

Abstract
Cysteine thiol modifications are increasingly recognized to occur under both physiological and pathophysiological conditions, making their accurate detection, identification, and quantification of growing importance. Amongst free cysteines, the bulk of modifications occur on a subset of cysteines that are more reactive. These exist as thiolate anions at physiological pH because of their surrounding electrostatic environment. Reagents with iodoacetamide active groups can be used to selectively label these reactive thiols with a high degree of selectivity. Thiol adducts can be detected by the failure to label with iodoacetamide or other reagents; restoration of labeling by specific reducing agents (eg. ascorbate or glutaredoxin), can be used to detect reversible S-nitroso and S-glutathione adducts. These adducts also may be detected with radiolabels and antibodies. S-glutathiolation in response to physiological stimuli may be detected in cells and tissues with glutathione ester labeled with biotin. Mass spectrometry can identify thiol modifications with precision, and with isotope-coded affinity tags, used to quantify modification of specific thiols. Combinations of these methods increase sensitivity and specificity, and enable quantification and precise identification of thiol modifications that occur under physiological and pathological conditions.
Introduction
Reactive oxygen species (ROS) including superoxide anion (O2-.), hydrogen peroxide (H2O2), and hydroxyl radical (OH.), and reactive nitrogen species (RNS) including nitric oxide (NO) and peroxynitrite (ONOO-) participate not only in normal physiological regulation, but also cellular dysfunction. The levels of ROS/RNS are normally tightly regulated by antioxidant defense mechanisms. Oxidative or nitrosative stress during aging and disease results from excessive generation of ROS/RNS and/or impaired antioxidant defenses. Although excess oxidants can directly affect other components of a cell including membrane lipids and nucleic acids, alteration of cellular homeostasis through post-translational modification of proteins is one of the major effects of oxidative and nitrosative stress [1]. Cysteine thiol groups of proteins are particularly susceptible to oxidation by ROS/RNS and other electrophilic molecules [2, 3]. Aside from the role that disulfides play in protein structure, modifications of reactive cysteine thiols can alter the function of proteins and may be involved in modulation of enzymatic activity. Thiols thus function not only in normal cell signaling via S-nitrosation [4], S-glutathiolation [5-9], or S-sulfenation [10, 11] but also may be irreversibly oxidized by aging and disease, and thus interfere with protein function [6, 8, 12].
Reactive thiols
The greater reactivity of a subset of all cysteine thiols is the key to understanding their participation in the regulation of protein function. The pKa of most protein thiols is approximately 8.5, and thus they are less reactive at intracellular pH. However, certain thiols have a lower pKa, which depends upon their local charge environment, and may exist as thiolate anions (cysteine-S-) at physiological pH. Because thiolate anions are more readily oxidized than are reduced thiols [13], they are more apt to react with ROS/RNS. As a result of these reactions, thiols may form sulfoxidation products, sulfenic (SOH), sulfinic (SO2H), and sulfonic (SO3H) acids, inter- or intra-protein disulfides (PrSSPr, PrSSPr’), or nitrosothiols (SNO, Figure 1). The complex chemistry of these reactions have been the subject of recent reviews [1, 4, 14] Protein disulfides, nitrosothiols, sulfenic acid and, in some cases, sulfinic acids [15], are reversible in the intracellular milieu, providing a mechanism for reversible changes in protein function, and therefore for cell regulation. Sulfenic acid may undergo further reversible reactions to form S-nitroso, S-glutathione, or S-sulfenamide modifications [10, 11]. Irreversible oxidation, to sulfonic acid for example, must be followed by protein degradation and re-synthesis to restore the protein to normal.
1.

Reactive thiolate anions are the sites of the most abundant cysteine modifications. Upper panel: A peptide containing a cysteine thiolate anion that can be oxidized to S-nitrosothiols (-SNO) or sulfenic, sulfinic, or sulfonic acids (SOxH) by various reactive oxygen and nitrogen species (nitric oxide, NO, hydrogen peroxide, H2O2, nitrogen dioxide, NO2, peroxynitrite, OONO-, hydroxyl radical, .OH). Lower panel: Reactive nitrogen and oxygen species (RNS/ROS) oxidize glutathione (GSH) to more reactive glutathione intermediates (glutathiyl radical, GS. ; glutathione sulfenic acid, GSOH; glutathione disulfide, GSSG; or S-nitrosoglutathione, GSNO) which can then form S-glutathione disulfide adducts with reactive thiolate anions.
In the presence of oxidants, protein thiols may also form mixed disulfides with glutathione (GSH). GSH is the most abundant low molecular weight thiol in cells consisting of a cysteine-containing tripeptide (glutamate-cysteine-glycine). The cysteine thiol in GSH has a pKa of 9.4 [16], near that of protein thiols. However, the thiol of GSH makes up for its relatively low reactivity by its high concentration in cells, which is in the millimolar range, and rapid enzymatic regeneration by glutathione reductase[17]. As a result of its reactions with ROS/RNS, GSH can form reactive intermediates including glutathione sulfenic acid (GSOH), glutathione disulfide (GSSG), and glutathiyl radicals, all of which can react with another GSH or protein thiols to form mixed disulfides. When the latter reactions occur at protein reactive thiols, they introduce a large protein glutathione adduct capable of altering protein function (Figure 1B). The term “S-glutathiolation” is used here rather than “S-glutathionylation” because no enzymatic catalysis, usually denoted by “-yl-”, has been defined for these reactions. S-glutathiolation is a reversible and regulated event, not only because it involves specific reactive cysteine residues, but also because it is enzymatically reduced by glutaredoxins, thioredoxins, and peroxiredoxins. In contrast to the lability of S-glutathione-protein adducts in cells, they are quite stable on recombinant proteins or when proteins are removed from the cellular environment. This attests to the influence of enzymatic reducing systems in cells. Thus, if these enzymatic reducing systems are inactivated by thiol modifiers such as N-ethyl maleimide, protein-glutathione adducts can be removed from cells and studied by separation and analytical techniques.
There now are many reports of proteins whose function is physiologically regulated by post-translational modifications of cysteine thiols [18]. The formation of H2O2 from O2.- generated from NADPH oxidase or other enzymatic sources stimulated by growth factors has been implicated as a mediator of S-glutathiolation in several cell types [7, 11, 19, 20]. In addition, ONOO-, arises in cells when both .NO and O2.- are present. Although its half-life is much shorter than H2O2, it is 2000 times more potent in oxidizing thiols than H2O2 (See for example, Figure 2).
2.

Labeling, pull-down and detection of protein thiolate anions. A. Iodoacetamide (IAM) binds selectively to protein thiolate anions at pH 6.5, form carboxymethyl adducts. After cell lysis in non-reducing buffer containing biotin tagged IAM (b-IAM, 0.2 - 1 mM), and removal of excess reagent with size exclusion columns, biotin-labeled proteins are collected on streptavidin-affinity beads. The protein is then cleaved with sodium dodecyl sulfate containing 5 M urea, and separated by SDS PAGE. B. Example of streptavidin-horse radish peroxidase (HRP) blot of whole cell lysate of bovine aortic endothelial cells exposed to ONOO- or H2O2 and labeled during lysis with b-IAM. Multiple proteins are detected with streptavidin-HRP, and very high concentrations of oxidants are required to detect decreased labeling. C. Immuno-blot of lysate of endothelial cells obtained after they were exposed to ONOO-, then pulled down and cleaved from streptavidin beads. Immuno-blot detection of p21ras that was pulled down on streptavidin beads shows that exposure of the cells to 100 μM ONOO- decreased b-IAM binding to p21ras by more than 50% [9], consistent with quantitative determinations made with ICAT labeling and mass spectrometry [48]. The higher concentration of ONOO- (1 mM) decreases labeling to a much greater degree.
An important question is whether regulation of protein function by thiol modification is specific or rather if it is a random event. It is not surprising that exposure of a purified protein to high concentrations of oxidants results in oxidization of many thiols, as well as other amino acid residues, and inhibition of protein function. It is hard to imagine how such a crude experiment could represent either physiological effects of low levels of oxidants or even, because proteins are constantly turning over, the effects of chronic, higher oxidant levels associated with aging or disease. However, during the last 10 years, a growing number of studies have shown that, 1) some proteins are regulated by physiological levels of oxidants generated during normal signaling events or under stress conditions under which levels of oxidants rise, 2) that the modification of protein structure by oxidants may be reversible, 3) that a high percentage of a specific cysteine residue in the protein may be modified, and 4) that mutant proteins lacking one specific cysteine residue may lack the modification and resist a change in enzymatic activity caused by ROS. In several cases such as glyceraldehyde phosphate dehydrogenase (GAPDH)[21], protein tyrosine phosphatase 1B [10, 11] sarcoplasmic reticulum calcium ATPase (SERCA) [8, 22], and p21ras [9, 23], nitrosothiol, glutathione, or sulfenamide adducts occur predominantly at one or more reactive thiols on the protein, whereas other thiols apparently do not participate to a significant extent, or their modification is not important to protein function. In addition, as in the case of carbonic anhydrase [24] and SERCA [8, 25], aging or disease are associated with irreversible sulfonic acid oxidation of a large fraction of a specific reactive cysteine thiol, preventing its further participation in reversible signaling events.
Because of the evident importance of regulation of protein function by thiol modifications, methods have been developed to 1) determine which proteins, and which cysteines within them, are most reactive, and 2) detect and identify the thiol modifications themselves. The first type of method provides the ability to screen for proteins not yet known to be regulated by thiol modifications. In the second, modified thiols are detected either by tags attached to the modification itself, as in the case of biotinylated glutathione, or indirectly by the failure of thiol reactive reagents to bind to otherwise occupied thiols [11, 12, 26]. Identifying the thiol modifications themselves is most accurately done with mass spectrometry, although as in the case of nitrosothiols, the modification in some cases may not be sufficiently stable to analyze. In this case, specific reduction of nitrosothiols under mild reductive conditions with ascorbate, followed by labeling of the freed thiol has been used to detect the modification indirectly [27, 28]. Mass spectrometric methods, labeling techniques and combinations of the two will be further addressed below.
In some cases, antibodies have been reported to react with S-nitrosothiol [29] and S-glutathione [23] protein adducts, or with sulfonic acid in specific sequences of proteins containing oxidized reactive cysteines, as in the case of PTP1B [30] or GAPDH [31]. These can provide some information particularly when the amount of protein available is limited, including by immunohistochemistry. Except in rare cases the S-nitrosothiol ([29, 31] and S-glutathione [23, 32] protein adduct antibodies have not been shown to stain specifically for the respective modification identified with a more precise analytical method, and it is not clear how an S-nitroso or S-glutathione adduct on a peptide could survive long enough to serve as an immunogen, or how immunogenicity survives formaldyde-induced cross-linking. However, staining with these antibodies may be prevented by reducing agents at least in some cases (see below) [23, 29, 32]. Therefore, use of these antibodies is recommended only with simultaneous verification by other methods such as labeling or mass spectrometry [23]. An antibody to the sulfonated active site cysteine residues in PTP1B was shown to be sequence specific. This antibody detected oxidation of the active site cysteine after treatment of cells with H2O2 or UV irradiation [30]. In addition, reversible modifications of the reactive thiols was demonstrated by first blocking free thiols with iodoacetic acid, then reducing the reversible modifications, and then oxidizing the newly exposed free thiols with pervanadate to the sulfonic acid form, which could then be detected with the antibody. While sequence specific antibodies are time consuming to prepare and serve for only one protein site, they represent a very sensitive technique to detect in vivo redox regulation of specific protein cysteine residues.
Free thiol labeling methods
Many thiol reactive probes have been used to quantify free protein thiols [33]. These include p-chloromercuribenzoate (PCMB), 5,5′-dithiobis (2-nitrobenzoic acid) (DTNB), hexyl)-3′-(2′-pyridyldithio)-propionamide (HPDP), N-ethyl maleimide (NEM), and iodoacetamide (IAM). All may inhibit protein function, and many studies have been done correlating the reaction of these probes with thiols and the effect on enzymatic activities. The reaction product of the first two reagents with protein thiols can be quantified spectrophotometrically, and because they react quantitatively with protein thiols, the reagents can be used to determine the number of free thiols on purified proteins or protein in cell lysates or fractions. This determination is useful also in assuring that these or other reagents are used in the required excess for quantitative labeling of free thiols. Because of the molecular size of the probes, they are somewhat selective for thiols on the surface of proteins, unless the protein is first denatured to allow access of the probe to thiols in the protein core. Selective surface labeling may be a disadvantage if one wants to determine the total number of free thiols. However, selective labeling of thiols that are accessible at the surface of native proteins can be an advantage because these tend to be the ones involved in physiological reactions. Methods for free thiol determination with PCMB or DTNB are available in standard analytical chemistry textbooks [34, 35]
N-ethyl maleimide, IAM and its analogue, iodoacetic acid (IAA) have been used extensively to detect free thiols. Each is available with various tags including radioactive or stable isotopes, biotin, and fluorophores. Biotin tags allow concentration of the labeled proteins on streptavidin (Figure 2) whereas radiolabels and fluorescent labels allow direct detection. Each of these probes can quantify free thiols, but PCMB and DTNB are, as mentioned above, easier to use for this purpose. As discussed below the tagged probes are more useful and sensitive for detecting and quantifying changes in free thiols.
NEM is highly reactive with free thiols over a wide range of pH, but because it can also react although at lower stoichiometry with lysines, methionine, histidine, and tyrosine, it is somewhat non-specific. IAA and IAM are less reactive and somewhat more specific for thiols than NEM. Because of the lower reactivity, the iodoacetates can differentially label reactive thiols particularly at pH 6.5-7.0. Although differential labeling of reactive and non-reactive thiols can be accomplished with iodoacetates in concentrations of 100-200 μM for 10-20 min at room temperature, quantitative labeling of reactive thiols can take hours and require concentrations in the millimolar range [36]. Protein labeling is fraught with difficulties, largely because of failure to achieve quantitative labeling and because protein denaturation results in exposure of additional thiols to labeling. Some investigators have succeeded in treating proteins with low concentrations of untagged NEM to rapidly react with, and thus block thiols on denatured protein and less reactive exposed thiols, and then to quantitatively label reactive thiols with higher concentrations of iodoacetates during a longer incubation [22, 36].
An example of the usefulness of IAM in quantitatively labeling reactive thiols is seen in Figure 2. Endothelial cell protein thiols were labeled with biotinylated IAM (BIAM) during cell lysis, and labeled proteins were affinity-purified with streptavidin, eluted with SDS and urea, separated by SDS-PAGE, transferred to PVDF membrane, and blotted with streptavidin-horseradish peroxidase (Figure 2B). Labeling of multiple proteins of various molecular weights is observed. There was little effect of treating the cells prior to lysis with ONOO- (100 μM), although increasing the concentration to 1 mM did decrease the labeling of many of the proteins. In contrast, the greater selectivity of labeling reactive thiols is seen in Figure 2C in which the streptavidin-purified proteins were separated by SDS-PAGE and immuno-blotted for p21ras, which has a free reactive surface thiol on cysteine-118. In this case, a significant decrease in IAM labeling is seen in lysates of cells exposed to ONOO- (100 μM), with a further decrease observed at 1 mM. One can assess the degree of oxidation of any number of oxidant-sensitive proteins in this manner, as long as one knows the identity of those proteins and has a good antibody towards them[12]. A word of caution therefore is warranted because generalized labeling methods such as those in Figure 2B may miss modification of reactive thiols because of the lack of sensitivity. This can be explained by the fact that proteins with reactive thiols that are modified to the greatest degree may be less abundant than those that are modified only to a small extent, and therefore altered labeling may not be detected.
Many improvements in labeling methods have been made. Development of newer avidin reagents, such as monovalent avidin, may offer some advantage because of decreased non-specific binding and easier protein elution. In addition, two dimensional (2-D) gel electrophoresis allows detection of a much larger number of labeled proteins, and this method has been used in conjunction with fluorescent tagged IAM labeling to identify proteins whose labeling changes in response to oxidant exposure [37]. In the cited study, IAM labeling was performed after blocking free thiols with NEM, followed by reducing thiols that had been reversibly oxidized, resulting in improved specificity and sensitivity to detect reversibly modified thiols. Protein spots whose staining was changed by exposing cells to oxidant were identified by MALDI-TOF mass spectrometry (see below).
As noted earlier, improved specificity for oxidant-sensitive thiol labeling can be obtained by labeling cell lysates at pH 6.5 at which unmodified reactive thiols exist as thiolate anions [12] that are selectively labeled by IAM [13]. An example of the selectivity for reactive thiols that is possible for IAM is shown in figure 3, in which only 25% of total IAM labeling of SERCA was shown to occur on a mutant protein lacking the most reactive cysteine-674, despite the fact that the protein has 14 free thiols[25]. Another improvement can be achieved by including a biotin-labeled protein as an internal standard in the cell lysate. This allows not only an accurate measure of the change in labeling, but also an estimate of the absolute number of IAM-labeled thiols in a particular protein [38].
3.
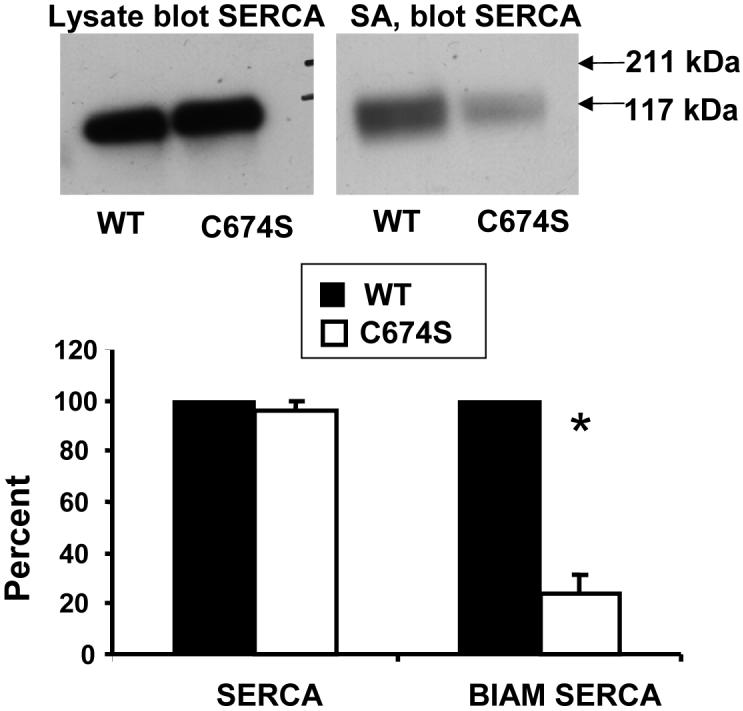
Biotin-IAM labeling of the reactive thiol of SERCA. B-IAM labeling was performed in lysates of HEK cells expressing wild-type (WT) or mutant human SERCA2b in which the reactive cysteine, cysteine-674, was mutated to serine (C674S). Protein for labeling was adjusted to contain equal amounts of SERCA, shown in the immuno-blot of the cell lysate on the left. The lysate was first exposed briefly to NEM (200 μM) to decrease non-specific labeling of partially denatured protein, and then labeled with b-IAM (1 mM, 30 min). The proteins pulled down with streptavidin (SA) beads were eluted, separated by SDS PAGE, and blotted for SERCA (right). The immuno-blot of labeled SERCA showed that cysteine-674 was responsible for approximately 75% of b-IAM labeling (bar graph). From reference [22].
S-nitrosation
S-nitrosothiol modifications are more labile than other modifications, although mass spectrometry detection of S-NO adducts is possible following extensive protein purification and preparation of a sample for mass spectrometry. While this has been commonly achieved for recombinant proteins, it is also possible to detect the S-NO adducts in proteins from tissues. For instance, in studies of SERCA purified from blood vessels exposed to NO gas, many cysteine containing peptides were identified which had an increase of 29 Da over the predicted mass, consistent with S-NO adducts (see reference [8] supplemental material). The detection of the adduct is not quantitative, though in the studies mentioned, the consistency with which the modified peptide masses were detected indicates that they were not minor contaminants.
Indirect labeling of S-NO adducts has been accomplished by blocking free thiols, reducing the S-NO adducts with ascorbate, and then labeling the freed thiols with a tagged thiol reagent such as fluorescent or biotin-tagged IAM or similar label [39]. The specificity of the procedure is therefore dependent on that of reduction of the S-NO by ascorbate, and the required sensitivity to detect the liberated free thiols. We have confirmed that millimolar concentrations of ascorbate do not reduce S-glutathione protein adducts, though they might be expected to reduce sulfenic acid modified thiols, and it is possible that sulfenic acid serves as a precursor of the S-NO adduct [32]. This technique has been used to detect S-nitrosated proteins in cultured cells and tissues exposed to exogenous or endogenous oxidants [27, 28, 40, 41]. A number of cautions have been published regarding the lack of specificity of the ascorbate reduction, which may give rise to false positive labeling even in preparations previously treated with high concentrations of reducing agents[42, 43]. Therefore, the procedure should be used in conjunction with other methods to confirm the SNO adducts.
S-glutathiolation
S-glutathiolated protein adducts occur transiently in cells, being reversed by glutaredoxin, thioredoxin, and peroxiredoxin family thiol reductases, but when removed from the intracellular milieu, the adduct is stable in millimolar concentrations of free GSH or ascorbate [9]. Although the reaction by which the S-glutathione adduct forms is a matter of some debate. The reactive cysteine may react with an oxidant to form a more reactive form, such as sulfenic acid, which then reacts with glutathione. Alternatively, a reactive glutathione intermediate (eg. GS., GSNO, GSSG.-) formed by oxidants reacts with the protein reactive cysteine [9]. Formation of one of the above glutathione reactive species might explain the relatively slow 15 to 30 minute time course of S-glutathiolation in response to a direct challenge of cells to a strong oxidant such as peroxynitrite which has a half life of seconds [9]. Because GSNO also induces intracellular protein S-glutathiolation [32], many previous studies that used this agent to initiate S-nitrosation, may have initiated protein modification and signaling via S-glutathiolation.
Detection of S-glutathiolated proteins was first done using radiolabeled cysteine that was incorporated into the glutathione tripeptide by γ-glutamyl cysteine synthase. The main disadvantage of using radiolabeled cysteine is the requirement to inhibit protein synthesis to prevent incorporation of the labeled cysteine into protein. Nevertheless, the technique was used to demonstrate S-glutathiolation of many proteins in cells as well as the kinetics of the formation of the protein adducts [44].
Biotin-labeled glutathione ester was developed by Sullivan et al. [45] as a convenient probe that is cell permeable and concentrated in cells due to cleavage of the ester by esterases. The biotinylated GSH (b-GSH) when adducted to reactive thiols, can be used to pull down the labeled proteins, after which they can be cleaved by reducing agents and analyzed by immuno-blot or mass spectrometry (Figure 4A). Figure 4B shows an example of a streptavidin horseradish peroxidase stain of proteins labeled with b-GSH in a smooth muscle cell lysate after stimulation with angiotensin II [7]. Importantly, dithiothreitol (DTT) added to the cell lysate cleaved most of the biotin-labeled GSH adduct, preventing the pull-down of the proteins. This control is an important one because of the existence native biotin containing proteins that resist such treatment. The method is optimally used to detect S-glutathiolation for known proteins in cells or isolated tissues such as the examples shown of p21ras and the sarcoplasmic reticulum calcium ATPase (Figure 4C and D) after the biotin-GSH labeled proteins are precipitated on streptavidin beads, cleaved with a reducing agent, and separated by SDS PAGE. The stability of the disulfide in the presence of ascorbate, which can reduce S-NO bonds, is demonstrated in Figure 4C).
4.

Biotin-tagged glutathione labeling of proteins in cells and tissues exposed to RNS/ROS. Biotin-tagged GSH is introduced into cells or tissues with the ester, which is cleaved by cellular esterases. RNS/ROS result in binding of the GSH adduct predominantly to protein thiolate anions. B. Streptavidin-HRP blot of lysates of rat aortic smooth muscle cells pre-labeled with biotin-tagged GSH and exposed to angiotensin II (Ang II). When cells were lysed in the absence of dithiothreitol (-DTT), multiple proteins were labeled in control cells (Cont), and the amount of labeling increased in cells exposed to Ang II. When the lysate was treated with DTT (10 mM, +DTT), much less streptavidin-HRP stained protein was detected. The remaining bands in lysates treated with DTT and that were unchanged by Ang II treatment are likely endogenous biotinylated proteins. From reference [7]. C. Immuno-blot for p21ras of avidin pull-down from lysates of bovine aortic endothelial cells prelabeled with biotin-tagged GSH and treated with ONOO- (100 μM, 5 min). ONOO- increased p21ras in the avidin affinity pull-down. DTT (1 mM), but not ascorbate (1 mM), added to the same cell lysate reduced the disulfide bond, cleaving the majority of the adduct from the protein. From [9]. D. Immuno-blot for SERCA of avidin pull-down from lysates of intact rabbit aortic rings pre-labeled with biotin-tagged GSH prior to exposure to nitric oxide (NO, 10 μM) during physiological contraction to phenylephrine. In normal rabbit aorta, NO increased the amount of SERCA labeled with the biotin-tagged GSH, whereas it did not increase labeling in atherosclerotic rabbit aorta in which it was demonstrated that the thiolate anionic site of binding was irreversibly oxidized. From [8].
Potential disadvantages of this technique include the fact that loading cells with the GSH ester results in as much as a two-fold increase in intracellular levels of GSH [9]. In addition, the biotin group introduces a rather large group on the glutamate relative to the size of the tripeptide that could affect binding kinetics. However, comparisons of results using radiolabeled and biotinylated probes are favorable [45], and the biotinylated tag provides significant ease in concentrating the labeled proteins.
The stoichiometry of S-glutathiolation is an important issue, and neither radiolabel nor biotin-tagged GSH methods indicate how much of a particular protein is S-glutathiolated, only that it is to some extent. However, estimates can be made of the change in free thiols accompanying S-glutathiolation by using labels that react with free thiols, such as biotinylated IAM. These estimates indicate that reversible thiol adducts can occupy a significant fraction (30-50%) of reactive thiols on proteins in cells exposed to NO or oxidants [8, 9]
As mentioned above, a commercially available anti-glutathione adduct antibody is available, and staining with this antibody is at least in some instances reversible with reducing agents. Immuno-precipitation of an S-glutathiolated protein, like p21ras, was required for detection of the adduct with the anti-GSH adduct antibody (Figure 5) [23]. The importance of showing that DTT can cleave the disulfide bond between glutathione and protein, and thereby prevent staining is re-emphasized.
5.

Detection of S-glutathiolation of p21ras with an antibody directed against S-glutathione protein adducts. Lysates were obtained from bovine aortic endothelial cells either under control conditions or after exposure to oxidized low density lipoproteins (oxLDL). p21ras was immunoprecipitated, separated by SDS PAGE, and after transfer to PVDF membranes, immuno-blotted anti-GSH adduct antibody. OxLDL increased the extent of immunoreactive S-glutathione adduct, and the detection of the adduct was prevented if dithiothreitol (DTT) was added the cell lysate consistent with reduction of the disulfide. From reference [23].
Mass spectrometry and proteomic identification of reactive cysteines
The principal shortcomings in using labeling methods in 1-D or 2-D gels in mass spectrometry are misidentification of the labeled protein and not knowing which cysteine of the protein in question is being labeled. The first problem occurs because many proteins co-migrate in 2-D gels. The identified protein, which is usually the most abundant within the spot, may not be the one whose thiol is labeled. Even if the identified protein is the one that is labeled, it is rarely possible to detect the residue and its modification due to its low abundance. The most definitive detection of modified cysteines requires purification of a protein of interest to an extent that allows the definitive identification of the protein from the masses of several of its peptides together with fragmentation studies to sequence one or more peptides. In matrix assisted laser desorption ionization (MALDI) mass spectrometry, peptide masses are matched to a protein database, and potential mass increments representing adducts to cysteines (or other modifications) can be queried from the database. Thus, increments in the expected peptide mass of 16, 32, or 64 Da indicate additions of oxygen, and 305 Da, that of glutathione. With current on-line databases, this is a cumbersome process, as only one or two modifications can be queried at a time, and the possibility of several potential modifications rapidly increases the complexity of the database search. The procedure can be improved considerably if a protein specific database consisting of possible proteolytic fragments with potential modifications is compared with the mass spectrum. Such a database can become large when one considers the many potential modifications, in addition to potential miscleavages of the protein by proteolytic enzymes. As an example, after immuno-precipitation, separation by SDS PAGE, and in-gel digestion of sarcoplasmic reticulum calcium ATPase (SERCA) purified from arteries exposed ex vivo to nitric oxide, several tryptic peptides containing cysteines had reproducible mass increments of 305 Da indicating S-glutathiolation. When diseased blood vessels were examined, increments of 48 Da were observed in cysteine containing peptides of SERCA, indicating sulfonic acid modification. Only rarely have sufficient amounts of this relatively abundant protein been purified to enable MS/MS sequencing. However, even without peptide fragmentation methods, confidence in identifying modifications can be increased when peptide masses matching the same modified cysteine are identified in more than one peptide, for instance with or without a proteolytic miscleavage, or when different peptide masses resulting from different cleavage sites, or masses with different charge states are matched to the modified peptide. When dealing with reversible modifications like S-glutathiolation, obtaining spectra after reduction can be used to verify that, as expected, the modified peak is no longer detected, as we were able to do in the case of p21ras purified from cells (Figure 6)[44]. If a peptide has more than one cysteine or more than one modification, for instance a methionine that can be oxidized, it sometimes is not possible to be definitive about the precise oxidized state of a cysteine unless MS/MS sequencing is possible. Some of these shortcomings can be overcome by thiol labeling combined with chromatographic separation and mass spectrometry (LC/MS), enabling sequencing and identification of the label on a particular cysteine.
6.

Detection by mass spectrometry of S-glutathione adducts on p21ras. Bovine aortic endothelial cells were treated with oxidized low density lipoproteins (LDL, 100 μg/mL, 1 h) and p21ras was immunoprecipitated from 1 mg of cell lysate protein under non-reducing conditions. p21ras was separated by SDS PAGE. Gels at left show immuno-precipitate stained with Coomassie blue (lane 1) and immuno-blot with anti-GSH antibody (lane 2) showing a prominent 21 kDa band. The protein in this band was digested in-gel with trypsin, and peptides were recovered and subjected to MALDI-TOF mass spectrometry. A peak in the mass spectrum corresponding to a tryptic peptide with a mass (953.32 Da) consistent with S-glutathiolated cysteine-118 (CDLAAR) was present, whose mass was 305 Da greater than the predicted mass of the peptide. When the lysate was treated with DTT, the mass spectrum peak was undetectable (right). In the same sample, a peak (1397.63 Da) representing another p21ras peptide that does not contain cysteine (QGVEDAFYTLVR, lower) appeared in the spectra of both non-reduced and reduced samples. From reference [23].
We have successfully used an approach to do this by labeling reactive cysteines on recombinant protein or in cells exposed to an oxidant stress with biotinylated glutathione ester. The biotin tag allows concentration of the glutathiolated proteins on monomeric avidin, release with excess biotin, and HPLC separation. First, the method was demonstrated using recombinant p21ras in which prominent labeling was observed on the reactive cysteine-118. In cells loaded with biotin GSH ester, the avidin purification step allowed concentration of S-glutathiolated proteins from cell lysates to a degree that allowed MS/MS identification of the tagged cysteine in many cases [46].
Because peptides differ in efficiency of ionization and detection, the sensitivity of their detection in a mass spectrometer also varies. This limits the ability to identify which thiols of cysteine-containing peptides are most affected by oxidants. Modification of a cysteine may result in different enzymatic cleavage products during proteolysis and other potential modifications make finding a particular modified peptide in a mass spectrum unpredictable. We have used one approach to overcome this by using an isotope-coded affinity tag (ICAT) that combines quantitative labeling by IAM with mass spectrometric measurement. An ICAT tag was used which was originally developed for quantitative measurements of protein abundance. The tag consists of an iodoacetamide thiol reactive moiety, an isotope tag containing 9- 12C or 9- 13C atoms, a biotin group allowing for concentration of labled peptides, and an acid cleavable linker. Labeling of proteins in two samples with the two tags of different mass, for example one control and the other oxidant exposed, is followed by mixing and proteolytic digestion of the entire mixture. The biotin-tagged peptides are pulled down by streptavidin, and the peptides cleaved by acid are separated by HPLC. The isotope-coded peptides elute simultaneously in the HPLC, and mass spectrometry detects peptide pairs with masses accounted for by the 9 Da difference in mass of the label. If in a sample exposed to oxidant the amount of labeled cysteine on a peptide is decreased because the cysteine thiol is otherwise occupied, the magnitude of the peak will be decreased accordingly. Thus, the ratio of the peak amplitudes provides an estimate of the change in the amount of a free thiol present in a particular peptide in the control sample (Figure 7). In practice, labeling can be done in high pH buffers to maximize labeling of all thiols regardless of pKa. Studies with the recombinant proteins, creatine phosphokinase and p21ras [47, 48], indicate that oxidants decrease labeling to a large extent at known reactive cysteines (Figure 7B), whereas labeling of non-reactive cysteines may be unchanged despite the same exposure (Figure 7C). In the case of p21ras, which was S-glutathiolated in the presence of ONOO- and GSH, less ICAT was bound to cysteines 118, 181, 184, and 186. Binding was restored by treatment of the protein with a reducing agent, consistent with occupation of the thiol by the reducible S-glutathione adduct [48]. This procedure can be used to screen for proteins with reactive cysteines by exposing a complex cell or tissue sample to oxidant. When we did this using a cardiac sarcoplasmic reticulum subcellular fraction, we were able to identify several known and many unknown protein thiols whose labeling was decreased by oxidant, thereby identifying proteins with reactive cysteines that may be important in physiological regulation [49].
7.

Use of isotope coded affinity tags (ICAT) for quantitation of cysteine thiol modifications. A. ICAT approach to quantitatively evaluate reversible and irreversible oxidative post-translational thiol modification. Protein samples with free reactive cysteine thiols (filled box) and less reactive thiols (open box) are exposed to peroxynitrite or GSSG or control buffer before labeling with ICAT reagent. Some of the reactive cysteine thiols are oxidized depending upon the level of the oxidant and oxidized thiols are designated as filled triangles (Ox). Following oxidation, labeling of the free thiols is performed, with light (filled keyhole) and heavy (open keyhole) ICAT tags. ICAT-labeled samples are mixed and digested with trypsin, followed by purification through HPLC cation exchange and avidin affinity cartridges. Affinity-captured peptides are analyzed by LC-MS and MS/MS. The oxidized reactive cysteines are not susceptible to labeling by ICAT reagent, and hence there is a decreased intensity in the mass spectrum peaks corresponding to the heavy-labeled peptide in the mass spectrum of the reactive cysteine-containing peptide (lower spectrum). For less reactive cysteines, the peptides in samples prepared under normal conditions or after oxidant exposure show equivalent signal intensity in the spectrum (upper). From the relative peak intensities of the MS of light and heavy ICAT-labeled peptides, the decrease of in thiol labeling and therefore the degree to which the cysteine is modified can be estimated. Identity of the peptide sequences can be derived from MS/MS analysis of these peptides. In order to evaluate whether the oxidation caused by oxidants such as ONOO- or GSSG is reversible or irreversible, the oxidized protein samples can be treated with reducing agent before labeling with heavy ICAT. From the LC-MS analysis, reversibility of the modifications caused by peroxynitrite or GSSG can be quantified. B. and C. Spectra of ICAT-labeled recombinant p21ras before and after exposure to ONOO- (100 μM). The spectrum in B shows that ONOO- decreases ICAT labeling of the known reactive cysteine-118, whereas no change in labeling occurred for the less reactive cysteine in the peptide shown in C. From reference [48].
An advantage of this ICAT technique is that identification of the cysteine and protein in question is usually unequivocal if MS/MS can confirm the sequence of the peptide and the site of the tag. Another major advantage is that the change in the thiol labeling, and thus the degree of reactivity of the thiol, is quantified. Thus, we were able to estimate that only 5-10% of identified proteins have cysteines whose labeling was decreased by oxidants by over 70% [49]. The major disadvantage of the technique is that the modified cysteine in the protein is not tagged, and therefore not purified by the streptavidin or available for examination. Consequently, it is not possible to identify what modification may be involved, only that the cysteine present in the control sample is not available for labeling by the ICAT in the test sample implying that it is modified. Furthermore, any potential modifications to the peptide not involving the cysteine would also remove it from the pair of labeled peptides. Because of these shortcomings, this method is best complemented by use together with other mass spectrometric techniques that provide identification of the cysteine modification or semi-quantitative labeling and blotting methods. Changes in the abundance of the protein also will change the amount of a labeled peptide. This factor can sometimes be controlled for by using the changes in a set of cysteine containing peptides of a particular protein that have been shown not to be affected acutely by oxidants. In the case of both creatine kinase and p21ras, some cysteines were shown to be remarkably resistant to oxidant-induced modification, making them and similar peptides candidates for normalizing for changes in protein abundance.
Another method worth mentioning is “top-down” mass spectrometry. Currently most suitable for highly purified proteins less than ∼50 kDa in size, this method can detect the intact protein ion. Fragmentation in the mass spectrometer provides the ability to track any modifications present in the protein in successively smaller fragments. Therefore, modifications of sufficient abundance will result in individual intact molecular ions, and precise identification of each modification during fragmentation. We used this approach to identify multiple oxidant-induced modifications in recombinant p21ras [50]. Direct exposure of the protein resulted in a highly complex mass spectrum, but exhaustive analysis was able to identify cysteine and tyrosine modifications as the most abundant species. Exposure of the protein to oxidants in the presence of glutathione resulted in identification of major molecular ions for the protein which was S-glutathiolated at the reactive cysteine-118 and the terminal cysteines [50].
Conclusions
Thiol modifications are increasingly being recognized to occur selectively on cysteines that are accessible on the protein surface and that are more reactive. Identifying these sites and quantitative assessment of their modification is increasingly recognized to be important for understanding cell signaling and function. Reversible adducts confer altered charge, structure, and activity of the modified protein. Irreversible oxidation is by its nature associated with protein degradation and re-synthesis, and interferes with reversible modifications normally occurring on the thiol.
Methods of detection and identification of oxidative post-translational modifications are developing rapidly. Although mass spectrometry can precisely identify modifications, the methods rely on the stability of the modifications during sample preparation, and it is currently difficult to quantify the extent of the modifications at any one site, except with special techniques. Methods utilizing cysteine labeling with tags that allow collection and concentration of the labeled protein are both semi-quantitative and relatively easily coupled with sensitive immuno-blotting methods to identify changes in the status of free thiols. When these assays are applied to intracellular proteins under physiological conditions, they may either perturb the intracellular milieu, or are indirect, relying on specific chemical reduction of a modification and subsequent labeling of the reduced thiol. For these reasons, using analytical methods helps to verify the modifications being detected. Finally, site-specific antibodies can be made against peptides containing cysteine sulfonic acid incorporated into peptides at sites of reactive thiols. The antibodies can be used to either detect the oxidized thiols, or, indirectly to detect reversible modifications after specific reduction and subsequent oxidation.
Acknowledgements
The authors were supported by National Institutes of Health grants R01 HL31607, R01 AG27080, P01 NIH HL 68758, P01 HL081738, the Boston University National Institutes of Health Cardiovascular Proteomics Center N01-HV-28178 and a predoctoral fellowship from the Institut de Recherches Servier (NC).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Stadtman ER, Berlett BS. Reactive oxygen-mediated protein oxidation in aging and disease. Drug Metab Rev. 1998;30:225–243. doi: 10.3109/03602539808996310. [DOI] [PubMed] [Google Scholar]
- [2].Ghezzi P, Bonetto V. Redox proteomics: identification of oxidatively modified proteins. Proteomics. 2003;3:1145–1153. doi: 10.1002/pmic.200300435. [DOI] [PubMed] [Google Scholar]
- [3].Cotgreave IA, Gerdes RG. Recent trends in glutathione biochemistry--glutathione-protein interactions: a molecular link between oxidative stress and cell proliferation? Biochem Biophys Res Commun. 1998;242:1–9. doi: 10.1006/bbrc.1997.7812. [DOI] [PubMed] [Google Scholar]
- [4].Stamler JS. Redox signaling: nitrosylation and related target interactions of nitric oxide. Cell. 1994;78:931–936. doi: 10.1016/0092-8674(94)90269-0. [DOI] [PubMed] [Google Scholar]
- [5].Klatt P, Lamas S. Regulation of protein function by S-glutathiolation in response to oxidative and nitrosative stress. Eur J Biochem. 2000;267:4928–4944. doi: 10.1046/j.1432-1327.2000.01601.x. [DOI] [PubMed] [Google Scholar]
- [6].Mallis RJ, Hamann MJ, Zhao W, Zhang T, Hendrich S, Thomas JA. Irreversible thiol oxidation in carbonic anhydrase III: protection by S-glutathiolation and detection in aging rats. Biol Chem. 2002;383:649–662. doi: 10.1515/BC.2002.067. [DOI] [PubMed] [Google Scholar]
- [7].Adachi T, Pimentel DR, Heibeck T, Hou X, Lee YJ, Jiang B, Ido Y, Cohen RA. S-glutathiolation of Ras mediates redox-sensitive signaling by angiotensin II in vascular smooth muscle cells. J Biol Chem. 2004;279:29857–29862. doi: 10.1074/jbc.M313320200. [DOI] [PubMed] [Google Scholar]
- [8].Adachi T, Weisbrod RM, Pimentel DR, Ying J, Sharov VS, Schoneich C, Cohen RA. S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat Med. 2004;10:1200–1207. doi: 10.1038/nm1119. [DOI] [PubMed] [Google Scholar]
- [9].Clavreul N, Adachi T, Pimental DR, Ido Y, Schoneich C, Cohen RA. S-glutathiolation by peroxynitrite of p21ras at cysteine-118 mediates its direct activation and downstream signaling in endothelial cells. Faseb J. 2006;20:518–520. doi: 10.1096/fj.05-4875fje. [DOI] [PubMed] [Google Scholar]
- [10].Salmeen A, Andersen JN, Myers MP, Meng TC, Hinks JA, Tonks NK, Barford D. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature. 2003;423:769–773. doi: 10.1038/nature01680. [DOI] [PubMed] [Google Scholar]
- [11].Lee SR, Kwon KS, Kim SR, Rhee SG. Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J Biol Chem. 1998;273:15366–15372. doi: 10.1074/jbc.273.25.15366. [DOI] [PubMed] [Google Scholar]
- [12].Kim JR, Yoon HW, Kwon KS, Lee SR, Rhee SG. Identification of proteins containing cysteine residues that are sensitive to oxidation by hydrogen peroxide at neutral pH. Anal Biochem. 2000;283:214–221. doi: 10.1006/abio.2000.4623. [DOI] [PubMed] [Google Scholar]
- [13].Winterbourn CC, Metodiewa D. Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radic Biol Med. 1999;27:322–328. doi: 10.1016/s0891-5849(99)00051-9. [DOI] [PubMed] [Google Scholar]
- [14].Ullrich V, Kissner R. Redox signaling: bioinorganic chemistry at its best. J Inorg Biochem. 2006;100:2079–2086. doi: 10.1016/j.jinorgbio.2006.09.019. [DOI] [PubMed] [Google Scholar]
- [15].Woo HA, Jeong W, Chang TS, Park KJ, Park SJ, Yang JS, Rhee SG. Reduction of cysteine sulfinic acid by sulfiredoxin is specific to 2-cys peroxiredoxins. J Biol Chem. 2005;280:3125–3128. doi: 10.1074/jbc.C400496200. [DOI] [PubMed] [Google Scholar]
- [16].Tajc SG, Tolbert BS, Basavappa R, Miller BL. Direct determination of thiol pKa by isothermal titration microcalorimetry. J Am Chem Soc. 2004;126:10508–10509. doi: 10.1021/ja047929u. [DOI] [PubMed] [Google Scholar]
- [17].Berndt C, Lillig CH, Holmgren A. Thiol-based mechanisms of the thioredoxin and glutaredoxin systems: implications for diseases in the cardiovascular system. Am J Physiol Heart Circ Physiol. 2007;292:H1227–1236. doi: 10.1152/ajpheart.01162.2006. [DOI] [PubMed] [Google Scholar]
- [18].Adachi T, Schoneich C, Cohen RA. S-glutathiolation in redox-sensitive signaling. Drug Discovery Today. 2005;2:39–46. [Google Scholar]
- [19].Rao GN, Berk BC. Active oxygen species stimulate vascular smooth muscle cell growth and proto-oncogene expression. Circ Res. 1992;70:593–599. doi: 10.1161/01.res.70.3.593. [DOI] [PubMed] [Google Scholar]
- [20].Sundaresan M, Yu ZX, Ferrans VJ, Irani K, Finkel T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270:296–299. doi: 10.1126/science.270.5234.296. [DOI] [PubMed] [Google Scholar]
- [21].Mohr S, Hallak H, de Boitte A, Lapetina EG, Brune B. Nitric oxide-induced S-glutathionylation and inactivation of glyceraldehyde-3-phosphate dehydrogenase. J Biol Chem. 1999;274:9427–9430. doi: 10.1074/jbc.274.14.9427. [DOI] [PubMed] [Google Scholar]
- [22].Ying J, Tong XY, Pimental DR, Weisbrod RM, Trucillo MP, Adachi T, Cohen RA. Cysteine-674 of the Sarco/Endoplasmic Reticulum Calcium ATPase Is Required for the Inhibition of Cell Migration by Nitric Oxide. Arterioscler Thromb Vasc Biol. 2007 doi: 10.1161/01.ATV.0000258413.72747.23. [DOI] [PubMed] [Google Scholar]
- [23].Clavreul N, Bachschmid MM, Hou X, Shi C, Idrizovic A, Ido Y, Pimentel D, Cohen RA. S-glutathiolation of p21ras by peroxynitrite mediates endothelial insulin resistance caused by oxidized low-density lipoprotein. Arterioscler Thromb Vasc Biol. 2006;26:2454–2461. doi: 10.1161/01.ATV.0000242791.28953.4c. [DOI] [PubMed] [Google Scholar]
- [24].Lii CK, Chai YC, Zhao W, Thomas JA, Hendrich S. S-thiolation and irreversible oxidation of sulfhydryls on carbonic anhydrase III during oxidative stress: a method for studying protein modification in intact cells and tissues. Arch Biochem Biophys. 1994;308:231–239. doi: 10.1006/abbi.1994.1033. [DOI] [PubMed] [Google Scholar]
- [25].Sharov VS, Dremina ES, Galeva NA, Williams TD, Schoneich C. Quantitative mapping of oxidation-sensitive cysteine residues in SERCA in vivo and in vitro by HPLC-electrospray-tandem MS: selective protein oxidation during biological aging. Biochem J. 2006;394:605–615. doi: 10.1042/BJ20051214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wu Y, Kwon KS, Rhee SG. Probing cellular protein targets of H2O2 with fluorescein-conjugated iodoacetamide and antibodies to fluorescein. FEBS Lett. 1998;440:111–115. doi: 10.1016/s0014-5793(98)01415-x. [DOI] [PubMed] [Google Scholar]
- [27].Jaffrey SR, Snyder SH. The biotin switch method for the detection of S-nitrosylated proteins. Sci STKE. 2001;2001:PL1. doi: 10.1126/stke.2001.86.pl1. [DOI] [PubMed] [Google Scholar]
- [28].Forrester MT, Foster MW, Stamler JS. Assessment and application of the biotin switch technique for examining protein S-nitrosylation under conditions of pharmacologically induced oxidative stress. J Biol Chem. 2007;282:13977–13983. doi: 10.1074/jbc.M609684200. [DOI] [PubMed] [Google Scholar]
- [29].Gow AJ, Chen Q, Hess DT, Day BJ, Ischiropoulos H, Stamler JS. Basal and stimulated protein S-nitrosylation in multiple cell types and tissues. J Biol Chem. 2002;277:9637–9640. doi: 10.1074/jbc.C100746200. [DOI] [PubMed] [Google Scholar]
- [30].Persson C, Sjoblom T, Groen A, Kappert K, Engstrom U, Hellman U, Heldin CH, den Hertog J, Ostman A. Preferential oxidation of the second phosphatase domain of receptor-like PTP-alpha revealed by an antibody against oxidized protein tyrosine phosphatases. Proc Natl Acad Sci U S A. 2004;101:1886–1891. doi: 10.1073/pnas.0304403101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Hara MR, Snyder SH. Nitric oxide-GAPDH-Siah: a novel cell death cascade. Cell Mol Neurobiol. 2006;26:527–538. doi: 10.1007/s10571-006-9011-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].West MB, Hill BG, Xuan YT, Bhatnagar A. Protein glutathiolation by nitric oxide: an intracellular mechanism regulating redox protein modification. Faseb J. 2006;20:1715–1717. doi: 10.1096/fj.06-5843fje. [DOI] [PubMed] [Google Scholar]
- [33].Wright SK, Viola RE. Evaluation of methods for the quantitation of cysteines in proteins. Anal Biochem. 1998;265:8–14. doi: 10.1006/abio.1998.2858. [DOI] [PubMed] [Google Scholar]
- [34].Gary E, Means REF. Chemical Modification of Proteins. Holden-Day, Inc; San Francisco: 1971. pp. 217–220. [Google Scholar]
- [35].Nohl H, Stolze K, Weiner LM. Noninvasive measurement of thiol levels in cells and isolated organs. Methods Enzymol. 1995;251:191–203. doi: 10.1016/0076-6879(95)51121-0. [DOI] [PubMed] [Google Scholar]
- [36].Yamashita T, Kawakita M. Reactive sulfhydryl groups of sarcoplasmic reticulum ATPase. II. Site of labeling with iodoacetamide and its fluorescent derivative. J Biochem (Tokyo) 1987;101:377–385. doi: 10.1093/oxfordjournals.jbchem.a121922. [DOI] [PubMed] [Google Scholar]
- [37].Baty JW, Hampton MB, Winterbourn CC. Detection of oxidant sensitive thiol proteins by fluorescence labeling and two-dimensional electrophoresis. Proteomics. 2002;2:1261–1266. doi: 10.1002/1615-9861(200209)2:9<1261::AID-PROT1261>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- [38].Landar A, Oh JY, Giles NM, Isom A, Kirk M, Barnes S, Darley-Usmar VM. A sensitive method for the quantitative measurement of protein thiol modification in response to oxidative stress. Free Radic Biol Med. 2006;40:459–468. doi: 10.1016/j.freeradbiomed.2005.08.046. [DOI] [PubMed] [Google Scholar]
- [39].Jaffrey SR. Detection and characterization of protein nitrosothiols. Methods Enzymol. 2005;396:105–118. doi: 10.1016/S0076-6879(05)96011-4. [DOI] [PubMed] [Google Scholar]
- [40].Yang Y, Loscalzo J. S-nitrosoprotein formation and localization in endothelial cells. Proc Natl Acad Sci U S A. 2005;102:117–122. doi: 10.1073/pnas.0405989102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Dudzinski DM, Igarashi J, Greif D, Michel T. The regulation and pharmacology of endothelial nitric oxide synthase. Annu Rev Pharmacol Toxicol. 2006;46:235–276. doi: 10.1146/annurev.pharmtox.44.101802.121844. [DOI] [PubMed] [Google Scholar]
- [42].Huang B, Chen C. An ascorbate-dependent artifact that interferes with the interpretation of the biotin switch assay. Free Radic Biol Med. 2006;41:562–567. doi: 10.1016/j.freeradbiomed.2006.03.006. [DOI] [PubMed] [Google Scholar]
- [43].Gladwin MT, Wang X, Hogg N. Methodological vexation about thiol oxidation versus S-nitrosation -- a commentary on “An ascorbate-dependent artifact that interferes with the interpretation of the biotin-switch assay”. Free Radic Biol Med. 2006;41:557–561. doi: 10.1016/j.freeradbiomed.2006.05.025. [DOI] [PubMed] [Google Scholar]
- [44].Fratelli M, Demol H, Puype M, Casagrande S, Eberini I, Salmona M, Bonetto V, Mengozzi M, Duffieux F, Miclet E, Bachi A, Vandekerckhove J, Gianazza E, Ghezzi P. Identification by redox proteomics of glutathionylated proteins in oxidatively stressed human T lymphocytes. Proc Natl Acad Sci U S A. 2002;99:3505–3510. doi: 10.1073/pnas.052592699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Sullivan DM, Wehr NB, Fergusson MM, Levine RL, Finkel T. Identification of oxidant-sensitive proteins: TNF-alpha induces protein glutathiolation. Biochemistry. 2000;39:11121–11128. doi: 10.1021/bi0007674. [DOI] [PubMed] [Google Scholar]
- [46].Clavreul N, Dauly C, Huang H, Sethuraman M, McComb M, Costello CE, Cohen RA.Detection and Identification of S-Glutathiolated Proteins in Endothelial Cells Exposed to Oxidants by a Biotin-labeling:Liquid Chromatographic and MS Method J Amer Soc Mass Spectrom 200617Suppl 1121S (ThP 560) [Google Scholar]
- [47].Sethuraman M, McComb ME, Heibeck T, Costello CE, Cohen RA. Isotope-coded affinity tag approach to identify and quantify oxidant-sensitive protein thiols. Mol Cell Proteomics. 2004;3:273–278. doi: 10.1074/mcp.T300011-MCP200. [DOI] [PubMed] [Google Scholar]
- [48].Sethuraman M, Clavreul N, Huang H, McComb ME, Costello CE, Cohen RA. Quantification of oxidative posttranslational modifications of cysteine thiols of p21ras associated with redox modulation of activity using isotope-coded affinity tags and mass spectrometry. Free Radic Biol Med. 2007;42:823–829. doi: 10.1016/j.freeradbiomed.2006.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Sethuraman M, McComb ME, Huang H, Huang S, Heibeck T, Costello CE, Cohen RA. Isotope-coded affinity tag (ICAT) approach to redox proteomics: identification and quantitation of oxidant-sensitive cysteine thiols in complex protein mixtures. J Proteome Res. 2004;3:1228–1233. doi: 10.1021/pr049887e. [DOI] [PubMed] [Google Scholar]
- [50].Zhao C, Sethuraman M, Clavreul N, Kaur P, Cohen RA, O’Connor PB. Detailed map of oxidative post-translational modifications of human p21ras using fourier transform mass spectrometry. Anal Chem. 2006;78:5134–5142. doi: 10.1021/ac060525v. [DOI] [PMC free article] [PubMed] [Google Scholar]