

Abstract
Protein aggregation into amyloid fibrils and protofibrillar aggregates is associated with a number of the most common neurodegenerative diseases. We have established, using a computational approach, that knowledge of the primary sequences of proteins is sufficient to predict their in vitro aggregation propensities. Here we demonstrate, using rational mutagenesis of the Aβ42 peptide based on such computational predictions of aggregation propensity, the existence of a strong correlation between the propensity of Aβ42 to form protofibrils and its effect on neuronal dysfunction and degeneration in a Drosophila model of Alzheimer disease. Our findings provide a quantitative description of the molecular basis for the pathogenicity of Aβ and link directly and systematically the intrinsic properties of biomolecules, predicted in silico and confirmed in vitro, to pathogenic events taking place in a living organism.
Author Summary
A wide range of diseases, including diabetes and common brain diseases of old age, are characterised by the deposition of protein in the affected tissues. Alzheimer disease, the most common neurodegenerative disorder, is caused by the aggregation and deposition of a peptide called Aβ in the brain. We have previously developed a computational procedure that predicts a particular peptide or protein's speed of aggregation in the test tube. Our goal was to test whether the speed of aggregate formation that we observe in the test tube is directly linked to the brain toxicity that we see in our fruit fly model of Alzheimer disease. We made flies that produce each of 17 variant forms of Aβ and show that the toxicity of each variant is closely linked to the tendency of each variant to form small soluble aggregates. Our computational procedure has previously been shown to be applicable to a wide range of different proteins and diseases, and so this demonstration that it can predict toxicity in an animal model system has implications for many areas of disease-related research.
A systematic analysis of Alzheimer disease amyloid β peptide variants inDrosophila brain demonstrates that their predicted propensity to form protofibrillar aggregates correlates best with toxicity.
Introduction
A wide range of proteins has been found to convert into extracellular amyloid fibrils, or amyloid-like intracellular inclusions, under physiological conditions [1,2]. Such proteins have largely been identified through their association with disease, although a number have been found to have beneficial physiological functions in organisms including, amongst others, bacteria [3], yeast [4], and humans [5]. Indeed, the ability to aggregate and assemble into amyloid-like fibrils has emerged as a common, and perhaps fundamental, property of polypeptide chains [1,6,7]. This discovery has stimulated extensive biophysical and mutational analysis of the underlying molecular determinants of amyloid fibril formation. These studies have resulted in the derivation of general models, based on physicochemical parameters, that both rationalise and predict the propensity of proteins to convert from their soluble forms into intractable amyloid aggregates in vitro [8–10].
The misfolding and aggregation of proteins in vivo, however, differ from similar processes taking place under in vitro experimental conditions, in that they occur in complex cellular environments containing a host of factors that are known to modulate protein aggregation and protect against any subsequent toxicity [11]. This difference between in vitro and in vivo experimental conditions represents a significant barrier to the development of a molecular understanding of protein aggregation in living systems and its consequences for disease. In this paper we describe the results of an approach designed to bridge this divide by expressing a range of mutational variants of Aβ42 in a Drosophila model of Alzheimer disease [12] and correlating their influence on the longevity and behaviour of the flies with their underlying physicochemical characteristics.
Results/Discussion
The expression of the Aβ42 peptide (coupled to a secretion signal peptide) in the central nervous system of Drosophila melanogaster results in both intracellular and extracellular deposition of Aβ42, along with neuronal dysfunction, revealed by abnormal locomotor behaviour and reduced longevity [12–14]. Learning and memory deficits are also observed in flies expressing Aβ42 and to a lesser extent in those expressing Aβ40. Importantly, the severity of the cognitive deficits is closely correlated with the magnitude of the locomotor and longevity phenotypes [14]. Our system, as with other recently developed invertebrate models of neurodegenerative disease, therefore produces clear, quantitative phenotypes that allow rapid and statistically robust assessments of the effects of mutations [15,16]. Using an algorithm described previously [8,10] we computed the intrinsic aggregation propensities (Z agg) of all 798 possible single point mutations of the Aβ42 peptide and also of the more toxic E22G Aβ42 peptide. A total of 17 mutational variants, with a wide range of aggregation propensities (Table 1), were then expressed throughout the central nervous system of Drosophila melanogaster, and their effects were compared to those of wild-type (WT) and E22G Aβ42 expression. The longevity of multiple lines of flies (n = 4–6 independent lines) for each variant was compared to that of flies expressing the WT or E22G Aβ42 peptide. This pooling of data from multiple independent lines for each Aβ42 mutant studied serves as a control for the potential variation in expression levels between transgene insertion sites. In addition, the locomotor ability of a representative selection of the Aβ42-variant-expressing flies was assessed to provide a measure of the early effects of the peptides on neuronal dysfunction. Examples of the results of this analysis are shown for four of the variants studied (Figure 1).
Table 1.
Predicted Aggregation Propensity and In Vivo Toxicity of the Aβ Variants Studied
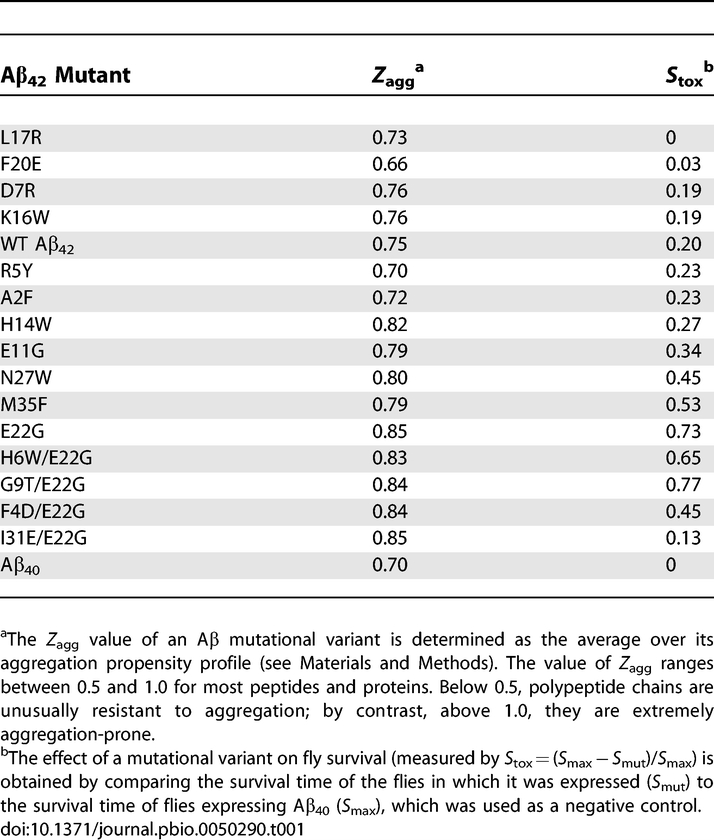
Figure 1. Correlation between Predicted Aggregation Propensity and In Vivo Effects of Aβ42 Mutants.

(A) Flies expressing F20E Aβ42 (blue line) live significantly longer (median survival 29 ± 1 d, n = 400, p < 0.0001) than flies expressing WT Aβ42 (red line) (median survival 24 ± 1 d, n = 100).
(B) Flies expressing I31E/E22G Aβ42 (blue line) show a dramatic increase in longevity (median survival = 27 ± 1 d, n = 600, p < 0.0001) compared to flies expressing E22G Aβ42 (red line) (median survival = 8 ± 1 d, n = 100).
(C) Flies expressing the F20E Aβ42 peptide (blue squares) have significantly improved locomotor ability (p < 0.001, n = 90 observations per line per time point) compared with flies expressing the WT Aβ42 peptide (red circles).
(D) Flies expressing the I31E/E22G Aβ42 peptide (blue squares) have significantly improved locomotor ability (p < 0.001, n = 90 observations per line per time point) compared to flies expressing E22G Aβ42 (red circles).
(E) Flies expressing E11G Aβ42 (green line) die significantly quicker (median survival 21 ± 1, n = 500, p < 0.0001) than those expressing WT Aβ42 (red line) (median survival 26 ± 1 d, n = 100).
(F) Flies expressing M35F Aβ42 (green line) die significantly more quickly (median survival = 15 ± 1, n = 500, p < 0.0001) than those expressing WT Aβ42 (red line) (median survival 26 ± 1 d, n = 100). Larger values of S tox indicate higher toxicity
Flies expressing the WT Aβ42 peptide have a median survival of 24 ± 1 d; flies expressing the E22G Aβ42 peptide associated with familial Alzheimer disease have a median survival of only 8 ± 1 d. In contrast, some of the peptide variants are less harmful. For example, flies expressing F20E Aβ42 have a median survival of 29 ± 1 d (Figure 1A), and flies expressing I31E/E22G Aβ42 peptide have a median survival of 27 ± 1 d (Figure 1B), representing substantial increases in longevity compared to WT and E22G Aβ42 flies. Furthermore, the longevity of these variants is comparable to that of flies expressing the Aβ40 peptide (median survival = 30 ± 1 d; Figure S1A and S1B), which has been previously shown to be non-toxic when expressed both in transgenic flies [12,13] and in transgenic mice [17]. F20E Aβ42 and I31E/E22G Aβ42 flies also have very significantly improved locomotor ability compared to WT and E22G Aβ42 flies (Figure 1C and 1D; Videos S1 and S2) and are comparable in locomotor performance to flies expressing the Aβ40 peptide (Figure S1C and S1D). We also analysed a range of Aβ42 variants that were more harmful than the WT peptide; for example, flies expressing the E11G or M35F variants of the Aβ42 peptide have significantly shorter lifespans than WT Aβ42 flies (median survival = 21 ± 1 and 15 ± 1 d, respectively; Figure 1E and 1F).
Quantitative analysis of all 17 Aβ variants studied reveals a highly statistically significant correlation between the propensity of a variant to aggregate (Z agg) and its effect on the survival of the flies (S tox) (Figure 2A; r = 0.75, p = 0.001). A significant correlation is also observed when we analyse the relationship between the predicted aggregation propensity (Z agg) of a representative selection of Aβ variants and their effects on mobility or locomotor performance (M tox) (Figure 2B; r = 0.65, p = 0.009). We have also verified that correlations exist between the measured aggregation rates (K agg) and both S tox and M tox for a representative selection of the Aβ42 variants, as we would expect from our predictions (Figure 3).
Figure 2. Correlation between In Vivo Toxicity and Aggregation Propensity (Z agg).

(A) There is a significant correlation between the propensities of the Aβ42 variants to aggregate (Z agg) and the relative survival of the flies (S tox; r = 0.75, p = 0.001).
(B) There is a similarly significant correlation between the propensities of Aβ42 variants to aggregate (Z agg) and the locomotor abilities of the flies (M tox; r = 0.65, p = 0.009).
In both panels the errors in the in vivo measurements (y-axis) are standard errors of the mean arising from the average of the independent lines tested for each variant. The errors in the predictions of aggregation propensity (Z agg) are also shown (x-axis).
Figure 3. Correlation between Measured Aggregation Rate (K agg) and both Longevity (S tox) and Locomotor Performance (M tox).

There is a significant correlation between neuronal dysfunction measured both by longevity (A) (S tox; r = 0.79, p = 0.017) and mobility (B) (M tox; r = 0.73, p = 0.03) and the rate of aggregation (K agg) in vitro (measured by thioflavin T fluorescence).
Whilst our analysis reveals a significant relationship between the aggregation propensity of Aβ42 and its effects on neuronal integrity in vivo, it has also uncovered a small number of variants that do not conform to this trend, most notably the I31E/E22G Aβ42 peptide. In order to determine the significance of such divergent behaviour for the origins of Aβ42 pathogenicity, we selected one peptide whose effects on the longevity and mobility of the flies is well predicted by its Z agg (F20E) and one whose effects did not correlate with its Z agg (I31E/E22G) and performed further analysis of their aggregation in vitro and in vivo.
The F20E mutation is predicted to reduce significantly the propensity of the Aβ42 peptide to aggregate (Table 1). Indeed, when we measure the rate of aggregation using thioflavin T fluorescence we find that F20E Aβ42 does aggregate significantly more slowly in vitro than the WT Aβ42 peptide (t 1/2 = 44 and 11 min, respectively; Figure 4A), in good accord with our predictions.
Figure 4. In Vitro and In Vivo Biochemical Analysis of F20E and I31E/E22G Aβ42 .

(A) F20E Aβ42 (blue squares) aggregates more slowly than WT Aβ42 (red circles), and both were found to have formed well-defined fibrils at the end point of this assay (Figure S3).
(B) Immunohistochemistry shows extensive Aβ42 deposition (brown staining) in the brain of WT-Aβ42--expressing flies at 20 d of age (arrows).
(C) In contrast, F20E Aβ42 flies show no evidence of Aβ42 deposition at 20 d of age.
(D) Both E22G (red circles) and I31E/E22G (blue squares) Aβ42 aggregate at similar rates as measured by Thioflavin T fluorescence.
(E) Aβ42 immunohistochemistry shows deposition throughout the cortex in the brain of E22G-Aβ42-expressing flies at 8 d of age (arrows). This deposition is also associated with the appearance of vacuoles (asterisks).
(F) Flies expressing I31E/E22G Aβ42 show extensive deposition of Aβ42 throughout their cortex (arrows). In contrast to (E), no evidence of neurodegeneration (vacuolation) is seen.
The in vivo aggregation of the F20E Aβ42 peptide is also significantly reduced compared to that of the WT Aβ42 peptide. Anti-Aβ42 immunohistochemistry using a C-terminal-specific antibody that binds an epitope (Aβ residues 35–42) [18] that does not include the residues being studied here, reveals progressive accumulation of Aβ42 in the brains of WT-Aβ42-expressing flies from 10 d of age, with extensive deposition being evident by day 20 (Figure 4B). In contrast to this behaviour, flies expressing F20E Aβ42 show no signs of Aβ42 deposition at day 20 (Figure 4C). Quantitative reverse transcription polymerase chain reaction (qRT-PCR) analysis of Aβ42 transcription levels was also carried out on WT Aβ42 and two independent lines of F20E Aβ42 fly brains to ensure that the reduced deposition and toxicity of the F20E Aβ42 peptide was not due to coincidentally lower transcription levels. In fact, the F20E Aβ42 transgene was transcribed at slightly higher levels than WT Aβ42 (Figure S2) in both lines tested.
That the F20E Aβ42 peptide does not form in vivo deposits, despite being able to form amyloid fibrils in vitro (albeit significantly more slowly than WT Aβ42) suggests that the F20E mutation reduces the aggregation propensity of Aβ42 sufficiently to allow cellular clearance mechanisms such as proteases (e.g., neprilysin) [13] to prevent its accumulation in vivo. We conclude, therefore, that the increased longevity and locomotor performance of F20E Aβ42 flies are indeed attributable to a measurable reduction in the aggregation propensity of this peptide in vivo, as predicted by our analysis.
In the case of the I31E/E22G Aβ42 variant there appears to be no correlation between its predicted aggregation propensity (which is very similar to that of the highly pathogenic E22G Aβ42 peptide; Table 1) and its effects on longevity and locomotor behaviour in the fly (Figure 1B and 1D). However, studies of the I31E/E22G and E22G Aβ42 peptides in vitro show that, as predicted by our algorithm, they aggregate at very similar rates (t 1/2 = 7 and 4 min, respectively; Figure 4D). Furthermore, anti-Aβ42 immunohistochemistry reveals similar levels of deposition in the brains of both E22G- and I31E/E22G-Aβ42-expressing flies at 8 d of age (Figure 4E and 4F) that cannot be accounted for by variations in transcription level as measured by qRT-PCR (Figure S2). Together these observations confirm that our predictions of aggregation propensity are accurate for these peptides in vivo as well as in vitro. To determine the consequences of peptide deposition on the integrity of the brain, we looked for the presence of vacuoles, which are a well-documented sign of neurodegeneration [19]. Despite comparable levels of deposition, the vacuoles seen in the brains of E22G-Aβ42-expressing flies are entirely absent from the brains of I31E/E22G-Aβ42-expressing flies. In this case, therefore, the relationship between the presence of Aβ42 deposits and the functional and anatomical integrity of the brain does not appear to hold.
This observation is reminiscent of the finding that there are cases in which the presence of Aβ plaques in the brains of elderly humans, and indeed in transgenic mouse models of Alzheimer disease, does not correlate with cognitive ability [20,21]. It has been proposed, in explanation of this finding, that the neuronal dysfunction and degeneration historically attributed to the presence of Aβ amyloid fibrils in the brains of patients with Alzheimer disease may in fact be caused by the concomitant presence of prefibrillar aggregates [22–24]. With this in mind, the unexpected in vivo effects of variants such as the I31E/E22G Aβ42 peptide prompted us to develop a second algorithm (see Materials and Methods) by analysing a set of data for which the rates of formation of protofibrils containing β-sheet structure have been reported [8]. This algorithm is able to predict the propensity of other polypeptides to form protofibrils. Whilst there are a few Aβ42 variants (including I31E/E22G Aβ42) whose global aggregation propensities (Zagg) do not correlate well with their in vivo effects on neuronal dysfunction (Figure 2), we find that the predicted propensities of these variants to form protofibrillar aggregates (Z tox) correlate very strongly with their in vivo effects (S tox, r = 0.83, p < 0.00001; M tox, r = 0.75, p = 0.001; Figure 5).
Figure 5. Propensity to Form Protofibrillar Aggregates (Z tox) as a Predictor of the Effects of Aβ42 in Flies.

(A) Z tox predicts more accurately (r = 0.83, p < 0.00001) than Z agg (Figure 2A) the relative longevity (S tox) of flies expressing different Aβ42 variants.
(B) Z tox predicts more accurately (r = 0.75, p = 0.001) than Z agg (Figure 2B) the relative locomotor ability (M tox) of flies expressing different Aβ42 variants.
The errors in S tox measurements (y-axis) are standard errors of the mean arising from the average of the independent lines of flies tested for each variant. The errors in the predictions of protofibril formation propensity (Z tox) are also shown (x-axis).
We propose, therefore, that the effects of all Aβ42 variants in the flies can be directly attributed to their effects on the intrinsic propensities to form deleterious protofibrillar aggregates. It is extremely interesting in this regard that a comparison, using electron microscopy, of the morphology of E22G and I31E/E22G Aβ42 aggregates formed under identical conditions reveals the presence of a significant quantity of protofibrils in the former and only well-defined fibrils in the latter (Figure S4). Furthermore, we propose that it is possible to predict accurately in silico the in vivo effects of the Aβ42 peptide from a knowledge only of the intrinsic physicochemical properties of its constituent amino acids. We believe that this approach to understanding the determinants of protein misfolding in vivo will be applicable to many other diseases as we have demonstrated previously that the physicochemical parameters that determine the aggregation propensity of Aβ also determine the aggregation behaviour of a wide range of both disease- and non-disease-related proteins [10,25].
It is also remarkable that, despite the fact that the intrinsic aggregation propensities of typical protein sequences vary by at least five orders of magnitude [25], we have been able to achieve profound alterations in the pathogenic effects of Aβ42 by increasing or decreasing its propensity to aggregate by less than 15%. This result suggests that proteins implicated in misfolding diseases are likely to be extremely close to the limit of their solubility under normal physiological conditions [26], and consequently the small alterations in their concentration, environment, or sequence, such as occur with genetic mutations [27] or with increasing age [23], are likely to be the fundamental origin of these highly debilitating and increasingly common conditions [28].
In conclusion, we have presented accurate, quantitative measurements of the relationships between the manifestations of neuronal dysfunction in a complex organism, such as locomotor deficits and reduced lifespan, and the fundamental physicochemical factors that determine the propensity of the Aβ42 peptide to aggregate into protofibrils. These results provide compelling evidence that, despite the presence within the cell of multiple regulatory mechanisms such as molecular chaperones and degradation systems [29], it is the intrinsic, sequence-dependent propensity of the Aβ42 peptide to aggregate to form protofibrillar aggregates that is the primary determinant of its pathological behaviour in living systems.
Materials and Methods
Generation of D. melanogaster expressing mutant Aβ 42 peptides.
Mutant Aβ42 expression constructs were produced by site-directed mutagenesis of the WT Aβ42 sequence in the pMT vector (Invitrogen) and were subcloned into the pUAST vector. Transgenic Drosophila expressing the desired Aβ42 variants were generated according to the procedures described by Crowther et al. [12].
Survival assays.
All survival assays were carried out as described previously [12]. Survival curves were calculated using Kaplan–Meier statistics, and differences between them analysed using the log rank method. All survival times in the text are given as median ± standard error of the median. For previously characterised control lines expressing either WT or E22G Aβ42, the survival of one representative line was measured. For each novel mutational variant of Aβ42, between four and six independent lines were analysed (n = 100 for each line) in order to control for variability in expression levels between individual lines due to the varying location of transgene insertion. The effect of a mutational variant on survival (S tox) was calculated by comparing the survival time of the flies in which it was expressed (S mut) to the survival of Aβ40-expressing flies (S max) used as a negative control in the same experiment: S tox = (S max − S mut)/S max.
Locomotor assays.
The locomotor ability of the flies was assessed in a 45-s negative geotaxis assay. Flies were placed in a plastic 25-ml pipette and knocked to the bottom of the pipette. The number reaching the top of the pipette (above the 25-ml line) and the number remaining at the bottom (below the 2-ml line) after 45 s was measured. The mobility index was calculated as (n top− n bottom + n total)/2n total. Two representative lines were tested for each novel mutant Aβ42 and one line for each previously characterised control (WT Aβ42 and E22G Aβ42). Three independent groups of 15 flies each were tested three times at each time point for each line. Differences between genotypes were analysed by ANOVA. The effect of each mutational variant on locomotor performance (M tox) was calculated by fitting the decline in mobility index over time to a straight line and then estimating the time at which each mutant line of flies had declined to a mobility index of 0.5.
Aβ 42 immunohistochemistry analysis.
Immunohistochemistry analysis was performed as described previously [12] on single representative lines for each genotype using the G2–11 anti-Aβ42 antibody (The Genetics Company). Representative lines of F20E- and I31E/E22G-Aβ42-expressing flies were chosen to have median survivals within 1 d of the combined median survival determined for each genotype.
Analysing the aggregation propensity of Aβ 42 mutants.
The propensity to form amyloid aggregates (Z agg) was calculated using an approach described previously [10]. Briefly, for a given protein, Z agg is obtained by averaging the propensities that are above zero in the aggregation profile. All the propensities are normalised into a variable that has an average of zero and a standard deviation that equals one (the normalisation is made using the propensities of a set of random sequences). In a profile there can be residues with a propensity larger than one, but these peaks are usually sparse and their contribution is diluted upon averaging. Consequently, sequences with an overall Z agg score larger than one are very rare. In order to calculate the propensity for forming protofibrillar aggregates (Z tox), we developed a method based on an equation containing the same physicochemical contributions used to calculate the propensity for fibrillar aggregation, but with specific weights determined using a set of experimentally determined protofibrillar aggregation rates for the protein acylphosphatase [30]. A Web server for calculating Z agg and Z tox is available at http://rd.plos.org/10.1371_journal.pbio.0050290_01.
Aβ peptide preparation for in vitro kinetic analysis of aggregation.
All peptides were dissolved in trifluoroacetic acid and sonicated for 30 s on ice. The trifluoroacetic acid was removed by lyophilization and the peptides were then dissolved in 1,1,1,3,3,3-hexafluoro-2-propanol and divided into aliquots that were dried by rotary evaporation at room temperature. The amount of peptide in the aliquots was determined by quantitative amino acid analysis.
In vitro kinetic analysis of Aβ 42 aggregation.
The peptides were dissolved at a concentration of 30 μM in 50 mM NaH2PO4 (pH 7.4) and incubated at 29 °C with continuous agitation. At regular time intervals, 5 μl of the peptide solution was removed and added to 100 μl of 20 μM thioflavin T in 50 mM Gly-NaOH (pH 8.5). Fluorescence intensity was measured at 440 nm excitation and 480 nm emission using BMG FLUOstar OPTIMA. The rate of aggregation (k) was determined by fitting the plot of fluorescence intensity versus time to a single exponential function y = q + Ae(−kt) [30], and t 1/2 was calculated using t 1/2 = ln2/k.
qRT-PCR.
Twenty flies expressing each variant of Aβ42 were collected at day 0 (i.e., on the day of eclosion) for each transgenic line to be analysed. The flies were then anaesthetised and decapitated, and the heads were collected and snap frozen in liquid N2. Total RNA was extracted from the fly heads using the Qiagen RNeasy mini kit with on-column genomic DNA digestion using DNAse 1. The concentration of total RNA purified for each line was measured using a NanoDrop spectrophotometer. One microgram of RNA was then subjected to reverse transcription using the Promega Reverse Transcription System with oligo dT primers. qRT-PCR was performed using a BioRad iCycler and Absolute QPCR SYBR Green Fluorescein Mix (ABgene). Each sample was analysed in triplicate and with both target gene (Aβ42) and control gene (RP49) primers in parallel. The primers for the Aβ42 PCR were directed to the 5′ end of the signal secretion peptide sequence and the 3′ end of the Aβ coding sequence: forward, GCATTCGTGAATTCATGGCGAGCAAAGT; reverse, TACTTCTAGATCCTCGAGTTACGCAATCAC. The RP49 primers were designed across an intron to avoid amplifying any residual genomic DNA contamination: forward, ATGACCATCCGCCCAGCATCAGG; reverse, ATCTCGCCGCAGTAAACG. Relative expression levels were calculated using the Livak method.
Supporting Information
The Aβ40 peptide has been previously demonstrated not to reduce lifespan or locomotor ability compared to non-transgenic flies when expressed in the Drosophila central nervous system [12]. The longevity and locomotor ability of a typical line of flies expressing the Aβ40 peptide under the control of the elav c155 promoter were assessed in parallel with those of the lines of flies expressing F20E and I31E/E22G Aβ42 as a negative control for the effects of expressing a less aggregation-prone peptide in the brain of the Drosophila.
(A) Flies expressing F20E Aβ42 (blue line) did not differ significantly in their longevity from flies expressing Aβ40 (red line).
(B) Flies expressing I31E/E22G Aβ42 (blue line) have slightly reduced longevity compared to Aβ40-expressing flies (red line).
(C and D) Flies expressing F20E or I31E/E22G Aβ42 (blue triangles) are indistinguishable in locomotor ability from flies expressing Aβ40 (red squares).
(723 KB EPS)
The level of Aβ42 mRNA present in each of two independent, representative lines of F20E- (F14 and F32) and I31E/E22G- (Isi68 and Isi51) Aβ42-expressing flies was compared to the level of Aβ42 mRNA in the brains of flies expressing WT and E22G Aβ42. All values are relative to the level of WT Aβ42 expression and normalised against the level of the housekeeping gene RP49 (see Materials and Methods).
(6263 KB EPS)
Samples were taken when the thioflavin T signal had reached a plateau for electron microscopic analysis. Aggregate solutions were placed on formvar-coated nickel grids and stained with uranyl acetate. WT Aβ42 (left) and F20E Aβ42 (right) show evidence of well-defined fibrils. Scale bar = 200 nm for both panels.
(2.3 MB TIF)
E22G and I31E/E22G Aβ42 were incubated for 24 h at room temperature (25 °C) without shaking in order to minimise disruption of the aggregates and so reveal any differences in morphology between the two samples. Aggregates were prepared as described in Materials and Methods. E22G Aβ42 forms both protofibrillar and fibrillar aggregates at this time (left). In stark contrast, I31E/E22G Aβ42 forms only well-defined fibrils (right). Scale bar = 500 nm for both panels.
(703 KB TIF)
This movie demonstrates the significantly greater locomotor ability of flies expressing F20E or L17R Aβ42 compared to that of flies expressing WT Aβ42.
(815 KB MOV)
This movie demonstrates the significantly greater locomotor ability of flies expressing F4D/E22G or I31E/E22G Aβ42 compared to that of flies expressing E22G Aβ42.
(1.0 MB MOV)
Acknowledgments
The work using Drosophila described in this paper was carried out in the University of Cambridge Department of Genetics, and we are extremely grateful to Professor M. Ashburner and Dr. S. Russell for providing the facilities for this study and for their invaluable advice. We would like to thank T. Sendall (Department of Medicine, University of Cambridge) for assistance with the fly work, and Dr. T. Rival (Department of Medicine, University of Cambridge) for advice on conducting the locomotor assays.
Abbreviations
- qRT-PCR
quantitative reverse transcription polymerase chain reaction
- WT
wild-type
Footnotes
Author contributions. LML, APP, MV, DAL, CMD and DCC conceived and designed the experiments. LML, ACB, APP, and IEW performed the experiments. LML, GGT, and ACB analyzed the data. GGT and DAL contributed reagents/materials/analysis tools. LML, FC, MV, DAL, CMD and DCC wrote the paper.
Funding. This work was supported by grants from the Medical Research Council/Engineering and Physical Sciences Research Council Discipline Bridging Fund (LML), Swiss National Science Foundation (grant PBZHA-112735 to GGT), Gates Cambridge Trust (APP), Swedish Research Council (ACB), Royal Society (MV), European Union (FC), Ministero dell'Università e della Ricerca, Italy (FC), Medical Research Council, United Kingdom (DAL and DCC), Papworth Hospital National Health Service Foundation Trust (DAL), Wellcome Trust (CMD), and Leverhulme Trust (MV and CMD).
Competing interests. The authors have declared that no competing interests exist.
References
- Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Folding proteins in fatal ways. Nature. 2003;426:900–904. doi: 10.1038/nature02264. [DOI] [PubMed] [Google Scholar]
- Barnhart MM, Chapman MR. Curli biogenesis and function. Annu Rev Microbiol. 2006;60:131–147. doi: 10.1146/annurev.micro.60.080805.142106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uptain SM, Lindquist S. Prions as protein-based genetic elements. Annu Rev Microbiol. 2002;56:703–741. doi: 10.1146/annurev.micro.56.013002.100603. [DOI] [PubMed] [Google Scholar]
- Fowler DM, Koulov AV, Alory-Jost C, Marks MS, Balch WE, et al. Functional amyloid formation within mammalian tissue. PLoS Biol. 2006;4:e6. doi: 10.1371/journal.pbio.0040006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fandrich M, Dobson CM. The behaviour of polyamino acids reveals an inverse side chain effect in amyloid structure formation. EMBO J. 2002;21:5682–5690. doi: 10.1093/emboj/cdf573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson CM. Protein misfolding, evolution and disease. Trends Biochem Sci. 1999;24:329–332. doi: 10.1016/s0968-0004(99)01445-0. [DOI] [PubMed] [Google Scholar]
- Chiti F, Stefani M, Taddei N, Ramponi G, Dobson CM. Rationalization of the effects of mutations on peptide and protein aggregation rates. Nature. 2003;424:805–808. doi: 10.1038/nature01891. [DOI] [PubMed] [Google Scholar]
- Fernandez-Escamilla AM, Rousseau F, Schymkowitz J, Serrano L. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat Biotechnol. 2004;22:1302–1306. doi: 10.1038/nbt1012. [DOI] [PubMed] [Google Scholar]
- Pawar AP, Dubay KF, Zurdo J, Chiti F, Vendruscolo M, et al. Prediction of “aggregation-prone” and “aggregation-susceptible” regions in proteins associated with neurodegenerative diseases. J Mol Biol. 2005;350:379–392. doi: 10.1016/j.jmb.2005.04.016. [DOI] [PubMed] [Google Scholar]
- Bukau B, Weissman J, Horwich A. Molecular chaperones and protein quality control. Cell. 2006;125:443–451. doi: 10.1016/j.cell.2006.04.014. [DOI] [PubMed] [Google Scholar]
- Crowther DC, Kinghorn KJ, Miranda E, Page R, Curry JA, et al. Intraneuronal Ab, non-amyloid aggregates and neurodegeneration in a Drosophila model of Alzheimer's disease. Neuroscience. 2005;132:123–135. doi: 10.1016/j.neuroscience.2004.12.025. [DOI] [PubMed] [Google Scholar]
- Finelli A, Kelkar A, Song HJ, Yang H, Konsolaki M. A model for studying Alzheimer's Abeta42-induced toxicity in Drosophila melanogaster. Mol Cell Neurosci. 2004;26:365–375. doi: 10.1016/j.mcn.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Iijima K, Liu HP, Chiang AS, Hearn SA, Konsolaki M, et al. Dissecting the pathological effects of human Abeta40 and Abeta42 in Drosophila: A potential model for Alzheimer's disease. Proc Natl Acad Sci U S A. 2004;101:6623–6628. doi: 10.1073/pnas.0400895101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilen J, Bonini NM. Drosophila as a model for human neurodegenerative disease. Annu Rev Genet. 2005;39:153–171. doi: 10.1146/annurev.genet.39.110304.095804. [DOI] [PubMed] [Google Scholar]
- Gidalevitz T, Ben-Zvi A, Ho KH, Brignull HR, Morimoto RI. Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science. 2006;311:1471–1474. doi: 10.1126/science.1124514. [DOI] [PubMed] [Google Scholar]
- Kim J, Onstead L, Randle S, Price R, Smithson L, et al. Abeta40 inhibits amyloid deposition In vivo. J Neurosci. 2007;27:627–633. doi: 10.1523/JNEUROSCI.4849-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ida N, Hartmann T, Pantel J, Schroder J, Zerfass R, et al. Analysis of heterogeneous A4 peptides in human cerebrospinal fluid and blood by a newly developed sensitive Western blot assay. J Biol Chem. 1996;271:22908–22914. doi: 10.1074/jbc.271.37.22908. [DOI] [PubMed] [Google Scholar]
- Buchanan RL, Benzer S. Defective glia in the drosophila brain degeneration mutant drop-dead. Neuron. 1993;10:839–850. doi: 10.1016/0896-6273(93)90200-b. [DOI] [PubMed] [Google Scholar]
- Westerman MA, Cooper-Blacketer D, Mariash A, Kotilinek L, Kawarabayashi T, et al. The relationship between Abeta and memory in the Tg2576 mouse model of Alzheimer's disease. J Neurosci. 2002;22:1858–1867. doi: 10.1523/JNEUROSCI.22-05-01858.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodart JC, Bales KR, Gannon KS, Greene SJ, DeMattos RB, et al. Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer's disease model. Nat Neurosci. 2002;5:452–457. doi: 10.1038/nn842. [DOI] [PubMed] [Google Scholar]
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, et al. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A. Opposing activities protect against age-onset proteotoxicity. Science. 2006;313:1604–1610. doi: 10.1126/science.1124646. [DOI] [PubMed] [Google Scholar]
- Townsend M, Shankar GM, Mehta T, Walsh DM, Selkoe DJ. Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: a potent role for trimers. J Physiol. 2006;572:477–492. doi: 10.1113/jphysiol.2005.103754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuBay KF, Pawar AP, Chiti F, Zurdo J, Dobson CM, et al. Prediction of the absolute aggregation rates of amyloidogenic polypeptide chains. J Mol Biol. 2004;341:1317–1326. doi: 10.1016/j.jmb.2004.06.043. [DOI] [PubMed] [Google Scholar]
- Tartaglia GG, Pechmann S, Dobson CM, Vendruscolo M. Life on the edge: a link between gene expression levels and aggregation rates of human proteins. Trends Biochem Sci. 2007;32:204–206. doi: 10.1016/j.tibs.2007.03.005. [DOI] [PubMed] [Google Scholar]
- Rovelet-Lecrux A, Hannequin D, Raux G, Le Meur N, Laquerriere A, et al. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet. 2006;38:24–26. doi: 10.1038/ng1718. [DOI] [PubMed] [Google Scholar]
- Dobson CM. An accidental breach of a protein's natural defenses. Nat Struct Mol Biol. 2006;13:295–297. doi: 10.1038/nsmb0406-295. [DOI] [PubMed] [Google Scholar]
- Tsubuki S, Takaki Y, Saido TC. Dutch, Flemish, Italian, and Arctic mutations of APP and resistance of Abeta to physiologically relevant proteolytic degradation. Lancet. 2003;361:1957–1958. doi: 10.1016/s0140-6736(03)13555-6. [DOI] [PubMed] [Google Scholar]
- Chiti F, Taddei N, Baroni F, Capanni C, Stefani M, et al. Kinetic partitioning of protein folding and aggregation. Nat Struct Biol. 2002;9:137–143. doi: 10.1038/nsb752. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The Aβ40 peptide has been previously demonstrated not to reduce lifespan or locomotor ability compared to non-transgenic flies when expressed in the Drosophila central nervous system [12]. The longevity and locomotor ability of a typical line of flies expressing the Aβ40 peptide under the control of the elav c155 promoter were assessed in parallel with those of the lines of flies expressing F20E and I31E/E22G Aβ42 as a negative control for the effects of expressing a less aggregation-prone peptide in the brain of the Drosophila.
(A) Flies expressing F20E Aβ42 (blue line) did not differ significantly in their longevity from flies expressing Aβ40 (red line).
(B) Flies expressing I31E/E22G Aβ42 (blue line) have slightly reduced longevity compared to Aβ40-expressing flies (red line).
(C and D) Flies expressing F20E or I31E/E22G Aβ42 (blue triangles) are indistinguishable in locomotor ability from flies expressing Aβ40 (red squares).
(723 KB EPS)
The level of Aβ42 mRNA present in each of two independent, representative lines of F20E- (F14 and F32) and I31E/E22G- (Isi68 and Isi51) Aβ42-expressing flies was compared to the level of Aβ42 mRNA in the brains of flies expressing WT and E22G Aβ42. All values are relative to the level of WT Aβ42 expression and normalised against the level of the housekeeping gene RP49 (see Materials and Methods).
(6263 KB EPS)
Samples were taken when the thioflavin T signal had reached a plateau for electron microscopic analysis. Aggregate solutions were placed on formvar-coated nickel grids and stained with uranyl acetate. WT Aβ42 (left) and F20E Aβ42 (right) show evidence of well-defined fibrils. Scale bar = 200 nm for both panels.
(2.3 MB TIF)
E22G and I31E/E22G Aβ42 were incubated for 24 h at room temperature (25 °C) without shaking in order to minimise disruption of the aggregates and so reveal any differences in morphology between the two samples. Aggregates were prepared as described in Materials and Methods. E22G Aβ42 forms both protofibrillar and fibrillar aggregates at this time (left). In stark contrast, I31E/E22G Aβ42 forms only well-defined fibrils (right). Scale bar = 500 nm for both panels.
(703 KB TIF)
This movie demonstrates the significantly greater locomotor ability of flies expressing F20E or L17R Aβ42 compared to that of flies expressing WT Aβ42.
(815 KB MOV)
This movie demonstrates the significantly greater locomotor ability of flies expressing F4D/E22G or I31E/E22G Aβ42 compared to that of flies expressing E22G Aβ42.
(1.0 MB MOV)