

Abstract
Flucloxacillin is often used for the treatment of serious infections due to sensitive staphylococci. The pharmacokinetic (PK)-pharmacodynamic (PD) breakpoint of flucloxacillin has not been determined by the use of population PK. Targets based on the duration of non-protein-bound concentrations above the MIC (fT>MIC) best correlate with clinical cure rates for beta-lactams. We compared the breakpoints for flucloxacillin between several dosage regimens. In a randomized, two-way crossover study, 10 healthy volunteers received 500 mg and 1,000 mg flucloxacillin as 5-min intravenous infusions. Drug concentrations were determined by high-pressure liquid chromatography. We used the programs WinNonlin for noncompartmental analysis and statistics and NONMEM for population PK and Monte Carlo simulation. We compared the probability of target attainment (PTA) for intermittent- and continuous-dosage regimens based on the targets of fT>MICs of ≥50% and ≥30% of the dosing interval. The clearance and the volume of distribution were very similar after the administration of 500 mg and 1,000 mg flucloxacillin. We estimated renal and nonrenal clearances of 5.37 liters/h (coefficient of variation, 19%) and 2.73 liters/h (33%). For near maximal killing (target, fT>MIC of ≥50%) flucloxacillin showed a robust (≥90%) PTA up to MICs of 0.75 to 1 mg/liter (PTA of 86% at 1 mg/liter) for a continuous or a prolonged infusion of 6 g/day. Short-term infusions of 6 g/day had a lower breakpoint of 0.25 to 0.375 mg/liter. The flucloxacillin PK was linear for doses of 500 mg and 1,000 mg. Prolonged and continuous infusion at a 66% lower daily dose achieved the same PK-PD breakpoints as short-term infusions. Prolonged infusion and continuous infusion are appealing options for the treatment of serious infections caused by sensitive staphylococci.
Flucloxacillin is an isoxazolyl penicillin and is active against many gram-positive bacteria, including penicillinase-producing staphylococci and streptococci, but not against methicillin-resistant Staphylococcus aureus (20). In the United Kingdom flucloxacillin remains the predominantly prescribed antistaphylococcal oral antibiotic (32). It is typically used for the treatment of skin and soft tissue infections and respiratory tract infections. For serious infections like endocarditis or osteomyelitis caused by methicillin-susceptible S. aureus (MSSA) it is administered intravenously as a slow injection, a short-term infusion, or a continuous infusion. It has been reported that limiting the use of glycopeptides could help to control the emergence of resistance to vancomycin (10, 21). Therefore, it might be preferable to use alternatives like flucloxacillin instead of glycopeptides against MSSA, if the use of the alternative grants a sufficient probability for a successful clinical outcome. In addition, flucloxacillin has been suggested to show more rapid killing of MSSA than vancomycin, and flucloxacillin levels usually do not need to be monitored (23).
The clinical outcome for treatment with beta-lactams is related to the proportion of the dosing interval for which non-protein-bound plasma concentrations exceed the MIC (fT>MIC) (12, 17). Attainment of the pharmacokinetic (PK)-pharmacodynamic (PD) target of an fT>MIC of ≥50% correlates best with the near maximal bactericidal activity of penicillins and is often used as a surrogate marker for successful clinical outcome (2, 11-13, 17). For drugs with short half-lives, like beta-lactams, prolonged and continuous infusions have been shown to achieve a longer fT>MIC than short-term infusions at the same daily dose (15, 18). Therefore, prolonged or continuous infusions may require lower daily doses compared to those required for intermittent treatment while achieving the same probability of target attainment (PTA) achieved with high-dose intermittent treatment.
Conditions like endocarditis and osteomyelitis usually require prolonged high-dose treatment. Continuous-infusion treatment can be managed at patients’ homes better than frequent intermittent infusions (every 6 h [q6h] or q4h) and has been reported to be efficacious for use for the completion of treatment for serious staphylococcal infections at patients’ homes (22, 23). Flucloxacillin solutions have been reported to be stable in water for injection for 7 days at 20 to 25°C (26). However, the optimal doses for continuous and prolonged infusion of flucloxacillin have not yet been determined by population PK and Monte Carlo simulation (MCS). In the absence of these data, some authors have reported that they administered by continuous infusion the same daily doses that they used for intermittent treatment (22, 23).
We used population PK and MCS to investigate the differences in the PK-PD target attainment between intermittent and continuous infusions. To study dosage regimens by MCS with various doses or other modes of administration, or both, it is important to know whether the clearance changes with the concentration in plasma at therapeutic concentrations. Therefore, our second objective was to compare the PK of flucloxacillin at two different dose levels.
MATERIALS AND METHODS
Study participants.
Ten healthy Caucasian subjects (five males and five females) participated in the study. Prior to entry into the study, all subjects were given a physical examination, electrocardiography, and laboratory tests, including urinalysis and screening for drugs of abuse. During the drug administration periods, the volunteers were encouraged to report any discomfort or adverse reactions and were closely observed by physicians. On each day of the study the subjects were asked to complete a questionnaire on their health status. The study was approved by the local ethics committee, and all subjects gave their written informed consent prior to starting the study.
Study design and drug administration.
The study was of a randomized, controlled, two-way crossover design. In each of the two study periods, each subject received a single dose of 500 mg or 1,000 mg flucloxacillin as a 5-min intravenous infusion. Food and fluid intakes were strictly standardized on each study day. The treatment periods were separated by a washout period of at least 4 days. The subjects were requested to abstain from consuming alcohol and caffeine-containing foods and beverages during the study periods.
Sampling schedule.
All blood samples were drawn from a forearm vein via an intravenous catheter placed on the forearm contralateral to the one used for drug administration. Blood samples were drawn immediately before the start of infusion; at the end of infusion; as well as at 5, 10, 15, 20, 25, 45, 60, 75, and 90 min and 2, 2.5, 3, 3.5, 4, 5, 6, 8, and 24 h after the end of the infusion. The samples were cooled in an ice-water bath before centrifugation. After centrifugation the plasma samples were immediately frozen and stored at −80°C until analysis. Urine samples were collected immediately before the start of the infusion, from the start of the infusion until 1 h after the end of the infusion, and at the following time intervals: 1 to 2, 2 to 3, 3 to 4, 4 to 5, 5 to 6, 6 to 8, 8 to 12, and 12 to 24 h after the end of the infusion. The urine samples were stored at 4°C during the collection period. The amount and pH of the urine were measured. Urine samples were immediately frozen and were stored at −80°C before analysis.
Determination of plasma and urine concentrations.
For determination of the flucloxacillin concentration in plasma, 100 μl of the sample was deproteinized with 200 μl acetonitrile containing the internal standard. After mixing of the solution and centrifugation at 15,000 rpm, 40 μl was injected onto a high-pressure liquid chromatography system. For determination of the flucloxacillin concentration in urine, 20 μl of the sample was diluted with 180 μl water. After the sample and water were mixed, 40 μl was injected onto the high-pressure liquid chromatography system. The flucloxacillin concentration was determined by using a reversed-phase column and a potassium dihydrogen phosphate (pH 6.2)-acetonitrile mobile phase at a flow of 2 ml/min. Flucloxacillin and the internal standard were detected at 220 nm.
The concentrations in the plasma and urine samples were measured against a plasma or urine calibration curve. The calibration row in plasma was prepared with a 10:1 dilution of tested drug-free plasma with a stock solution of flucloxacillin to obtain the highest calibration level. The other calibration levels were obtained by 1:1 dilution of the highest calibration level or a level of higher concentration with drug-free plasma. The calibration curve for urine was prepared with a 10:1 dilution of tested drug-free urine with a stock solution to obtain the highest calibration level. The other calibration levels were obtained by 1:1 dilution of the highest calibration level or a higher concentration with drug-free urine. For the control of inter- and intra-assay variations, spiked quality controls in plasma and urine were prepared by adding defined volumes of the stock solution of flucloxacillin or the spiked control of a higher concentration to defined volumes of tested drug-free plasma or urine.
No interferences were observed in plasma and urine for flucloxacillin or the internal standard. Calibration was performed by linear regression. The linearity of the flucloxacillin calibration curves in plasma and urine was demonstrated from 0.500 to 250 mg/liter and 5.00 to 400 mg/liter, respectively. The quantification limits were identical to the lowest calibration levels. The interday precision and the analytical recovery of the spiked quality control standards of flucloxacillin in human plasma ranged from 4.1 to 7.7% and from 84.9 to 106.0%, respectively. The interday precision and the analytical recovery of the spiked quality control standards of flucloxacillin in human urine ranged from 3.3 to 5.1% and from 100.0 to 103.0%, respectively. The intraday precision and the analytical recovery of the spiked quality control standards of flucloxacillin in human plasma ranged from 3.9 to 7.0% and from 85.1 to 107.0%, respectively. The intraday precision and the analytical recovery of the spiked quality control standards of flucloxacillin in human urine ranged from 2.9 to 5.0% and from 99.0 to 103.2%, respectively.
Pharmacokinetics. (i) Noncompartmental analysis.
The maximum concentration in plasma for each subject was read directly from the plasma concentration-time curves. The area under the plasma concentration-time curve for each subject was calculated by using the linear up/log down method, as implemented in WinNonlin Professional (version 4.0.1) (30). This algorithm uses the linear trapezoidal rule when concentrations are increasing or constant and the log-trapezoidal rule when concentrations are decreasing.
(ii) Population PK analysis.
We considered models with one, two, or three disposition compartments and (i) first-order elimination, (ii) mixed-order (Michaelis-Menten) elimination, or (iii) parallel first-order and mixed-order elimination. The differential equations of our final model are as follows:
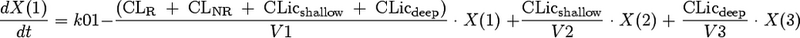 |
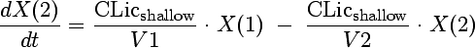 |
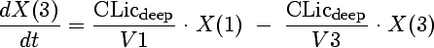 |
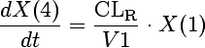 |
where X(1), X(2), and X(3) are the amounts of flucloxacillin in the central, shallow peripheral, and deep peripheral compartments, respectively; X(4) is the cumulative amount excreted unchanged in urine; CLR is renal clearance; CLNR is nonrenal clearance; CLicshallow is the intercompartmental clearance between the central and the shallow peripheral compartment; and CLicdeep is the intercompartmental clearance between the central and the deep peripheral compartment. Drug input was described by a time-delimited zero-order input process with rate k01. The analytical solution to these linear differential equations, as implemented in NONMEM (ADVAN 11 and TRANS 4), was used for estimation.
Competing models were discriminated by their predictive performance, as assessed by visual predictive checks and the use of the NONMEM objective function and residual plots.
For the visual predictive check, we simulated the plasma and urine profiles for 10,000 subjects by using the competing models. From these data we calculated the median, the nonparametric 80% prediction interval (10th to 90th percentiles), and the nonparametric 50% prediction interval (25th to 75th percentiles) for the predicted concentrations in plasma and the amounts in urine. These prediction interval lines were then overlaid on the original raw data. If the model described the data correctly, then 20% of the observed data should fall outside the 80% prediction interval at each time point and 50% of the data should fall outside the interquartile range. We compared the median predicted concentrations and the prediction intervals with the raw data and tested whether the median and the prediction intervals mirrored the central tendency and the variability of the raw data for the respective model.
(iii) Individual PK model.
We estimated the between-subject variability (BSV) for all parameters except the duration of the zero-order input and intercompartmental clearances. We assumed a log-normal distribution for the PK parameters and used a full variance-covariance matrix to describe the variability of the PK parameters as well as their pairwise correlations. The NONMEM program estimates BSV as variance. For more convenient interpretation, we report the square root of the variance for BSV, as the square root of the variance is an approximation of the apparent coefficient of variation of a normal distribution on a log scale.
(iv) Observation model.
We described the residual unidentified variability by using a combined additive and proportional error model for the concentrations in plasma and the amounts excreted in urine.
(v) Computation.
We used the first-order conditional estimation method with the interaction estimation option in NONMEM (version V, release 1.1; NONMEM Project Group, University of California, San Francisco) (6) for all population PK modeling and WinNonlin Professional (version 4.0.1; Pharsight Corp., Mountain View, CA) (30) for noncompartmental analysis and equivalence statistics.
(vi) MCS.
We used fT>MIC for at least 30% or 50% of the dosing interval as PK-PD targets for flucloxacillin. An fT>MIC of ≥50% is the target for the near maximal bactericidal activity of penicillins, and an fT>MIC of ≥30% is the target for bacteriostasis (2, 11, 12, 14, 17). We calculated the PTA within the MIC range from 0.0625 to 16 mg/liter, and as protein binding was not measured in the current study, we used the protein binding of 96% for flucloxacillin that has been reported in the literature (7, 8).
We compared three dosage regimens at a daily dose of 6 g: (i) continuous infusion, (ii) prolonged (4 h) infusion of 2 g q8h, and (iii) a short-term (0.5 h) infusion of 1.5 g q6h. In addition, we simulated 12 g flucloxacillin daily as 0.5- h infusions of 2 g q4h. We simulated the plasma concentration time profiles of 10,000 virtual subjects for each dosage regimen at steady-state in absence of residual error with NONMEM. The fT>MIC values and the PTA were calculated by linear interpolation between simulated concentrations (frequent sampling) with Perl scripts, written by J. Bulitta. These Perl scripts were validated for all dosage regimens studied for this and several other studies.
We used the population mean PK parameter estimates and the variance-covariance matrix representing the BSV estimated by NONMEM for MCS and assumed that the dose, duration of infusion, and timing of infusion had no variability. We used the clearance and volume notation of our population PK model during both estimation and MCS and assumed a normal distribution of the PK parameters on a log scale, as described above.
We derived the PTA for each of the two targets by calculating the fraction of subjects who attained the target at each MIC. The PK-PD breakpoint was defined as the highest MIC for which the PTA was at least 90%. To put these values into clinical perspective, the expectation value for the population PTA, i.e., the expected population PTA for a specific dosage regimen and a specific population of microorganisms, was calculated.
Statistical analysis.
We tested the noncompartmental parameter estimates for differences between treatments (500 mg and 1,000 mg flucloxacillin). We used analysis of variance statistics on a log scale and an α level of significance of 0.05.
RESULTS
Demographics.
All 10 subjects completed the study. The median weight was 71 kg (range, 52 to 83 kg), the median height was 178 cm (range, 165 to 190 cm), and the median age was 25 years (range, 23 to 34 years). The subjects had normal renal and hepatic functions.
Noncompartmental analysis.
The flucloxacillin concentrations in plasma and the amounts in urine after infusion of 500 mg and 1,000 mg are shown in Fig. 1. Table 1 shows the results of the noncompartmental analysis. The peak concentrations and the areas under the curve were dose linear. All other PK parameters were very similar at both dose levels.
FIG. 1.

Average ± standard deviation profiles of flucloxacillin in healthy volunteers after 5-min infusions of 500 mg and 1,000 mg flucloxacillin.
TABLE 1.
Pharmacokinetic parameters for 500 mg and 1,000 mg flucloxacillin from noncompartmental analysis
| Parameter | Geometric mean (% CV)
|
Point estimate (90% confidence interval) for 1,000 mg/500 mg | P value | |
|---|---|---|---|---|
| 500 mg | 1,000 mg | |||
| Total body clearance (liters/h) | 8.16 (21) | 8.18 (20) | 100 (94-107) | 0.94 |
| Renal clearance (liters/h) | 5.37 (22) | 5.29 (22) | 99 (90-108) | 0.774 |
| Nonrenal clearance (liters/h) | 2.72 (30) | 2.79 (33) | 103 (88-119) | 0.766 |
| Fraction excreted unchanged in urine | 0.66 (10) | 0.65 (12) | 98 (93-104) | 0.62 |
| Volume of distribution at steady state (liters) | 9.63 (15) | 9.97 (17) | 104 (96-112) | 0.44 |
| Peak plasma concentration (mg/liter) | 86.8 (13) | 167 (16) | 192 (175-211) | <0.01 |
| Terminal half-life (h) | 1.40 (26) | 1.62 (25) | 116 (87-153) | 0.363 |
| Mean residence time (h) | 1.18 (19) | 1.22 (14) | 103 (97-110) | 0.364 |
Population PK.
The three-compartment model had a 200-point better objective function and a better predictive performance than the two-compartment model. A one-compartment model was inappropriate for our data set. The parameter estimates for the three-compartment model are shown in Table 2, and the variance-covariance matrix is shown in Table 3. Figure 2 shows the visual predictive checks for this model. There was no indication that clearance decreased for the increase in dose from 500 mg to 1,000 mg. The distributions of subjects with lower and higher individual estimates for the 500-mg dose than for the 1,000-mg dose were five and five for total clearance; five and five for renal clearance; and five and five for nonrenal clearance. Thus, there was no trend of any saturation of clearance at these dose levels. Three-compartment models with mixed-order or parallel first-order and mixed-order elimination yielded no significant improvement in the objective function compared to that achieved with the model with first-order elimination, and the parameter estimates of models with saturable elimination indicated no or negligible saturation at the dose levels studied. We selected the three-compartment model with first-order elimination as our final model, as it had excellent predictive performance and as the models with saturable elimination neither improved the objective function significantly nor improved the predictive performance.
TABLE 2.
Population parameter estimates
| Parametera | Unit | Estimated valueb |
|---|---|---|
| CLTc | liters h−1 | 8.10 |
| CLR | liters h−1 | 5.37 (19) |
| CLNR | liters h−1 | 2.73 (33) |
| Vssc | liters | 9.57 |
| V1 | liters | 4.79 (17) |
| V2 | liters | 2.61 (37) |
| V3 | liters | 2.17 (15) |
| CLicshallowd | liters h−1 | 15.3 |
| CLicdeepd | liters h−1 | 1.23 |
| Tk0 (fixed) | min | 5 |
| CVC | % | 9.4 |
| SDC | mg liter−1 | 0.155 |
| CVAU | % | 20.9 |
| SDAU | mg | 1.04 |
CLT, total clearance; CLR, renal clearance; CLNR, nonrenal clearance; Vss, volume of distribution at steady-state; V1, volume of distribution for the central compartment; V2, volume of distribution for the shallow peripheral compartment; V3, volume of distribution for the deep peripheral compartment; CLicshallow, intercompartmental clearance between the central and the shallow peripheral compartment; CLicdeep, intercompartmental clearance between the central and the deep peripheral compartment; Tk0, duration of zero-order input (not estimated); CVC, proportional residual error component for the plasma concentrations; SDC, additive residual error component for the plasma concentrations; CVAU, proportional residual error component for the amounts excreted in urine; SDAU, additive residual error component for the amounts excreted in urine.
The values for the structural PK parameters are geometric means (between-subject coefficients of variation).
Derived from parameter estimates (not estimated).
No between subject variability included for distributional clearance.
TABLE 3.
Variance-covariance matrix (on natural log scale) for flucloxacillina
| Parameter | Variance-covariance
|
||||
|---|---|---|---|---|---|
| CLR | CLNR | V1 | V2 | V3 | |
| CLR | 0.0343 | ||||
| CLNR | 0.0124 | 0.112 | |||
| V1 | 0.00488 | 0.00551 | 0.0282 | ||
| V2 | 0.0415 | 0.0641 | 0.00168 | 0.138 | |
| V3 | 0.0172 | 0.00804 | 0.0175 | 0.0351 | 0.023 |
Log-normal distributions of the PK parameters were assumed. We did not include the variability of the intercompartmental clearances in our parameter variability model, as estimation of these variance and covariance terms did not significantly improve the objective function and did not improve the predictive performance shown in Fig. 2. See footnote a of Table 2 for the definitions of the parameters.
FIG. 2.

Visual predictive check for concentrations in plasma and amounts excreted unchanged in urine: The plots show the raw data, the 80% prediction interval (10th to 90th percentile), and the interquartile range (25th to 75th percentile). Ideally, 50% of the raw data should fall inside the interquartile range at each time point and 80% of the raw data should fall inside the 80% prediction interval. The coefficient of correlation for the observed versus the individual predicted concentrations was 0.987.
MCSs.
We compared the PTA versus MIC profiles for three dosage regimens with a daily dose of 6 g and for one dosage regimen at a daily dose of 12 g at two PK-PD targets (Fig. 3). The PK-PD breakpoints for the targets of fT>MIC of ≥50% (near maximal bactericidal activity of penicillins) and fT>MIC of ≥30% (bacteriostasis) are shown in Table 4.
FIG. 3.

Probabilities of target attainment for different dosage regimens and PK-PD targets of flucloxacillin. Symbols: ▴, daily dose of 6 g flucloxacillin, continuous infusion; □, daily dose of 6 g flucloxacillin, 2 g q8h as a 4-h infusion; ⋄, daily dose of 6 g flucloxacillin, 1.5 g q6h as a 0.5-h infusion; •, daily dose of 12 g flucloxacillin, 2 g q4h as a 0.5-h infusion.
TABLE 4.
Breakpoints of various dosage regimens for flucloxacillin
| Dosage regimen | Daily dose (g) | Breakpoint (mg/liter) for fT>MIC of:
|
|
|---|---|---|---|
| ≥50% | ≥30% | ||
| 2 g q8h as 4-h infusions | 6 | 0.75 (86a) | 1.5 |
| 6 g as continuous infusion | 6 | 0.75 (86a) | 0.75 (86a) |
| 1.5 g q6h as 0.5-h infusions | 6 | 0.25 (79b) | 0.75 (83a) |
| 2 g q4h as 0.5-h infusions | 12 | 1 | 2 |
Values in parentheses are the PTA (in percent) at 1 mg/liter.
Value in parentheses is the PTA (in percent) at 0.375 mg/liter.
DISCUSSION
Flucloxacillin is indicated for the treatment of infections due to susceptible gram-positive organisms, including beta-lactamase-producing staphylococci and streptococci. It is used intravenously for the treatment of serious infections caused by MSSA. In a randomized comparison trial with flucloxacillin (1 g q6h) and teicoplanin (6 mg/kg q12h for three doses and then the same dose given once daily) for the treatment of burn wound infections due to gram-positive pathogens, no significant differences in the clinical and the microbiological success rates between flucloxacillin and teicoplanin were found (33). However, the rate of the emergence of resistance to glycopeptides is increasing, and restriction of their use has been reported to be helpful in controlling vancomycin-resistant enterococci (10, 21). Therefore, it may be preferable to use alternatives like flucloxacillin against MSSA whenever possible in order to postpone and limit the use of glycopeptides.
The PK of flucloxacillin was studied by Nauta and Mattie (29) during a 3-h infusion of 1.2 g/h (loading dose, 1 g) in healthy volunteers. They reported average total, renal, and nonrenal clearances of 7.4, 5.3, and 2.1 liters/h, respectively. Adam et al. (1) found an average total clearance of 7.1 liters/h in healthy volunteers after 1 g flucloxacillin was given as a 5-min intravenous infusion. Our results are in good agreement with those clearances.
For beta-lactams like flucloxacillin, the clinical and microbiological outcomes best correlate with fT>MIC (12, 17). At the same daily dose, prolonged or continuous infusions achieve a longer fT>MIC than intermittent short-term infusions. This is especially evident for drugs with short half-lives, like beta-lactams. Consequently, Drusano (18) proposed the use of prolonged infusions to optimize the PTAs of carbapenems.
We used population PK analysis and MCS to compare the PTAs of short-term, prolonged, and continuous infusions for flucloxacillin. MCS was shown to be a very useful tool for rational dose selection for phase II/III clinical trials (19). Compared to healthy volunteers, patients often have lower clearances and larger volumes of distribution, which result in higher average plasma concentrations and longer elimination half-lives. Both these alterations of the PK in patients increase the PTA. Thus, our estimates for the PTA in healthy volunteers are conservative estimates for the PTA in patients. A higher PTA for hospitalized patients compared to that for healthy volunteers has also been shown by Lodise et al. (24) for piperacillin given in combination with tazobactam.
To determine the PTA at various dose levels and for different modes of administration, it is important to know whether the clearance changes with the plasma concentration at therapeutic concentrations. Therefore, we compared the PK of flucloxacillin at two dose levels in a crossover study with healthy volunteers.
Our noncompartmental analysis showed no differences in the clearance or the volume of distribution between the low and the high dose (Table 1). A three-compartment population PK model with linear renal and nonrenal elimination showed excellent predictive performance, and there was no indication for a decrease in the total, renal, or nonrenal clearance with an increase in the dose from 500 mg to 1,000 mg. Therefore, we used this model, estimated from plasma and urine data at both dose levels for MCS, to compare the PTA profiles with the MIC profiles for four dosage regimens (Fig. 3). The target in our MCS was based on the non-protein-bound plasma concentrations. A range of 94.6 to 96.2% for the plasma protein binding of flucloxacillin has been reported (7, 9, 31). These values correspond to a rather wide range for the non-protein-bound fraction of 3.8 to 5.4%. This is a difference of 42% (5.4 versus 3.8%) for the non-protein-bound concentrations. To account for this situation, we chose a relatively high protein binding of 96%, which has also been reported by Bergan (7), as this choice results in conservative (low) non-protein-bound plasma concentrations.
For serious infections, doses of up to 8 g flucloxacillin per day (2 g given three to four times daily) are recommended both in the United Kingdom (4) and in Germany (5). As intravenous flucloxacillin is more likely to be given for serious infections than for uncomplicated infections, we chose for our simulations a daily dose of 6 g, which is within the dose range recommended for the treatment of serious infections. Our MCS with the bacteriostasis target (fT>MIC ≥ 30%) showed that prolonged infusion had a higher PTA than continuous infusion and short-term infusion at the same daily dose (Table 4). For the target near maximal killing (fT>MIC ≥ 50%), prolonged infusion and continuous infusion both had a three times higher PK-PD breakpoint than short-term infusion at the same daily dose of 6 g. Thus, prolonged infusion achieved a PTA that was higher than or similar to the PTAs of the other two regimens for both targets.
The different ranking of the three dosage regimens at the same daily dose of 6 g for the two targets can be explained as follows: the breakpoint for continuous infusion at steady state is independent of the chosen target, as for any one patient fT>MIC can be only 0% or 100%. The profile for short-term infusion shows a rather pronounced peak without a plateau, and the profile for prolonged infusion shows a flat peak with a rather broad plateau. Subsequently, short-term infusions reach very high concentrations for a short period of time, and prolonged infusions reach moderately high concentrations, but for a longer period of time. We determined for flucloxacillin that short-term (0.5 h) infusions had the highest PTA for targets up to fT>MIC of ≥20%, prolonged (4-h) infusions performed best for targets between 20% and 50%, and continuous infusion had the highest PTA for longer targets (55% and above). Continuous infusion was superior to short-term infusions for targets of 30% and above (Fig. 4).
FIG. 4.

Unbound concentrations and median and prediction interval (5th to 95th percentile) of the fT>MIC at steady state in healthy volunteers after a 30-min infusion of 1.5 g q6h (continuous line, marker •), after a 4-h infusion of 2 g q8h (dashed line, marker ▪), and after continuous infusion of 6 g per day (dashed line, marker ♦) (bottom left panel) and after a 30-min infusion of 2 g q4h (continuous line, marker ▪) (bottom right panel). We assumed a fixed protein binding of 96% for the simulations. The curve for the 4-h infusion was shifted to the right by 6% in the MIC for easier identification of the corresponding prediction intervals (5th to 95th percentile). The 5th and 95th percentiles for the continuous infusion were omitted for clarity. After continuous infusion, more than 99% of the subjects had an fT>MIC of 100% at an MIC of 0.75 mg/liter.
The PK-PD breakpoints need to be compared to the MICs encountered in clinical practice to put these results into clinical perspective. The MIC90s of flucloxacillin for MSSA are usually reported to be ≤0.5 mg/liter (22, 27, 28, 33). For an MIC of 0.5 mg/liter, continuous infusion and prolonged infusion of 6 g/day had PTAs of more than 99%, whereas short-term infusion of 6 g/day reached a PTA of only 46%, based on the target for near maximal killing. At an MIC of 1.0 mg/liter, a short-term infusion of 2 g every 4 h had a PTA of more than 90% for the target fT>MIC of ≥50% (Table 4; Fig. 3).
The most relevant target depends on the clinical situation of the patient. For patients with uncomplicated infections and an intact immune system, it might be sufficient to achieve bacteriostasis. However, for serious infections the near maximal bactericidal activity of the antibiotic might be required (17). As flucloxacillin is also available for oral treatment, intravenous dosing is more relevant for patients with serious infections.
As flucloxacillin showed no saturation in clearance, doubling of the dose will yield concentrations twice as high and, therefore, PK-PD breakpoints twice as high. The PK-PD breakpoints for an MCS with a daily dose of 12 g will be twice as high as the PK-PD breakpoints for 6 g/day shown in Table 4, as flucloxacillin exhibited linear pharmacokinetics. As the PK-PD breakpoint of the short-term infusion q6h of 6 g/day was 0.25 mg/liter, a daily dose of about 12 g would be required to reach a breakpoint of 0.5 mg/liter for near maximal killing. However, 4 g/day as a continuous or a prolonged infusion would be sufficient to reach a breakpoint of 0.5 mg/liter.
The MIC90 of flucloxacillin is typically ≤0.5 mg/liter for MSSA. Even without data on the distribution of the flucloxacillin MIC for MSSA, it is still possible to calculate the expectation value for the population PTA and an MIC90 of 0.5 mg/liter. The PTA-versus-MIC profile (Fig. 3) for continuous or prolonged infusion of 4 g/day with a PK-PD breakpoint of 0.5 mg/liter can be roughly simplified to assuming a PTA of 100% for all MICs ≤ 0.5 mg/liter and a PTA of 0% for all MICs above 0.5 mg/liter. For an MIC90 of 0.5 mg/liter, 90% of the patients (PTA = 100% for MICs ≤ 0.5 mg/liter) will achieve the target (corresponding to the 90th percentile of the MIC distribution) and 10% of the patients (PTA = 0% for MICs > 0.5 mg/liter) will not achieve the target. Therefore, the expectation value for the population PTA will be at least 90% for prolonged or continuous infusion of 4 g/day and an MIC90 of 0.5 mg/liter. As described above, a dose of 12 g/day given as short-term infusions q6h would be required to reach a PK-PD breakpoint of 0.5 mg/liter and an expectation value of about 90% for an MIC90 of 0.5 mg/liter. This means that a 66% lower daily dose is sufficient for continuous or prolonged infusion compared to the dose required for short-term infusion q6h in order to reach the same expectation value for a successful clinical outcome.
Besides a cost reduction for drug acquisition secondary to the dose reduction for prolonged or continuous infusion, a lower risk of adverse events from flucloxacillin treatment is probably a major advantage of prolonged and continuous infusion. Cholestatic jaundice and hepatitis rarely occur in relation to flucloxacillin intake, with the risk factors being the patient's age, preexisting hepatic impairment, and long-term use (>14 days [3]). In a population-based case-control study, de Abajo et al. (16) identified a dose dependency for these adverse effects. It remains to be determined whether high peak concentrations, total exposure, or trough concentrations contribute to the frequency of adverse events for flucloxacillin. However, prolonged and continuous infusions allow a significant dose reduction and avoid high peak concentrations, while they achieve the same PTAs as short-term infusion.
Whether prolonged or continuous infusion would be used depends on the clinical situation of the patient. The continuous infusion of flucloxacillin may be used for the home treatment of MSSA infections that require high-dose intravenous treatment over prolonged time periods. This procedure has been shown to be safe, convenient, and effective for the follow-up treatment of serious staphylococcal infections (e.g., sepsis, endocarditis, and osteomyelitis) and cellulitis (22, 23), and flucloxacillin solutions have also been reported to have sufficient stability at room temperature (26). However, in the hospital setting and with patients receiving multiple intravenous drugs, one of the major drawbacks of continuous infusion is the need for an additional infusion line to prevent incompatibilities with other drugs and the resulting increase of the risk for line infections (25).
In conclusion, we found that the PK of flucloxacillin is linear for the doses of 500 mg and 1,000 mg administered as 5-min intravenous infusions. For near maximal killing (target, an fT>MIC of ≥50%) flucloxacillin showed a robust (≥90%) PTA up to MICs of 0.75 to 1 mg/liter (PTA of 86% at 1 mg/liter) for prolonged infusions or a continuous infusion of 6 g/day. Short-term infusions of 6 g/day given q6h had a lower breakpoint of 0.25 to 0.375 mg/liter (PTA of 79% at 0.375 mg/liter). For an MIC90 of 0.5 mg/liter, which is typically found for flucloxacillin against MSSA, prolonged and continuous infusion at a daily dose of 4 g had an expectation value for the population PTA of at least 90%. To achieve the same expectation value, a daily dose of 12 g given as short-term infusions given q6h would be required. This dose reduction from 12 g/day for short-term infusion to 4 g/day for prolonged infusion and continuous infusion might be a considerable advantage in terms of the risk for adverse events and drug acquisition costs. Future comparative clinical trials are warranted to show whether prolonged and continuous infusions of flucloxacillin at lower dose levels achieve similar or better clinical success rates than short-term infusion.
Footnotes
Published ahead of print on 18 June 2007.
REFERENCES
- 1.Adam, D., P. Koeppe, and H. D. Heilmann. 1983. Pharmacokinetics of amoxicillin and flucloxacillin following the simultaneous intravenous administration of 4 g and 1 g, respectively. Infection 11:150-154. [DOI] [PubMed] [Google Scholar]
- 2.Ambrose, P. G., S. M. Bhavnani, C. M. Rubino, A. Louie, T. Gumbo, A. Forrest, and G. L. Drusano. 2007. Pharmacokinetics-pharmacodynamics of antimicrobial therapy: it's not just for mice anymore. Clin. Infect. Dis. 44:79-86. [DOI] [PubMed] [Google Scholar]
- 3.Anonymous. 2005. British National Formulary 49. British Medical Association and Royal Pharmaceutical Society of Great Britain, London, United Kingdom.
- 4.Anonymous. 2007, posting date. Electronic Medicines Compendium (eMC). Datapharm Communications Ltd., Surrey, United Kingdom.
- 5.Anonymous. 2007, posting date. Rote Liste. Arzneimittelinformationen für Deutschland. Rote Liste Service GmbH, Frankfurt am Main.
- 6.Beal, S. L., A. J. Boeckmann, L. B. Sheiner, and NONMEM Project Group. 1999. NONMEM users guides, version 5 ed. University of California at San Francisco.
- 7.Bergan, T. 1978. Penicillins. Antibiot. Chemother. 25:1-122. [DOI] [PubMed] [Google Scholar]
- 8.Bergan, T., A. Engeset, W. Olszewski, N. Ostby, and R. Solberg. 1986. Extravascular penetration of highly protein-bound flucloxacillin. Antimicrob. Agents Chemother. 30:729-732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bergeron, M. G., J. L. Brusch, M. Barza, and L. Weinstein. 1976. Bactericidal activity and pharmacology of flucloxacillin. Am. J. Med. Sci. 271:13-20. [DOI] [PubMed] [Google Scholar]
- 10.Bonten, M. J., S. Slaughter, A. W. Ambergen, M. K. Hayden, J. van Voorhis, C. Nathan, and R. A. Weinstein. 1998. The role of “colonization pressure” in the spread of vancomycin-resistant enterococci: an important infection control variable. Arch. Intern. Med. 158:1127-1132. [DOI] [PubMed] [Google Scholar]
- 11.Craig, W. A. 2002. Pharmacodynamics of antimicrobials: general concepts and applications, p. 1-22. In C. H. Nightingale, T. Murakawa, and P. G. Ambrose (ed.), Antimicrobial pharmacodynamics in theory and clinical practice. Marcel Dekker, New York, NY.
- 12.Craig, W. A. 1998. Pharmacokinetic/pharmacodynamic parameters: rationale for antibacterial dosing of mice and men. Clin. Infect. Dis. 26:1-10. [DOI] [PubMed] [Google Scholar]
- 13.Craig, W. A., and D. Andes. 1996. Pharmacokinetics and pharmacodynamics of antibiotics in otitis media. Pediatr. Infect. Dis. J. 15:255-259. [DOI] [PubMed] [Google Scholar]
- 14.Craig, W. A., S. Ebert, and Y. Watanabe. 1993. Differences in time above MIC required for efficacy of beta-lactams in animal infection models, abstr. 86. Abstr. 33rd Intersci. Conf. Antimicrob. Agents Chemother. American Society for Microbiology, Washington, DC.
- 15.Craig, W. A., and S. C. Ebert. 1992. Continuous infusion of beta-lactam antibiotics. Antimicrob. Agents Chemother. 36:2577-2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Abajo, F. J., D. Montero, M. Madurga, and L. A. Garcia Rodriguez. 2004. Acute and clinically relevant drug-induced liver injury: a population based case-control study. Br. J. Clin. Pharmacol. 58:71-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Drusano, G. L. 2004. Antimicrobial pharmacodynamics: critical interactions of ‘bug and drug.’ Nat. Rev. Microbiol. 2:289-300. [DOI] [PubMed] [Google Scholar]
- 18.Drusano, G. L. 2003. Prevention of resistance: a goal for dose selection for antimicrobial agents. Clin. Infect. Dis. 36:S42-S50. [DOI] [PubMed] [Google Scholar]
- 19.Drusano, G. L., S. L. Preston, C. Hardalo, R. Hare, C. Banfield, D. Andes, O. Vesga, and W. A. Craig. 2001. Use of preclinical data for selection of a phase II/III dose for evernimicin and identification of a preclinical MIC breakpoint. Antimicrob. Agents Chemother. 45:13-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.GlaxoSmithKline. 2005. Flucloxacillin product information (Floxapen). GlaxoSmithKline, Uxbridge, Middlesex, United Kingdom.
- 21.Gould, I. M. 1999. A review of the role of antibiotic policies in the control of antibiotic resistance. J. Antimicrob. Chemother. 43:459-465. [DOI] [PubMed] [Google Scholar]
- 22.Howden, B. P., and M. J. Richards. 2001. The efficacy of continuous infusion flucloxacillin in home therapy for serious staphylococcal infections and cellulitis. J. Antimicrob. Chemother. 48:311-314. [DOI] [PubMed] [Google Scholar]
- 23.Leder, K., J. D. Turnidge, T. M. Korman, and M. L. Grayson. 1999. The clinical efficacy of continuous-infusion flucloxacillin in serious staphylococcal sepsis. J. Antimicrob. Chemother. 43:113-118. [DOI] [PubMed] [Google Scholar]
- 24.Lodise, T. P., Jr., B. Lomaestro, K. A. Rodvold, L. H. Danziger, and G. L. Drusano. 2004. Pharmacodynamic profiling of piperacillin in the presence of tazobactam in patients through the use of population pharmacokinetic models and Monte Carlo simulation. Antimicrob. Agents Chemother. 48:4718-4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lomaestro, B. M., and G. L. Drusano. 2005. Pharmacodynamic evaluation of extending the administration time of meropenem using a Monte Carlo simulation. Antimicrob. Agents Chemother. 49:461-463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lynn, B. 2002. Recent work on parenteral penicillins. J. Hosp. Pharmacy 29:183-194. [Google Scholar]
- 27.Meyer, B., S. Ahmed el Gendy, G. Delle Karth, G. J. Locker, G. Heinz, W. Jaeger, and F. Thalhammer. 2003. How to calculate clearance of highly protein-bound drugs during continuous venovenous hemofiltration demonstrated with flucloxacillin. Kidney Blood Press. Res. 26:135-140. [DOI] [PubMed] [Google Scholar]
- 28.Mouton, J. W., H. P. Endtz, J. G. den Hollander, N. van den Braak, and H. A. Verbrugh. 1997. In-vitro activity of quinupristin/dalfopristin compared with other widely used antibiotics against strains isolated from patients with endocarditis. J. Antimicrob. Chemother. 39(Suppl. A):75-80. [DOI] [PubMed] [Google Scholar]
- 29.Nauta, E. H., and H. Mattie. 1975. Pharmacokinetics of flucloxacillin and cloxacillin in healthy subjects and patients on chronic intermittent haemodialysis. Br. J. Clin. Pharmacol. 2:111-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pharsight. 2002. WinNonlin user's guide. Pharsight, Mountain View, CA.
- 31.Roder, B. L., N. Frimodt-Moller, F. Espersen, and S. N. Rasmussen. 1995. Dicloxacillin and flucloxacillin: pharmacokinetics, protein binding and serum bactericidal titers in healthy subjects after oral administration. Infection 23:107-112. [DOI] [PubMed] [Google Scholar]
- 32.Russmann, S., J. A. Kaye, S. S. Jick, and H. Jick. 2005. Risk of cholestatic liver disease associated with flucloxacillin and flucloxacillin prescribing habits in the UK: cohort study using data from the UK General Practice Research Database. Br. J. Clin. Pharmacol. 60:76-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Steer, J. A., R. P. Papini, A. P. Wilson, D. A. McGrouther, and N. Parkhouse. 1997. Teicoplanin versus flucloxacillin in the treatment of infection following burns. J. Antimicrob. Chemother. 39:383-392. [DOI] [PubMed] [Google Scholar]