

Abstract
We aimed to correlate pathologic findings with MTM1 mutation type in a series of molecularly defined XLMTM cases. Clinical data from 15 XLMTM patients and their corresponding 16 muscle biopsies were studied. All patients were infants (range: 6 to 217 days old) when initially biopsied. The proportion of myofibers with central nuclei did not correlate with clinical outcome, however, morphometric studies showed that survivors had larger myofiber diameters in infancy than those who died (10.4±3.9 μm versus 8.9±3 μm; p<0.001). As a corollary, patients with MTM1 missense mutations had larger myofiber diameters (11.1±4 μm), than those with truncation/deletion mutations (8.6±2.7 μm) (controls 11.7±2.5 μm) (p<0.0001). These data indicate that differences in myofiber size correlate with MTM1 mutation type and patient outcome. Failure to attain and/or maintain myofiber size, along with fiber type perturbations and the misplacement of myofiber nuclei and other organelles, are important components of XLMTM muscle pathology.
Keywords: centronuclear myopathy, morphometry, MTM1, myotubularin, X-linked myotubular myopathy
INTRODUCTION
X-linked myotubular myopathy (XLMTM) is a severe congenital myopathy that primarily affects males at birth. XLMTM is the best studied of the centronuclear myopathies (CNM) and it is characterized by an increased proportion of hypotrophic myofibers with centrally placed nuclei on muscle biopsy (1–4). Patients are born with severe generalized hypotonia and weakness, and respiratory difficulty that typically requires ventilatory support. In the past, it was thought that the majority of XLMTM patients die in infancy due to respiratory insufficiency (4). Recently, however, it has been recognized that the prognosis may not be so grave, since some patients become independent of the ventilator, and others may only require periodic ventilation such as during sleep. Some patients may even survive to adulthood (5–7).
XLMTM is due to mutations of the MTM1 gene, which encodes myotubularin, a phosphatidylinositol phosphatase (8, 9). Myotubularin is the archetypical member of a family of conserved proteins that characteristically possess a number of sequence motifs including a protein tyrosine phosphatase domain (PTP) and a SET-protein interacting domain (SID) (10, 11). Truncating mutations are generally associated with severe myopathy and early death (12, 13), but some patients with distal C-terminal truncations may have a mild clinical course (14). In contrast, missense mutations are generally associated with a more favorable clinical course, although changes in the PTP or SID motifs can be associated with severe disease (12, 14–16). Much of what we know about myofiber pathology in XLMTM is based on reports of single patients or small series that predate the identification of MTM1, and often included autosomal CNMs (3, 5, 17–28). Therefore, in this study, we sought to better establish the pathologic changes of XLMTM by examining muscle morphology from XLMTM patients with identified MTM1 mutations.
MATERIALS AND METHODS
Enrollment of XLMTM patients was facilitated by the AHB laboratory web site (www.childrenshospital.org/research/beggs) and by referrals from North American neuromuscular clinics. A total of 19 XLMTM patients were enrolled with informed consent and assigned a unique identification number. MTM1 mutation analysis was performed either by our laboratory or by commercial diagnostic laboratories using DNA sequencing of genomic PCR products. The study genetic counselor (JB) collected clinical information by contacting the patient’s neurologist or other referring physician, who completed a questionnaire that included information regarding pregnancy, birth, developmental history, the nature of a patient’s disabilities and disease course. Pathology materials were available for study from 15 of the 19 patients and were acquired by the study neuropathologist (CRP) who requested biopsy material from the pathology department that performed the diagnostic services, which included frozen sections stained with hematoxylin and eosin (H&E) and some histochemical stains. Material used in the evaluation of muscle ultrastructure, i.e., electron micrograph prints and/or grids were available from nine biopsies.
Morphometry was performed on frozen sections by measuring all of the myofibers in consecutive, non-overlapping 400x high-powered fields (hpf) that spanned the largest dimension of the biopsy section on the slide. The smallest dimension passing in a plane through the center of an individual myofiber, in cross section, was recorded (29, 30) using a Nikon Optiphot-2 microscope outfitted with Bioquant Nova version 5.50.8MR morphometry software (Bioquant Image Analysis Corp. Nashville, TN). Three histologically unremarkable, H&E stained frozen quadriceps sections, from age-matched patients with no neuromuscular disease were used as controls. The total number of myofibers counted and mean diameter were recorded. Data were plotted as frequency histograms using Microsoft Excel. Centrally nucleated myofibers were counted in consecutive non-overlapping 400x hpfs that spanned the largest dimension across H&E stained sections. Percent myofibers with central nuclei/hpf were represented as box plots using SigmaPlot 4.00 from Systat Software Inc. (Richmond, California). Chi-square test, t-test and correlation analysis were performed using Intercooled STATA 8 from Stata Corporation (College Station, Texas). Statistical significance was defined as p<0.05 in all tests.
RESULTS
MTM1 mutation status and clinical findings
Eight patients had mutations that truncate or internally delete myotubularin (truncation/deletion group), while seven had missense mutations (Table 1). Three patients had the R69C mutation (93-1, 171-1, 169-1) one of these patients was biopsied twice (171-1a and b). The early clinical period was similar with all patients presenting with severe hypotonia and weakness at birth. Ventilation or supplemental oxygen was required at birth in all of the patients except one (171-1), who had a missense mutation. Four patients (92-1, 206-1, 504-2 and 93-1) were born prematurely (<37 gestational weeks) ranging from 30 to 35 gestational weeks. Five of the eight patients with truncation/deletion mutations, died by six years of age (range five weeks to six years) (Table 1), three of these patients (92-1, 462-1, 592-1) died by three months of age when supportive care was redirected because of ventilator-dependency. The cause of death for the two other patients included hemorrhage from hepatic peliosis (504-2), a recognized complication of XLMTM (7), and unexpected death during sleep (113-1) (Table 1). Two patients with missense changes died one (798-1) had a mutation that involved the SID motif, and he succumbed to pneumonia at five months of age, while another (205-1) died at 2 years of age of unknown cause (Table 1).
Table 1.
Age at biopsy, biopsy site, MTM1 mutation, clinical course, and morphometry data in 15 XLMTM patients.
| ID | Age at Biopsy | Biopsy Site | MTM1 Mutation | Clinical Course | H&E Morphometry Mean±SD (μm) |
|---|---|---|---|---|---|
| 92-1 | 15 d | RQ | c.1467delG
pLys490ArgfsX12 |
Died, 7.5w | 8.7±2.5 |
| 113-1 | 21 d | NR | c.445_678del
p.Pro149-Pro226del loss of exons 7 & 8 |
Died, 2y 3m | 9.5±2.5 |
| 206-1 | 1 m | LQ | c.232-1G>A involves AG acceptor site in intron 4 | Stable, 3 y11m | 7.9±1.7 |
| 262-1 | 7m 1w | RQ | c.444G>C loss of exon 6 | Stable, 8y | ND |
| 447-1 | 6 d | RQ | c.961_962delTT
p.leu321lysfsX10 |
Stable, 4y 1m | 8.3±2.3 |
| 462-1 | 22 d | LQ | c.601delT
p.Leu201PhefsX49 |
Died, 5w | 6.1±1.8 |
| 504-2 | 11 d | LB | c.593_594insA
p.Tyr198X |
Died, 6y 2m | 10.3±2.6 |
| 592-1 | 1m 3w | NR | c.1304_1306delCTC
p.Pro436del |
Died, 3m | 9.7±2.4 |
| 93-1 | 1 m | NR | c.205C>T
p.Arg69Cys |
Stable, 16m | 9.9±2.6 |
| 169-1 | 3m 1w | NR | c.205C>T
p.Arg69Cys |
Stable, 11y 11m | 10.5±2.8 |
| 171-1a | 8 d | LQ | c.205C>T
p.Arg69Cys |
Stable, 14y 10m | 14.9±3.8 |
| 171-1b | 3.3 y | RQ | - | - | 12.0±6.1 |
| 189-1 | <9 m | NR | c.679G>A
p.Val227Met |
Stable, 18y | 8.7±2.8 |
| 205-1 | 1 y | RQ | c.1262G>A
p.ArgR421Gln |
Died, 2y | 9.7±2.9 |
| 765-1 | 2 m | LD | c.779A>C
p.Tyr260Ser |
Stable, 23y | 14.3±3.9 |
| 798-1 | 29 d | RQ | c.1406A>G
p.His469Arg |
Died, 5m | 9.7±3.5 |
Abbreviations: d, days; H&E, hematoxylin and eosin; LB, left biceps; LD, left deltoid; LQ, left quadriceps; m, months, ND, not determined since only fixed H&E stained sections were available; NR, not recorded; RQ, right quadriceps; w, weeks; y, years. For reference, quadriceps myofiber size in unaffected infants is 10–12 μm (31, 32). MTM1 reference sequence NM_000252, with cDNA sequence numbered relative to A of initiator methionine.
The remaining patients are alive with stable disease at ages ranging from 16 months to 23 years (Table 1). Two survivors with truncating mutations are approximately four years old (447-1, 206-1) and both require 24 hour ventilatory support. Only one patient in the truncation/deletion group achieved independent respiration and he is also the only member of this group to walk (262-1). In contrast, four patients with missense mutations require no ventilatory support or supplemental oxygen (169-1, 171-1, 189-1, 765-1), but another has been ventilator-dependent since birth (93-1). Three patients with missense mutations are ambulatory (171-1, 189-1, 765-1).
Biopsy findings
Myopathology
H&E stained sections from all biopsies showed markedly increased variation in myofiber size with numerous small rounded and scattered larger myofibers harboring central nuclei (Figure 1A, B, C). Histopathological examination of all biopsies side-by-side revealed that some had smaller myofibers than others, and occasionally, extremely small myofibers were present (compare Figure 1A and B). Despite the small myofiber size no significant degenerative or regenerative changes were seen (Figure 1A, B, C). Type 1 predominance was noted in four of seven biopsies; that included one patient from the truncation/deletion group (592-1, 79.1% type 1 fibers) and three with missense mutations (169-1, 62.2% type 1 fibers; 171-1a, 62.2% type 1 fibers; 798-1, 72.2% type 1 fibers). No biopsy showed definite type 2 predominance. Frequency histograms showed small type 1 and type 2 fibers with type 1 fibers being more severely affected (data not shown). Electron microscopy showed some myofibers with redundant basal lamina, suggesting that these small myofibers may be atrophic (Figure 1D).
Figure 1.
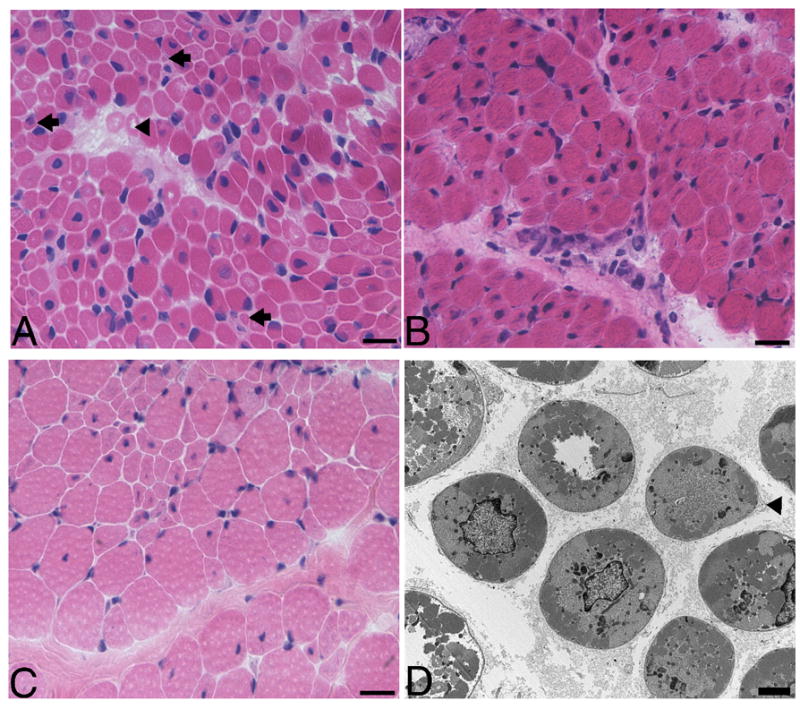
(A) H&E stained frozen section of the quadriceps biopsy performed at 6 days of age from patient 447-1, who has an MTM1 mutation that truncates myotubularin shows scattered centrally nucleated hypotrophic myofibers with the characteristic appearance of myotubes (arrowhead). Note the presence of many extremely small myofibers (arrows). (B) H&E stained frozen sections from patient 171-1a with R69C missense mutation and had a quadriceps biopsy at 8 days. Note that there are no extremely small myofibers like those depicted in A. (C) The contralateral quadriceps was biopsied in patient 171-1b at 3.3 years and it shows a population of considerably larger fibers. (D) Electron micrographs show small rounded myofibers with central nuclei and glycogen, which was qualitatively similar whether a patient had an MTM1 missense mutation or truncation/deletion mutation. Note the redundant basal lamina suggestive of atrophy (arrowhead). The bar is 20 μm in A, B and C and 2 μm in D.
Morphometry
A mean of 639 myofibers (range 268–880) were morphometrically studied in H&E stained sections, from 15 biopsies. Histograms revealed a shift in myofiber size distribution towards smaller values in all patients (Figure 2 A, B) with a range of myofiber size from 6.1±1.8 to 14.9±3.8 μm, which is in keeping with previous reports (17, 18, 20, 21, 26–28). Myofiber size is normally expected to increase with age (31, 32). However, in these biopsies, regression analysis did not show a correlation between mean fiber size and patient age at biopsy in days (r2=0.05; p=0.45). In addition, the patient who was biopsied twice (171-1) showed a decrease in myofiber size, from 14.9±3.8 μm to 12.0±6.1 μm, in the three year interval between biopsies (p<0.0001) (Figure 2A). This prompted us to test potential relationships between myofiber size and outcome, and between myofiber size and MTM1 mutation type. One patient (205-1) was biopsied later, at one year of age, and he was excluded. There was no significant difference in age at initial biopsy between the missense (mean 98 days, range 8 days to less than 9 months) and truncation/deletion (mean 47 days, range 6 days to 7 months) groups (p=0.37) or between patients who died (mean 25 days, range 11 days to 51 days) and those who survived (mean 102 days, 6 days to less than 9 months) (p=0.17). The relationships of myofiber size with MTM1 mutation type and outcome were statistically analyzed using biopsies taken from patients who were less than one year of age at the time of biopsy, because we felt that at this time myofiber size would be less influenced by potential age-related degenerative changes.
Figure 2.
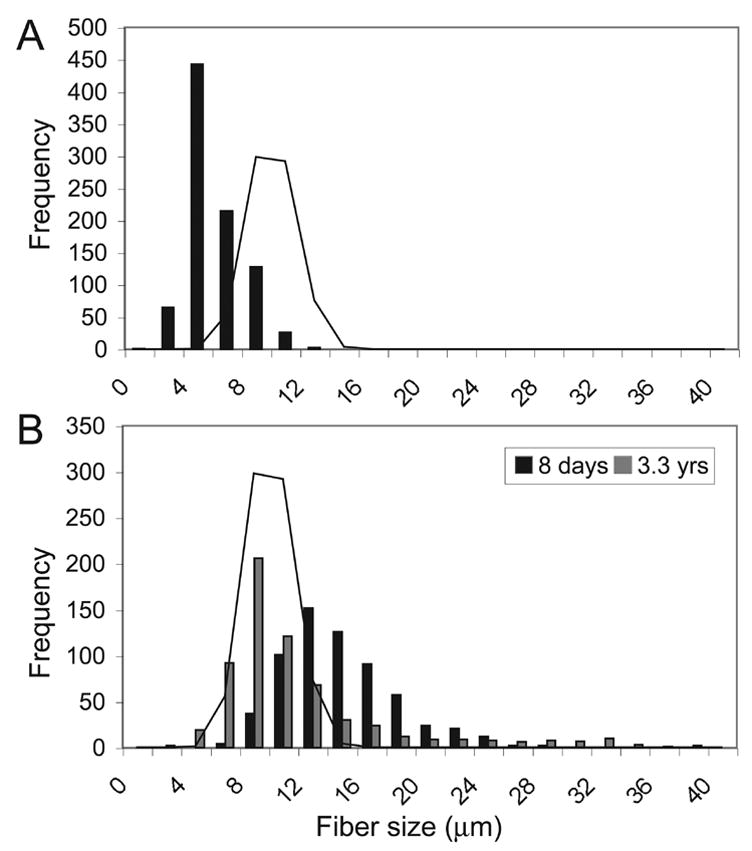
(A) Histogram of morphometry data from patient 462-1, shows a shift toward smaller sized fibers with a mean myofiber size of 6.1±1.8 μm. (B) Histogram from patient 171-1 comparing morphometry data from quadriceps biopsies taken at 8 days of age (black) and at 3.3 years of age (grey). There is an overall decrease in mean myofiber size, from 14.9±3.8 μm to 12.0±6.1 μm, in the interval between biopsies (p<0.0001). Both biopsies contain some large myofibers, but this population is far more prevalent in the second biopsy. Solid line indicates average frequency distribution for a 12 day old normal control biopsy.
Despite overlapping distributions, mean myofiber size differed significantly among those who died (8.9±3 μm), those who survived (10.4±3.9 μm) and controls (11.7±2.5 μm; normal 10–12 μm)(F = 566, p < 0.0001) (Table 1) (31, 32). Patients who died had significantly smaller fibers than survivors (mean difference = 1.5μm, p<0.001) or controls (mean difference = 2.8 μm, p<0.001) and survivors had smaller fibers than controls (mean difference = 1.3 μm, p<0.001). Mean fiber size was 11.1±4 μm in patients with missense mutations and 8.6±2.7 μm in the truncation/deletion mutation group (F = 1024, p < 0.0001) (Table 1) (31, 32). Bonferroni test showed that patients with truncation/deletion mutations had significantly smaller fibers than those with missense mutations (mean difference = 2.5 μm, p<0.001) and controls (mean difference = 3.2μm, p<0.001). In contrast, the difference in mean myofiber size between the missense mutation and control groups was small (mean difference = 0.7μm, p<0.001).
Although these intergroup differences in fiber size were statistically significant, mean myofiber size did not always correlate with outcome on a case-by-case basis. For example, patient 189-1 who is now an ambulatory, ventilator-independent adult had a mean myofiber diameter of only 8.7 μm, while patient 92-1 who had myofibers of similar size, died in infancy. However, the three patients with R69C mutations showed some variability in their clinical courses, the severity of which, correlated with myofiber size. Patient 171-1, who had the largest mean myofiber size of the R69C patients (14.9±3.8 μm), started walking at 2.5 years, and has breathed independently since birth. In contrast, patient 93-1 had the smallest mean myofiber size (9.9±2.6 μm) and he has required 24 hour ventilatory support since birth, and is non-ambulatory, although he is only 16 months old. Patient 169-1 who had an intermediate mean myofiber size (10.5±2.8μm) has an intermediate clinical course; he breathes independently since the first week of life, but has never walked.
Central Nuclei
Myofibers with central nuclei were counted in a mean of 11 hpf/biopsy (range 7–16 hpf/biopsy). The mean percent of central nuclei/hpf was elevated in all of the biopsies (range 4.7%–39.7%), some of which showed wide variability in the number of central nuclei from field-to-field (Figure 3). The mean percent of myofibers with central nuclei was 19.2% in patients with missense mutations (range 6.5%–39.7%), and 11.1% in those with truncation/deletion mutations (range 4.7%–17.1%) (p<0.0001) (Figure 3). There was no correlation with survival (mean 15.5% in survivors versus 13.8% in those who died; p=0.32). The mean percent of central nuclei increased with patient age (p=0.029), although the correlation was not very strong (r2= 0.34) and this increase was probably influenced by two patients who were biopsied at more advanced ages (171-1b, 205-1). The mean percentage of central nuclei increased, from 18.1% to 32.5%, in the interval between biopsies in the patient who was biopsied twice (Figure 3).
Figure 3.
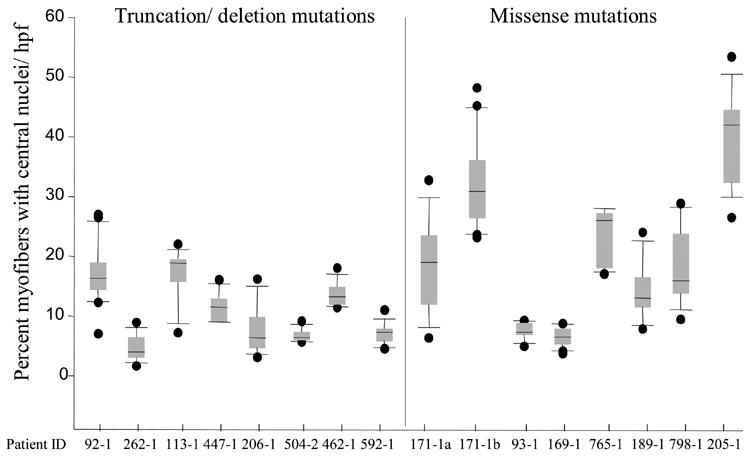
Box plots of percent myofibers with central nuclei/hpf. The mean percent of myofibers with central nuclei ranged from 4.7% to 39.7% with wide variability from field-to-field in some biopsies. The black horizontal line in each box denotes the mean. The top of the box denotes the 75th percentile, while the bottom indicates the 25th percentile. The top bar shows the 95th percentile and the bottom bar shows the 5th percentile. The dots outside of the bars represent outlying data points.
DISCUSSION
This study examined the relationship between histopathologic changes in muscle biopsies and clinical findings in a series of XLMTM patients with confirmed MTM1 mutations. These data are relevant as most of our knowledge of XLMTM pathology comes from studies that predate the identification of MTM1, and some of these studies also included patients with autosomal CNM (3, 5,17–28). We confirm that patients with missense mutations can have a relatively favorable clinical course, while those with truncations tend to have more severe disease (12, 13) and further show that MTM1 mutation type and patient outcome is correlated with mean myofiber size. The early clinical course in XLMTM patients is rather uniform, but in time a divergence occurs with survivors becoming independent of the ventilator, while those who remain ventilator-dependent often die in early childhood. This study indicates that differences in myofiber size could, at least in some patients, underlie part of this divergence in the early clinical course. Patients with larger myofibers tended to have better outcomes, often had missense mutations, and frequently, were able to respire on their own. In contrast, patients who died, tended to have smaller myofibers, often had truncation/deletion mutations and were frequently ventilator-dependent.
Two patients (765-1, 171-1a) with favorable outcomes had mean myofiber sizes that were larger than controls (31, 32). Although, since this is a retrospective study, we can not conclusively rule out that some or even all of this size variation may be due to variation in processing techniques, we feel it is unlikely since both of these, and all the control biopsies, were prepared in the same neuropathology laboratory at Children’s Hospital Boston using identical protocols. Even though myofiber size correlated with clinical course in the three unrelated R69C patients, other patients with comparable mean myofiber sizes had dramatically different outcomes. Therefore, caution is warranted in regard to applying myofiber size as a means of predicting an individual’s outcome and it is unlikely that myofiber size could be utilized as an independent prognostic indicator on a case-by-case basis. The size of our cohort is clearly a limitation of this study. XLMTM is a rare disease so designing sufficiently powered studies that could account for other differences among patients that could potentially impact biopsy findings, such as prematurity, clinical management, or other genetic factors, is difficult. As the presence of small myofibers is not specific to XLMTM, it would be interesting to see if similar studies of other myopathies also detect a correlation between fiber size and outcome.
These data and studies in the Mtm1 knockout mouse (33) support the idea that a component of the pathogenesis of XLMTM may be due to an inability to attain or maintain proper myofiber size. Regression analysis failed to show the expected age-dependent increase in myofiber size as seen in normal growth (31, 32), and, in fact, the patient who was biopsied twice had an overall decrease in mean myofiber size in the interval between biopsies. XLMTM patients with missense mutations had fiber sizes that were comparable to that of controls, or even larger in some cases, possibly due to residual myotubularin activity in these patients (34). Truncating and deletion mutations are more severe MTM1 alterations that likely impair the synthesis of functional myotubularin, and this may account for the smaller fiber sizes in these patients. How myotubularin operates in maintaining myofiber size is unknown, but these observations suggest that a role in growth factor signaling and/or response is a possibility.
The proportion of myofibers with central nuclei reported here is consistent with previous studies (17, 18, 20, 25) and it appeared to increase with age, as previously observed in XLMTM patients (25) and in the Mtm1 knockout mouse (33). Since increases in the proportion of centrally nucleated myofibers is a nonspecific finding that can vary by MTM1 mutation type, muscle type (20), patient age (25), and from microscopic field-to-field (20), designating a particular threshold of central nuclei as diagnostic of XLMTM (or other CNM) is not advisable, especially in light of a recently reported patient with a documented MTM1 mutation with no increase in central nuclei on biopsy (35).
Pathologically, XLMTM is characterized by small myofibers, the misplacement of organelles, and fiber type alterations. Although we found a relationship between myofiber size and disease severity, which correlated with MTM1 mutation type, it is clear that other environmental and/or genetic factors must also play a role in affecting clinical outcome. Larger myofiber size was associated with a better outcome in this study, and although these data can not by nature, show causality, this finding merits further investigation, and suggests that the consideration of therapeutic interventions aimed at increasing myofiber size may be warranted (36).
Acknowledgments
The authors thank Mr. Howard Mulhern and Ms. Lena Liu for their expertise and help with electron microscopy and histochemistry, respectively, and Ms. Elizabeth Taylor for assistance with clinical data collection. We are indebted to all of the patients and referring physicians who kindly provided the biopsy materials and clinical information. This work was supported by National Institute of Health grants RO1 AR44435 and PO1 NS40828, from the NIAMS and NINDS, and especially by generous ongoing support from the Joshua Frase Foundation, the Lee and Penny Anderson Family Foundation, and the Muscular Dystrophy Association (to AHB). CRP is supported by KO8 NS049095-01A1 from the National Institute of Neurological Disorders and Stroke.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Laporte J, Hu LJ, Kretz C, Mandel JL, Kioschis P, Coy JF, et al. A gene mutated in X-linked myotubular myopathy defines a new putative tyrosine phosphatase family conserved in yeast. Nat Genet. 1996;13:175–182. doi: 10.1038/ng0696-175. [DOI] [PubMed] [Google Scholar]
- 2.Pierson CR, Tomczak K, Agrawal P, Moghadaszadeh B, Beggs AH. X-linked myotubular and centronuclear myopathies. J Neuropathol Exp Neurol. 2005;64:555–564. doi: 10.1097/01.jnen.0000171653.17213.2e. [DOI] [PubMed] [Google Scholar]
- 3.Spiro AJ, Shy GM, Gonatas NK. Myotubular myopathy. Persistence of fetal muscle in an adolescent boy. Arch Neurol. 1966;14:1–14. doi: 10.1001/archneur.1966.00470070005001. [DOI] [PubMed] [Google Scholar]
- 4.Wallgren-Pettersson C, Clarke A, Samson F, Fardeau M, Dubowitz V, Moser H, et al. The myotubular myopathies: differential diagnosis of the X linked recessive, autosomal dominant, and autosomal recessive forms and present state of DNA studies. J Med Genet. 1995;32:673–679. doi: 10.1136/jmg.32.9.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barth PG, Van Wijngaarden GK, Bethlem J. X-linked myotubular myopathy with fatal neonatal asphyxia. Neurology. 1975;25:531–536. doi: 10.1212/wnl.25.6.531. [DOI] [PubMed] [Google Scholar]
- 6.Herman GE, Kopacz K, Zhao W, Mills PL, Metzenberg A, Das S. Characterization of mutations in fifty North American patients with X-linked myotubular myopathy. Hum Mutat. 2002;19:114–121. doi: 10.1002/humu.10033. [DOI] [PubMed] [Google Scholar]
- 7.Herman GE, Finegold M, Zhao W, de Gouyon B, Metzenberg A. Medical complications in long-term survivors with X-linked myotubular myopathy. J Pediatr. 1999;134:206–214. doi: 10.1016/s0022-3476(99)70417-8. [DOI] [PubMed] [Google Scholar]
- 8.Blondeau F, Laporte J, Bodin S, Superti-Furga G, Payrastre B, Mandel JL. Myotubularin, a phosphatase deficient in myotubular myopathy, acts on phosphatidylinositol 3-kinase and phosphatidylinositol 3-phosphate pathway. Hum Mol Genet. 2000;9:2223–2229. doi: 10.1093/oxfordjournals.hmg.a018913. [DOI] [PubMed] [Google Scholar]
- 9.Taylor GS, Maehama T, Dixon JE. Inaugural article: myotubularin, a protein tyrosine phosphatase mutated in myotubular myopathy, dephosphorylates the lipid second messenger, phosphatidylinositol 3-phosphate. Proc Natl Acad Sci U S A. 2000;97:8910–8915. doi: 10.1073/pnas.160255697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wishart MJ, Dixon JE. PTEN and myotubularin phosphatases: from 3-phosphoinositide dephosphorylation to disease. Trends Cell Biol. 2002;12:579–585. doi: 10.1016/s0962-8924(02)02412-1. [DOI] [PubMed] [Google Scholar]
- 11.Laporte J, Bedez F, Bolino A, Mandel JL. Myotubularins, a large disease-associated family of cooperating catalytically active and inactive phosphoinositides phosphatases. Hum Mol Genet. 2003;(12 Spec No 2):R285–292. doi: 10.1093/hmg/ddg273. [DOI] [PubMed] [Google Scholar]
- 12.Biancalana V, Caron O, Gallati S, Baas F, Kress W, Novelli G, et al. Characterisation of mutations in 77 patients with X-linked myotubular myopathy, including a family with a very mild phenotype. Hum Genet. 2003;112:135–142. doi: 10.1007/s00439-002-0869-1. [DOI] [PubMed] [Google Scholar]
- 13.McEntagart M, Parsons G, Buj-Bello A, Biancalana V, Fenton I, Little M, et al. Genotype-phenotype correlations in X-linked myotubular myopathy. Neuromuscul Disord. 2002;12:939–946. doi: 10.1016/s0960-8966(02)00153-0. [DOI] [PubMed] [Google Scholar]
- 14.Laporte J, Biancalana V, Tanner SM, Kress W, Schneider V, Wallgren-Pettersson C, et al. MTM1 mutations in X-linked myotubular myopathy. Hum Mutat. 2000;15:393–409. doi: 10.1002/(SICI)1098-1004(200005)15:5<393::AID-HUMU1>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 15.de Gouyon BM, Zhao W, Laporte J, Mandel JL, Metzenberg A, Herman GE. Characterization of mutations in the myotubularin gene in twenty six patients with X-linked myotubular myopathy. Hum Mol Genet. 1997;6:1499–1504. doi: 10.1093/hmg/6.9.1499. [DOI] [PubMed] [Google Scholar]
- 16.Yu S, Manson J, White S, Bourne A, Waddy H, Davis M, et al. X-linked myotubular myopathy in a family with three adult survivors. Clin Genet. 2003;64:148–152. doi: 10.1034/j.1399-0004.2003.00118.x. [DOI] [PubMed] [Google Scholar]
- 17.Ambler MW, Neave C, Singer DB. X-linked recessive myotubular myopathy: II. Muscle morphology and human myogenesis. Hum Pathol. 1984;15:1107–1120. doi: 10.1016/s0046-8177(84)80305-6. [DOI] [PubMed] [Google Scholar]
- 18.Ambler MW, Neave C, Tutschka BG, Pueschel SM, Orson JM, Singer DB. X-linked recessive myotubular myopathy: I. Clinical and pathologic findings in a family. Hum Pathol. 1984;15:566–574. doi: 10.1016/s0046-8177(84)80011-8. [DOI] [PubMed] [Google Scholar]
- 19.Heckmatt JZ, Sewry CA, Hodes D, Dubowitz V. Congenital centronuclear (myotubular) myopathy. A clinical, pathological and genetic study in eight children. Brain. 1985;108:941–964. doi: 10.1093/brain/108.4.941. [DOI] [PubMed] [Google Scholar]
- 20.Helliwell TR, Ellis IH, Appleton RE. Myotubular myopathy: morphological, immunohistochemical and clinical variation. Neuromuscul Disord. 1998;8:152–161. doi: 10.1016/s0960-8966(98)00010-8. [DOI] [PubMed] [Google Scholar]
- 21.Oldfors A, Kyllerman M, Wahlstrom J, Darnfors C, Henriksson KG. X-linked myotubular myopathy: clinical and pathological findings in a family. Clin Genet. 1989;36:5–14. doi: 10.1111/j.1399-0004.1989.tb03360.x. [DOI] [PubMed] [Google Scholar]
- 22.Sarnat HB. Vimentin and desmin in maturing skeletal muscle and developmental myopathies. Neurology. 1992;42:1616–1624. doi: 10.1212/wnl.42.8.1616. [DOI] [PubMed] [Google Scholar]
- 23.Sarnat HB. Myotubular myopathy: arrest of morphogenesis of myofibres associated with persistence of fetal vimentin and desmin. Four cases compared with fetal and neonatal muscle. Can J Neurol Sci. 1990;17:109–123. doi: 10.1017/s0317167100030304. [DOI] [PubMed] [Google Scholar]
- 24.Sarnat HB, Roth SI, Jimenez JF. Neonatal myotubular myopathy: neuropathy and failure of postnatal maturation of fetal muscle. Can J Neurol Sci. 1981;8:313–320. doi: 10.1017/s0317167100043444. [DOI] [PubMed] [Google Scholar]
- 25.Sasaki T, Shikura K, Sugai K, Nonaka I, Kumagai K. Muscle histochemistry in myotubular (centronuclear) myopathy. Brain Dev. 1989;11:26–32. doi: 10.1016/s0387-7604(89)80005-1. [DOI] [PubMed] [Google Scholar]
- 26.Sawchak JA, Sher JH, Norman MG, Kula RW, Shafiq SA. Centronuclear myopathy heterogeneity: distinction of clinical types by myosin isoform patterns. Neurology. 1991;41:135–140. doi: 10.1212/wnl.41.1.135. [DOI] [PubMed] [Google Scholar]
- 27.Silver MM, Gilbert JJ, Stewart S, Brabyn D, Jung J. Morphologic and morphometric analysis of muscle in X-linked myotubular myopathy. Hum Pathol. 1986;17:1167–1178. doi: 10.1016/s0046-8177(86)80423-3. [DOI] [PubMed] [Google Scholar]
- 28.van Wijngaarden GKFP, Bethlem J, Meijer AE. Familial “myotubular” myopathy. Neurology. 1969;19:901–908. doi: 10.1212/wnl.19.9.901. [DOI] [PubMed] [Google Scholar]
- 29.Brooke MH, Engel WK. The histographic analysis of human muscle biopsies with regard to fiber types. 4. Children’s biopsies. Neurology. 1969;19:591–605. doi: 10.1212/wnl.19.6.591. [DOI] [PubMed] [Google Scholar]
- 30.Brooke MH, Engel WK. The histographic analysis of human muscle biopsies with regard to fiber types. 3. Myotonias, myasthenia gravis, and hypokalemic periodic paralysis. Neurology. 1969;19:469–477. doi: 10.1212/wnl.19.5.469. [DOI] [PubMed] [Google Scholar]
- 31.Oertel G. Morphometric analysis of normal skeletal muscles in infancy, childhood and adolescence. An autopsy study J Neurol Sci. 1988;88:303–313. doi: 10.1016/0022-510x(88)90227-4. [DOI] [PubMed] [Google Scholar]
- 32.Vogler C, Bove KE. Morphology of skeletal muscle in children. An assessment of normal growth and differentiation. Arch Pathol Lab Med. 1985;109:238–242. [PubMed] [Google Scholar]
- 33.Buj-Bello A, Laugel V, Messaddeq N, Zahreddine H, Laporte J, Pellissier JF, et al. The lipid phosphatase myotubularin is essential for skeletal muscle maintenance but not for myogenesis in mice. Proc Natl Acad Sci U S A. 2002;99:15060–15065. doi: 10.1073/pnas.212498399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Laporte J, Kress W, Mandel JL. Diagnosis of X-linked myotubular myopathy by detection of myotubularin. Ann Neurol. 2001;50:42–46. doi: 10.1002/ana.1033. [DOI] [PubMed] [Google Scholar]
- 35.de Goede CG, Kelsey A, Kingston H, Tomlin PI, Hughes MI. Muscle biopsy without centrally located nuclei in a male child with mild X-linked myotubular myopathy. Dev Med Child Neurol. 2005;47:835–837. doi: 10.1017/S0012162205001763. [DOI] [PubMed] [Google Scholar]
- 36.Patel K, Amthor H. The function of Myostatin and strategies of Myostatin blockade-new hope for therapies aimed at promoting growth of skeletal muscle. Neuromuscul Disord. 2005;15:117–126. doi: 10.1016/j.nmd.2004.10.018. [DOI] [PubMed] [Google Scholar]