

Abstract
The multidrug resistance proteins P-glycoprotein (P-gp), breast cancer resistance protein (Bcrp), and multidrug resistance-associated protein 2 (Mrp2) are the three major canalicular transport proteins responsible for the biliary excretion of most drugs and metabolites. Previous in vitro studies demonstrated that P-gp transported macrolide antibiotics, including spiramycin, which is eliminated primarily by biliary excretion. Bcrp was proposed to be the primary pathway for spiramycin secretion into breast milk. In the present study, the contributions of P-gp, Bcrp, and Mrp2 to the biliary excretion of spiramycin were examined in single-pass perfused livers of male C57BL/6 wild-type, Bcrp-knockout, and Mrp2-knockout mice in the presence or absence of GF120918 (GW918), a P-gp and Bcrp inhibitor. Spiramycin was infused to achieve steady-state conditions, followed by a washout period, and parameters governing spiramycin hepatobiliary disposition were recovered by using pharmacokinetic modeling. In the absence of GW918, the rate constant governing spiramycin biliary excretion was decreased in Mrp2− knockout mice (0.0013 ± 0.0009 min−1) relative to wild-type mice (0.0124 ± 0.0096 min−1). These data are consistent with the ∼8-fold decrease in the recovery of spiramycin in the bile of Mrp2-knockout mice and suggest that Mrp2 is the major canalicular transport protein responsible for spiramycin biliary excretion. Interestingly, biliary recovery of spiramycin in Bcrp-knockout mice was increased in both the absence and presence of GW918 compared to wild-type mice. GW918 significantly decreased the rate constant for spiramycin biliary excretion and the rate constant for basolateral efflux of spiramycin. In conclusion, the biliary excretion of spiramycin in mice is mediated primarily by Mrp2 with a modest P-gp component.
Spiramycin is used to treat respiratory infections (8, 17). Although spiramycin is associated with fewer severe adverse reactions than other macrolide antibiotics (e.g., erythromycin) (17), cases of spiramycin-induced cholestatic hepatitis and liver injury have been reported (4, 18). The systemic clearance of spiramycin is 1.14 ± 0.27 liters/min in humans (5); renal clearance of spiramycin in humans is low (0.144 ± 0.047 liters/min) relative to nonrenal clearance (0.887 ± 0.096 liters/min) (5). Spiramycin is not extensively metabolized, and biliary excretion is the primary route of spiramycin elimination (2, 7).
Biliary excretion of xenobiotics is mediated primarily by ATP-dependent transport proteins located on the canalicular membrane of hepatocytes, namely, P-glycoprotein (P-gp, encoded by Abcb1), breast cancer resistance protein (Bcrp, encoded by Abcg2), and multidrug resistance-associated protein 2 (Mrp2, encoded by Abcc2) (3). Intracellular accumulation of spiramycin was reduced in murine fibroblast cells transfected with the human MDR1 gene (15); cellular concentrations of spiramycin in MDR1-transfected cells were one-third of that observed in nontransfected cells. Although spiramycin exhibits high penetration into many tissues, it is undetectable in rat or monkey brain (21, 23), which implies that P-gp, Bcrp, or possibly Mrp2 limits spiramycin central nervous system exposure (6, 12).
Given the importance of biliary clearance in the elimination of spiramycin, the present study was designed to elucidate the roles of canalicular transport proteins mediating spiramycin biliary excretion.
MATERIALS AND METHODS
Chemicals.
Spiramycin and rosamicin were purchased from Sigma-Aldrich (St. Louis, MO). Spiramycin was a mixture of spiramycin I (92.6%), II (0.4%), and III (1.1%). Rosamicin had a purity of 98%. The P-gp and Bcrp inhibitor GF120918, N-(4-[2-(1,2,3,4-tetrahydro-6,7-dimethoxy-2-isoquinolinyl)ethyl]-phenyl)-9,10-dihydro-5-methoxy-9-oxo-4-acridine carboxamide] (abbreviated as GW918), was a gift from GlaxoSmithKline (Research Triangle Park, NC). All other reagents were of analytical grade and were readily available from commercial sources.
Animals.
Male C57BL/6 (wild-type), Abcc2−/− (Mrp2-knockout), and Abcg2−/− (Bcrp-knockout) (abbreviated B6, Mrp2KO, and BcrpKO, respectively) mice (24 to 27 g) were gifts from Eli Lilly and Company. Further details regarding the background, generation, and breeding of these mice have been described by Nezasa et al. (14). Mice were housed in an alternating 12-h light-dark cycle with free access to rodent chow and water. All of the animal procedures were approved by The Institutional Animal Care and Use Committee at the University of North Carolina at Chapel Hill.
Single-pass liver perfusion.
Briefly, mice were anesthetized with ketamine and xylazine (140 and 8 mg/kg given intraperitoneally) and maintained at 37°C. The gallbladder was cannulated with PE-10 tubing. The portal vein and inferior vena cava between the liver and the heart were cannulated with a 20G catheter, and the inferior vena cava below the liver was ligated between the kidney and the liver. The liver was perfused in situ in a single-pass manner via the portal vein with Krebs-Henseleit buffer containing 5 μM taurocholate (5-ml/min flow rate, 37°C, buffer saturated with 95% O2 and 5% CO2). The liver was allowed to equilibrate during the 15-min preperfusion period. Subsequently, the liver was perfused for 15 min to achieve steady-state conditions with buffer containing 1 μM spiramycin and 10 μM GW918 or vehicle (dimethyl sulfoxide), followed by a 25-min washout phase during which the liver was perfused with blank buffer. Perfusate outflow was collected in toto at designated time points throughout the 40-min experiment. Bile was collected in toto at 7-min intervals.
Sample analysis.
To prepare samples for analysis, perfusate and bile were diluted 11- and 250-fold, respectively, with methanol-water (50:50 [vol/vol]). Rosamicin was used as the internal standard. Spiramycin and rosamicin concentrations were quantified by reversed-phase high performance liquid chromatography with detection by tandem mass spectrometry (Applied Biosystems API 4000 triple quadrupole with Turbo-IonSpray interface; MDS Sciex, Foster City, CA). After a 10-μl sample injection, analytes were eluted from an Aquasil column (C18, 5 μm, 50 by 2.1 mm; Thermo Electron, Bellafonte, PA) using the following gradient (mobile phase A = 1% formic acid in water; mobile phase B = 1% formic acid in methanol): for 0 to 0.8 min, hold at 30% mobile phase B; for 0.8 to 3.5 min, linear gradient to 85% mobile phase B; for 3.5 to 4 min, hold at 85% mobile phase B; for 4 to 4.1 min, linear gradient to 30% mobile phase B; for 4.1 to 5 min, hold at 30% mobile phase B, with a flow rate of 0.75 ml/min; and for 0.8 to 5 min directed to mass spectrometer. Spiramycin (m/z 843.6→540.4) and rosamicin (m/z 582.4→158.1) were detected in positive ion mode by using multiple reaction monitoring. Consistent with a previous report (19), the spiramycin was stable when stored at 4°C, and assay variations were within acceptable ranges (within ±18% for bile samples and ±13% for perfusate samples) with a lower limit of quantitation of 10 ng/ml.
Pharmacokinetic modeling.
Compartmental modeling by nonlinear least squares regression was used to characterize the hepatobiliary disposition of spiramycin in perfused livers. Various two- and three-compartment models were developed to describe the spiramycin outflow perfusate rate and biliary excretion rate data. The goodness of fit of each model was evaluated based on visual inspection, residual distribution, coefficients of variation on the parameter estimates, the condition number, and Akaike's information criterion (1). The compartment model that best described the data is shown in Fig. 1. The first liver compartment (L1) represented the hepatocellular space, which quickly equilibrates with perfusate. The second (L2) and third (L3) liver compartments were used to describe the hepatocellular distribution and sequestration of spiramycin. Metabolism was assumed not to occur (2, 8). Differential equations derived from the model scheme in Fig. 1 were as follows:
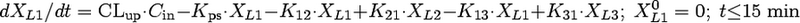 |
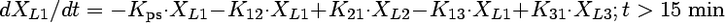 |
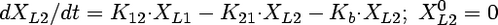 |
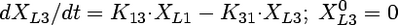 |
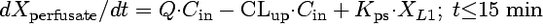 |
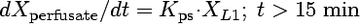 |
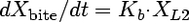 |
where CLup is the hepatic uptake clearance, Kps is the first-order rate constant governing the basolateral excretion of spiramycin from the liver back into perfusate, Kb is the first-order rate constant governing the biliary excretion of spiramycin, and Kij represents the first-order rate constant governing the movement of spiramycin between liver compartments “i” and “j”, respectively; and XLi represents the mass of spiramycin in liver compartment “Li”. Xperfusate and Xbile represent the mass of spiramycin in perfusate and bile, respectively. Simulations of cumulative biliary excretion profiles were conducted.
FIG. 1.

Model scheme depicting spiramycin disposition in the single-pass perfused liver. Q, flow rate; Cin, concentration of spiramycin in inflow perfusate; Cout, concentration of spiramycin in outflow perfusate; CLup, uptake clearance into the liver; Kps, first-order rate constant for basolateral excretion; XL1, XL2, and XL3, amount of spiramycin in liver compartments 1, 2, and 3, respectively; K12 and K21, first-order rate constants for distribution between liver compartments 1 and 2, respectively; K13 and K31, first-order rate constants for distribution between liver compartments 1 and 3, respectively; Kb, first-order rate constant for biliary excretion; Xbile, amount of spiramycin in bile.
Data analysis.
All data are reported as mean ± standard deviation (SD) (n = three animals per group). Statistical significance (P < 0.05) was assessed on raw or log-transformed data, as appropriate to address unequal variance, by two-way analysis of variance with mouse type (Mrp2KO, BcrpKO, or B6) and treatment (vehicle or GW918) as the independent variables.
RESULTS
Recovery of spiramycin.
The total percent recoveries of spiramycin in outflow perfusate and bile at the end of the liver perfusion were 90.5% ± 11.7%, 82.4% ± 12.6%, and 86.5% ± 15.8% of the dose for B6, Mrp2KO, and BcrpKO mice, respectively. There were no significant differences in the total recoveries of spiramycin in B6, Mrp2KO, and BcrpKO mice. The total recovery of spiramycin was not significantly altered by coinfusion of GW918. Spiramycin was concentrated in the bile of B6 and BcrpKO mice. The maximum spiramycin concentrations in bile were 8.65 ± 3.43, 3.45 ± 2.15, and 19.1 ± 2.6 μg/ml, respectively, in control B6, Mrp2KO, and BcrpKO mice; during coinfusion of GW918, the maximum spiramycin concentrations in bile were 7.82 ± 9.12, 1.77 ± 0.74, and 14.0 ± 5.8 μg/ml, respectively. In the absence of GW918, the recovery of spiramycin in the bile of Mrp2KO mice was 19 and 12% of that in B6 and BcrpKO mice, respectively. In the presence of GW918, the recovery of spiramycin in the bile of Mrp2KO mice was 17 and 10% of that in B6 and BcrpKO mice, respectively.
Cumulative excretion of spiramycin in perfusate and bile during the washout phase was analyzed and expressed as a percentage of spiramycin liver content after the 15-min infusion of spiramycin (Table 1). Cumulative biliary excretion of spiramycin decreased ∼8-fold in Mrp2KO and increased approximately 54% in BcrpKO mice compared to B6 mice (Table 1). GW918 significantly decreased the cumulative amount of spiramycin recovered in bile during the 25-min washout phase (Table 1).
TABLE 1.
Spiramycin recovery in perfusate and bile and pharmacokinetic parameter estimates governing spiramycin disposition in single-pass perfused livers from B6, Mrp2KO, and BcrpKO micea
| Parameter | B6 | Mrp2KO | BcrpKO |
|---|---|---|---|
| Spiramycin recovery (% of liver content at end of infusion) | |||
| Perfusate | |||
| Vehicle | 32.5 ± 4.8 | 29.5 ± 6.0 | 28.1 ± 5.6 |
| GW918 | 34.1 ± 4.5 | 24.0 ± 4.8 | 25.2 ± 2.3 |
| Bileb,c | |||
| Vehicle | 9.15 ± 4.15 | 1.21 ± 1.23 | 14.1 ± 2.0 |
| GW918 | 4.36 ± 0.48 | 0.58 ± 0.26 | 8.28 ± 0.80 |
| Pharmacokinetic parameter estimates | |||
| CLup (ml/min) | |||
| Vehicle | 4.63 ± 0.37 | 5.15 ± 0.23 | 4.55 ± 0.44 |
| GW918 | 4.64 ± 0.20 | 4.70 ± 0.14 | 5.03 ± 0.05 |
| Kps (min−1)c | |||
| Vehicle | 0.488 ± 0.178 | 0.420 ± 0.079 | 0.453 ± 0.047 |
| GW918 | 0.317 ± 0.041 | 0.373 ± 0.034 | 0.311 ± 0.014 |
| K12 (min−1) | |||
| Vehicle | 0.0427 ± 0.0151 | 0.0501 ± 0.0317 | 0.0441 ± 0.0200 |
| GW918 | 0.0240 ± 0.0110 | 0.0410 ± 0.0080 | 0.0209 ± 0.0040 |
| K21 (min−1) | |||
| Vehicle | 0.1021 ± 0.0380 | 0.1434 ± 0.1261 | 0.1261 ± 0.0431 |
| GW918 | 0.1012 ± 0.0407 | 0.0679 ± 0.0240 | 0.0933 ± 0.0100 |
| Kb(min−1)b,c | |||
| Vehicle | 0.0124 ± 0.0096 | 0.0013 ± 0.0009 | 0.0270 ± 0.0153 |
| GW918 | 0.0106 ± 0.0011 | 0.0004 ± 0.0001 | 0.0183 ± 0.0054 |
| K13 (min−1) | |||
| Vehicle | 0.1066 ± 0.0618 | 0.2064 ± 0.0894 | 0.1669 ± 0.0781 |
| GW918 | 0.1457 ± 0.0253 | 0.2123 ± 0.0674 | 0.2021 ± 0.0113 |
| K31 (min−1) | |||
| Vehicle | 0.0009 ± 0.0001 | <0.0001 | 0.0022 ± 0.0013 |
| GW918 | 0.0007 ± 0.0012 | <0.0001 | 0.0007 ± 0.0002 |
Values are mean ± SD (n = 3 per group).
P < 0.05, among mouse groups.
P < 0.05, GW918 vs vehicle.
Pharmacokinetic modeling and simulations.
The model scheme depicted in Fig. 1 best described the data. Representative fits of the model to spiramycin biliary excretion rate and outflow perfusate rate versus time data are shown in Fig. 2. Pharmacokinetic parameter estimates are reported in Table 1. The uptake clearance (CLup) approximated the perfusate flow rate (5 ml/min). The basolateral efflux rate constant (Kps) was not altered by knockout of Mrp2 or Bcrp but was significantly decreased by GW918. The biliary excretion rate constant (Kb) was decreased in knockout relative to wild-type mice. In the absence of GW918, Kb in Mrp2KO mice was decreased 10-fold compared to B6 mice (0.0124 ± 0.0096 min−1 versus 0.0013 ± 0.0009 min−1). Although the biliary excretion rate constant in BcrpKO mice was highly variable, it was >2-fold that in B6 mice (0.0124 ± 0.0096 min−1 versus 0.0270 ± 0.0153 min−1). GW918 decreased the biliary excretion rate constant. Based on simulations, the pharmacokinetic model accurately described the biliary excretion of spiramycin (Fig. 3). No statistically significant differences were observed in intercompartmental rate constants between the perfused livers from wild-type and knockout mice in the absence or presence of GW918.
FIG. 2.

Representative fit of the compartmental model (Fig. 1) to spiramycin outflow perfusate rate (○, ng/min) and biliary excretion rate (•, ng/min/g liver) data.
FIG. 3.

Cumulative biliary excretion of spiramycin in mice (▵, B6; ▴, B6+GW918; ○, Mrp2KO; •, Mrp2KO+GW918; □, BcrpKO; ▪, BcrpKO+GW918). Symbols represent means ± the SD (n = three animals per group). Lines represent the simulation of cumulative biliary excretion of spiramycin (in nanograms) with the compartmental model (Fig. 1) and mean parameter estimates (Table 1).
DISCUSSION
Previous pharmacokinetic studies indicated that biliary excretion is the major route of spiramycin elimination (2). Three major canalicular transport proteins (P-gp, Bcrp, and Mrp2) have been implicated in the biliary transport of spiramycin (6, 10, 12, 15, 21). Elucidation of the transport proteins mediating spiramycin biliary excretion is important for the prediction of potential spiramycin-related drug-drug interactions and may contribute to our understanding of the mechanisms of biliary excretion of related macrolide antibiotics.
The importance of Mrp2 in the biliary excretion of organic anions is well documented in Mrp2-deficient rats, Mrp2KO mice, and humans with Dubin-Johnson syndrome (9, 16). In the present study, knockout of Mrp2 caused a 10-fold decrease in the rate constant governing spiramycin biliary excretion, which clearly demonstrated that Mrp2 is the major transport protein responsible for the canalicular excretion of spiramycin into bile. Individuals with impaired Mrp2 function may be susceptible to spiramycin-induced liver injury. Cases of spiramycin-induced cholestatic hepatitis and liver injury have been reported (4, 18); elevated bilirubin levels were observed in these patients.
Bcrp was identified as an antibiotic transporter by using transfected cell lines and BcrpKO mice (13). Unlike Mrp2 and P-gp, Bcrp can transport nitrofurantoin with high efficiency, and the biliary excretion of nitrofurantoin was negligible in BcrpKO mice (13). Milk concentrations of nitrofurantoin were decreased 80-fold in BcrpKO mice; it was postulated that spiramycin is a Bcrp substrate based on its high milk/plasma concentration ratio in mice (10). Surprisingly, BcrpKO mice did not exhibit a decrease in the biliary excretion rate constant of spiramycin compared to wild-type mice. On the contrary, the average biliary excretion rate constant of spiramycin in BcrpKO mice was ∼2-fold higher than in B6 mice in the present study, suggesting that knockout of Bcrp might have indirectly altered the hepatobiliary disposition of spiramycin. This phenomenon also was observed in our previous study in which the biliary excretion of the Mrp2 probe substrate 5(and 6)-carboxy-2′,7′-dichlorofluorescein was significantly increased in BcrpKO mice (14). This increase may be due to stimulation of Mrp2 activity because of the accumulation of Bcrp substrates in the hepatocytes of these knockout mice and/or to increased trafficking of Mrp2 to the canalicular membrane (14).
P-gp was the first mammalian protein identified that transported macrolide antibiotics (20, 22, 24), including spiramycin, in MDR1-expressing cell lines (15). Although GW918 significantly decreased the biliary excretion of spiramycin, based on our present observations, the role of P-gp in the biliary excretion of spiramycin is modest compared to Mrp2. Interestingly, GW918 significantly decreased the rate constant for basolateral efflux of spiramycin consistent with a previous report that GW918 inhibited the basolateral excretion of the opioid peptide [d-Pen2,d-Pen5]-enkephalin (11).
Collectively, multiple transport systems are involved in the biliary clearance of spiramycin in mice. Mrp2 is the major transport protein responsible for the biliary excretion of spiramycin; P-gp serves as a backup transport system in the absence of Mrp2. Bcrp may exert an indirect influence on the biliary excretion of spiramycin.
Acknowledgments
We thank Nicole White and Ken-ichi Nezasa for their expertise in study conduct.
This study was funded by National Institutes of Health grant R01 GM41935 and an Eli Lilly and Company research grant. Knockout mice were created by Deltagen, Inc., San Carlos, CA.
Footnotes
Published ahead of print on 18 June 2007.
REFERENCES
- 1.Akaike, H. 1976. An information criterion. Math Sci. 14:5-9. [Google Scholar]
- 2.Brook, I. 1998. Pharmacodynamics and pharmacokinetics of spiramycin and their clinical significance. Clin. Pharmacokinet. 34:303-310. [DOI] [PubMed] [Google Scholar]
- 3.Chandra, P., and K. L. R. Brouwer. 2004. The complexities of hepatic drug transport: current knowledge and emerging concepts. Pharm. Res. 21:719-735. [DOI] [PubMed] [Google Scholar]
- 4.Denie, C., J. Henrion, M. Schapira, A. Schmitz, and F. R. Heller. 1992. Spiramycin-induced cholestatic hepatitis. J. Hepatol. 16:386. [DOI] [PubMed] [Google Scholar]
- 5.Frydman, A. M., Y. Le Roux, J. F. Desnottes, P. Kaplan, F. Djebbar, A. Cournot, J. Duchier, and J. Gaillot. 1988. Pharmacokinetics of spiramycin in man. J. Antimicrob. Chemother. 22:93-103. [DOI] [PubMed] [Google Scholar]
- 6.Graff, C. L., and G. M. Pollack. 2004. Drug transport at the blood-brain barrier and the choroid plexus. Curr. Drug Metab. 5:95-108. [DOI] [PubMed] [Google Scholar]
- 7.Inoue, A., and T. Deguchi. 1982. The pharmacokinetic studies on spiramycin and acetylspiramycin in rats. Jpn. J. Antibiot. 35:1998-2004. [PubMed] [Google Scholar]
- 8.Jain, R., and L. H. Danziger. 2004. The macrolide antibiotics: a pharmacokinetic and pharmacodynamic overview. Curr. Pharm. Des. 10:3045-3053. [DOI] [PubMed] [Google Scholar]
- 9.Jansen, P. L., W. H. Peter, and W. H. Lamers. 1985. Hereditary chronic conjugated hyperbilirubinemia in mutant rats caused by defective hepatic anion transport. Hepatology 5:573-579. [DOI] [PubMed] [Google Scholar]
- 10.Jonker, J. W., G. Merino, S. Musters, A. E. van Herwaarden, E. Bolscher, E. Wagenaar, E. Mesman, T. C. Dale, and A. H. Schinkel. 2005. The breast cancer resistance protein BCRP (ABCG2) concentrates drugs and carcinogenic xenotoxins into milk. Nat. Med. 11:127-129. [DOI] [PubMed] [Google Scholar]
- 11.Hoffmaster, K. A., M. J. Zamek-Gliszczynski, G. M. Pollack, and K. L. R. Brouwer. 2004. Hepatobiliary disposition of the metabolically stable opioid peptide [d-Pen2,d-Pen5]-enkephalin (DPDPE): pharmacokinetic consequences of the interplay between multiple transport systems. J. Pharmacol. Exp. Ther. 311:1203-1210. [DOI] [PubMed] [Google Scholar]
- 12.Loscher, W., and H. Potschka. 2005. Blood-brain barrier active efflux transporters: ATP-binding cassette gene family. NeuroRx 21:86-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Merino, G., J. W. Jonker, E. Wagenaar, A. E. van Herwaarden, and A. H. Schinkel. 2005. The breast cancer resistance protein (BCRP/ABCG2) affects pharmacokinetics, hepatobiliary excretion, and milk secretion of the antibiotic nitrofurantoin. Mol. Pharmacol. 67:1758-1764. [DOI] [PubMed] [Google Scholar]
- 14.Nezasa, K., X. Tian, M. J. Zamek-Gliszczynski, N. J. Patel, T. J. Raub, and K. L. R. Brouwer. 2006. Altered hepatobiliary disposition of 5 (and 6)-caboxy-2′,7′-dichlorofluorescein in Abcg2 (Bcrp1) and Abcc2 (Mrp2) knockout mice. Drug Metab. Dispos. 34:718-723. [DOI] [PubMed] [Google Scholar]
- 15.Nichterlein, T., M. Kretschmar, A. Schadt, A. Meyer, A. Wildfeuer, H. Laufen, and H. Hof. 1998. Reduced intracellular activity of antibiotics against Listeria monocytogenes in multidrug resistant cells. Int. J. Antimicrob. Agents 10:119-125. [DOI] [PubMed] [Google Scholar]
- 16.Nies, A. T., and D. Keppler. 2007. The apical conjugate efflux pump ABCC2 (MRP2). Pflugers Arch. 453:643-659. [DOI] [PubMed] [Google Scholar]
- 17.Rubinstein, E., and N. Keller. 1998. Spiramycin renaissance. J. Antimicrob. Chemother. 42:572-576. [DOI] [PubMed] [Google Scholar]
- 18.Saab, Y. B., and M. Mroueh. 2002. Spiramycin-induced liver injury. Ann. Pharmacother. 36:1972. [DOI] [PubMed] [Google Scholar]
- 19.Sagan, C., A. Salvador, D. Dubreuil, P. P. Poulet, D. Duffaut, and I. Brumpt. 2005. Simultaneous determination of metronidazole and spiramycin I in human plasma, saliva and gingival crevicular fluid by LC-MS/MS. J. Pharm. Biomed. Anal. 38:298-306. [DOI] [PubMed] [Google Scholar]
- 20.Saito, H., Y. Fukasawa, Y. Otsubo, K. Yamada, H. Sezaki, and S. Yamashita. 2000. Carrier-mediated transport of macrolide antimicrobial agents across Caco-2 cell monolayers. Pharm. Res. 17:761-765. [DOI] [PubMed] [Google Scholar]
- 21.Schoondermark-Van de Ven, E., J. Galama, W. Camps, T. Vree, F. Russel, J. Meuwissen, and W. Melchers. 1994. Pharmacokinetics of spiramycin in the rhesus monkey: transplacental passage and distribution in tissue in the fetus. Antimicrob. Agents Chemother. 38:1922-1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schuetz, E. G., Y. Yasuda, K. Arimori, and J. D. Schuetz. 1998. Human MDR1 and mouse mdr1a P-glycoprotein alter the cellular retention and disposition of erythromycin, but not of retinoic acid or benzo(a)pyrene. Arch. Biochem. Biophys. 350:340-347. [DOI] [PubMed] [Google Scholar]
- 23.Shi, X. G., Y. M. Sun, Y. F. Zhang, and D. F. Zhong. 2004. Tissue distribution of bitespiramycin and spiramycin in rats. Acta Pharmacol. Sin. 25:1396-1401. [PubMed] [Google Scholar]
- 24.Takano, M., R. Hasegawa, T. Fukuda, R. Yumoto, J. Nagai, and T. Murakami. 1998. Interaction with P-glycoprotein and transport of erythromycin, midazolam, and ketoconazole in Caco-2 cells. Eur. J. Pharm. 358:289-294. [DOI] [PubMed] [Google Scholar]