

Abstract
Brecanavir, a novel tyrosyl-based arylsulfonamide, high-affinity, human immunodeficiency virus type 1 (HIV-1) protease inhibitor (PI), has been evaluated for anti-HIV activity in several in vitro assays. Preclinical assessment of brecanavir indicated that this compound potently inhibited HIV-1 in cell culture assays with 50% effective concentrations (EC50s) of 0.2 to 0.53 nM and was equally active against HIV strains utilizing either the CXCR4 or CCR5 coreceptor, as was found with other PIs. The presence of up to 40% human serum decreased the anti-HIV-1 activity of brecanavir by 5.2-fold, but under these conditions the compound retained single-digit nanomolar EC50s. When brecanavir was tested in combination with nucleoside reverse transcriptase inhibitors, the antiviral activity of brecanavir was synergistic with the effects of stavudine and additive to the effects of zidovudine, tenofovir, dideoxycytidine, didanosine, adefovir, abacavir, lamivudine, and emtricitabine. Brecanavir was synergistic with the nonnucleoside reverse transcriptase inhibitor nevirapine or delavirdine and was additive to the effects of efavirenz. In combination with other PIs, brecanavir was additive to the activities of indinavir, lopinavir, nelfinavir, ritonavir, amprenavir, saquinavir, and atazanavir. Clinical HIV isolates from PI-experienced patients were evaluated for sensitivity to brecanavir and other PIs in a recombinant virus assay. Brecanavir had a <5-fold increase in EC50s against 80% of patient isolates tested and had a greater mean in vitro potency than amprenavir, indinavir, lopinavir, atazanavir, tipranavir, and darunavir. Brecanavir is by a substantial margin the most potent and broadly active antiviral agent among the PIs tested in vitro.
Human immunodeficiency virus (HIV) protease is an essential enzyme required for viral proliferation. HIV protease inhibitors (PIs) are among the most potent and effective antiretrovirals and are considered essential components of successful combination therapy or highly active antiretroviral therapy (HAART) to treat HIV disease. Current guidelines include an initial treatment option that specifies the use of two nucleoside reverse transcriptase (RT) inhibitors (NRTIs) with a ritonavir (RTV)-boosted PI (5). However, emergence of viral resistance to HIV PIs and cross-resistance between members of the PI class are some of several major factors linked to the failure of the clinical management of HIV disease. Additionally, treatment regimens composed of several RT inhibitors and PIs have produced complex patterns of compound interaction and cross-resistance among drug classes (2). The development of compounds that are active against PI-resistant strains of HIV appears needed to assure optimal treatment of PI-experienced patients. Moreover, recent evidence indicates the transmission of drug-resistant HIV strains to treatment-naive patient populations (1, 6, 15), and this has implications for the treatment regimens selected for recently diagnosed patients. It is also essential that new antiretrovirals interact favorably with other components of the complex HAART regimens.
In an attempt to overcome the challenge of cross-resistance within the PI class, novel inhibitors need to be designed that target critical sites on the protease enzyme that are not targeted by currently used PIs. The goal of drug discovery should involve the identification of inhibitors of a broad spectrum of mutant HIV-1 strains, which are compatible with existing treatments and select unique protease mutations which remain susceptible to other PIs.
Our research has identified novel inhibitors of the HIV protease that possess potent activities against a variety of clinically relevant mutant HIV strains, are compatible with other anti-HIV agents, and have unique resistance profiles. Recently we described a series of novel arylsulfonamide PIs with potent anti-HIV activities against both wild-type and drug-resistant viral strains (18). Additional structural modifications that introduced a tyrosine moiety into the P1 position on the series have led to exceptionally potent compounds. In this article, we describe the in vitro activities of brecanavir (BCV) (Fig. 1), a tyrosyl peptidomimetic with low nanomolar activities against both wild-type and PI-resistant HIV, additive to synergistic activities in combination with other antiretrovirals, and having a unique in vitro resistance profile.
FIG. 1.

Structure of BCV.
(Results of this study were presented in part at the 2nd International AIDS Society Conference on HIV Pathology and Treatment, Paris, France, 13 to 16 July 2003.)
MATERIALS AND METHODS
Compounds.
BCV (GW640385), (3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-yl (1S,2R)-[(1,3-benzodioxol-5-yl-sulfonyl)(isobutyl)amino]-2-hydroxy-1-{4-[(2- methyl-1,3-thiazol-4-yl)methoxy]benzyl}propylcarbamate (Fig. 1), was synthesized at GlaxoSmithKline (Research Triangle Park, NC). Zidovudine (ZDV), abacavir (ABC), stavudine (d4T), dideoxycytidine (ddC), didanosine (ddI), nevirapine (NVP), delavirdine (DLV), lamivudine (3TC), darunavir (DRV), adefovir (ADV), and amprenavir (APV) were synthesized by GlaxoSmithKline. The nucleotide prodrug tenofovir disoproxil fumarate was purchased in the pharmacy, and the active drug substance, (R)-9-(2-phosphonylmethoxypropyl)-adenine (tenofovir [TFV]), was isolated in the Medicinal Chemistry Department at GlaxoSmithKline. The PI tipranavir (TPV) was isolated from the commercial formulation, Aptivus. The marketed PIs indinavir (IDV), lopinavir (LPV), nelfinavir (NFV), RTV, atazanavir (ATV), and saquinavir (SQV) were obtained from the Medicinal Chemistry Department at GlaxoSmithKline.
Cell lines and primary cell cultures.
MT-4 cells, a human T-cell leukemia virus type 1-transformed human T-cell line (19), were obtained from long-term cultures of samples supplied by B. Larder and maintained as previously described (3). HeLa-CD4-LTR-β-gal cells (obtained from Michael Emerman through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH) were maintained in Dulbecco's modified Eagle's medium with 10% fetal bovine serum (FBS) (catalog no. SH30070.03; HyClone) and under the selective pressure of Geneticin (catalog no. 10131-035; Invitrogen) and hygromycin B (catalog no. 10687-010; Invitrogen). Normal donor peripheral blood mononuclear cells (PBMCs) were isolated from random buffy coats (35 to 40 ml of elutriated whole blood in anticoagulant from HIV-negative donors) received from the American Red Cross, Carolina Division. PBMCs were isolated by density gradient centrifugation over lymphocyte separation medium (Mediatech catalog no. 25-072-CL) and stimulated by the addition of 5 μg/ml phytohemagglutinin (PHA) (catalog no. L9017; Sigma) for 24 to 48 h (3).
Virus strains.
HIV-1 strain IIIB was derived from cell-free supernatants of cultures of the chronically infected cell line H93B (H9/human T-cell leukemia virus type 1 strain IIIB). HIV-1 strain HXB2, derived from the molecular clone pHXB2-D (8), was obtained from B. Larder. High-titer HIV-1 Ba-L was purchased from Advanced Biotechnologies and expanded in PHA-activated PBMCs (see above), and titers in PBMCs were determined. Viral input was determined by selecting a dilution that gave a RT signal within the linear range of the assay and a signal-to-background ratio of 20 to 30.
MT-4 cell assay.
Antiviral HIV activity and compound-induced cytotoxicity were measured in parallel by means of a methanethiosulfonate tetrazolium reagent (MTS)-based procedure in the HTLV-1-transformed cell line MT-4. Aliquots of the test compounds were serially diluted in RPMI 1640 medium (catalog no. 22400; Invitrogen), 10% [vol/vol] FBS, and 10 μg/ml gentamicin (catalog no. 15750-060; Invitrogen) in 96-well plates. Exponentially growing MT-4 cells were harvested and centrifuged at 192 × g for 10 min. Cell pellets were resuspended in fresh medium (RPMI 1640, 20% [vol/vol] FBS, 20% [vol/vol] interleukin-2 [catalog no. 801017; Zeptometrix], and 10 μg/ml gentamicin) to a density of 5 × 105 cells/ml. Cell aliquots were infected by the addition of HIV-1 strain IIIB, diluted to give a viral inoculum of 100 × 50% tissue culture infective doses per well. A similar cell aliquot was diluted with medium to provide a mock-infected control. Cell infection was allowed to proceed for 1 h at 37°C in a tissue culture incubator with humidified 5% CO2 atmosphere. After incubation, the virus-treated cell suspensions were diluted sixfold with fresh medium, and 125 μl of the cell suspension was added to each well of the plate containing prediluted compound. Plates were then placed in a tissue culture incubator at 37°C with humidified 5% CO2 for 5 days. HIV-induced cytopathic effects were assessed by the CellTiter96 MTS staining method (catalog no. G3581; Promega, Madison, WI). The optical density at 492 nm was measured by using a microplate absorbance reader (catalog no. 20-300; Tecan, Research Triangle Park, NC).
Modified MT-4 cell assay to assess combination antiviral activity.
For combination testing, aliquots of BCV were serially diluted vertically in a 96-well master assay plate in RPMI 1640 medium, 10% [vol/vol] FBS, and 10 μg/ml gentamicin. Approved HIV inhibitors were diluted horizontally across separate master assay plates. Checkerboard-style dilutions were arranged by combining aliquots from both the horizontally and vertically diluted master plates into daughter plates, so that every concentration of BCV was tested in the presence and absence of every concentration of the approved HIV inhibitor. Anti-HIV activity tests were performed in a minimum of triplicate assays of each combination. Cell infection, incubation, and MTS staining were carried out by the same methods used in the standard MT-4 cell assay.
Antiviral assay in the presence of human serum and human serum proteins.
Exponentially growing MT-4 cells were harvested and centrifuged at 400 × g for 5 min at room temperature, and the cell pellet was resuspended in RPMI 1640 medium, 10% [vol/vol] FBS, supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin (catalog no. 15140-122; Invitrogen), and 2 mM l-glutamine (catalog no. 25030-081; Invitrogen) in the presence or absence of human serum or human serum proteins. The MT-4 cell suspension was batch infected with HIV-1 HXB2 at a multiplicity of infection of 0.0001 (100 50% tissue culture infective doses per 1 × 106 cells) in a volume of 0.5 ml and a cell density of 9.6 × 107 cells/ml. Infection was allowed to proceed for 1 h at 37°C in a tissue culture incubator with a humidified atmosphere of 5% CO2 in air. Following infection, cells were diluted in corresponding protein-supplemented medium and plated on a 96-well plate containing serially diluted BCV, DRV, or TPV in the presence of 10% or 40% FBS, 10% to 40% human serum (catalog no. H1388; Sigma, St. Louis, MO), 40 mg/ml human serum albumin (catalog no. A8763; Sigma, St. Louis, MO), 1 mg/ml α1-acid glycoprotein (catalog no. G9885; Sigma, St. Louis, MO), or combinations of protein treatments. The final MT-4 cell density was 4 × 104 cells per well in a volume of 200 μl. After 5 days of incubation, the 50% effective concentration (EC50) was determined by a cell viability assay using the MTS reagent with optical density measured by a Vmax microplate reader (Molecular Devices, Sunnyvale, CA). The aim of these measurements was to determine the effective concentration of drug in human serum, generally considered to be the concentration of free drug in solution (21). It is not practical to measure the effect of drug in 100% human serum due to cellular toxicity, but concentrations up to 40% can be used. Extrapolation of the data to 100% serum is necessary for in vitro-to-in vivo scaling. Extrapolation can be done with greater confidence if we can predict with certainty a linear relationship between the concentration of free drug and the total serum protein concentration. If the total, free, and bound concentrations of drug are represented as T, F, and B, respectively, P is the total concentration of protein capable of binding drug, and Pf is unbound protein, then.
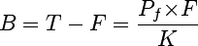 |
(1) |
where K is the dissociation constant for binding. In serum the concentrations of binding proteins range from 20 μM (α1-acid glycoprotein) to ∼500 μM (human serum albumin) (21), and the inhibitors used are present in concentrations ∼1,000-fold lower. Thus, P is ≫B, and in equation 1 we can substitute Pf = P, which gives
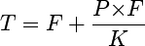 |
(2) |
Therefore, the total concentration of drug required for the maintenance of a fixed concentration of free drug is a linear function of the protein concentration and predicts a linear increase of the EC50 with the serum concentration. Such a linear relationship has been demonstrated at total drug concentrations below 100 nM (Fig. 2). Thus, since equation 2 is theoretically valid and experimentally verified over the practical range of measurement, we believe that it can be used to extrapolate the experimental data to determine the EC50 at 100% human serum.
FIG. 2.

Effect of human serum on the antiviral activity of BCV (closed circles) or darunavir (filled squares) in vitro.
PBMC assay.
PHA-stimulated PBMCs were pelleted, washed once with PBS and resuspended to 8 × 106 cells/ml in RPMI 1640 medium with 20% (vol/vol) FBS, 10% (vol/vol) interleukin 2, and 10 μg/ml gentamicin, and 50 μl was distributed to 96-well tissue culture plates. Compounds were serially diluted in medium in fourfold increments at two times the final concentration. Fifty microliters of diluted compound was transferred to the PBMCs and placed in a humidified incubator at 37°C, 5% CO2, for 1 h. An additional 60 μl of diluted compound was transferred to a separate 96-well plate containing 60 μl of appropriately diluted HIV-1 Ba-L and thoroughly mixed. One hundred microliters of this mixture was transferred into the PBMC-compound mixture and placed in a humidified incubator at 37°C, 5% CO2, for 7 days. On day 7 of the assay, 50 μl of culture supernatant was transferred to a new 96-well plate. The RT levels in the supernatants were measured by the method of Schwartz et al. (23).
Drug combinations (deviations from dose-wise additivity).
EC50s were calculated by curve fitting data to the Hill equation (12), using a nonlinear least-squares curve-fitting program based on the Marquardt-Levenberg algorithm (16). The interaction of each pair of compound combinations was analyzed by the methods described by Selleseth et al. (24), which provide an estimation of the strength of any interaction and of its statistical significance. Synergy and antagonism are defined as deviations from dosewise additivity, which results when two drugs interact as if they were the same drug. Values for average deviation from additivity in the range of −0.1 to −0.2 indicate weak synergy and values between −0.2 and −0.5 indicate strong synergy of interaction. Conversely, values of +0.1 to +0.2 indicate that a weak antagonism exists between the treatments.
HeLa-CD4 MAGI antiviral assay.
Compound anti-HIV-1 activity was determined in HeLa-CD4-LTR-β-gal (14) by the method of Ferris et al. (7).
HIV protease enzymology.
Inhibition constant (Ki) values were determined from a continuous fluorescence activity assay for HIV-1 protease or calculated from the bimolecular rate constants (k1) for association of enzyme with inhibitor and the values for the first-order rate constants (k−1) for the dissociation of enzyme and inhibitor (10).
Phenotypic susceptibilities of 94 viruses obtained from PI-experienced patients.
One hundred nine viruses obtained from PI treatment-experienced patients were studied. Ninety-four of these viruses were selected based on the presence of major protease resistance-associated mutations (RAMS) at residues 30 (n = 3), 32 (n = 9), 33 (n = 31), 46 (n = 60), 47 (n = 6), 48 (n = 10), 50 (V, n = 5; L, n = 5), 82 (n = 48), 84 (n = 36), 88 (n = 7), and 90 (n = 44) (Table 1). Viruses were selected so that single, double, triple and multiple mutations of different combinations were included. Sequence analysis and drug susceptibility testing were performed at Monogram Biosciences, Inc., South San Francisco, CA. Viral DNA sequences were determined by a thermocycling method using fluorescent dye-labeled dideoxynucleotide chain terminator chemistry. Resistance-associated mutations were classified based on International AIDS Society resistance tables (13). The mean percent inhibition of each drug concentration was determined and used to calculate the EC50. The n-fold change (FC) in drug susceptibility was determined by comparing the EC50 for the subject virus to the EC50 for the drug-sensitive reference virus containing the protease and RT sequences of the NL4-3 strain of HIV-1.
TABLE 1.
Major HIV-1 protease mutations associated with PI resistance identified in clinical-isolate viruses
| Mutation | No. of isolates with mutationa | % Viruses with mutation |
|---|---|---|
| D30N | 3 | 3 |
| V32I | 9 | 9 |
| L33F or I | 31 | 33 |
| M46I or L | 60 | 64 |
| I47V | 6 | 6 |
| G48S or V | 10 | 10 |
| I50V or L | 10 | 10 |
| V82A or C or I or T or F or S | 48 | 51 |
| I84A or V | 36 | 38 |
| N88D or G or S or T | 7 | 7 |
| L90M | 44 | 47 |
Ninety-four isolates were analyzed.
RESULTS
Activity of BCV against wild-type HIV.
The in vitro anti-HIV activities of BCV against wild-type laboratory strains of HIV-1 were evaluated with acutely infected MT-4 cells, human PBMCs, or HeLa-CD4 cells (Table 2). In assays with MT-4 cells acutely infected with HIV-1 that utilize the CXCR4 coreceptor, namely, strain IIIB or the molecular clone HXB2, BCV produced equivalent EC50s of 0.44 or 0.53 nM, respectively. The anti-HIV-1 activity of BCV was also characterized with human PBMCs isolated from noninfected donors. BCV inhibited HIV-1 strain IIIB and the strain Ba-L, with subnanomolar activities of 0.2 and 0.42 nM, respectively. The cell culture 50% inhibitory concentration values (CCIC50s) for BCV were determined with a small panel of uninfected B and T cells (data not shown). For MT-4, Molt-4, IM-9, or U-937 cells, the CCIC50 for BCV was greater than 25 μM, the highest concentration tested. These data, in conjunction with the EC50 of 0.44 nM obtained with MT-4 cells with wild-type HIV-1, estimate the in vitro selectivity index (CCIC50/EC50) at >55,000 for BCV. In assays with HeLa-CD4 cells and HIV-1 strain HXB2, BCV was inactive up to the limit of solubility (approximately 50 μM). Since this assay relies on only one round of infection, inhibitors of HIV-1 protease are uniformly inactive.
TABLE 2.
Activities of BCV against wild-type HIV
| Virus strain | Cell type | Assay readout (n)a | Anti-HIV EC50 (nM)b
|
|
|---|---|---|---|---|
| Mean | SD | |||
| HIV-1 IIIB | MT-4 | CPE (8) | 0.44 | 0.19 |
| HIV-1 HXB2 | MT-4 | CPE (13) | 0.53 | 0.11 |
| HIV-1 IIIB | PBMC | RT (3) | 0.2 | 0.05 |
| HIV-1 Ba-L | PBMC | RT (8) | 0.42 | 0.07 |
| HIV-1 HXB2 | HeLa-CD4 | β-Gal (2) | >90,000 | NA |
CPE, HIV-induced cytopathic effect; RT, measurement of HIV RT activity in assay supernatants; β-Gal, β-galactosidase activity readout utilizing the Tropix assay system. n, number of determinations.
Values are means ± standard deviations except in the case of HIV-1 Ba-L, where values are geometric means ± standard deviations; NA, not applicable.
Antiviral activity in human PBMCs.
A comparison of the anti-HIV-1 strain Ba-L activities of BCV with several PIs in human PBMCs is shown in Table 3. BCV was significantly more potent (P < 0.0001) than any of the other PIs tested (range, 6.7- to 720-fold). BCV was 720-fold more potent than the nonpeptidomimetic, TPV, 130-fold more potent than APV, 85-fold more potent than either NFV, 10-fold more potent than ATV, and 6.7-fold more potent than the structurally related PI, DRV.
TABLE 3.
Anti-HIV-1 Ba-L activities of BCV and approved PIs in PBMC
| Compound | Anti-HIV-1 Ba-L EC50 (nM)a
|
||
|---|---|---|---|
| Mean ± SD | Lower 95% CI | Upper 95% CI | |
| BCV | 0.42 ± 0.15b | 0.31 | 0.57 |
| ATV | 4.4 ± 1.9 | 3.1 | 6.3 |
| NFV | 36 ± 23 | 21 | 62 |
| APV | 57 ± 20 | 43 | 76 |
| TPV | 304 ± 50 | 260 | 350 |
| DRV | 2.8 ± 1.3 | 1.8 | 4.2 |
Values are geometric mean ± standard deviations for eight determinations; CI, 95% confidence interval.
Significantly different from each of the other mean EC50s (P < 0.0001).
Antiviral activity in the presence of human serum and human serum proteins.
The effect of human serum on the anti-HIV activity of BCV, DRV, or TPV in MT-4 cells was estimated by determining the ratio of EC50s observed in the absence or presence of various concentrations of human serum. These data, shown in Table 4, indicate that BCV maintains approximately sevenfold-greater antiviral potency than DRV across a concentration range of 0 to 40% added human serum. In contrast, the BCV-to-TPV potency ratio increased from 58-fold in the absence of added serum to approximately 170-fold at 40% human serum. Extrapolation of the data for the n-fold shifts in potencies of the agents as if tested in the presence of 100% human serum indicates that the potency ratio of BCV to DRV would be maintained, while the BCV-to-TPV potency ratio would be further amplified.
TABLE 4.
Effect of human serum on antiviral activity of BCV, DRV, or TPV in MT-4 cells
| % FBS | % Human serum | Anti-HIV-1 HXB2 EC50 of compound, nM (FS)a
|
||
|---|---|---|---|---|
| BCV | DRV | TPV | ||
| 10 | 0 | 0.7 (1) | 5.6 (1) | 40.9 (1) |
| 40 | 0 | 1.1 (1.6) | 11 (1.9) | NT |
| 10 | 10 | 1.4 (1.9) | 11 (2.2) | 310 (7.7) |
| 10 | 20 | 2.2 (2.2) | 15 (2.9) | 507 (12.4) |
| 10 | 30 | 2.6 (3.6) | 19 (3.7) | 580 (14.1) |
| 10 | 40 | 3.8 (5.2) | 22 (4.3) | 660 (16) |
Values are means from two determinations; FS, n-fold shift, the ratio of the EC50 in the presence of human serum to the EC50 with 10% FBS; NT, not tested. Extrapolated n-fold shifts at 100% human serum (means ± standard errors of the means) are as follows: for BCV, 11 ± 2.9; for DRV, 9.3 ± 1.9; for TPV, 39 ± 6.5.
The effect of human serum albumin (HSA) or α1-acid glycoprotein (AAG) on the potency of brecanavir or DRV was determined by methods similar to those used in experiments testing the effects of human serum (Table 5). HSA (40 mg/ml) and/or 1 mg/ml AAG resulted in a 3.7- or 7.6-fold increase in the EC50 of BCV, respectively. The combination of HSA and AAG resulted in an 8.2-fold increase in the EC50 of BCV. For DRV, there was a 1.3- or 8.8-fold increase in EC50 when tested with either HSA or AAG alone, respectively. When tested in the presence of both HSA and AAG, there was an 8.7-fold increase in the EC50 for DRV. Since the concentrations chosen were those reported for human serum, these results are in excellent agreement with values obtained by addition of whole serum (Table 4).
TABLE 5.
Effect of α1-acid glycoprotein or human serum albumin on antiviral activity of BCV or DRV
| % FBS | Concn of α1-acid glycoprotein (mg/ml) | Concn of human serum albumin (mg/ml) | Anti-HIV-1 HXB2 EC50 of compound, nM (FS)a
|
|
|---|---|---|---|---|
| BCV | DRV | |||
| 10 | 0 | 0 | 0.7 (1) | 5.6 (1) |
| 10 | 1 | 0 | 5.5 (7.6) | 49 (8.8) |
| 10 | 1 | 40 | 5.9 (8.2) | 49 (8.7) |
| 10 | 0 | 40 | 2.7 (3.7) | 7.5 (1.3) |
| 40 | 0 | 0 | 1.1 (1.6) | 10.8 (1.9) |
Values are means from two determinations; FS, n-fold shift, the ratio of the EC50 in the presence of α1-acid glycoprotein or human serum albumin to the EC50 with 10% FBS.
Combination antiviral activity in MT-4 cells.
Table 6 depicts the values for the deviation from additivity for combinations of BCV and currently approved anti-HIV-1 drugs. These effects are further detailed in the graphical presentation of the isobolograms for the interaction of BCV with the other agents (Fig. 3). In the current series of experiments, tests where BCV was used as both the horizontally diluted and vertically diluted agent (sham combination) show that no artifactual synergistic or antagonistic effects were seen. In combination testing with NRTIs (Table 6; Fig. 3b), the activity of BCV was additive to the effects of ZDV, ABC, 3TC, TFV, ddI, and ddC. BCV was synergistic with d4T. With NNRTIs (Table 6; Fig. 3c), the activity of BCV was additive to the effects of EFV but was synergistic with those of DLV and NVP. As expected, combination testing of BCV with the PIs (Table 6; Fig. 3a) APV, SQV, IDV, RTV, NFV, LPV, and ATV resulted in additive anti-HIV effects.
TABLE 6.
Inhibition of HIV-1 strain IIIB by BCV in combination with other approved anti-HIV agents in MT-4 cells
| Compound | Deviation from additivitya
|
Interaction with BCV | ||
|---|---|---|---|---|
| Avg | SEM | P(t) | ||
| BCV | −0.068 | 0.060 | 0.169 | Additive |
| ZDV | −0.050 | 0.040 | 0.120 | Additive |
| d4T | −0.330 | 0.080 | 0.008 | Synergistic |
| TFV | 0.005 | 0.061 | 0.470 | Additive |
| ddC | 0.005 | 0.037 | 0.450 | Additive |
| ddI | −0.030 | 0.036 | 0.210 | Additive |
| ADV | −0.092 | 0.138 | 0.261 | Additive |
| ABC | −0.050 | 0.040 | 0.150 | Additive |
| 3TC | 0.080 | 0.072 | 0.160 | Additive |
| FTC | −0.226 | 0.144 | 0.089 | Additive |
| EFV | −0.100 | 0.060 | 0.080 | Additive |
| NVP | −0.260 | 0.085 | 0.015 | Synergistic |
| DLV | −0.190 | 0.076 | 0.027 | Synergistic |
| IDV | −0.004 | 0.062 | 0.480 | Additive |
| LPV | −0.020 | 0.047 | 0.380 | Additive |
| NFV | 0.034 | 0.057 | 0.290 | Additive |
| RTV | 0.070 | 0.047 | 0.100 | Additive |
| APV | 0.040 | 0.060 | 0.290 | Additive |
| SQV | 0.055 | 0.056 | 0.190 | Additive |
| ATV | −0.095 | 0.067 | 0.088 | Additive |
Values are means ± standard errors of the means from at least three determinations. P(t), probability, as determined by the t test, that the deviation from additivity is actually equal to zero.
FIG. 3.

Isobolograms of the inhibition of HIV-1 by BCV in combination with several marketed anti-HIV agents in MT-4 cells. (a) In combination with PIs (PIs); (b) in combination with NRTIs; (c) in combination with NNRTIs. FIC, fractional inhibitory concentration.
Phenotypic susceptibilities of 94 viruses obtained from PI-experienced patients.
The mean antiviral potency of BCV was greater than those of APV, IDV, LPV, ATV, TPV, and DRV based on testing of viruses from 94 treatment-experienced patients (Fig. 4; Table 7). The mean number of major PI RAMs per isolate was 2.8 (range, 0 to 6; median, 3), and the mean number of total protease mutations was 16 (range, 1 to 27; median, 17).
FIG. 4.

Phenotypic susceptibilities to atazanavir, amprenavir, tipranavir, darunavir, and BCV of 94 PI-resistant clinical isolate viruses obtained from PI-experienced patients.
TABLE 7.
Anti-HIV-1 EC50s of several PIs against 94 clinical-isolate viruses
| Compound | Anti-HIV-1 EC50, nM, mean ± SEM (range) | Mean FCa | No. of viruses for which FC was:
|
|
|---|---|---|---|---|
| <3 | <10 | |||
| BCV | 0.88 ± 0.21 (0.1-15) | 6.6 | 58 | 85 |
| APV | 190 ± 23 (1.2-1,300) | 13.7 | 24 | 51 |
| IDV | 220 ± 30 (3.2-1,500) | 26 | 23 | 39 |
| LPV | 230 ± 34 (0.7-1,450) | 43 | 21 | 40 |
| ATV | 47 ± 7.5 (0.8-500) | 26 | 20 | 34 |
| TPV | 190 ± 24 (7-1,080) | 2.4 | 77 | 89 |
| DRV | 4.6 ± 1.5 (0.1-104) | 7.2 | 60 | 87 |
Mean fold change (FC) in EC50 from that for the drug-sensitive reference wild-type virus, HIV-1 strain NL4-3. Reference virus compound sensitivities (EC50) were 0.18, 15.3, 9.8, 4.4, 1.7, 76.2, and 0.78 nM for BCV, APV, IDV, LPV, ATV, TPV, and DRV, respectively.
Approximately 80% (75 out of 94) of all isolates tested were fully susceptible to BCV, based on an FC in EC50s of <5 compared to results with the reference wild-type virus, HIV-1 strain NL4-3. BCV was more than 200-fold more potent than APV, IDV, LPV, or TPV and more than 50-fold more potent than ATV against all of the 94 isolates. BCV was more than fivefold more potent than the structurally related PI, DRV. In addition to the absolute potency advantage for BCV, the mean FC for BCV was lower than for all other PIs except for TPV and DRV, where BCV showed a similar FC.
In general, the viruses with more protease mutations resulted in larger increases in EC50s than viruses with fewer protease mutations. The 10 patient viruses with the highest FC with BCV (mean FC = 41.9; range, 8.9 to 112; median, 13) had a mean of 4.4 major PI RAMs (range, 3 to 6; median, 4) per isolate and a mean of 17.8 total protease mutations (range, 12 to 22; median, 17) per isolate. Two isolates that had an FC for BCV of >100 had genotypes containing the BCV-associated resistance mutations, M46I and A71V, that were identified by in vitro resistance selection with HIV-1 strain HXB2 (26). Against isolates containing 0 to 3 major PI RAMs, BCV had <3 FC compared to the reference virus and with isolates containing 4 or 5 RAMs had FCs of 10 or 48, respectively (Fig. 5). Against isolates containing 0 to 2 BCV-associated mutations identified during in vitro serial virus passage (26), there was a 1.5- to 6.2-fold change in the activity of BCV (BCV EC50 range = 0.1 to 10.2 nM) compared to that of the wild-type reference virus.
FIG. 5.

Relationship between number of major protease resistance mutations and n-fold change of BCV activity. Number of isolates studied per group was 2, 13, 19, 34, 20, 5, or 1 for isolate groups having 0, 1, 2, 3, 4, 5, or 6 PI resistance mutations per isolate, respectively.
DISCUSSION
PI-based HAART regimens are demonstrably potent therapies enabling long-lasting suppression of HIV-associated disease, prolongation of life, and reduction of morbidity in an increasing patient population (22). However, the clinical effectiveness of current PI therapies has been hindered by, among other factors, difficulty in adherence to a high pill burden in dosing regimens, leading to inadequate virus suppression, which correlates with the development of viral escape mutants and disease progression (11). The development of new PIs with strong activities against drug-resistant HIV would provide a tremendous clinical benefit to patients with limited therapeutic options.
Recently we documented our efforts to optimize the arylsulfonamide scaffold of PIs (17). This work resulted in the discovery of BCV, which exhibited exceptionally high potency (Ki = 15 fM) against the wild-type HIV protease enzyme. In a competitive binding assay (10), BCV was found to be 2,000-fold more potent than APV (APV Ki = 36 pM) (10) and 10-fold more potent than DRV (DRV Ki = 147 fM) (unpublished results). We also determined the Ki value of amprenavir by inhibition of enzyme activity using a fluorescent peptide substrate (10) and found the Ki value (57 pM) to be very similar to the value determined with the competitive binding assay. Another newer PI, TPV, was reported to have a Ki similar to that of amprenavir (8 pM) (25). This value was also determined with a fluorescent peptide substrate and suggests that TPV is several orders of magnitude weaker than BCV.
In the present study, we have shown that BCV has subnanomolar potency against several wild-type laboratory HIV strains in both MT-4 cells and PBMCs. Compared to published anti-HIV strain IIIB activities in MT-4 cells, BCV is more than 270- or 850-fold more potent than the PIs APV or TPV, respectively (20), and more than 10-fold more potent than the structurally similar PI, DRV (4). BCV also potently suppressed the virus strain Ba-L, with nearly equivalent activity seen with strains IIIB and HXB2. A comparison of the activity of BCV with those of other PIs against strain Ba-L in PBMCs showed that BCV was significantly more potent than any of the other PIs tested.
Like other PIs, BCV was not active against HIV in the HeLa-CD4 cell assay. The absence of activity in the HeLa-CD4 cell assay is a common characteristic of PIs, since this assay has only a single round of infection and has little or no production of progeny viral particles, where PIs would be active during maturation.
No cellular toxicity was observed in several cell lines up to the highest concentration of BCV tested (25 μM), which indicates a high selectivity index of >55,000.
The in vitro interaction of BCV with other antivirals was determined with MT-4 cells. In combination with the marketed anti-HIV-1 agents, the activity of BCV was found to be additive to or synergistic with the activities of the other agents. It is not clear why results of in vitro tests of BCV in combination with one member of a drug class would be different from those with others in the same drug class. For example, activity of BCV in combination with EFV was additive, whereas that in combinations with either NVP or DLV was synergistic. The differences seen in the interaction of BCV and various members of the NNRTI class may be a matter of degree. By our definition of synergy, a value of −0.1 for average deviation from additivity would indicate a weak synergy for the combination. For combinations of EFV and BCV, the value of −0.1 achieves the minimum value that would indicate synergy, but it fails to achieve statistical significance (P = 0.08). The greatest value of in vitro combination studies is to identify combinations that result in antagonistic antiviral effects and exclude such combinations from clinical consideration. There are many other important factors that also must be considered in the selection of combination therapeutics, including pharmacokinetics, interactions with drug-metabolizing enzymes, and toxicities. These factors can be evaluated only in clinical trials.
BCV is approximately 98% bound to serum proteins (9). Despite this high level of protein binding, BCV retains potent activity when antiviral assessments are performed in the presence of physiological levels of human proteins. In the absence of added human serum, the in vitro potency of DRV was eightfold less than the potency of BCV. In the presence of 40% human serum, the highest concentration tested, the in vitro potency of DRV was 5.8-fold less than the potency of BCV. Protein binding often increases the half-life of the drug by reducing free concentrations that are available for metabolism or excretion. However, protein binding and the resultant reduction of free drug also attenuate the pharmacology of the drug (i.e., antiviral activity in this case). Therefore, these current in vitro studies may serve as a means to estimate the antiviral effect of total drug concentrations determined in vivo in patient plasma.
In testing against clinical isolate viruses derived from PI-experienced patients, BCV has shown greater in vitro potency than APV, IDV, LPV, ATV, TPV, and DRV and a lower FC than all other PIs tested except for a FC similar to those of TPV and DRV. Due to the small scale of the clinical isolate data set, it was difficult to ascribe BCV resistance of the clinical isolates to specific mutational patterns.
In summary, the present study demonstrates the following in vitro preclinical properties of BCV. (i) It has potent subnanomolar in vitro antiretroviral activity against several laboratory HIV strains and is significantly more potent than other PIs tested against viruses using the CCR5 coreceptor. (ii) In the presence of added human serum or serum proteins, BCV retains a potency advantage over other PIs. (iii) It is additive or synergistic when tested in combination with other antiretrovirals. (iv) It exhibited mean subnanomolar potencies against a panel of viruses from PI-experienced patients.
Acknowledgments
This study was funded by GlaxoSmithKline.
We acknowledge Monogram Biosciences, Inc., South San Francisco, CA, for performing the sequence analysis and Phenosense assays. We also acknowledge Steve Novick for assistance in data analysis.
Footnotes
Published ahead of print on 9 July 2007.
REFERENCES
- 1.Ammaranond, P., P. Cunningham, R. Oelrichs, K. Suzuki, C. Harris, L. Leas, A. Grulich, D. A. Cooper, and A. D. Kelleher. 2003. Rates of transmission of antiretroviral drug resistant strains of HIV-1. J. Clin. Virol. 26:153-161. [DOI] [PubMed] [Google Scholar]
- 2.Boden, D., and M. Markowitz. 1998. Resistance to human immunodeficiency virus type 1 protease inhibitors. Antimicrob. Agents Chemother. 42:2775-2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Daluge, S. M., D. J. M. Purifoy, P. M. Savina, M. H. St. Clair, N. R. Parry, I. K. Dev, P. Novak, K. M. Ayers, J. E. Reardon, G. B. Roberts, J. A. Fyfe, M. R. Blum, D. R. Averett, R. E. Dornsife, B. A. Domin, R. Ferone, D. A. Lewis, and T. A. Krenitsky. 1994. 5-Chloro-2′,3′-dideoxy-3′-fluorouridine (935U83), a selective anti-human immunodeficiency virus agent with an improved metabolic and toxicological profile. Antimicrob. Agents Chemother. 38:1590-1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Meyer, S., H. Azijn, D. Surleraux, D. Jochmans, A. Tahri, R. Pauwels, P. Wigerinck, and M. de Bethune. 2005. TMC114, a novel human immunodeficiency virus type 1 protease inhibitor active against protease inhibitor-resistant viruses, including a broad range of clinical isolates. Antimicrob. Agents Chemother. 49:2314-2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Department of Health and Human Services Panel, A Working Group of the Office of AIDS Research Advisory Council. 10 October 2006, posting date. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. http://www.aidsinfo.nih.gov/guidelines.
- 6.Dumans, A. T., M. A. Soares, D. Pieniazek, M. L. Kalish, V. De Vroey, K. Hertogs, and A. Tanuri. 2002. Prevalence of protease and reverse transcriptase drug resistance mutations over time in drug-naïve human immunodeficiency type 1-positive individuals in Rio de Janeiro, Brazil. Antimicrob. Agents Chemother. 46:3075-3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferris, R. G., R. J. Hazen, G. B. Roberts, M. H. St. Clair, J. H. Chan, K. R. Romines, G. A. Freeman, J. H. Tidwell, L. T. Schaller, J. R. Cowen, S. A. Short, K. L. Weaver, D. W. Selleseth, K. R. Moniri, and L. Boone. 2005. Antiviral activity of GW678248, a novel benzophenone nonnucleoside reverse transcriptase inhibitor. Antimicrob. Agents Chemother. 49:4046-4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fisher, A. G., E. Collalti, L. Ratner, R. C. Gallo, and F. Wong-Staal. 1985. A molecular clone of HTLV-III with biological activity. Nature 316:262-265. [DOI] [PubMed] [Google Scholar]
- 9.Ford, S. L., Y. S. Reddy, M. T. Anderson, S. C. Murray, P. Fernandez, D. S. Stein, and M. A. Johnson. 2006. Single-dose safety and pharmacokinetics of brecanavir, a novel human immunodeficiency virus protease inhibitor. Antimicrob. Agents Chemother. 50:2201-2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hanlon, M. H., D. J. T. Porter, E. S. Furfine, A. Spaltenstein, H. L. Carter, D. Danger, A. Y. L. Shu, I. W. Kaldor, J. F. Miller, and V. A. Samano. 2004. Inhibition of wild-type and mutant human immunodeficiency virus type 1 proteases by GW0385 and other arylsulfonamides. Biochemistry 43:14500-14507. [DOI] [PubMed] [Google Scholar]
- 11.Harrigan, P. R., R. S. Hogg, W. W. Y. Dong, B. Yip, B. Wynhoven, J. Woodward, C. J. Brumme, Z. L. Brumme, T. Mo, C. S. Alexander, and J. S. G. Montaner. 2005. Predictors of HIV drug-resistance mutations in a large antiretroviral-naïve cohort initiating triple antiretroviral therapy. J. Infect. Dis. 191:339-347. [DOI] [PubMed] [Google Scholar]
- 12.Hill, A. V. 1910. The possible effects of the aggregation of the molecules of haemoglobin on its dissociation curves. J. Physiol. (London) 40:4-8. [Google Scholar]
- 13.Johnson, V. A., F. Brun-Vezinet, B. Clotet, B. Conway, D. R. Kuritzkes, D. Pillay, J. Schapiro, A. Telenti, and D. Richman. 2005. Update of the drug resistance mutations in HIV-1: 2005. Top. HIV Med. 13:51-57. [PubMed] [Google Scholar]
- 14.Kimpton, J., and M. Emerman. 1992. Detection of replication-competent and pseudotyped human immunodeficiency virus with a sensitive cell line on the basis of activation of an integrated β-galactosidase gene. J. Virol. 66:2232-2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Little, S. J., E. S. Daar, R. T. D'Aquila, P. H. Keiser, E. Connick, J. M. Whitcomb, N. S. Hellmann, C. J. Petropoulos, L. Sutton, J. A. Pitt, E. S. Rosenberg, R. A. Koup, B. D. Walker, and D. D. Richman. 1999. Reduced antiretroviral drug susceptibility among patients with primary HIV infection. JAMA 282:1142-1149. [DOI] [PubMed] [Google Scholar]
- 16.Mager, M. E. 1972. Data analysis in biochemistry and biophysics. Academic Press, New York, NY.
- 17.Miller, J. F., C. W. Andrews, M. Brieger, E. S. Furfine, M. R. Hale, M. H. Hanlon, R. J. Hazen, I. Kaldor, E. W. McLean, D. Reynolds, D. M. Sammond, A. Spaltenstein, R. Tung, E. M. Turner, R. X. Xu, and R. G. Sherrill. 2006. Ultra-potent P1 modified arylsulfonamide HIV protease inhibitors: the discovery of GW0385. Bioorg. Med. Chem. Lett. 16:1788-1794. [DOI] [PubMed] [Google Scholar]
- 18.Miller, J. F., E. S. Furfine, M. H. Hanlon, R. J. Hazen, J. A. Ray, L. Robinson, V. Samano, and A. Spaltenstein. 2004. Novel arylsulfonamides possessing sub-picomolar HIV protease activities and potent anti-HIV activity against wild-type and drug-resistant viral strains. Bioorg. Med. Chem. Lett. 14:959-963. [DOI] [PubMed] [Google Scholar]
- 19.Miyoshi, I., I. Kubonishi, S. Yoshimoto, T. Akagi, Y. Ohtsuki, Y. Shiraishi, K. Nagata, and Y. Hinuma. 1981. Type C virus particles in a cord T-cell line derived by co-cultivating normal cord leukocytes and human leukaemic T cells. Nature 294:770-771. [DOI] [PubMed] [Google Scholar]
- 20.Molla, A., H. Mo, S. Vasavanonda, L. Han, C. T. Lin, A. Hsu, and D. J. Kempf. 2002. In vitro antiviral interaction of lopinavir with other protease inhibitors. Antimicrob. Agents Chemother. 46:2249-2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Molla, A., S. Vasavanonda, G. Kumar, H. L. Sham, M. Johnson, B. Grabowski, J. F. Denissen, W. Kohlbrenner, J. J. Plattner, J. M. Leonard, D. W. Norbeck, and D. J. Kempf. 1998. Human serum attenuates the activity of protease inhibitors toward wild-type and mutant human immunodeficiency virus. Virology 250:255-262. [DOI] [PubMed] [Google Scholar]
- 22.Palella, F. J., K. M. Delaney, A. C. Moorman, M. O. Loveless, J. Fuhrer, G. A. Satten, D. J. Aschman, S. D. Holmberg, and the HIV Outpatient Study Investigators. 1998. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. N. Engl. J. Med. 338:853-860. [DOI] [PubMed] [Google Scholar]
- 23.Schwartz, O., Y. Henin, V. Marechal, and L. Montagnier. 1988. A rapid and simple colorimetric test for the study of anti-HIV agents. AIDS Res. Hum. Retrovir. 4:441-448. [DOI] [PubMed] [Google Scholar]
- 24.Selleseth, D. W., C. L. Talarico, T. Miller, M. W. Lutz, K. K. Biron, and R. J. Harvey. 2003. Interactions of 1263W94 with other antiviral agents in inhibition of human cytomegalovirus replication. Antimicrob. Agents Chemother. 47:1468-1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thaisrivongs, S., and J. W. Strohbach. 1999. Structure-based discovery of tipranavir disodium (PNU-140690E): a potent, orally bioavailable, nonpeptidic HIV protease inhibitor. Biopolymers 51:51-58. [DOI] [PubMed] [Google Scholar]
- 26.Yates, P. J., R. Hazen, M. St. Clair, L. Boone, M. Tisdale, and R. C. Elston. 2006. In vitro development of resistance to human immunodeficiency virus protease inhibitor GW640385. Antimicrob. Agents Chemother. 50:1092-1095. [DOI] [PMC free article] [PubMed] [Google Scholar]