

Abstract
Leishmania are dimorphic protozoan parasites that live as flagellated forms in the gut of their sandfly vector and as aflagellated forms in their mammalian hosts. Although both parasite forms can infect macrophages and dendritic cells, they elicit distinct responses from mammalian cells. Amastigotes are the parasites forms that persist in the infected host; they infect cells recruited to lesions and disseminate the infection to secondary sites. In this review I discuss studies that have investigated the mechanisms that Leishmania amastigotes employ to harness the host cell’s response to infection. It should be acknowledged that our understanding of the mechanisms deployed by Leishmania amastigotes to modulate the host cell’s response to infection is still rudimentary. Nonetheless, the results show that amastigote interactions with mammalian cells promote the production of anti-inflammatory cytokines such as IL-10 and TGF-ß while suppressing the production of IL-12, superoxide and nitric oxide. An underlying issue that is considered is how these parasites that reside in sequestered vacuolar compartments target host cell processes in the cytosol or the nucleus; does this occur through the release of parasite molecules from parasitophorous vacuoles or by engaging and sustaining signaling pathways throughout the course of infection?
Keywords: Protozoan, Leishmania, Macrophages, Dendritic cells, Amastigote, Superoxide, Nitric oxide, IL-12
1. Introduction
Leishmania parasites are dimorphic organisms that live and replicate in the gut of sandflies as flagellated forms (promastigote) or as aflagellated forms (amastigotes) in mammalian cells. In the mammalian host these parasites preferentially infect phagocytic cells, primarily macrophages and dendritic cells. It is, however, well documented that within lesions and in the skin, other cell types including neutrophils and fibroblast cells have also been shown to be infected (Bogdan et al., 2000a; Laskay et al., 2003). Several factors determine the outcome of the interactions between mammalian cells and Leishmania parasites. One of these factors is the Leishmania species. A startling example of an outcome that is species-dependent is the development of morphologically distinct parasitophorous vacuoles (PVs). Whereas parasites of the Leishmania mexicana complex (L. mexicana, Leishmania amazonensis and Leishmania pifanoi) reside in communal PVs that become increasingly distended, parasites of the Leishmania donovani complex (L. donovani, Leishmania infantum and Leishmania chagasi) for the most part reside in tight individual PVs from where daughter cells segregate into their own PVs. Such differences in the outcome of the interactions between parasites and macrophages is a powerful reminder that Leishmania spp. are diverse organisms with significant differences at the genetic level, which result in differences at multiple points in their interactions with hosts cells. The reader is encouraged to consult a recent review by McMahon and Alexander (2004) for a more extensive discussion of the diversity of Leishmania and the host immune response to these organisms.
Both the promastigote and the amastigote forms of Leishmania can initiate infections. When infections are initiated with promastigotes, these parasites transform within PVs into amastigotes over a period of 24 to 72 h. Thereafter, in infected cells and infected hosts, infections are sustained by amastigote forms. Amastigotes can persist in cells for many days if not weeks. There are differences in the host cell response following interactions with either promastigote or amastigote forms. For example, studies with several Leishmania species have shown that whereas macrophages produce superoxide in response to infection with promastigotes, much lower levels of superoxide are produced in response to infection with amastigotes (Pearson et al., 1983; Channon et al., 1984; Pham et al., 2005). Such differences in the host response are to be expected since these parasite forms express stage-specific molecules that can serve as virulence factors. A notable example of a stage specific virulence factor is the lipophosphoglycan (LPG) molecule which is expressed on the surface of the promastigote form, but is minimally expressed on the amastigote form (Igoutz and McConville, 2001). LPG, especially the variant expressed by L. donovani parasites, has been shown to exert a dominant effect on numerous processes in the host cell in infections initiated with promastigotes (Descoteaux and Turco, 2002). The majority of studies that have investigated the host cell response to Leishmania have evaluated early macrophage responses initiated by promastigotes. Some reports, though fewer in number, have assessed infections that were initiated with amastigote forms. Since these two parasite forms have been shown to elicit differing host cell response, this review will focus primarily on host cell events modulated by amastigote forms of Leishmania. Wherever possible, the most recent studies that delve into the mechanisms that determine the host response to infection with amastigotes will be considered. The reader is encouraged to consult several excellent recent reviews in which infections initiated with promastigote forms were considered (Basu and Ray, 2005; Gregory and Olivier, 2005; Olivier et al, 2005).
2. Parasite entry mechanisms
The signaling pathways that mediate the host cell responses activated by infection with amastigotes are undoubtedly dependent on the internalization receptor(s) engaged by Leishmania amastigotes. There is general agreement that amastigotes gain entry into cells by phagocytosis, however it is known that amastigotes can infect non-phagocytic cells as well. Although the mechanism of entry of these parasites into non-phagocytic cells is poorly understood, the implication is that these parasites can exploit multiple mechanisms to gain entry into cells. For infection of phagocytic cells, several studies have shown that internalization of Leishmania amastigotes, especially in the in vivo setting, is mediated by Fc and complement receptors (Guy and Belosevic, 1993; Peters et al., 1995; Love et al., 1998; Kima et al., 2000). Phagocytosis mediated by Fc and complement receptors has been studied extensively and many of the signaling intermediates that are recruited upon the engagement of each of these receptors are known (Underhill and Ozinsky, 2002). Mosser and colleagues have shown that, similar to the case with other opsonized particles, internalization of amastigotes through the Fc receptor results in macrophage release of anti-inflammatory cytokines such as IL-10 (Kane and Mosser, 2001; Miles et al., 2005). Given the detailed understanding of Fc receptor signaling, the finding by Lodge and Descoteaux (2007) that even in the absence of antibody opsonization, L. donovani amastigotes were internalized through a Rac1-dependent mechanism, was a surprise. Whereas Rac1 has been shown to be required for Fc receptor mediated phagocytosis, many reports have shown that it is not activated during phagocytosis mediated by CR3 (Caron and Hall, 1998; Underhill and Ozinsky, 2002). Based on that knowledge, a requirement for Rac1 in the entry of non-opsonized amastigotes into macrophages would therefore suggest that even when amastigotes are internalized by phagocytic cells, these parasites appear to engage unconventional interactions that might condition the early host cell response to infection.
Interestingly, Morehead et al. (2003) had earlier found that internalization of L. amazonensis amastigotes into Chinese hamster ovary cells was Rac1-dependent only when parasites were opsonized with antibodies and entry was Fc receptor-mediated. Otherwise, under non-opsonic conditions parasite entry was Rac1-independent. These are contradictory observations that can be explained by the fact that different cell types and Leishmania species were used in both studies. Nonetheless, these results might be an indication that the uptake of amastigotes involves complex interactions that can yield unexpected outcomes. One cautionary note to bear in mind while evaluating such studies comes from a recent study that showed that contrary to the previous studies that had found that Rac1 was only involved in Fc receptor-mediated entry, other receptors, notably CR3, can also engage Rac1 (Hall et al., 2006).
Another molecule that has been implicated in the internalization of Leishmania amastigotes is phosphatidylserine (PS). Wanderley et al. (2006) and de Freitas Balanco et al. (2001) showed that L. amazonensis amastigotes obtained from infected tissue display PS on their surface. Acquisition and display of PS is described as apoptotic mimicry by these parasites. When Leishmania parasites were internalized via the recently described PS receptors on macrophages (Fadock et al., 2000), the macrophages were induced to secrete IL-10 and TGFβ (Fig. 1). One of the intriguing aspects of that observation was that cytokine production by macrophages was dependent on the density of PS on parasites. PS density on parasites was in turn dependent on ill-defined characteristics of parasite lesions from where the tissue-derived parasites were obtained. This would suggest a scenario in which parasites in a host exhibit varying surface characteristics, which would in turn result in the elicitation of varied host cell responses.
Fig. 1.
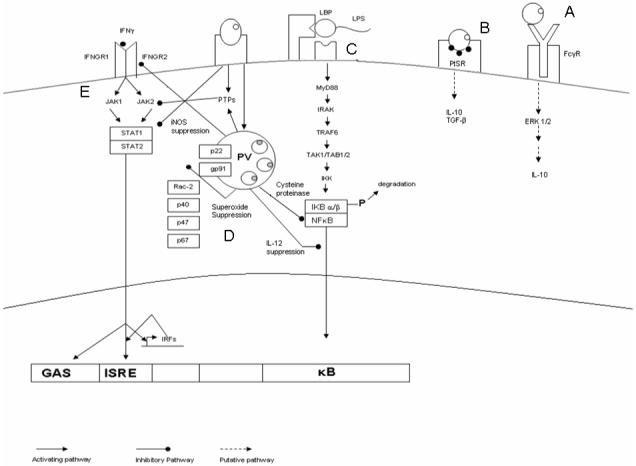
Schematic diagram showing pathways and processes targeted or engaged by Leishmania amastigotes. Amastigotes in tissue are opsonized with antibodies or they acquire and display phosphatidylserine (PS). (A and B) These parasites can be phagocytosed via the Fc receptor (FcγR) or the PS receptor (PtSR), respectively. Engagement of both of these receptors leads to the production of anti-inflammatory cytokines. (C) Leishmania amastigotes can suppress lipopolysaccharide (LPS) induced IL-12 production. Either acting through cysteine peptidases or by yet undefined effectors, these parasites degrade NFκB and prevent the translocation of functional dimers into the nucleus, which blocks the secretion of IL-12 in response to LPS. (D) Amastigotes within parasitophorous vacuoles (PVs) inhibit the assembly of the multi-subunit nicotinamide adenine dinucleotide phosphate (NADPH) -oxidase on the PV membrane (PVM) and by so doing they suppress the production of superoxide. (E) Leishmania parasites block the response to IFNγ at multiple points, which results in the suppression of nitric oxide (NO) production: upon internalization and during the course of infection, they can induce the activation of phosphotyrosine phosphatases (PTPs) that target Jak2; infection can downregulate the expression of IFNγ receptors (IFNGR1 and IFNGR2); infection can induce the degradation of STAT1.
A novel internalization strategy not previously implicated in the internalization of Leishmania parasites was recently described by Rodriguez et al. (2006). They found that the promastigote forms of L. chagasi can be internalized via a caveolin-mediated mechanism. Caveolin-mediated entry involves the participation of lipid rafts enriched with cholesterol that direct particles into intracellular compartment that may sometimes exhibit delayed interactions with lysosomes (Pelkmans et al., 2001). It is interesting that Lodge and Descoteaux (2006), using L. donovani amastigotes, found that infection perturbed lipid rafts. Although this would seem contradictory to the involvement of lipid rafts in the L. chagasi study, it nonetheless suggests some interaction of amastigotes with lipid raft structures that might ultimately affect intracellular signaling.
Dendritic cells are another important host cell for Leishmania parasites. They have been implicated in the dissemination of Leishmania parasites (Moll et al., 1993). The dendritic cell designation actually includes cells of different origin including lymphoid, myeloid and plasmacytoid (Shortman and Liu, 2002). Generation of dendritic cells from mouse bone marrow cells or human peripheral blood usually results in the expansion of dendritic cells of myeloid origin. These cells differ phenotypically and functionally from skin dendritic cells, which include Langerhans cells and dermal dendritic cells (Shortman and Liu, 2002). Leishmania amastigote entry into dendritic cells, which occurs without induction of maturation, has been characterized as ‘silent entry’ (Bennett et al., 2001; Brandonisio et al., 2004). Although amastigote entry does not induce dendritic cell maturation, infected dendritic cells can be induced to undergo maturation in response to exogenous stimuli (Prina et al., 2004). This would suggest that the parasite’s effects on the cell are mostly localized.
The silent entry of amastigotes into dendritic cells might depend on the receptor engaged by these parasite forms. Colmenares et al. (2002) identified the C-type lectin, ICAM-3-grabbing nonintegrin (DC-SIGN) as the putative receptor on dendritic cells for L. pifanoi and L. infantum amastigotes. These molecules are parasite species-restricted since another C-type lectin; L-SIGN was shown to be a receptor for L. infantum and not L. pifanoi (Colmenares et al., 2004). Internalization of particles by these C-type lectins does not result in activation of dendritic cells. Apparently, amastigotes avoid the activation of dendritic cells, not by elaborating virulence factors that deactivate host cell processes, but by selective interaction with a surface receptor that does not result in cell activation. Uptake of antibody opsonized amastigotes resulted in the induction of dendritic cell maturation (Prina et al, 2004).
3. Modulation of signaling in response to infection
Beyond the parasite’s interactions at the host cell membrane, it engages signaling pathways that help condition the host cell’s responses. Studies that have assessed the early response of macrophages to amastigote infection by assessing global changes in phosphorylation have shown that amastigote infection deactivates or silences the macrophage. However, studies that have focused on the parasite’s modulation of specific signaling pathways have yielded mixed and somewhat contradictory results. In light of observations from other systems that had shown that changes in tyrosine phosphorylation regulates several biological processes in cells, studies aimed at evaluating the effect of Leishmania infection on macrophage signaling assessed global changes in tyrosine phosphorylation after infection with Leishmania amastigotes. Martiny et al. (1999) showed that infection with L. amazonensis amastigotes resulted in reduced levels of tyrosine phosphorylation of several proteins tracked by Western blotting. This was in contrast to a previous study by Martiny et al. (1996) that had shown that infection with the promastigote form of L. amazonensis induced the appearance of tyrosine phosphorylated proteins. Another study that used L. amazonensis amastigotes as well, found that the amount and the pattern of protein tyrosine phosphorylation observed during amastigote uptake by macrophages was reduced compared with that observed during IgG-erythrocyte phagocytosis (Love et al., 1998). Furthermore, a comparable reduction in overall tyrosine phosphorylation was not found during the uptake of heat-killed amastigotes, implying that the actions of live amastigotes must have led to the widespread dephosphorylation.
Although it was not determined what mechanisms were engaged by L. amazonensis amastigotes that resulted in the widespread tyrosine dephosphorylation, several recent studies have shown that Leishmania infection induces the activation of phosphotyrosine phosphatases of which the Src homology 2 domain containing tyrosine phosphatase (SHP-1) has been the most extensively studied (Forget et al., 2001). Although most of the in vitro studies that have assessed the role of SHP-1 were initiated with promastigote forms of L. major and L. donovani, in vivo studies where amastigotes are exclusively found, have established a prominent role for these phosphatases in the conditioning of the host cell response by infecting parasites. Since the involvement of SHP-1 during parasite infection has been more fully addressed in the context of parasite suppression of nitric oxide release in response to IFNγ, it will be considered below.
A few studies have investigated the effect of Leishmania amastigote infection on mitogen activated protein (MAP) kinases. MAP kinases are important signal transduction intermediates that are activated in response to stress and other stimuli, which then mediate several cellular processes, including cell division, differentiation, cell survival/apoptosis, gene expression, motility and metabolism. Since it is likely that infection alters some of these processes, it is instructive to assess the functional state of MAP kinases in infected cells. One of the molecules that were dephosphorylated by L. amazonensis amastigote infection in the study by Martiny et al. (1999) was the extracellular signal regulated protein kinase 1 (ERK1). The authors showed that there was a steady decrease in ERK as the infection progressed. Unfortunately, it was not determined whether the loss in ERK phosphorylation was the result of selective degradation of this molecule or the action of a phosphatase activated by infection.
A recent study by Cameron et al. (2004) also found that in L. mexicana amastigote-infected cells activated with lipopolysaccharide (LPS) there was selective degradation of the MAP kinases JNK and ERK. By using mutant parasites in which the cysteine peptidase b gene was deleted (Δcpb) as well as inhibitors to cysteine peptidases, they proposed that parasite cysteine peptidases mediated the degradation of these MAP kinases. That study did not specifically address the phosphorylation status of the MAP kinases. In a more recent study, Yang et al. (2007) reported that infection with L. amazonensis amastigotes results in the activation of the MAP kinase ERK1/2. These authors showed that activation of ERK1/2 was required for the parasite-induced secretion of IL-10 by the infected macrophage. The observation in this later study that infection with L. amazonensis amastigotes results in ERK1/2 activation obviously contradicts the results of Martiny et al. (1999) and somewhat the study of Cameron et al. (2004) where infections with L. amazonensis and L. mexicana amastigotes were assessed, respectively. Such discrepancies are difficult to reconcile, although they might reflect the fact macrophages from different anatomical sites (peritoneal cavity [Martiny et al. study] versus bone marrow [Yang et al. study]) might respond differently. It is also likely that the conditions of parasite culture might influence the outcome of experimental infections. Nonetheless, it does appear that infection with amastigotes modulates MAP kinase signaling that might result in suppression of pro-inflammatory signals while promoting the secretion of IL-10. Hopefully future studies will help clarify which other biological processes are affected by the parasite’s effects on MAP kinases.
4. Suppression of superoxide production
Phagocytic uptake of particles mediated by some opsonic and non-opsonic receptors results in superoxide production (Underhill and Ozinsky, 2002). However, several studies have shown that unlike the situation with promastigote forms, macrophage uptake of amastigote forms of several Leishmania species does not activate superoxide production (Pearson et al., 1983; Channon et al., 1984; Pham et al., 2005). Furthermore, amastigote infection can suppress superoxide production elicited by otherwise potent activators of superoxide production (Pham et al., 2005). Superoxide is the product of the multisubunit nicotinamide adenine dinucleotide phosphate (NADPH) oxidase enzyme complex. This complex contains the membrane-bound cytochrome b558, which is composed of at least two polypeptides (gp91phox and p22phox) and two non-identical heme groups that are associated with gp91phox (Nauseef, 2004). The gp91phox subunit is synthesized as a 58-kDa polypeptide and, after limited glycosylation in the endoplasmic reticulum, becomes a 65-kDa molecule. Thereafter, it traffics through the trans-Golgi network, where it is additionally glycosylated, acquires heme and emerges as a molecule of 91 kDa (Yu et al., 1999; DeLeo et al., 2000; Nauseef, 2004). This processing or maturation of gp91phox increases its affinity for the other membrane resident subunit, p22phox. Four additional components of the NADPH oxidase enzyme, p40, p47, p67 and Rac2 are mostly found in the cytosol and associate with the membrane-bound components upon activation (Nauseef, 2004). Assembly of this enzyme complex on the target membrane is essential for the local release of optimal amounts of superoxide. Several intracellular pathogens have evolved strategies that target distinct points in the assembly of the NADPH oxidase complex (Banarjee et al., 2000; Gallois et al., 2001). The absence of superoxide production elicited by infection could be either an example of evasion or an example of parasite-induced suppression. A couple of reports have considered the molecular mechanism(s) that Leishmania deploy to suppress superoxide production. In studies with L. pifanoi amastigotes, Pham et al. (2005) showed that assembly of the functional NADPH oxidase enzyme did not occur on PVs that harbored amastigote forms, which was in contrast to vacuoles that harbored promastigote forms. Western blot analysis of gp91phox on PV-enriched fractions isolated from infected cells suggested that only the immature form of gp91phox, p65, which forms an unstable association with p22phox, was present on PV membranes. This finding was reminiscent of observations with succinyl acetone that had shown that when this pharmacological agent was added to cells, it caused a block in gp91phox maturation by increasing heme oxygenase I (HO-1), the rate limiting enzyme in heme degradation (Taille et al., 2004; Otterbein et al., 2000). Pham et al. (2005) then considered whether infection with Leishmania amastigotes might similarly induce HO-1. Indeed, HO-1 is induced by Leishmania infection. Furthermore, pharmacological compounds that inhibit heme degradation and HO-1 activity reversed the suppression of superoxide release by Leishmania amastigote-infected cells.
Lodge and Descoteaux (2006) also found that infection with L. donovani amastigotes do not trigger superoxide production by macrophages. However, they proposed that the inability of the NADPH oxidase enzyme complex to assemble on the PV was the result of defective phosphorylation of one of the cytoplasmic subunits, p47. Apparently, infection of macrophages with L. donovani amastigote inhibits protein kinase c (PKC) activity, which has been shown to be required for the phosphorylation of p47 (Fontanye et al., 2002). Amastigotes, like promastigotes, have been shown to inhibit PKC activity (Turco, 1999). However, unlike promastigotes where LPG has been shown to mediate the interactions with PKC, no specific molecules on amastigotes have been identified that function similarly in targeting PKC. Future studies will hopefully identify the parasite molecules on amastigotes that target PKC. The different mechanisms proposed for how Leishmania amastigotes suppress superoxide activation might be another example of the different outcomes from the interaction of different Leishmania species with macrophages. Nonetheless, these studies show that the absence of superoxide production during amastigote infection of macrophages is the result of parasite-induced suppression, via mechanisms that take advantage of existent host cell processes. The positive aspect of these findings is that parasite-induced suppression of superoxide production can be reversed with appropriate pharmacological agents.
5. Suppression of nitric oxide production
Although there is compelling evidence from murine models of leishmaniasis that nitric oxide (NO) plays a significant role in limiting experimental Leishmania infections (Murray and Nathan, 1999; Bogdan et al., 2000b), a few reports have shown that Leishmania infection of macrophages can block NO production. NO in macrophages is the product of the inducible NO synthetase gene (iNOS), which is induced by IFNγ. As discussed earlier, Wanderley et al., (2006) showed that L. amazonensis amastigotes obtained from infected tissue display PS. When these parasites are internalized via PS receptors on macrophages, the macrophages are induced to secrete IL-10 and TGFβ, which in turn block the induction of iNOS, therefore inhibiting the production of NO. It is quite likely that this strategy for parasite uptake can contribute significantly to the persistence of the infection.
Several studies have shown that Leishmania-infected macrophages are unresponsive to IFNγ induction of NO production. Binding of IFN-γ to its receptor on the macrophage surface, results in the dimerization of α and ß receptor units and their phosphorylation. This then initiates a signaling cascade that involves the phosphorylation of the receptor kinases, Janus kinases (Jak)1 and 2, and beyond that the signal transducer and activator of transcription (Stat) 1, which translocates into the nucleus to activate transcription of some IFNγ-responsive genes (Blanchette et al., 2003). Both promastigote and amastigote forms of Leishmania have been shown to selectively inhibit IFNγ activation of Jak 1 and 2 as well as Stat1 (Nandan and Reiner, 1995; Blanchette et al., 1999). Leishmania parasites apparently target multiple points in the IFNγ signaling cascade. A few reports have shown that infection results in expression of fewer IFNγ receptors on the infected cell surface and reduced phosphorylation of the remaining receptors (Fig. 1) (Nandan and Reiner, 1995; Ray et al., 2000). Using mice genetically altered to lack SHP-1, Olivier and colleagues have shown that activation of this phosphatase during infection blocks Jak2 signaling (Blanchette et al., 1999; Forget et al., 2001). More recently, Forget et al. (2005) showed that beyond the effects of Leishmania infection on the phosphorylation status of signaling intermediates in the IFNγ signaling cascade, infection with these parasites also resulted in proteosome-mediated degradation of Stat1. The authors reported that most of the Leishmania species tested (L. major, L. donovani and L. mexicana) exhibited this capacity to induce the degradation of Stat1. Since these parasites reside in PVs with different characteristics, it is uncertain which signals are transmitted from PVs or which parasite proteins access the cytosol to mediate the activities described above. Studies on a parasite-derived protein that has been shown to bind to SHP-1 are discussed below.
There is some evidence that Leishmania infection of humans cells results in limited production of nitric oxide (Gantt et al. 2001). The issue of whether limited NO production is a result of parasite evasion or whether parasites actively suppress the production of NO is just as relevant here. In the hamster model of visceral leishmaniasis where the progression of disease is comparable to that observed in humans, it has been shown that there is also defective production of NO in response to Leishmania infection (Melby et al., 2001). However, recent studies (Perez et al., 2005) have shown that this defect might be an epigenetic phenomenon, which stems from the fact that expression of the iNOS gene appears to be uncoupled from other IFNγ responsive genes. Given the similarities in the iNOS gene organization in hamsters and humans, it was suggested that a similar epigenetic mechanism might explain the reduced NO production of human cells in response to Leishmania infection (Perez et al., 2005).
6. Inhibition of IL-12 production
A TH1 response is required for control of leishmaniasis (Reiner and Locksley, 1995; Scott, 2003). The development of a TH1 response is dependent on the presence of IL-12 at the initiation of the immune response (Scott and Trinchieri, 1997). However, given the critical role of IL-12, it is significant that many studies have found that infection of macrophages with either the amastigote or promastigote forms of Leishmania does not activate macrophages to produce IL-12 (Carrera et al., 1996; Weinheber et al., 1998; Belkaid et al., 1999). Rodriguez-Sosa et al. (2001) reported that susceptibility of most mouse strains to L. mexicana is due to the inability of infected cells to produce IL-12, rather than a defect in IL-12 responsiveness. Carrera et al. (1996) and Belkaid et al. (1999) showed that the suppression of IL-12 is selective since infection induced the production of most other cytokines that were assayed. Furthermore, not only do infected cells not produce IL-12 but Leishmania infection suppresses the production of IL-12 by infected macrophages in response to LPS, an otherwise potent activator of IL-12.
IL-12 is composed of two covalently linked glycosylated chains, p40 and p35, which form the biologically active p70 heterodimer (Scott and Trinchieri, 1997). The p35 gene is ubiquitously expressed by most cells whereas the p40 gene is primarily expressed by phagocytic cells, most efficiently in response to microbial agents and their products (Ma and Trinchieri, 2001). There are both positive and negative inducers of IL-12 (Ma and Trinchieri, 2001). Whereas IFNγ is a positive inducer of IL-12 production, phagocytic receptor co-ligation (Fc and complement receptors, for example), engagement of G protein-coupled receptors and IL-10 negatively regulate IL-12 production (D’Andrea et al., 1993; Marth and Kelsall, 1997; Wagoneer et al., 2005). Thus far, it does not appear that Leishmania suppression of IL-12 production is mediated by IL-10 or G protein-coupled receptors (Carrera et al., 1996; Kima, unpublished data). Since both parasite forms inhibit IL-12 production, let us first consider the mechanisms that have been proposed for inhibition of IL-12 by promastigote forms. McDowell and Sacks (1999) proposed that given the inhibitory effects of Leishmania on stimuli that rely on Jak and Stat signaling, interference with this signaling pathway might be responsible for the inhibition of IL-12 production. However, no studies have formally shown that IL-12 is produced in the absence of this pathway. Studies with L. donovani promastigotes had implicated the engagement of the MAP kinase ERK in the suppression of IL-12 production (Feng et al., 1999). This effect was shown to be mediated by the LPG expressed on the parasite surface. However, there are contradictory observations that have found that infection with L. donovani promastigote does not result in the activation of ERK (Prive and Descoteaux, 2000), thus calling into question the role of ERK in the suppression of IL-12 by these parasites. In studies with L. amazonensis parasites, we have found that although ERK and the other MAP kinases are activated by infection with the promastigote form, inhibition of the activation of these MAP kinases does not reverse suppression of IL-12 during infection (unpublished data). Instead, inhibition of signaling through phosphoinositol 3 kinase (PI3K) and the downstream kinase protein kinase B (PKB, Akt) relieves IL-12 suppression induced by infection. On this point, there is evidence from other systems that PI3K/Akt signaling suppresses the production of IL-12 (Martin et al., 2003). Activated Akt (phospho-Akt) can translocate into the nucleus where it interacts with targets that are ill defined (Medema et al., 2000).
It is not known whether infections with Leishmania amastigotes employ the same mechanism for the suppression of IL-12 as the promastigote form. LPG expressed on the promastigote surface, which had been implicated in the suppression of IL-12, is minimally expressed on amastigotes. Instead, amastigotes express free glycoinositolglycolipids, also called GIPLs, at high levels as lyso alkyl or alkyacyl-GPI anchored lipids, and other lipids acquired from the host (McConville and Blackwell, 1991). Some studies have speculated that these lipids might indeed mediate the ‘avoidance of activation of macrophages’ and possibly the suppression of IL-12 production in cells infected with Leishmania amastigotes (Zufferey et al., 2003; Naderer et al., 2004). However, it has been difficult to evaluate the role of these lipids in events within the infected cell. One limitation of the Leishmania system is that gene-targeted knockouts are performed in the promastigote form. Since the synthetic pathways utilized in the synthesis of these lipids are also important for the synthesis of GIPLs that are expressed in the promastigote stage, knockout parasites have been found to inefficiently transform into the infective metacyclic stage and are therefore not viable inside macrophages (Zhang et al., 2005). Recently, studies with parasites that were engineered not to express sphingolipids by deletion of the palmitoyl transferase gene (spt-/-) (the first enzyme in the de novo SL biosynthesis pathway responsible for synthesis of the sphingo base) provided an opportunity to test the role of these lipids in amastigote survival in macrophages, including the suppression of IL-12 production and macrophage activation (Zhang et al., 2005). However, the authors found that the spt-/- knockout parasites were able to salvage the synthesis of inositol phosphoryl ceramides, which complicated the analysis of the role of this class of lipids in amastigotes. Nonetheless, they concluded that although these lipids might be required for survival in infected macrophages, they are not primarily responsible for the avoidance of activation of macrophages.
Studies by Cameron et al. (2004) using L. mexicana amastigotes implicated cysteine peptidase as the effectors of the inhibition of LPS induced IL-12 production by parasite infected cells. LPS induced production of IL-12 is mediated by NF-κB signaling (Murphy et al., 1995). The NF-κB family is composed of five members: NF-κB1 (p50), NF-κB2 (p52), RelA (p65), RelB and c-Rel (Ghosh et al., 1998). Engagement of NF-κB signaling results in the translocation of NF-κB dimers into the nucleus where they bind DNA and turn on responsive genes. Cameron et al. proposed that parasite cysteine peptidases expressed within the infected cell partially degraded NF-κB subunits, which resulted in inefficient translocation of these dimers into the nucleus and abolition of their capacity to bind DNA (Fig. 1). The Leishmania amastigote strategy described by Cameron et al. for inhibiting NF-κB appears to be a novel. Other pathogens are also able to inhibit NF-κB signaling. For example, Toxoplasma gondii parasites inhibit NF-κB by inhibiting the phosphorylation of p65/RelA, which blocks the translocation of NF-κB dimers into the nucleus (Shapira et al., 2005).
Another study showed that although L. major amastigotes induced the degradation of NF-κB subunits in phorbol myristate acetate (PMA)-activated human cells, the inhibitory effect of the parasite is selectively on the p50/p65 heterodimer (Guizani-Tabbane et al., 2004). The translocation of p50/c-Rel to the nucleus in PMA-treated cells infected with amastigotes was unabated. In addition, the authors found that parasite internalization was not required for inhibition p50/p65 translocation. The results of this study implied that parasites can engage an endogenous host cell signaling pathway that mediates the selective degradation of some NF-κB dimers while permitting nuclear translocation of dimers that are inhibitory (May and Ghosh, 1997). More studies are needed in this area.
7. How do Leishmania amastigotes alter processes in the host cell cytosol or nucleus many hours or days after parasite internalization?
Most studies that have assessed the host cell response to Leishmania infection have mainly evaluated events that occur within the first 24 h of infection. From the point of parasite contact with mammalian cells, parasites are sequestered from the host cell cytosol by a membrane barrier that undergoes a change in composition as the PV matures. It is from within these PVs that parasites engage signaling pathways and target host cell processes. As discussed above, some signaling pathways, which are selectively engaged by the choice of receptors at the initiation of infection, either convey neutral signals into the cell or engage signals that mediate the suppression of selective processes. One question that is still unresolved is how are signals that modulate host cell processes sustained once the parasite PV is formed? We have found that in infections with L. amazonensis promastigotes, which were shown to activate PI3K signaling (Ruhland et al., 2007) PVs display the lipid products of PI3K through at least 12 h p.i. The consequence of this is that PVs become sites for the activation of PKB/Akt, a downstream kinase in the PI3K signaling pathway (unpublished data). Although PI3K signaling plays a significant role in promoting infected cell survival at early times after infection, it apparently plays a minimal role in the survival of infections that are older than 24 h (Ruhland et al., 2007). This would imply that other pathways are engaged by amastigote forms that continue to promote cell survival. It is of interest to determine the identities of the signaling mechanisms that promote survival of older infected cells that harbor replicating parasites. Also, it would be of interest to determine whether other signaling pathways can be initiated from molecules expressed on PVs.
In addition to, or in place of, PVs being sites from where signaling is initiated, it is likely that parasite-derived molecules secreted into the PV can access the host cell cytosol and beyond that the nucleus, and target host cell processes therein. Thus far, the best example of a parasite molecule that can access the host cell cytosol is the parasite-derived elongation factor 1α (EF-1α) that localizes in the cytosol of 16-18 h-old L. donovani promastigote-infected cells (Nandan and Reiner, 2005). Amastigotes also secrete EF-1α. EF-1α has been shown to bind to and activate SHP-1, which in turn inactivates Jak2. There is only circumstantial evidence that other parasite-derived molecules traverse the PV membrane to gain access to the host cell cytosol. As discussed earlier, the cysteine peptidases have been implicated in the degradation of NFκB subunits and MAP kinases (Cameron et al., 2004). To be in position to perform these activities, these peptidases would have to traverse the PV membrane and access the host cell cytosol. Mottram el al. (2004) recently proposed a model by which these cysteine peptidases might access the cell cytosol. In their model, vesicles containing these peptidases bud off the PV and their contents are eventually released into the cell cytosol. It is not known whether the cargo of such vesicles would be selective. The A2 genes are amastigote-specific genes that are found in L. donovani but not L. major (Zhang and Matlashewski, 2001; Zhang et al., 2003). Ectopic expression of A2 in L. major enhances the survival of the transgenic parasites in the spleen and confers to A2-expressing L. major the capacity to visceralize (Zhang and Matlashewski, 2001). How do the A2 proteins achieve this? Is their localization limited to the PV? Similar queries can be made of the rest of the amastigote-specific genes. Answers to questions such as these will greatly advance our understanding of the biology of Leishmania in infected cells
8. Concluding Remarks
Amastigote survival within infected cells is undoubtedly dependent on the outcome of the parasite’s interaction with the host cell at multiple points. In the early stages of infection, these parasites must either avoid inappropriate activation of their host cell or elaborate mechanisms that inactivate anti-parasitic effectors at the time of infection. The internalization receptors engaged by the parasite during cell entry could obviously play a determining role here. In the in vivo setting, parasites have to reduce pro-inflammatory signals such as cytokines or chemokines emanating from the infected cell, which can promote anti-leishmanial responses in their vicinity. Alternatively, it would be prudent for the parasite to sequester parasite molecules from accessing antigen presentation pathways and therefore limit T cell recognition of the infected cell (not discussed). Beyond that, parasites must promote survival of the infected cell in a milieu that is rich with immunologic activity. Finally, parasites have to acquire nutrients from the host cell (not discussed). Our current understanding of the mechanisms that Leishmania parasites deploy to modulate these processes is incomplete. The evidence that is currently available suggests that Leishmania parasites selectively engage and exploit host cell processes from the point of parasite entry and promote the release of anti-inflammatory cytokines. It is not known how they sustain their effects on these processes from within PVs through the course of infection. There is, however, emerging evidence that parasite-derived molecules such as cysteine peptidases might exert functions beyond PVs in infected cells. Studies that demonstrate the existence of parasite molecules beyond PVs and that attempt to establish their function would contribute significantly to our understanding of the biology of Leishmania parasites in infected cells.
Acknowledgements
Thanks to Dr. Howard Johnson for reading the manuscript and Dr Aaron Ruhland for help with the artwork. This work was supported by a grant from the NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alexandre Fontayne, Pham My-Chan Dang, Marie-Anne Gougerot-Pocidalo, Jamel El Benna. Phosphorylation of p47phox Sites by PKCα, βII, δ, and ζ: Effect on Binding to p22phox and on NADPH Oxidase Activation. Biochemistry. 2002:7743–7750. [Google Scholar]
- Banerjee R, Anguita J, Fikrig E. Cutting edge: infection by the agent of human granulocytic ehrlichiosis prevents the respiratory burst by down-regulating gp91phox. J. Immunol. 2000;164:3946–3949. doi: 10.4049/jimmunol.164.8.3946. [DOI] [PubMed] [Google Scholar]
- Basu MK, Ray M. Macrophage and Leishmania: an unacceptable coexistence. Crit Rev Microbiol. 2005;31:145–54. doi: 10.1080/10408410591005101. [DOI] [PubMed] [Google Scholar]
- Belkaid Y, Butcher B, Sacks DL. Analysis of cytokine production by inflammatory mouse macrophages at the single-cell level: selective impairment of IL-12 induction in Leishmania-infected cells. Eur J Immunol. 1998;28:1389–400. doi: 10.1002/(SICI)1521-4141(199804)28:04<1389::AID-IMMU1389>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Bennett CL, Misslitz A, Colledge L, Aebischer T, Blackburn CC. Silent infection of bone marrow-derived dendritic cells by Leishmania mexicana amastigotes. Eur J Immunol. 2001;31:876–83. doi: 10.1002/1521-4141(200103)31:3<876::aid-immu876>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- Blanchette J, Racette N, Faure R, Siminovitch KA. Olivier M. Leishmania-induced increases in activation of macrophage SHP-1 tyrosine phosphatase are associated with impaired IFN-gamma-triggered JAK2 activation. Eur J Immunol. 1999;29:3737–44. doi: 10.1002/(SICI)1521-4141(199911)29:11<3737::AID-IMMU3737>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Blanchette J, Jaramillo M, Olivier M. Signalling events involved in interferon-gamma-inducible macrophage nitric oxide generation. Immunology. 2003;108:513–22. doi: 10.1046/j.1365-2567.2003.01620.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdan C, Donhauser N, Doring R, Rollinghoff M, Diefenbach A, Rittig MG. Fibroblasts as host cells in latent leishmaniosis. J Exp Med. 2000a;191:2121–30. doi: 10.1084/jem.191.12.2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdan C, Rollinghoff M, Diefenbach A. The role of nitric oxide in innate immunity Immunol. Rev. 2000b;173:17–26. doi: 10.1034/j.1600-065x.2000.917307.x. [DOI] [PubMed] [Google Scholar]
- Brandonisio O, Spinelli R, Pepe M. Dendritic cells in Leishmania infection. Microbes Infect. 2004;6:1402–9. doi: 10.1016/j.micinf.2004.10.004. [DOI] [PubMed] [Google Scholar]
- Cameron P, McGachy A, Anderson M, Paul A, Coombs GH, Mottram JC, Alexander J, Plevin R. Inhibition of lipopolysaccharide-induced macrophage IL-12 production by Leishmania mexicana amastigotes: the role of cysteine peptidases and the NF-kappaB signaling pathway. J Immunol. 2004;173:3297–304. doi: 10.4049/jimmunol.173.5.3297. [DOI] [PubMed] [Google Scholar]
- Carrera L, Gazzinelli RT, Badolato R, Hieny S, Muller W, Kuhn R, Sacks DL. Leishmania promastigotes selectively inhibit interleukin 12 induction in bone marrow-derived macrophages from susceptible and resistant mice. J Exp Med. 1996;183:515–26. doi: 10.1084/jem.183.2.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caron E, Hall A. Identification of two distinct mechanisms of phagocytosis controlled by different Rho GTPases. Science. 1998;282:1717–21. doi: 10.1126/science.282.5394.1717. [DOI] [PubMed] [Google Scholar]
- Channon JY, Roberts MB, Blackwell JM. A study of the differential respiratory burst activity elicited by promastigotes and amastigotes of Leishmania donovani in murine resident peritoneal macrophages. Immunology. 1984;53:345–55. [PMC free article] [PubMed] [Google Scholar]
- Colmenares M, Corbi AL, Turco SJ, Rivas L. The dendritic cell receptor DC-SIGN discriminates among species and life cycle forms of Leishmania. J Immunol. 2004;172:1186–90. doi: 10.4049/jimmunol.172.2.1186. [DOI] [PubMed] [Google Scholar]
- Colmenares M, Puig-Kröger A, Pello OM, Corbi AL, Rivas L. Dendritic cell (DC)-specific intercellular adhesion molecule 3 (ICAM-3)-grabbing nonintegrin (DC-SIGN, CD209), a C-type surface lectin in human DCs, is a receptor for Leishmania amastigotes. J. Biol. Chem. 2002;277:36766–36769. doi: 10.1074/jbc.M205270200. [DOI] [PubMed] [Google Scholar]
- D’Andrea A, Aste-Amezaga M, Valiante NM, Ma X, Kubin M, Trinchieri G. Interleukin 10 (IL-10) inhibits human lymphocyte interferon γ-production by suppressing natural killer cell stimulatory factor/IL-12 synthesis in accessory cells. J. Exp. Med. 1993;178:1041–1048. doi: 10.1084/jem.178.3.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Freitas Balanco JM, Moreira ME, Bonomo A, Bozza PT, Amarante-Mendes G, Pirmez C, Barcinski MA. Apoptotic mimicry by an obligate intracellular parasite downregulates macrophage microbicidal activity. Curr Biol. 2001;11:1870–1873. doi: 10.1016/s0960-9822(01)00563-2. [DOI] [PubMed] [Google Scholar]
- DeLeo FR, Burritt JB, Yu L, Jesaitis AJ, Dinauer MC, Nauseef WM. Processing and maturation of flavocytochrome b558 include incorporation of heme as a prerequisite for heterodimer assembly. J. Biol. Chem. 2000;275:13986–13993. doi: 10.1074/jbc.275.18.13986. [DOI] [PubMed] [Google Scholar]
- Descoteaux A, Turco SJ. Functional aspects of the Leishmania donovani lipophosphoglycan during macrophage infection. Microbes Infect. 2002;4:975–81. doi: 10.1016/s1286-4579(02)01624-6. [DOI] [PubMed] [Google Scholar]
- Fadok VA, Bratton DL, Rose DM, Pearson A, Ezekewitz RAB, Henson PM. A new receptor for phosphatidylserine-specific clearance of apoptotic cells. Nature. 2000;405:85–90. doi: 10.1038/35011084. [DOI] [PubMed] [Google Scholar]
- Feng GJ, Goodridge HS, Harnett MM, Wei XQ, Nikolaev AV, Higson AP, Liew FY. Extracellular signal-related kinase (ERK) and p38 mitogen-activated protein (MAP) kinases differentially regulate the lipopolysaccharide-mediated induction of inducible nitric oxide synthase and IL-12 in macrophages: Leishmania phosphoglycans subvert macrophage IL-12 production by targeting ERK MAP kinase. J Immunol. 1999;163:6403–6412. [PubMed] [Google Scholar]
- Forget G, Siminovitch KA, Brochu S, Rivest S, Radzioch D, Olivier M. Role of host phosphotyrosine phosphatase SHP-1 in the development of murine leishmaniasis. Eur J Immunol. 2001;31:3185–96. doi: 10.1002/1521-4141(200111)31:11<3185::aid-immu3185>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Forget G, Gregory DJ, Olivier M. Proteasome-mediated degradation of STAT1alpha following infection of macrophages with Leishmania donovani. J Biol Chem. 2005;280:30542–9. doi: 10.1074/jbc.M414126200. [DOI] [PubMed] [Google Scholar]
- Liew F-Y. Extracellular signal-related kinase (ERK) and p38 mitogen-activated protein (MAP) kinases differentially regulate the lioplysaccharide-mediated induction of inducible nitric oxide synthase and IL-12 in macrophages: Leishmania phosphoglycans subvert macrophage IL-12 production by targeting ERK MAP kinase. J. Immunol. 1999;163:6403. [PubMed] [Google Scholar]
- Gallois A, Klein JR, Allen LA, Jones BD, Nauseef WM. Salmonella pathogenicity island 2-encoded type III secretion system mediates exclusion of NADPH oxidase assembly from the phagosomal membrane. J Immunol. 2001;166:5741–8. doi: 10.4049/jimmunol.166.9.5741. [DOI] [PubMed] [Google Scholar]
- Gantt KR, Goldman TL, McCormick ML, Miller MA, Jeronimo SM, Nascimento ET, Britigan BE, Wilson ME. Oxidative responses of human and murine macrophages during phagocytosis of Leishmania chagasi. J Immunol. 2001;167:893–901. doi: 10.4049/jimmunol.167.2.893. [DOI] [PubMed] [Google Scholar]
- Ghosh S, May MJ, Kopp EB. NF-κB and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- Gregory DJ, Olivier M. Subversion of host cell signalling by the protozoan parasite Leishmania. Parasitology. 2005;130(Suppl):S27–35. doi: 10.1017/S0031182005008139. [DOI] [PubMed] [Google Scholar]
- Guizani-Tabbane L, Ben-Aissa K, Belghith M, Sassi A, Dellagi K. Leishmania major amastigotes induce p50/c-Rel NF-kappa B transcription factor in human macrophages: involvement in cytokine synthesis. Infect Immun. 2004;72:2582–9. doi: 10.1128/IAI.72.5.2582-2589.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy RA, Belosevic M. Comparison of receptors required for entry of Leishmania major amastigotes into macrophages. Infect. Immun. 1993;61:1553–1558. doi: 10.1128/iai.61.4.1553-1558.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall AB, Gakidis MA, Glogauer M, Wilsbacher JL, Gao S, Swat W, Brugge JS. Requirements for Vav guanine nucleotide exchange factors and Rho GTPases in FcgammaR- and complement-mediated phagocytosis. Immunity. 2006;24:305–16. doi: 10.1016/j.immuni.2006.02.005. [DOI] [PubMed] [Google Scholar]
- He J, Gurunathan S, Iwasaki A, Ash-Shaheed B, Kelsall BL. Primary role for Gi protein signaling in the regulation of interleukin 12 production and the induction of T helper cell type 1 responses. J Exp Med. 2000;191:1605–1610. doi: 10.1084/jem.191.9.1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilgoutz SC, McConville MJ. Function and assembly of the Leishmania surface coat. Int J Parasitol. 2001;31:899–908. doi: 10.1016/s0020-7519(01)00197-7. [DOI] [PubMed] [Google Scholar]
- Jebbari H, Stagg AJ, Davidson RN, Knight SC. Leishmania major promastigotes inhibit dendritic cell motility in vitro. Infect Immun. 2002;70:1023–1026. doi: 10.1128/iai.70.2.1023-1026.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane MM, Mosser DM. The role of IL-10 in promoting disease progression in leishmaniasis. J Immunol. 2001;166:1141–1147. doi: 10.4049/jimmunol.166.2.1141. [DOI] [PubMed] [Google Scholar]
- Kima PE, Constant SL, Hannum L, Colmenares M, Lee KS, Haberman AM, Shlomchik MJ, McMahon-Pratt D. Internalization of Leishmania mexicana complex amastigotes via the Fc receptor is required to sustain infection in murine cutaneous leishmaniasis. J Exp Med. 2000;191:1063–1068. doi: 10.1084/jem.191.6.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskay T, van Zandbergen G, Solbach W. Neutrophil granulocytes--Trojan horses for Leishmania major and other intracellular microbes? Trends Microbiol. 2003;11:210–4. doi: 10.1016/s0966-842x(03)00075-1. [DOI] [PubMed] [Google Scholar]
- Lodge R, Descoteaux A. Phagocytosis of Leishmania donovani amastigotes is Rac1 dependent and occurs in the absence of NADPH oxidase activation. Eur J Immunol. 2006;36:2735–44. doi: 10.1002/eji.200636089. [DOI] [PubMed] [Google Scholar]
- Love DC, Mentink Kane M, Mosser DM. Leishmania amazonensis: the phagocytosis of amastigotes by macrophages. Exp Parasitol. 1998;88:161–71. doi: 10.1006/expr.1998.4232. [DOI] [PubMed] [Google Scholar]
- Ma X, Trinchieri G. Regulation of interleukin-12 production in antigen-presenting cells. Adv Immunol. 2001;79:55–92. doi: 10.1016/s0065-2776(01)79002-5. [DOI] [PubMed] [Google Scholar]
- Marth T, Kelsall BL. Regulation of interleukin-12 by complement receptor 3 signaling. J. Exp. Med. 1997;185:1987–1995. doi: 10.1084/jem.185.11.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema RH, Kops GJ, Bos JL, Burgering BM. AFX-like Forkhead transcription factors mediate cell-cycle regulation by ras and PKB through p27kip1. Nature. 2000;404:782–787. doi: 10.1038/35008115. [DOI] [PubMed] [Google Scholar]
- Martin M, Schifferle RE, Cuesta N, Vogel SN, Katz J, Michalek SM. Role of the phosphatidylinositol 3 kinase-Akt pathway in the regulation of IL-10 and IL-12 by Porphyromonas gingivalis lipopolysaccharide. J Immunol. 2003;171:717–725. doi: 10.4049/jimmunol.171.2.717. [DOI] [PubMed] [Google Scholar]
- Martiny A, Vannier-Santos MA, Borges VM, Meyer-Fernandes JR, Assreuy J, Cunha e Silva NL, de Souza W. Leishmania-induced tyrosine phosphorylation in the host macrophage and its implication to infection. Eur J Cell Biol. 1996;71:206–15. [PubMed] [Google Scholar]
- Martiny A, Meyer-Fernandes JR, de Souza W, Vannier-Santos MA. Altered tyrosine phosphorylation of ERK1 MAP kinase and other macrophage molecules caused by Leishmania amastigotes. Mol Biochem Parasitol. 1999;1021:1–12. doi: 10.1016/s0166-6851(99)00067-5. [DOI] [PubMed] [Google Scholar]
- May MJ, Ghosh S. Rel/NF-κB and IκB proteins: an overview Semin. Cancer Biol. 1997;8:63–73. doi: 10.1006/scbi.1997.0057. [DOI] [PubMed] [Google Scholar]
- McConville MJ, Blackwell JM. Developmental changes in the glycosylated phosphatidylinositols of Leishmania donovani. Characterization of the promastigote and amastigote glycolipids. J Biol Chem. 1991;266:15170–15179. [PubMed] [Google Scholar]
- McDowell MA, Sacks DL. Inhibition of host cell signal transduction by Leishmania: observations relevant to the selective impairment of IL-12 responses. Curr Opin Microbiol. 1999;2:438–43. doi: 10.1016/S1369-5274(99)80077-0. [DOI] [PubMed] [Google Scholar]
- McMahon-Pratt D, Alexander J. Does the Leishmania major paradigm of pathogenesis and protection hold for New World cutaneous leishmaniases or the visceral disease? Immunol Rev. 2001;201:206–24. doi: 10.1111/j.0105-2896.2004.00190.x. [DOI] [PubMed] [Google Scholar]
- Miles SA, Conrad SM, Alves RG, Jeronimo SM, Mosser DM. A role for IgG immune complexes during infection with the intracellular pathogen Leishmania. J Exp Med. 2005;201:747–54. doi: 10.1084/jem.20041470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moll H, Fuchs H, Blank C, Rollinghoff M. Langerhans cells transport Leishmania major from the infected skin to the draining lymph node for presentation to antigen-specific T cells. Eur J Immunol. 1993;23:1595–601. doi: 10.1002/eji.1830230730. [DOI] [PubMed] [Google Scholar]
- Morehead J, Coppens I, Andrews NW. Opsonization modulates Rac-1 activation during cell entry by Leishmania amazonensis. Infect Immun. 2002;70:4571–80. doi: 10.1128/IAI.70.8.4571-4580.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mottram JC, Coombs GH, Alexander J. Cysteine peptidases as virulence factors of Leishmania. Curr Opin Microbiol. 2004;7:375–81. doi: 10.1016/j.mib.2004.06.010. [DOI] [PubMed] [Google Scholar]
- Murphy TL, Cleveland MG, Kulesza P, Magram J, Murphy KM. Regulation of interleukin 12 p40 expression through an NF-kappa B half-site. Mol Cell Biol. 1995;1995:10–5258. doi: 10.1128/mcb.15.10.5258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray HW, Nathan CF. Macrophage microbicidal mechanisms in vivo: reactive nitrogen versus oxygen intermediates in the killing of intracellular visceral Leishmania donovani. J. Exp. Med. 1999;189:741–746. doi: 10.1084/jem.189.4.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naderer T, Vince JE, McConville MJ. Surface determinants of Leishmania parasites and their role in infectivity in the mammalian host. Curr Mol Med. 2004;4:649–65. doi: 10.2174/1566524043360069. [DOI] [PubMed] [Google Scholar]
- Nandan D, Reiner NE. Attenuation of gamma interferon-induced tyrosine phosphorylation in mononuclear phagocytes infected with Leishmania donovani: selective inhibition of signaling through Janus kinases and Stat1. Infect Immun. 1995;63:4495–500. doi: 10.1128/iai.63.11.4495-4500.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandan D, Reiner NE. Leishmania donovani engages in regulatory interference by targeting macrophage protein tyrosine phosphatase SHP-1. Clin Immunol. 2005;114:266–77. doi: 10.1016/j.clim.2004.07.017. [DOI] [PubMed] [Google Scholar]
- Nauseef WM. Assembly of the phagocyte NADPH oxidase. Histochem. Cell Biol. 2004;122:277–291. doi: 10.1007/s00418-004-0679-8. [DOI] [PubMed] [Google Scholar]
- Olivier M, Gregory DJ, Forget G. Subversion mechanisms by which Leishmania parasites can escape the host immune response: a signaling point of view. Clin Microbiol Rev. 2005;18:293–305. doi: 10.1128/CMR.18.2.293-305.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otterbein LE, Choi AM. Heme oxygenase: colors of defense against cellular stress. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1029–1037. doi: 10.1152/ajplung.2000.279.6.L1029. [DOI] [PubMed] [Google Scholar]
- Pearson RD, Harcus JL, Roberts D, Donowitz GR. Differential survival of Leishmania donovani amastigotes in human monocytes. J Immunol. 1983;131:1994–9. [PubMed] [Google Scholar]
- Pelkmans L, Kartenbeck J, Helenius A. Caveolar endocytosis of simian virus 40 reveals a new two-step vesicular-transport pathway to the ER. Nat Cell Biol. 2001;3:473–483. doi: 10.1038/35074539. [DOI] [PubMed] [Google Scholar]
- Perez LE, Chandrasekar B, Saldarriaga OA, Zhao W, Arteaga LT, Travi BL, Melby PC. Reduced nitric oxide synthase 2 (NOS2) promoter activity in the Syrian hamster renders the animal functionally deficient in NOS2 activity and unable to control an intracellular pathogen. J Immunol. 2006;176:5519–28. doi: 10.4049/jimmunol.176.9.5519. [DOI] [PubMed] [Google Scholar]
- Peters C, Aebischer T, Stierhof YD, Fuchs M, Overath P. The role of macrophage receptors in adhesion and uptake of Leishmania mexicana amastigotes. J Cell Sci. 1995;108:3715–24. doi: 10.1242/jcs.108.12.3715. [DOI] [PubMed] [Google Scholar]
- Pham NK, Mouriz J, Kima PE. Leishmania pifanoi amastigotes avoid macrophage production of superoxide by inducing heme degradation. Infect Immun. 2005;73:8322–33. doi: 10.1128/IAI.73.12.8322-8333.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponte-Sucre A, Heise D, Moll H. Leishmania major lipophosphoglycan modulates the phenotype and inhibits migration of murine Langerhans cells. Immunology. 2001;104:462–7. doi: 10.1046/j.1365-2567.2001.01333.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prina E, Abdi SZ, Lebastard M, Perret E, Winter N, Antoine JC. Dendritic cells as host cells for the promastigote and amastigote stages of Leishmania amazonensis: the role of opsonins in parasite uptake and dendritic cell maturation. J Cell Sci. 2004;117:315–25. doi: 10.1242/jcs.00860. [DOI] [PubMed] [Google Scholar]
- Prive C, Descoteaux A. Leishmania donovani promastigotes evade the activation of mitogen-activated protein kinases p38, c-Jun N-terminal kinase, and extracellular signal-regulated kinase-1/2 during infection of naive macrophages. Eur J Immunol. 2000;30:2235–2244. doi: 10.1002/1521-4141(2000)30:8<2235::AID-IMMU2235>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Ray M, Gam AA, Boykins RA, Kenney RT. Inhibition of interferon-gamma signaling by Leishmania donovani. J Infect Dis. 2000;181:1121–8. doi: 10.1086/315330. [DOI] [PubMed] [Google Scholar]
- Reiner SL, Locksley RM. The regulation of immunity to Leishmania major. Annu Rev Immunol. 1995;13:151–77. doi: 10.1146/annurev.iy.13.040195.001055. [DOI] [PubMed] [Google Scholar]
- Rodriguez NE, Gaur U, Wilson ME. Role of caveolae in Leishmania chagasi phagocytosis and intracellular survival in macrophages. Cell Microbiol. 2006;8:1106–20. doi: 10.1111/j.1462-5822.2006.00695.x. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Sosa M, Monteforte GM, Satoskar AR. Susceptibility to Leishmania mexicana infection is due to the inability to produce IL-12 rather than lack of IL-12 responsiveness. Immunol Cell Biol. 2001;79:320–2. doi: 10.1046/j.1440-1711.2001.01014.x. [DOI] [PubMed] [Google Scholar]
- Ruhland A, Leal N, Kima PE. Leishmania promastigotes activate PI3K/Akt signalling to confer host cell resistance to apoptosis. Cell Microbiol. 2007;9:84–96. doi: 10.1111/j.1462-5822.2006.00769.x. [DOI] [PubMed] [Google Scholar]
- Scott P. Development and regulation of cell-mediated immunity in experimental leishmaniasis. Immunol Res. 2003;27:489–98. doi: 10.1385/IR:27:2-3:489. [DOI] [PubMed] [Google Scholar]
- Scott P, Trinchieri G. IL-12 as an adjuvant for cell-mediated immunity. Semin Immunol. 1997;9:285–91. doi: 10.1006/smim.1997.0084. [DOI] [PubMed] [Google Scholar]
- Shapira S, Harb OS, Margarit J, Matrajt M, Han J, Hoffmann A, Freedman B, May MJ, Roos DS, Hunter CA. Initiation and termination of NF-kappaB signaling by the intracellular protozoan parasite Toxoplasma gondii. J Cell Sci. 2005;118(Pt 15):3501–3508. doi: 10.1242/jcs.02428. [DOI] [PubMed] [Google Scholar]
- Shortman K, Liu YJ. Mouse and human dendritic cell subtypes. Nat. Rev. Immunol. 2002;2:151–161. doi: 10.1038/nri746. [DOI] [PubMed] [Google Scholar]
- Taillé C, El-Benna J, Lanone S, Dang MC, Ogier-Denis E, Aubier M, Boczkowski J. Induction of heme oxygenase-1 inhibits NAD(P)H oxidase activity by down-regulating cytochrome b558 expression via the reduction of heme availability. J. Biol. Chem. 2004;279:28681–28688. doi: 10.1074/jbc.M310661200. [DOI] [PubMed] [Google Scholar]
- Turco SJ. Adversarial relationship between the Leishmania lipophosphoglycan and protein kinase C of host macrophages. Parasite Immunol. 1999;21:597–600. doi: 10.1046/j.1365-3024.1999.00266.x. [DOI] [PubMed] [Google Scholar]
- Underhill DM, Ozinsky A. Phagocytosis of microbes: complexity in action. Annu Rev Immunol. 2002;20:825–52. doi: 10.1146/annurev.immunol.20.103001.114744. [DOI] [PubMed] [Google Scholar]
- Waggoner SN, Cruise MW, Kassel R, Hahn YS. gC1q receptor ligation selectively down-regulates human IL-12 production through activation of the phosphoinositide 3-kinase pathway. J Immunol. 2005;175:4706–4714. doi: 10.4049/jimmunol.175.7.4706. [DOI] [PubMed] [Google Scholar]
- Wanderley JL, Moreira ME, Benjamin A, Bonomo AC, Barcinski MA. Mimicry of apoptotic cells by exposing phosphatidylserine participates in the establishment of amastigotes of Leishmania (L) amazonensis in mammalian hosts. J Immunol. 2006;176:1834–1839. doi: 10.4049/jimmunol.176.3.1834. [DOI] [PubMed] [Google Scholar]
- Weinheber N, Wolfram M, Harbecke D, Aebischer T. Phagocytosis of Leishmania mexicana amastigotes by macrophage leads to a sustained suppression of IL-12 production. Eur. J. Immunol. 1998;28:2467–2477. doi: 10.1002/(SICI)1521-4141(199808)28:08<2467::AID-IMMU2467>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Yang Z, Mosser DM, Zhang X. Activation of the MAPK, ERK, following Leishmania amazonensis infection of macrophages. J Immunol. 2007;178:1077–85. doi: 10.4049/jimmunol.178.2.1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, DeLeo FR, Biberstine-Kinkade KJ, Renee J, Nauseef WM, Dinauer MC. Biosynthesis of flavocytochrome b558. gp91(phox) is synthesized as a 65-kDa precursor (p65) in the endoplasmic reticulum. J. Biol. Chem. 1999;274:4364–4369. doi: 10.1074/jbc.274.7.4364. [DOI] [PubMed] [Google Scholar]
- Zhang K, Hsu FF, Scott DA, Docampo R, Turk J, Beverley SM. Leishmania salvage and remodelling of host sphingolipids in amastigote survival and acidocalcisome biogenesis. Mol Microbiol. 2005;55:1566–78. doi: 10.1111/j.1365-2958.2005.04493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang WW, Mendez S, Ghosh A, Myler P, Ivens A, Clos J, Sacks DL, Matlashewski G. Comparison of the A2 gene locus in Leishmania donovani and Leishmania major and its control over cutaneous infection. J Biol Chem. 2003;278:35508–15. doi: 10.1074/jbc.M305030200. [DOI] [PubMed] [Google Scholar]
- Zufferey R, Allen S, Barron T, Sullivan DR, Denny PW, Almeida IC, Smith DF, Turco SJ, Ferguson MA, Beverley SM. Ether phospholipids and glycosylinositolphospholipids are not required for amastigote virulence or for inhibition of macrophage activation by Leishmania major. J Biol Chem. 2003;278:44708–44718. doi: 10.1074/jbc.M308063200. [DOI] [PubMed] [Google Scholar]
- Zhang WW, Matlashewski G. Characterization of the A2-A2rel gene cluster in Leishmania donovani: involvement of A2 in visceralization during infection. Mol Microbiol. 2001;39:935–948. doi: 10.1046/j.1365-2958.2001.02286.x. [DOI] [PubMed] [Google Scholar]