

Abstract
Lopinavir (LPV)-ritonavir has demonstrated durable antiviral activity in human immunodeficiency virus type 1 (HIV-1)-infected antiretroviral-naïve and protease inhibitor (PI)-experienced patients. However, information on LPV activity against HIV-2 and the patterns of mutations in HIV-2 in response to selection by LPV is limited. The activity of LPV against three strains of HIV-2 was assessed and compared to activity against a reference HIV-1 strain. LPV demonstrated activity similar to that observed against HIV-1 in two HIV-2 strains (HIV-2MS and HIV-2CBL-23) tested. On the other hand, approximately 10-fold-reduced susceptibility was observed with the third HIV-2 strain, HIV-2CDC310319. Passage of HIV-2MS with increasing concentrations of LPV selected mutations V47A and D17N in the HIV-2 protease gene. The introduction of both 17N and 47A either individually or together into HIV-2ROD molecular infectious clones showed that the single V47A substitution in HIV-2 resulted in a substantial reduction in susceptibility to LPV. In contrast, this mutant retained wild-type susceptibility to other PIs and appeared to be hypersusceptible to atazanavir and saquinavir.
Lopinavir (LPV) is a human immunodeficiency virus (HIV) protease inhibitor (PI) that is approximately 10-fold more potent than the PI ritonavir (RTV) against HIV type 1 (HIV-1) in the presence of 50% human serum (50% effective concentration [EC50] of ca. 0.1 μM) (24). LPV is coformulated with low-dose RTV (LPV/r), which results in enhanced LPV plasma levels by virtue of decreased LPV metabolism through RTV inhibition of intestinal and hepatic cytochrome P450-3A4. When used in combination with reverse transcriptase (RT) inhibitors, LPV/r has demonstrated potent and durable virologic efficacy in HIV-1-infected antiretroviral-naïve patients over 7 years of therapy (6) as well as in PI-experienced patients (3). However, lower rates of response to LPV/r have been observed in those PI-experienced patients whose baseline viral isolates displayed susceptibility to LPV reduced by 10-fold or greater (14). Lower rates of response to LPV/r in HIV-1 have been associated with the presence of specific resistance mutations in the HIV-1 protease gene (L10F/I/R/V, K20M/R, L24I, M46I/L, F53L, I54L/T/V, L63P, A71I/L/T/V, V82A/F/T, I84V, and L90M) (13). In addition to these mutations, the emergence of the I47A mutation (a two-step mutation in HIV-1 of I47→I47V→I47A), together with V32I, has recently been described both in vitro and in HIV-1-infected patients during LPV/r therapy (4, 7, 8, 11, 14).
Western Africa is an area where HIV-2 infection is endemic, and HIV-2 infection has been noted in East Asia, Western Europe, and North America (15, 25). The optimal treatment of HIV-2 infection remains uncertain (16). Nucleoside RT inhibitors have typically demonstrated activity against HIV-2 and appear to select resistance mutations similar to those selected in HIV-1 (22). In contrast, currently available nonnucleoside RT inhibitors are much less active against HIV-2 than against HIV-1, likely due to the presence of residues in wild-type HIV-2 RT (such as RT 188L), which confer markedly decreased susceptibility to existing nonnucleoside RT inhibitors in HIV-1 (9, 28). Limited information on the activity of PIs against HIV-2 is currently available. Studies have found that saquinavir (SQV), RTV, and indinavir (IDV) generally have comparable in vitro activities against wild-type HIV-2 and HIV-1 isolates, while amprenavir (APV) appears to be less active against HIV-2 than HIV-1 (27). The in vitro activities of neither LPV nor darunavir (DRV) against HIV-2 have been fully described. The mechanisms of development of resistance of HIV-2 to PIs are even less well characterized. Of note, the sequence of wild-type HIV-2 protease contains amino acids that, in the context of HIV-1, are typically associated with significant PI resistance. These include the amino acids 10V/I, 32I, 46I, 47V, 71V, and 82I, all present in >85% of HIV-2 isolates (18). The effect of these sequence differences on resistance patterns in HIV-2 and their implications for the genetic barrier to resistance development in vivo have not been elucidated.
In this study, the activities of LPV and other PIs against three different wild-type HIV-2 strains (HIV-2MS, HIV-2CBL-23, and HIV-2CDC310319) were quantified and compared to the activity against a well-characterized wild-type HIV-1 strain (HIV-1NL4-3). In addition, using in vitro passage of HIV-2MS, we selected variants with reduced susceptibility to LPV and defined the genotypic basis for the observed resistant phenotype. Finally, by using site-directed mutagenesis, we characterized the effects of the critical amino acid substitutions in HIV-2 protease on the viral replication capacity and susceptibility to LPV and other PIs.
MATERIALS AND METHODS
Viruses, cells, and antiviral agents.
The following reagents were obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: HIV-1NL4-3 from Malcolm Martin (1), U937/HIV-2MS from Phyllis Kanki (12), HIV-2CBL-23/H9 from Robin Weiss (23), HIV-2CDC310319 from Stefan Wiktor and Mark Rayfield (17), 293 cells from Andrew Rice, CEM174 cells from Beatrice Han and George Shaw, and MT-4 cells from Douglas Richman. pSL-3 is an infectious molecular clone of HIV-2ROD (5) obtained through Beatrice Hahn. PIs were synthesized at Abbott Laboratories according to standard procedures.
MT4 cells were grown in RPMI 1640 medium (Sigma) supplemented with penicillin-streptomycin and 10% fetal bovine serum (FBS) (Invitrogen). Peripheral blood mononuclear cells (PBMCs) were freshly isolated from human donors using a standard protocol (4). These cells were grown in RPMI 1640 medium with penicillin-streptomycin and 20% FBS. After 3 days of stimulation with phytohemagglutinin (5 μg/ml), they were maintained in phytohemagglutinin-free medium supplemented with 20% FBS, penicillin-streptomycin, and interleukin-2 (50 U/ml). All cells were maintained at 37°C and 5% CO2 and split 1:10 every 3 to 5 days.
Determination of antiviral activity of PIs.
HIV-2MS and HIV-1NL4-3 replicated well in MT4 cells and produced a cytopathic effect. The antiviral activity of PIs in these strains was determined in MT4 cells using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Sigma) assay. In contrast, HIV-2CBL-23 and HIV-2CDC310319 replicated poorly in MT4 cells (data not shown). The antiviral activity of PIs in these strains was evaluated in PBMCs, with viral replication monitored by RT activity. All of the HIV-2MS mutant populations generated by selection with LPV replicated well in MT4 cells but failed to produce a cytopathic effect. Therefore, the susceptibility of the HIV-2MS mutant populations was monitored by RT activity.
The susceptibility of each virus to a PI was determined in three independent experiments. For each experiment, MT4 cells or PBMCs were infected with a virus at a concentration of 50 to 100 pg RNA for 1 h, washed twice to remove unadsorbed virus, and seeded in a 96-well plate at 100 μl/well. PI (solubilized in dimethyl sulfoxide solution, 1% final volume; Sigma) was then added in a series of half-log dilutions to media in triplicates. Two control cultures were routinely included: a virus-infected cell control culture treated in a manner identical to that of test cultures but without the addition of PI and an uninfected cell control culture maintained in the absence of PI or virus. Plates were incubated for 5 days in a CO2 incubator at 37°C, at which time viral replication was assessed by either RT activity or cytopathic effects.
For the RT activity assay, cell-free culture supernatants were collected on day 5, and the levels of RT were determined by a Lenti-RT activity assay (Cavidi Tech, Uppsala, Sweden). Percent inhibition of antigen production was determined as follows: (RT level in test well/RT level in infected control well) × 100.
For cytopathic effect assays, a stock solution of MTT (4 μg/ml in phosphate-buffered saline; Invitrogen) was added to all wells at 25 μl per well on day 5. Plates were incubated with MTT for 4 h and then treated with 20% sodium dodecyl sulfate plus 0.2 N HCl at 50 μl per well to lyse the cells. After an overnight incubation, optical density (OD) was measured by reading the plates at 570/650-nm wavelengths on a Bio-Tek microtiter plate reader. Percent reduction in cytopathic effect was determined as follows: [(OD test well − OD infected control well)/(OD uninfected control well − OD infected control well)] × 100.
The EC50 was then calculated by nonlinear regression analysis using Prism (GraphPad Software, Inc.). Reported EC50s are mean results of three independent experiments.
The susceptibility of each HIV-2ROD mutant clone to a PI was determined in triplicate using CEM174 cells. For each experiment, equal amounts of HIV-2ROD wild-type or mutant virus were used (100 pg RT). After incubation for 2 h at 37°C, cells were washed to remove unadsorbed virus and seeded in a 96-well plate at 100 μl/well (2 × 105 cells/ml). PI compounds (solubilized in dimethyl sulfoxide solution, 1% final volume; Sigma) were serially diluted 1:4 in RPMI at a 2× final concentration and plated at 100 μl per well. Two control cultures were included as described above, and plates were incubated for 7 days in a CO2 incubator at 37°C, at which time viral replication was assessed by RT activity. EC50 was calculated as described above, and resistance values (n-fold) were determined by dividing the EC50 for HIV-2ROD mutants by the EC50 for HIV-2ROD.
Generation of reduced susceptibility to LPV in HIV-2.
MT4 cells were infected with HIV-2MS at a multiplicity of infection of 0.003 for 2 h, washed, and then cultured in the presence of LPV. Viral replication was monitored every 3 to 5 days by determinations of levels of HIV-2 p27 antigen (Abbott Laboratories, Chicago, IL) in the culture supernatant as well as by observations for any cytopathic effects present in the cultures. The concentration of LPV used in the initial passage was 10 nM. Cells were passaged every 5 days regardless of virus production. When p27 antigen was undetectable (antibody assay resulted in an OD of <1), cells were split 1:2, 1:3, or 1:4, depending on confluence, and drug was added to the culture to bring the LPV concentration back to the level prior to the split. When p27 antigen was detectable (antibody assay resulted in an OD of >1), the culture supernatant was frozen at −80°C, and cells were washed and stored at −20°C for subsequent analysis. One aliquot of culture supernatant from the previous passage was then used to infect fresh MT4 cells. Following each passage when p27 antigen levels were detectable, the drug concentration was increased by 1.5- to 6-fold.
Sequencing of HIV-2 protease and gag cleavage sites.
RNA from culture supernatant was extracted using a QIAamp Viral RNA Mini kit (QIAGEN). The protease-coding region was amplified by RT-PCR using a OneStep RT-PCR kit (QIAGEN) and primers specific to HIV-2: 5′-AGACAGGGCTGCTGGAAATGTG-3′ (primer 1) and 5′-GGTCCATCTTTCCCTGGCTTCAAC-3′ (primer 2). The RT-PCR consisted of reverse transcription at 50°C for 30 min; denaturation at 95°C for 15 min; 40 amplification cycles of 94°C for 45 s, 55°C for 45 s, and 72°C for 2 min; and a final elongation step at 72°C for 10 min. Similarly, the gag p7/p1 and p1/p6 cleavage site region was amplified by RT-PCR using the following primers specific to HIV-2: 5′-AGAAGRCAGGGMTGCTGGAARTG-3′ (primer 1) and 5′-CCTGTGTCTAGTAACACTTCTACTGGCTGACC-3′ (primer 2). The RT-PCR consisted of reverse transcription at 50°C for 30 min; denaturation at 95°C for 15 min; 30 amplification cycles of 94°C for 30 s, 63°C for 30 s, and 72°C for 1 min; and a final elongation step at 72°C for 10 min. The amplified products were purified and sequenced by automated sequencing using a BigDye Terminator v3.1 cycle sequencing kit (Applied Biosystems) on an ABI Prism 3100 sequencer (Applied Biosystems). Individual clones were created from the amplified product using an Invitrogen TOPO cloning kit and were then isolated and sequenced in the same manner.
Generation of site-directed mutants.
Plasmid DNA of pSL-3 was used as a parental template DNA to introduce mutations of amino acids 17N and 47A in the protease of HIV-2ROD. A site-directed point mutation was introduced by using the QuikChange II XL site-directed mutagenesis kit (Stratagene). Primer G17NROD-F (5′-GTAGTCACAGCATACATTGAGAATCAGCCAGTAGAAGTCTTG-3′) and its complement primer, G17NROD-R, were used for the generation of the G17N mutant. Primer V47AROD-F (5′-CAATATAGCCCAAAAATAGCAGGGGGAATAGGGGGATTC-3′) and its complement primer, V47AROD-R, were used for the generation of the V47A mutant. The G17N/V47A double mutant was constructed by using the G17N mutant as the template and primer pair V47AROD-F and V47AROD-R for site-directed mutagenesis. The introduced mutations were always confirmed by sequencing of the complete protease gene.
Determination of replication capacity.
To generate infectious virus stocks of HIV-2ROD and its site-directed mutants, 293T cells (2.4 × 106 cells in 100-mm cell culture dishes) were transfected with plasmid DNA of pSL-3 or mutant constructs pSL-3/G17N, pSL-3/V47A, and pSL-3/G17N-V47A, respectively. The transfection was introduced by using Fugene6 reagent (Roche) according to the manufacturer's instructions. Virus-containing culture supernatants were harvested 3 days later, filtered, and stored at −80°C. The presence of viruses in the supernatant was determined by measuring the RT activity with a Lenti-RT activity assay kit.
In order to evaluate virus replication capacity, an equal amount of HIV-2ROD or mutant viruses (100 pg of RT) was used to infect CEM174 cells. After adsorption for 2 h at 37°C, the infected cells were washed and resuspended in medium, and the plate was incubated at 5% CO2 and 37°C. Each assay was performed in triplicate, and supernatants from each culture were collected at various days after infection. Virus replication was determined by measuring RT levels in culture supernatants. Differences in growth kinetics for HIV-2ROD and mutants were analyzed by comparing the absorbance values at OD405 for various time points.
RESULTS
Activity of PIs in HIV-2.
The in vitro susceptibilities of three HIV-2 isolates (HIV-2MS, HIV-2CBL-23, and HIV-2CDC310319) and HIV-1NL4-3 to several PIs in clinical use (APV, ATV, DRV, IDV, LPV, NFV, RTV, and SQV) are shown in Table 1. The activities of LPV, SQV, and IDV in HIV-2MS and HIV-2CBL-23 were similar to those in HIV-1NL4-3, with a less-than-threefold change in the EC50 in all cases. ATV and NFV demonstrated slightly lower activity in HIV-2MS and HIV-2CBL-23 than in HIV-1NL4-3, with four- and threefold increases in EC50 values in HIV-2MS and eight- and fivefold increases in HIV-2CBL-23, respectively. The activity of RTV was reduced by 8-fold in HIV-2MS and 11-fold in HIV-2CBL-23 compared to HIV-1NL4-3. Similarly, the activity of APV was reduced 13-fold in both HIV-2MS and HIV-2CBL-23 compared to HIV-1NL4-3. DRV activity was reduced sevenfold in HIV-2CBL-23 compared to the activity in HIV-1NL4-3.
TABLE 1.
Activity of protease inhibitors in HIV-2a
| Protease inhibitor | EC50 (nM) (fold change compared to HIV-1)
|
|||
|---|---|---|---|---|
| HIV-1NL4-3 | HIV-2MS | HIV-2CBL-23 | HIV-2CDC310319 | |
| APV | 72 ± 30 | 900 ± 63 (12.5) | 939 ± 166 (13.0) | 674 ± 79 (9.2) |
| ATV | 5 ± 2 | 20 ± 1 (4.0) | 39 ± 12 (7.8) | 110 ± 34 (22.0) |
| DRV | 12 ± 5 | ND | 83 ± 31 (6.9) | 155 ± 52 (12.9) |
| IDV | 38 ± 8 | 22 ± 8 (0.5) | 33 ± 8 (0.8) | 108 ± 6 (2.6) |
| LPV | 18 ± 3 | 15 ± 7 (0.8) | 12 ± 4 (0.7) | 180 ± 71 (10.0) |
| NFV | 18 ± 12 | 48 ± 23 (2.7) | 83 ± 17 (4.6) | 389 ± 66 (21.6) |
| RTV | 46 ± 15 | 349 ± 165 (7.6) | 514 ± 172 (11.2) | 665 ± 98 (14.5) |
| SQV | 12 ± 1 | 5 ± 1 (0.4) | 8 ± 2 (0.7) | 68 ± 24 (5.7) |
Each EC50 value represents the mean of at least three determinations. ND, not determined.
HIV-2CDC31019 differed from HIV-2MS and HIV-2CBL-23 in having generally lower susceptibility to most PIs tested. IDV demonstrated the smallest decrease in susceptibility (threefold compared to that of HIV-1NL-43), whereas ATV had the highest (22-fold).
In order to confirm that EC50 data are comparable across cell cultures and assays, the LPV EC50 for HIV-2MS was assessed in both MT4 cells (by MTT and RT assays) and PBMCs (by RT assay). The average of at least three experiments showed that the LPV EC50 for HIV-2MS in MT4 cells was 15 ± 7 nM with the MTT assay and 6 ± 1 nM with the RT assay, whereas in PBMCs, it was 15 ± 6 nM with the RT assay, demonstrating no substantial differences between types of cell cultures or assays. The same series of experiments performed with the HIV-1NL4-3 virus also showed similarity across cell lines and assays.
Sequencing of the HIV-2 protease regions.
In order to assess the relationship of amino acid variation in protease to susceptibility to PIs, both the protease-coding region and gag cleavage sites p7/p1 and p1/p6 from each HIV-2 strain were amplified, sequenced, and compared across HIV-2 and HIV-1 isolates. The results of this analysis are provided in Fig. 1. Of note, all three isolates of HIV-2 had specific amino acid changes in the protease gene compared to HIV-1 at positions known or suspected to contribute to decreased PI susceptibility in HIV-1. These changes included 10V, 20V, 32I, 36I, 46I, 47V, 71V, and 82I (10). Compared to HIV-2MS and HIV-2CBL-23, HIV-2CDC310319 had six unique protease substitutions: 7R, 12K, 64V, 67V, 91N, 92S, and 99F. In the gag cleavage sites, HIV-2CDC310319 had only one unique substitution (S451T) compared to HIV-2MS and HIV-2CBL-23. However, none of these substitutions has previously been identified as conferring decreased susceptibility to PIs in HIV-1.
FIG. 1.

Alignment of the protease amino acid sequence (A) and gag p7/p1 and p1/p6 cleavage sites (B) from the HIV-2 isolates. The dots represent homology to the reference strain HIV-1NL4-3. A letter indicates an amino acid substitution relative to the reference. Underlined amino acids are those associated with a decreased susceptibility to PIs in HIV-1, and arrows point to the positions where mutations were observed after LPV selection in the current study.
Selection for decreased susceptibility to LPV in HIV-2 by in vitro passage.
To select HIV-2 with decreased susceptibility to LPV in vitro, MT4 cells were infected with HIV-2MS and serially passaged in the presence of increasing concentrations of LPV beginning with 10 nM LPV (passage 1). Cultures were tested for virus production every 3 to 5 days with the HIV-2 p27 assay and passaged under increasing concentrations of LPV as shown in Fig. 2. The concentration of LPV was increased to 1,000 nM by passage 18. Phenotypic analysis of virus at passage 18 revealed 34-fold-decreased susceptibility to LPV compared to baseline virus (Table 2).
FIG. 2.
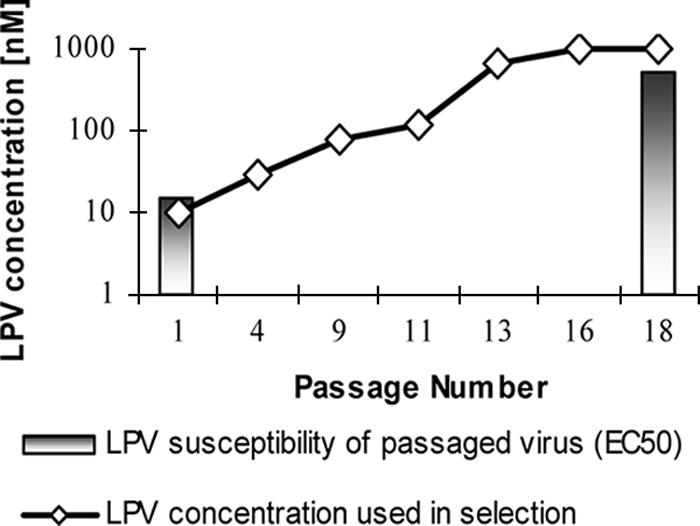
Concentration of LPV used in selection of LPV resistance in HIV-2MS. The line shows the concentration of LPV in nM versus passage number, and the bars show the EC50 of LPV versus the passaged virus stock at passages 1 and 18.
TABLE 2.
Genotype and phenotype of LPV-passaged HIV-2MSa
| Passage | LPV concn (nM) | No. of positive clones/total no. of clones tested
|
EC50 (nM) (fold change) | |||
|---|---|---|---|---|---|---|
| WT | D17N | V47A | D17N/ V47A | |||
| 1 | 1 | 8/8 | 0/8 | 0/8 | 0/8 | 15 (1) |
| 11 | 120 | 12/23 | 7/23 | 4/23 | 0/23 | ND |
| 18 | 1,000 | 5/22 | 5/22 | 1/22 | 11/22 | 515 (34) |
WT, wild type; ND, not determined.
Sequence analysis of LPV-selected HIV-2 clones.
To examine the protease sequence changes in LPV-selected HIV-2MS, proviral DNA sequences from infected cells were cloned and sequenced for passages 1, 11, and 18. Clonal sequencing of the protease and p7/p1 and p1/p6 gag cleavage site coding region of these virus populations revealed two developing mutations, G17N and V47A (Table 2). The wild-type sequence was observed in all eight of the clones obtained from passage 1. By passage 11, two single protease mutations, D17N and V47A, had emerged and were present in 7 and 4 of 23 clones sequenced, respectively. No clone contained both mutations at this time point. In contrast, at passage 18, when LPV concentrations reached 1,000 nM, half of the clones sequenced showed the double mutation D17N/V47A. Interestingly, 5 of the 23 clones sequenced at passage 18 still carried the wild-type sequence at these two amino acids, 5 contained the single mutation D17N, and 1 clone had the single V47A amino acid substitution. The five clones carrying wild-type sequence at positions 17 and 47 contained no other amino acid substitutions.
Effect of protease 17N and 47A on PI susceptibility and replicative fitness in HIV-2.
To investigate the relationship between the amino acid changes in passaged HIV-2MS and the decreased susceptibility to LPV, a series of molecular clones was constructed in HIV-2ROD containing each mutation individually and one containing the double mutation. Note that while HIV-2MS carries D at protease amino acid position 17, all other HIV-2 (including HIV-2ROD) and HIV-1 strains in this study contain G at this position, so this mutation will be referred to as G17N.
PI EC50 determinations show that the single mutation V47A and the double mutation G17N/V47A exhibit approximately 10-fold-reduced susceptibility to LPV (Table 3). The single G17N mutation did not affect the susceptibility of the virus to LPV or to any of the other PIs either as a sole mutation or on a background of V47A. HIV-2ROD clones containing either the V47A or the double G17N/V47A mutation maintained wild-type susceptibility to DRV, IDV, NFV, and APV. However, these mutants also displayed hypersusceptibility to ATV and SQV, with both compounds having approximately 10-fold-greater activity in these mutants than in HIV-2ROD.
TABLE 3.
Activity of protease inhibitors in HIV-2ROD and site-directed mutantsa
| Protease inhibitor | EC50 (nM) (fold change compared to HIV-2ROD)
|
|||
|---|---|---|---|---|
| HIV-2ROD | G17N | V47A | G17N/V47A | |
| ATV | 34 ± 3 | 20 ± 1 (0.6) | 5 ± 0.5 (0.1) | 3 ± 0.5 (0.1) |
| APV | 855 ± 55 | 583 ± 28 (0.7) | 630 ± 118 (0.7) | 404 ± 20 (0.5) |
| DRV | 9 ± 2 | 6 ± 0.4 (0.7) | 10 ± 1 (1.1) | 8 ± 0.3 (0.9) |
| IDV | 65 ± 5 | 52 ± 5 (0.8) | 34 ± 5 (0.5) | 24 ± 1 (0.5) |
| LPV | 35 ± 2 | 28 ± 1 (0.8) | 340 ± 15 (9.7) | 260 ± 25 (7.4) |
| NFV | 281 ± 32 | 199 ± 9 (0.7) | 177 ± 17 (0.6) | 100 ± 34 (0.4) |
| RTV | 421 ± 10 | 310 ± 23 (0.7) | 115 ± 6 (0.3) | 84 ± 5 (0.2) |
| SQV | 3.6 ± 0.6 | 2.2 ± 0.1 (0.6) | 0.2 ± 0.2 (0.1) | 0.2 ± .07 (0.1) |
Each EC50 value represents the mean of three determinations.
The replication capacity of each of the clones, as measured by RT activity assay, is shown in Fig. 3. Both the V47A single and G17N/V47A double mutants grew slightly more slowly than HIV-2ROD.
FIG. 3.
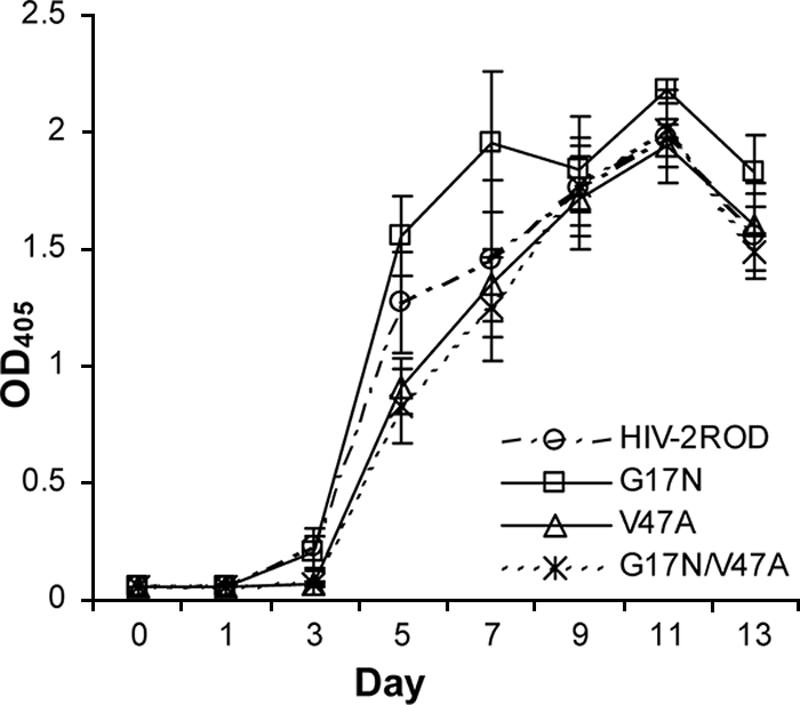
Replication capacity of HIV-2ROD and site-directed mutants. Replication capacity was measured by OD405 (OD at 405 nm).
DISCUSSION
In this study, we have shown that HIV-1 PIs demonstrate various activities against HIV-2. LPV showed substantial activity in both HIV-2MS and HIV-2CBL-23, with EC50 values similar to the EC50 value in HIV-1NL4-3. However, HIV-2CDC310319 displayed approximately 10-fold-reduced susceptibility to LPV. IDV, SQV, and NFV had similar activities in HIV-1NL4-3, HIV-2MS, and HIV-2CBL23, whereas APV and RTV had significantly reduced activity against these two HIV-2 strains. These results concur with data from previous comparisons of these compounds against HIV-2ROD and HIV-2EHO (27).
The mechanism of the generally reduced susceptibility of HIV-2CDC310319 to PIs is unknown. However, it is noteworthy that several amino acid changes that contribute to reduced susceptibility to PIs in HIV-1 were identified in the sequence of all three HIV-2 strains, including 10I, 20V, 32I, 36I, 46I, 47V, 71V, and 82I. Any additional mutations superimposed on this preexisting genotype may result in a significant reduction in PI activity against HIV-2.
An analysis of the p7/p1 and p1/p6 gag cleavage sites did not reveal any further insight into this reduced susceptibility. HIV-2MS, HIV-2CBL23, and HIV-2CDC310319 all carry five amino acid differences from the HIV-1 sequence in these regions: N432G, I437G, R444K, G446R, and L449P. To our knowledge, none of the changes at these sites has previously been associated with PI resistance (2, 26). HIV-2CDC310319 carries two changes in the gag cleavage sites (K436F and S451T) not seen in either HIV-2MS or HIV-2CBL23. In HIV-1, the accumulation of different mutations at K436 was associated with PI treatment (26), and we cannot rule out the possibility that this change may confer reduced susceptibility to PIs. In addition, HIV-2CBL23 carries a change (A431V) that has been associated with PI exposure both in vitro (4) and in vivo (2, 26), particularly in the presence of the M46I PI resistance mutation in the protease gene (a substitution present in the sequences of all three HIV-2 strains studied here). However, this combination does not appear to confer reduced susceptibility to LPV in HIV-2CBL23.
The passage of the HIV-2MS strain in the presence of increasing concentrations of LPV selected two protease mutations, 17N and 47A. Sequencing of the gag cleavage sites p7/p1 and p1/p6 revealed no mutations at passage 18 compared to the wild type, eliminating this area as a possible source of additional resistance in this study. In HIV-1, protease 47A has been demonstrated to produce decreased susceptibility to LPV but not to other PIs. While the genetic change to 47A is a two-step process (I4→I47V→I47A) in HIV-1, most wild-type HIV-2 strains appear to have Val at position 47 of protease instead of Ile. Thus, only a single mutation is needed to develop 47A in HIV-2, a lower genetic barrier than in HIV-1. Our data confirm the ease of in vitro selection of this mutation by LPV in HIV-2.
Protease 47A demonstrated reduced susceptibility to LPV when introduced into an HIV-2ROD molecular clone, confirming the importance of this amino acid change with regard to LPV susceptibility. Protease 47A also appeared to confer 10-fold-increased susceptibility (hypersusceptibility) to ATV and SQV, suggesting the potential utility of these PIs in HIV-2 with this drug resistance mutation. These data are consistent with observations of HIV-1 in which 47A appeared to confer reduced susceptibility specific to LPV, with hypersusceptibility to SQV observed (8). In contrast, the introduction of protease 17N into HIV-2ROD (with or without the presence of 47A) did not appear to affect the potency of any of the PI compounds tested or the replication capacity of the virus. Whether the presence of 17N on a background of 47A would result in decreased LPV susceptibility or an increased replication capacity in HIV-2MS (which differs from HIV-2ROD in 10% of amino acids in the protease gene) would need to be investigated further once an HIV-2MS molecular clone becomes available.
While no controlled clinical trials of LPV/r for the treatment of HIV-2 have been performed, limited case reports suggesting clinical antiviral activity of LPV/r against HIV-2 have been published (16, 19, 20). However, the failure of LPV/r has also been described relatively frequently in these reports, consistent with the hypothesis that the high genetic barrier noted with LPV/r treatment of HIV-1 may be compromised by the presence of PI resistance-associated mutations in wild-type HIV-2 (21). In addition, the presence of protease 47V in wild-type HIV-2 implies that a single nucleotide change would result in 47A, producing clinically significant LPV resistance. Recent reports suggest that this may be a relatively common pathway for the emergence of LPV resistance in HIV-2-infected patients treated with LPV/r (21).
In conclusion, our in vitro studies suggest variable activity of PIs against HIV-2. Notably, the presence of several mutations in wild-type HIV-2 protease generally associated with PI resistance in HIV-1 along with the ease of selection of 47A (a one-step process from 47V in HIV-2) will likely result in a lower genetic barrier to clinically significant PI resistance and may compromise the response to therapy. However, other PIs, including SQV and ATV, appear to maintain potent activity against viral isolates that have developed the 47A mutation after LPV exposure and may be appropriate considerations for therapy of select patients with LPV-resistant HIV-2 infection.
Acknowledgments
This work was supported in part by a grant from the National Institutes of Health (GM 065057).
Footnotes
Published ahead of print on 18 June 2007.
REFERENCES
- 1.Adachi, A., H. E. Gendelman, S. Koenig, T. Folks, R. Willey, A. Rabson, and M. A. Martin. 1986. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 59:284-291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bally, F., R. Martinez, S. Peters, P. Sudre, and A. Telenti. 2000. Polymorphism of HIV type 1 gag p7/p1 and p1/p6 cleavage sites: clinical significance and implications for resistance to protease inhibitors. AIDS Res. Hum. Retrovir. 16:1209-1213. [DOI] [PubMed] [Google Scholar]
- 3.Benson, C. A., S. G. Deeks, S. C. Brun, R. M. Gulick, J. J. Eron, H. A. Kessler, R. L. Murphy, C. Hicks, M. King, D. Wheeler, J. Feinberg, R. Stryker, P. E. Sax, S. Riddler, M. Thompson, K. Real, A. Hsu, D. Kempf, A. J. Japour, and E. Sun. 2002. Safety and antiviral activity at 48 weeks of lopinavir/ritonavir plus nevirapine and 2 nucleoside reverse-transcriptase inhibitors in human immunodeficiency virus type 1-infected protease inhibitor-experienced patients. J. Infect. Dis. 185:599-607. [DOI] [PubMed] [Google Scholar]
- 4.Carrillo, A., K. D. Stewart, H. L. Sham, D. W. Norbeck, W. E. Kohlbrenner, J. M. Leonard, D. J. Kempf, and A. Molla. 1998. In vitro selection and characterization of human immunodeficiency virus type 1 variants with increased resistance to ABT-378, a novel protease inhibitor. J. Virol. 72:7532-7541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clavel, F., M. Guyader, D. Guetard, M. Salle, L. Montagnier, and M. Alizon. 1986. Molecular cloning and polymorphism of the human immune deficiency virus type 2. Nature 324:691-695. [DOI] [PubMed] [Google Scholar]
- 6.da Silva, B., F. McMillan, C. Hicks, J. Eron, P. Wolfe, R. Gulick, M. Glesby, M. Thompson, C. Benson, A. C. White, M. Albrecht, H. Kessler, K. Niemi, K. King, D. Calhoun, M. King, G. Hanna, and S. Brun. 2005. Seven year follow-up of a lopinavir/ritonavir (LPV/r)-based regimen in antiretroviral (ARV)-naïve subjects, abstr. PE7.9/3. Abstr. 10th Eur. AIDS Conf.
- 7.de Mendosza, C., L. Valer, L. Bacheler, T. Pattery, A. Corral, and V. Soriano. 2006. Prevalence of the HIV-1 protease 147A in clinical practice and association with lopinavir resistance. AIDS 20:1071-1073. [DOI] [PubMed] [Google Scholar]
- 8.Friend, J., N. Parkin, T. Liegler, J. N. Martin, and S. G. Deeks. 2004. Isolated lopinavir resistance after virological rebound of a ritonavir/lopinavir-based regimen. AIDS 18:1965-1966. [DOI] [PubMed] [Google Scholar]
- 9.Isaka, Y., S. Miki, S. Kawauchi, A. Suyama, H. Sugimoto, A. Adachi, T. Miura, M. Hayami, O. Yoshie, T. Fujiwara, and A. Sato. 2001. A single amino acid change at Leu-188 in the reverse transcriptase of HIV-2 and SIV renders them sensitive to non-nucleoside reverse transcriptase inhibitors. Arch. Virol. 146:743-755. [DOI] [PubMed] [Google Scholar]
- 10.Johnson, V. A., F. Brun-Vezinet, B. Clotet, D. R. Kuritzkes, D. Pillay, J. M. Schapiro, and D. D. Richman. 2006. Update of the drug resistance mutations in HIV-1: fall 2006. Top. HIV Med. 14:125-130. [PubMed] [Google Scholar]
- 11.Kagan, R. M., M. D. Shenderovich, P. N. Heseltine, and K. Ramnarayan. 2005. Structural analysis of an HIV-1 protease I47A mutant resistant to the protease inhibitor lopinavir. Protein Sci. 14:1870-1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kanki, P., F. Barin, and M. Essex. 1988. Antibody reactivity to multiple HIV-2 isolates, abstr. 1659. Abstr. Int. Conf. AIDS.
- 13.Kempf, D., J. Isaacson, M. King, S. Brun, J. Sylte, B. Richards, B. Bernstein, R. Rode, and E. Sun. 2002. Analysis of the virological response with respect to baseline viral phenotype and genotype in protease inhibitor-experienced HIV-1-infected patients receiving lopinavir/ritonavir therapy. Antivir. Ther. 7:165-174. [PubMed] [Google Scholar]
- 14.Mo, H., M. S. King, K. King, A. Molla, S. Brun, and D. J. Kempf. 2005. Selection of resistance in protease inhibitor-experienced, human immunodeficiency virus type 1-infected subjects failing lopinavir- and ritonavir-based therapy: mutation patterns and baseline correlates. J. Virol. 79:3329-3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mukhopadhyay, C., G. Nath, A. K. Gulati, and S. C. Mohapatra. 2001. Prevalence of HIV among low and high risk population of eastern part of northern India. J. Commun. Dis. 33:136-142. [PubMed] [Google Scholar]
- 16.Mullins, C., G. Eisen, S. Popper, A. D. Sarr, J. L. Sankale, J. J. Berger, S. B. Wright, H. R. Chang, G. Coste, T. P. Cooley, P. Rice, P. R. Skolnik, M. Sullivan, and P. J. Kanki. 2004. Highly active antiretroviral therapy and viral response in HIV type 2 infection. Clin. Infect. Dis. 38:1771-1779. [DOI] [PubMed] [Google Scholar]
- 17.Owen, S. M., D. Ellenberger, M. Rayfield, S. Wiktor, P. Michel, M. H. Grieco, F. Gao, B. H. Hahn, and R. B. Lal. 1998. Genetically divergent strains of human immunodeficiency virus type 2 use multiple coreceptors for viral entry. J. Virol. 72:5425-5432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pieniazek, D., M. Rayfield, D. J. Hu, J. N. Nkengasong, V. Soriano, W. Heneine, C. Zeh, S. M. Agwale, C. Wambebe, L. Odama, and S. Z. Wiktor. 2004. HIV-2 protease sequences of subtypes A and B harbor multiple mutations associated with protease inhibitor resistance in HIV-1. AIDS 18:495-502. [DOI] [PubMed] [Google Scholar]
- 19.Rodes, B., J. Sheldon, C. Toro, V. Jimenez, M. A. Alvarez, and V. Soriano. 2006. Susceptibility to protease inhibitors in HIV-2 primary isolates from patients failing antiretroviral therapy. J. Antimicrob. Chemother. 57:709-713. [DOI] [PubMed] [Google Scholar]
- 20.Rodes, B., C. Toro, V. Jimenez, and V. Soriano. 2005. Viral response to antiretroviral therapy in a patient coinfected with HIV type 1 and type 2. Clin. Infect. Dis. 41:e19-e21. [DOI] [PubMed] [Google Scholar]
- 21.Rodes, B., C. Toro, J. A. Sheldon, V. Jimenez, K. Mansinho, and V. Soriano. 2006. High rate of proV47A selection in HIV-2 patients failing lopinavir-based HAART. AIDS 20:127-129. [DOI] [PubMed] [Google Scholar]
- 22.Rodes, B., A. Holguin, V. Soriano, M. Dourana, K. Mansinho, F. Antunes, and J. Gonzalez-Lahoz. 2000. Emergence of drug resistance mutations in human immunodeficiency virus type 2-infected subjects undergoing antiretroviral therapy. J. Clin. Microbiol. 38:1370-1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schulz, T. F., D. Whitby, J. G. Hoad, T. Corrah, H. Whittle, and R. A. Weiss. 1990. Biological and molecular variability of human immunodeficiency virus type 2 isolates from The Gambia. J. Virol. 64:5177-5182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sham, H. L., D. J. Kempf, A. Molla, K. C. Marsh, G. N. Kumar, C. M. Chen, W. Kati, K. Stewart, R. Lal, A. Hsu, D. Betebenner, M. Korneyeva, S. Vasavanonda, E. McDonald, A. Saldivar, N. Wideburg, X. Chen, P. Niu, C. Park, V. Jayanti, B. Grabowski, G. R. Granneman, E. Sun, A. J. Japour, J. M. Leonard, J. J. Plattner, and D. W. Norbeck. 1998. ABT-378, a highly potent inhibitor of the human immunodeficiency virus protease. Antimicrob. Agents Chemother. 42:3218-3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shanmugam, V., W. M. Switzer, J. N. Nkengasong, G. Garcia-Lerma, T. A. Green, E. Ekpini, M. Sassan-Morokro, F. Antunes, K. Manshino, V. Soriano, S. Z. Wiktor, and W. Heneine. 2000. Lower HIV-2 plasma viral loads may explain differences between the natural histories of HIV-1 and HIV-2 infections. J. Acquir. Immune Defic. Syndr. 24:257-263. [DOI] [PubMed] [Google Scholar]
- 26.Verheyen, J., E. Litau, T. Sing, M. Däumer, M. Balduin, M. Oette, G. Fätkenheuer, J. K. Rockstroh, U. Schuldenzucker, D. Hoffman, H. Pfister, and R. Kaiser. 2006. Compensatory mutations at the HIV cleavage sites p7/p1 and p1/p6-gag in therapy-naïve and therapy-experienced patients. Antivir. Ther. 11:879-887. [PubMed] [Google Scholar]
- 27.Witvrouw, M., C. Pannecouque, W. M. Switzer, T. M. Folks, E. De Clercq, and W. Heneine. 2004. Susceptibility of HIV-2, SIV and SHIV to various anti-HIV-1 compounds: implications for treatment and postexposure prophylaxis. Antivir. Ther. 9:57-65. [PubMed] [Google Scholar]
- 28.Witvrouw, M., C. Pannecouque, K. Van Laethem, J. Desmyter, E. De Clercq, and A. M. Vandamme. 1999. Activity of non-nucleoside reverse transcriptase inhibitors against HIV-2 and SIV. AIDS 13:1477-1483. [DOI] [PubMed] [Google Scholar]