Guzman E, Flechner L, Su AI, Hogenesch JB, Montminy M. Genome-wide analysis of CREB target genes reveals a core promoter requirement for cAMP responsiveness. Mol Cell. 2003;11:1101–1108. doi: 10.1016/s1097-2765(03)00134-5. [DOI] [PubMed] [Google Scholar]
- Deogracias R, Espliguero G, Iglesias T, Rodriguez-Pena A. Expression of the neurotrophin receptor trkB is regulated by the cAMP/CREB pathway in neurons. Mol Cell Neurosci. 2004;26:470–480. doi: 10.1016/j.mcn.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Marin C, Rey MJ, Ribalta T, Goutan E, Blanco R, Tolosa E, Marti E. BDNF and full-length and truncated TrkB expression in Alzheimer disease. Implications in therapeutic strategies. J Neuropathol Exp Neurol. 1999;58:729–739. doi: 10.1097/00005072-199907000-00007. [DOI] [PubMed] [Google Scholar]
- Fumagalli F, Racagni G, Riva MA. The expanding role of BDNF: a therapeutic target for Alzheimer’s disease? Pharmacogenomics Journal. 2006;6:8–15. doi: 10.1038/sj.tpj.6500337. [DOI] [PubMed] [Google Scholar]
- Fumagalli F, Racagni G, Riva MA. Shedding light into the role of BDNF in the pharmacotherapy of Parkinson’s disease. Pharmacogenomics Journal. 2006;6:95–104. doi: 10.1038/sj.tpj.6500360. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Carnahan J, Greenberg ME. Requirement for BDNF in activity-dependent survival of cortical neurons. Science. 1994;263:1618–1623. doi: 10.1126/science.7907431. [DOI] [PubMed] [Google Scholar]
- Hashimoto K, Shimizu E, Iyo M. Critical role of brain-derived neurotrophic factor in mood disorders. Brain Res Brain Res Rev. 2004;45:104–114. doi: 10.1016/j.brainresrev.2004.02.003. [DOI] [PubMed] [Google Scholar]
- Holsinger RMD, Schnarr J, Henry P, Castelo VT, Fahnestock M. Quantitation of BDNF mRNA in human parietal cortex by competitive reverse transcription-polymerase chain reaction: decreased levels in Alzheimer’s disease. Mol Brain Res. 2000;76:347–354. doi: 10.1016/s0169-328x(00)00023-1. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci. 2001;24:677–736. doi: 10.1146/annurev.neuro.24.1.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Ann Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Kingsbury TJ, Murray PD, Bambrick LL, Krueger BK. Ca2+-dependent regulation of trkB expression in neurons. J Biol Chem. 2003;278:40744–40748. doi: 10.1074/jbc.M303082200. [DOI] [PubMed] [Google Scholar]
- Korte M, Griesbeck O, Gravel C, Carroll P, Staiger V, Thoenen H, Bonhoeffer T. Virus-mediated gene transfer into hippocampal CA1 region restores long-term potentiation in brain-derived neurotrophic factor mutant mice. Proc Natl Acad Sci USA. 1996;93:12547–12552. doi: 10.1073/pnas.93.22.12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laub F, Aldabe R, Friedrich V, Jr, Ohnishi S, Yoshida T, Ramirez F. Developmental expression of mouse Krüppel-like transcription factor KLF7 suggests a potential role in neurogenesis. Dev Biol. 2001;233:305–318. doi: 10.1006/dbio.2001.0243. [DOI] [PubMed] [Google Scholar]
- Laub F, Lei L, Sumiyoshi H, Kajimura D, Dragomir C, Smaldone S, Puche AC, Petros TJ, Mason C, Parada LF, Ramirez F. Transcription factor KLF7 is important for neuronal morphogenesis in selected regions of the nervous system. Mol Cell Biol. 2005;25:5699–5711. doi: 10.1128/MCB.25.13.5699-5711.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TI, Young RA. Regulation of gene expression by TBP-associated proteins. Genes & Dev. 1998;12:1398–1408. doi: 10.1101/gad.12.10.1398. [DOI] [PubMed] [Google Scholar]
- Lei L, Ma L, Nef S, Thai T, Parada LF. mKlf7, a potential transcriptional regulator of TrkA nerve growth factor receptor expression in sensory and sympathetic neurons. Development. 2001;128:1147–1158. doi: 10.1242/dev.128.7.1147. [DOI] [PubMed] [Google Scholar]
- Lei L, Laub F, Lush M, Romero M, Zhou J, Luikart B, Klesse L, Ramirez F, Parada LF. The zinc finger transcription factor Klf7 is required for TrkA gene expression and development of nociceptive sensory neurons. Genes and Dev. 2005;19:1354–1364. doi: 10.1101/gad.1227705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35:605–623. doi: 10.1016/s0896-6273(02)00828-0. [DOI] [PubMed] [Google Scholar]
- Maniatis T, Goodbourn S, Fischer JA. Regulation of inducible and tissue-specific gene expression. Science. 1987;236:1237–1245. doi: 10.1126/science.3296191. [DOI] [PubMed] [Google Scholar]
- Matsumoto N, Laub F, Aldabe R, Zhang W, Ramirez F, Yoshida T, Terada M. Cloning the cDNA for a new human zinc finger protein defines a group of closely related Kruppel-like transcription factors. J Biol Chem. 1998;273:28229–28237. doi: 10.1074/jbc.273.43.28229. [DOI] [PubMed] [Google Scholar]
- Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nature Rev Mol Cell Biol. 2001;2:600–609. doi: 10.1038/35085068. [DOI] [PubMed] [Google Scholar]
- McAllister AK, Katz LC, Lo DC. Neurotrophins and synaptic plasticity. Annu Rev Neurosci. 1999;22:295–318. doi: 10.1146/annurev.neuro.22.1.295. [DOI] [PubMed] [Google Scholar]
- Murer MG, Yan Q, Raisman-Vozari R. Brain-derived neurotrophic factor in the control human brain, and in Alzheimer’s disease and Parkinson’s disease. Prog Neurobiol. 2001;63:71–124. doi: 10.1016/s0301-0082(00)00014-9. [DOI] [PubMed] [Google Scholar]
- Nagappan G, Lu B. Activity-dependent modulation of the BDNF receptor TrkB: mechanisms and implications. Trends Neurosci. 2005;28:464–471. doi: 10.1016/j.tins.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM. Neurobiology of depression. Neuron. 2002;34:13–25. doi: 10.1016/s0896-6273(02)00653-0. [DOI] [PubMed] [Google Scholar]
- Patterson SL, Abel T, Deuel TAS, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- Pombo PMG, Barettino D, Espliguero G, Metsis M, Iglesias T, Rodriguez-Pena A. Transcriptional repression of neurotrophin receptor trkB by thyroid hormone in the developing rat brain. J Biol Chem. 2000;275:37510–37517. doi: 10.1074/jbc.M006440200. [DOI] [PubMed] [Google Scholar]
- Quinn PG, Wong TW, Magnuson MA, Shabb JB, Granner DK. Identification of basal and cyclic AMP regulatory elements in the promoter of the phosphoenolpyruvate carboxykinase gene. Mol Cell Biol. 1988;8:3467–3475. doi: 10.1128/mcb.8.8.3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson LM, Kerppola TK, Vendrell M, Luk D, Smeyne RJ, Bocchiaro C, Morgan JI, Curran T. Regulation of c-fos expression in transgenic mice requires multiple interdependent transcription control elements. Neuron. 1995;14:241–252. doi: 10.1016/0896-6273(95)90282-1. [DOI] [PubMed] [Google Scholar]
- Saifudeen Z, Dipp S, Fan H, El-Dahr SS. Combinatorial control of the bradykinin B2 receptor promoter by p53, CREB, KLF-4, and CBP: implications for terminal nephron differentiation. Am J Physiol Renal Physiol. 2005;288:F899–909. doi: 10.1152/ajprenal.00370.2004. [DOI] [PubMed] [Google Scholar]
- Shieh PB, Hu S-C, Bobb K, Timmusk T, Ghosh A. Identification of a signaling pathway involved in calcium regulation of BDNF expression. Neuron. 1998;20:727–740. doi: 10.1016/s0896-6273(00)81011-9. [DOI] [PubMed] [Google Scholar]
- Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron. 1998;20:709–726. doi: 10.1016/s0896-6273(00)81010-7. [DOI] [PubMed] [Google Scholar]
- Turner J, Crossley M. Mammalian krüppel-like transcription factors: more than just a pretty finger. Trends Biochem Sci. 1999;24:236–240. doi: 10.1016/s0968-0004(99)01406-1. [DOI] [PubMed] [Google Scholar]
- West AE, Chen WG, Dalva MB, Dolmetsch RE, Kornhauser JM, Shaywitz AJ, Takasu MA, Tao X, Greenberg ME. Calcium regulation of neuronal gene expression. Proc Natl Acad Sci USA. 2001;98:11024–11031. doi: 10.1073/pnas.191352298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z, Dudek H, Miranti CK, Greenberg ME. Calcium influx via the NMDA receptor induces immediate early gene transcription by a MAP kinase/ERK-dependent mechanism. J Neurosci. 1996;16:5425–5436. doi: 10.1523/JNEUROSCI.16-17-05425.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, Gottschalk W, Chow A, Wilson RI, Schnell E, Zang K, Wang D, Nicoll RA, Lu B, Reichardt LF. The role of brain-derived neurotrophic factor receptors in the mature hippocampus: modulation of long-term potentiation through a presynaptic mechanism involving TrkB. J Neurosci. 2000;20:6888–6897. doi: 10.1523/JNEUROSCI.20-18-06888.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Odom DT, Koo S-H, Conkright MD, Canettieri G, Best J, Chen H, Jenner R, Herbolsheimer E, Jacobsen E, Kadam S, Ecker JR, Emerson B, Hogenesch JB, Unterman T, Young RA, Montminy M. Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc Natl Acad Sci USA. 2005;102:4459–4464. doi: 10.1073/pnas.0501076102. [DOI] [PMC free article] [PubMed] [Google Scholar]