

Abstract
Bevirimat [3-O-(3′,3′-dimethylsuccinyl)betulinic acid] is the first in a new class of anti-human immunodeficiency virus (HIV) drugs that inhibit viral maturation by specifically blocking cleavage of the Gag capsid (CA) precursor, CA-SP1, to mature CA protein, resulting in defective core condensation and release of immature noninfectious virions. Four cohorts of six HIV-infected adults, with CD4 counts of >200 and plasma viral loads of 5,000 to 250,000 transcripts/ml and not currently receiving antiretroviral therapy, were randomized to receive a single oral dose of placebo, 75, 150, or 250 mg of bevirimat. Thirty blood samples for drug concentrations and 20 HIV RNA measures were collected from each subject over a 20-day period. Candidate pharmacokinetic/pharmacodynamic models were fit to individual subjects by maximum likelihood followed by Bayesian estimation; model discrimination was by corrected Akaike's Information Criterion. The bevirimat pharmacokinetics was well described by an oral two-compartment linear model (r2, 0.98), with a mean (percent coefficient of variation) half-life of 60.3 (13.6) h and apparent oral clearance of bevirimat from the plasma compartment of 0.17 (18) liters/h. HIV RNA was modeled as being produced in infected CD4 cells, with bevirimat inhibiting infection of new CD4 cells thru a Hill-type function (r2, 0.87). Single oral doses of bevirimat were well tolerated and demonstrated a dose-dependent reduction in viral load. The average maximum reduction from baseline following the 150- and 250-mg doses was greater than 0.45 log10, with individual patients having reductions of greater than 0.7 log10. No bevirimat resistance mutations were detected during the course of the study.
Significant progress has been made in the treatment of human immunodeficiency virus (HIV) infection, due to the availability of distinct classes of antiretrovirals that inhibit HIV replication by different mechanisms of action. Highly active antiretroviral therapy is now the standard of care, with medication regimens for newly diagnosed patients comprising a combination of two nucleoside analogues and either a protease inhibitor or efavirenz (1). However, current therapy is limited by toxicity and the eventual development of resistance, which in numerous cases may result in resistance to an entire class of drugs. Other factors, such as drug interactions and barriers to adherence, make current therapies less than ideal. Clearly, new agents with novel mechanisms of action are needed in order to more effectively manage patients, address and prevent resistance, and provide alternative therapies for those patients who fail or cannot tolerate approved antiretrovirals.
Bevirimat [3-O-(3′,3′-dimethylsuccinyl)betulinic acid] is the first agent in a new class of antiretrovirals that inhibit viral maturation (Fig. 1). Bevirimat targets a late step in the Gag processing cascade, namely, the release of the capsid protein (CA/p24) from the capsid precursor (CA-SP1/p25). This disruption to Gag protein processing results in defective core condensation and the release of noninfectious virus particles, blocking the spread of the infection to new cells (7). Bevirimat has demonstrated potent in vitro and in vivo activities, with an in vitro 90% inhibitory concentration of 22.1 ng/ml (37.8 nM), and retains activity against viruses that are resistant to other classes of antiretrovirals (7).
FIG. 1.

Molecular structure of bevirimat.
Phase I healthy volunteer studies have characterized the single- and multiple-dose pharmacokinetics of bevirimat when administered orally (9, 10). Following oral administration, bevirimat is well absorbed, well tolerated, and has a long half-life of approximately 55 h in healthy volunteers. Bevirimat is metabolized via hepatic glucuronidation, mediated primarily by UGT1A3, and bevirimat does not interact with the hepatic cytochrome P450 enzyme system. Based on its novel mechanism of action, long half-life, low probability of drug-drug interactions, and demonstrated potency in preclinical models (14), bevirimat is a promising candidate for further investigation in humans.
Because HIV treatment involves the use of drugs in combination therapy, the clinical development of new antiretroviral drugs offers a unique challenge for early dose selection. The goal of a phase II trial is to rapidly select one or possibly two potential doses to study in larger phase III trials. However, coadministering a new drug with two additional active drugs makes it difficult to attribute any of the observed pharmacological activity to the new agent. Therefore, abbreviated monotherapy viral dynamic studies are necessary to evaluate potential doses for future clinical trials. Unfortunately, the clinical outcomes of interest in HIV infection are typically measured in terms of weeks to months, while the development of resistance with monotherapy occurs much more rapidly. Therefore, the usual compromise is to study a new agent as monotherapy for 7 to 14 days during the acute phase of HIV type 1 (HIV-1) RNA decline. We have previously demonstrated that the integration of pharmacokinetic/pharmacodynamic modeling and simulation is a useful tool for identifying effective doses in abbreviated viral dynamic studies (13). The goal of the current study was to develop a pharmacokinetic/pharmacodynamic model suitable for the evaluation of the antiviral activity of bevirimat in HIV-infected patients not receiving concurrent antiretroviral therapy.
MATERIALS AND METHODS
Study design.
This was a randomized, double-blind, placebo-controlled study in HIV-infected adults with a CD4 count greater than 200 cells/mm3 and plasma viral load between 5,000 and 250,000 copies/ml. Patients were either antiretroviral naïve or had not been receiving active therapy for a minimum of 4 weeks prior to enrollment in the study. Patients were otherwise required to be in good health, with normal renal and hepatic function. Genotyping for mutations associated with antiretroviral resistance was conducted at baseline and at the end of the study.
The primary objective of the study was to assess the antiretroviral activity of single oral doses of bevirimat in HIV-1-infected patients not receiving active therapy. The secondary objective was to characterize the safety and pharmacokinetics/pharmacodynamics of bevirimat.
Patients were randomly assigned to receive bevirimat (75, 150, or 250 mg) or placebo. Blood samples for determination of bevirimat concentrations were collected at 0, 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 18, 24, 36, and 48 h after oral dosing of bevirimat or placebo and on days 3, 4, 5, 6, 7, 8, 9, 10, 12, 14, 16, 18, 20, and 24. Plasma viral load (HIV RNA) was determined at screening, just prior to the dose of bevirimat, and at 6, 12, 18, 24, 36, and 48 h after dosing. Additional viral load samples were taken on days 3, 4, 5, 6, 7, 8, 9, 10, 12, 14, 16, 18, 20, and 24.
Determination of bevirimat plasma concentrations.
Bevirimat plasma concentrations were measured using reverse-phase high-performance liquid chromatography assays with tandem mass spectrometric detection over the calibration ranges of 20.0 to 16,000 ng/ml (75-mg dose) and 100 to 60,000 ng/ml (150- and 250-mg doses). For the lower concentration range, heparinized plasma samples (100 μl) were treated with 50 μl of internal standard (1.50-μg/ml solution of 2,2-dimethylsuccinyl-4-dihydrobetulinic acid ester [DSD] in acetonitrile with 0.1% acetic acid) followed by 450 μl of cold, acidified acetonitrile to precipitate plasma proteins. After digestion and vortexing, the samples were centrifuged at 4°C and 13,000 × g for 10 min. The supernatant (500 μl) was evaporated to dryness with nitrogen at 30°C and the residue reconstituted in 80% methanol in 0.1% acetic acid. Samples were maintained at 4°C in the autosampler, and a 30-μl aliquot was injected into the liquid chromatography-tandem mass spectrometry system. For the higher concentration range, plasma samples (100 μl) were treated in a similar manner except that 10 μl of supernatant was directly injected without evaporation and reconstitution.
For both concentration ranges, prepared samples were chromatographed over a Luna C18 (2) high-performance liquid chromatography column (2.0 mm by 50 mm; 3.0-μm particles; part 00B-4251-B0; Phenomenex, Torrance, CA) maintained at 40°C using a mobile phase consisting of 83% methanol in 75 mM ammonium acetate buffer. The mobile-phase flow rate was 0.2 ml/min. The resulting retention times for bevirimat and DSD were approximately 5.0 and 6.5 min, respectively. Bevirimat and DSD were detected using a triple quadrupole mass spectrometer (model API 365; Applied Biosystems, Foster City, CA), with multiple reaction monitoring (bevirimat, 583.3 to 455.2 m/z; DSD, 585.3 to 457.3 m/z).
Linearity was observed over both calibration curve ranges. The overall accuracy (percent analytical recovery) and precision (percent coefficient of variation [CV]) of the assay were determined from the plasma quality control samples that were analyzed during the analysis of the study samples. For the 20.0- to 16,000-ng/ml calibration range, the accuracy and precision were calculated as 97.7% (92.2 to 102%) and 6.78% (4.87 to 7.81%), respectively. For the 100- to 60,000-ng/ml calibration range, the accuracy and precision were calculated as 98.1% (93.0 to 104%) and 5.34% (3.92 to 7.92%), respectively. The quantitation limit of the assays was 20.0 ng/ml and 100 ng/ml, respectively, for the lower and upper curve ranges. Bevirimat has demonstrated stability in heparinized human plasma for 95 days when stored at −70°C (unpublished data).
Pharmacokinetic/pharmacodynamic modeling methods.
The plasma concentration and viral load data were analyzed by the fitting of candidate pharmacokinetic and pharmacodynamic models to the data using a maximum likelihood estimation (ADAPT II). The processes of model development for the pharmacokinetic and pharmacodynamic models were similar. The data were evaluated graphically, and candidate models were fit to the data. Discrimination between and selection of a final model were based on simplicity and Hurvich and Tsai's corrected Akaike's Information Criterion (5). Initially, the pharmacokinetic model was developed without consideration of pharmacodynamics. Following selection of the final pharmacokinetic model, the pharmacokinetic parameters were held constant while the pharmacodynamic model was developed for the viral load data. The initial pharmacodynamic model evaluated was that described previously by Perelson and colleagues (11, 12). The final pharmacokinetic and pharmacodynamic parameters were determined in each individual subject by fitting both models simultaneously by maximum a posteriori Bayesian estimation, with the initial maximum likelihood parameter estimates serving as the a priori Bayesian prior parameter set. The Bayesian prior estimates were updated twice during the course of the final analysis.
Resistance testing.
The protease and reverse transcriptase genes were sequenced and mutations identified using the LabCorp GenoSure testing method (Laboratory Corporation of America, Research Triangle Park, NC).
Changes to the gag CA-SP1 cleavage site were also evaluated. Viral RNA was purified from patient plasma using the QIAamp Mini viral RNA purification kit (QIAGEN). gag cDNA was synthesized by reverse transcription-PCR using the StrataScript first-strand synthesis system (Stratagene) for reverse transcription, followed by amplification of double-stranded DNA using the PicoMaxx high-fidelity PCR master mix (Stratagene). A final product of approximately 1 kb was purified using the MinElute PCR purification kit (QIAGEN). Both strands of the DNA product were sequenced by the University of Pittsburgh DNA sequencing core facility.
RESULTS
Clinical study results.
Twenty-four adult male HIV-infected patients completed the study, of which 18 were antiretroviral naïve and 4 were treatment experienced. The treatment-experienced patients were randomized to each of the four dose groups. The mean (and standard deviation [SD]) age was 40.7 (7.6) years, and the mean (SD) CD4 cell count at baseline was 466 (154). The dosing groups were well matched with respect to demographics, with no significant differences noted, with the exception of the placebo group being younger than the 75-mg dose group, having a mean of 33.0 years versus 45.3 years (P = 0.02, analysis of variance). None of the other groups differed significantly from any other by age. The time from diagnosis of HIV infection ranged from 0.8 to 17.2 years, with a mean of 5.2 years. Only two patients harbored virus mutations associated with drug resistance at baseline. One patient (150-mg dose group) had virus with the M184V mutation, which is associated with nucleoside reverse transcriptase inhibitor resistance, and the K103N mutation, which is associated with nonnucleoside reverse transcriptase inhibitor (NNRTI) resistance. One patient in the 250-mg dose group had virus with the K103N and Y181C mutations, both of which are associated with NNRTI resistance, as well as the L90M mutation, which is associated with protease inhibitor resistance. Other patients had preexisting mutations/polymorphisms that confer minimal or no resistance when present alone (i.e., V77I, M36I, and A71V).
Bevirimat was well tolerated at all studied doses, with no significant treatment-related adverse effects, no serious adverse events, and no deaths reported. The mean (SD) change in CD4 cell count 10 days after bevirimat for the placebo, 75-, 150-, and 250-mg doses was +123 (91), −15 (142), +121 (191), and +104 (128), respectively. These changes in CD4 cell counts were not statistically significant by dose or over the course of the study period.
Single oral doses of bevirimat gave up to a 0.72-log10 reduction in viral load in HIV-infected patients. Of the 12 patients at the 150- and 250-mg dose levels, 8 (67%) had greater than a 0.3-log10 reduction, and 5 (42%) had greater than a 0.5-log10 reduction. Of particular interest, the two patients with baseline resistance to approved drugs had robust responses to bevirimat, with reductions of 0.53 log10 (150-mg dose) and 0.73 log10 (250-mg dose). The by-dose mean reduction in viral load from baseline is illustrated in Fig. 2. The mean (SD) maximum observed log reduction in viral load demonstrated by dose was −0.15 (0.17), −0.14 (0.09), −0.47 (0.28), and −0.46 (0.17) log10 HIV RNA copies/ml for the placebo, 75-mg, 150-mg, and 250-mg doses, respectively (P < 0.05, analysis of variance). The data on patients who received the 75-mg dose did not differ from placebo and there was significantly less activity than in patients who received either the 150-mg or 250-mg dose of bevirimat.
FIG. 2.

Mean reduction in viral load following bevirimat treatment by dose.
Pharmacokinetic/pharmacodynamic results.
The final pharmacokinetic/pharmacodynamic model is illustrated in Fig. 3. The pharmacokinetic model was a linear two-compartment model, with oral doses absorbed following a lag time from an absorption compartment. This model fit the data well, with an overall r2 of 0.98 (Fig. 4). The mean concentration-time profile is shown in Fig. 5, and a summary of the pharmacokinetic parameters is reported in Table 1. The pharmacokinetics of bevirimat in this HIV-infected patient population were similar to previous reports in healthy volunteers, with a long half-life of approximately 60 h and low variability in oral clearance (CV of 18.2%). The pharmacokinetics were linear, with no change in oral clearance over the studied dosage range. The area under the concentration-time curve and the maximum drug concentration in plasma appeared to increase proportionally with increasing doses. An example of the fit of the pharmacokinetic model to a typical patient is shown in Fig. 6.
FIG. 3.

Pharmacokinetic/pharmacodynamic model for single-dose bevirimat. Abbreviations: Abs, site of absorption of an oral dose; TLag, delay prior to the onset of oral absorption; Ka, rate constant for oral absorption; Vp and Vc, apparent volumes of distribution for the peripheral (tissue) and central (plasma) compartments; CLd, distributional clearance of bevirimat between the peripheral and central compartments; CLt/F, apparent oral clearance of bevirimat from the plasma compartment; G, production of uninfected CD4 cells; Kd, death rate constant of uninfected CD4 cells; Ki, infectivity rate constant; Kd*, death rate constant of infected CD4 cells; c, clearance rate of virions.
FIG. 4.

Goodness of fit of the pharmacokinetic (above) and pharmacodynamic (below) models for all bevirimat (PA-457) concentration and viral load data. The solid line represents the line of identity. Drug concentration units are ng/ml; viral load units are log10 HIV RNA copies/ml. The r2 values for drug concentrations and viral load are 0.99 and 0.87, respectively.
FIG. 5.

Mean concentration-time pharmacokinetic profiles for 75, 150, and 250 mg of bevirimat (PA-457) administered as a single oral dose to HIV-infected patients. Error bars represent standard deviations.
TABLE 1.
Pharmacokinetic parameters of single-dose bevirimat in HIV-infected patientsa
| Dose (mg) | Vss/F (liters) | CLd/F (liters/h) | CLt/F (liters/h) | Tlag (h) | Ka (1/h) | Half-life (h) |
|---|---|---|---|---|---|---|
| 75 | 13.9 (13.7) | 0.48 (14.3) | 0.17 (28.6) | 0.18 (49.3) | 3.8 (46.8) | 63.9 (12.1) |
| 150 | 12.0 (3.8) | 0.48 (13.2) | 0.16 (8.0) | 0.20 (6.5) | 3.4 (48.8) | 55.1 (9.8) |
| 250 | 14.0 (18.5) | 0.45 (24.6) | 0.17 (15.0) | 0.15 (11.4) | 3.7 (30.0) | 62.1 (15.1) |
| All | 13.3 (14.9) | 0.47 (17.0) | 0.17 (18.2) | 0.17 (30.5) | 3.6 (40.3) | 60.3 (13.6) |
Data are reported as the mean with the percent CV in parentheses.
bVss/F, steady-state volume of distribution; CLd/F, distributional clearance; CLt/F, oral clearance; Tlag, lag time; Ka, rate constant for oral absorption.
FIG. 6.

Example fit of the pharmacokinetic (above) and pharmacodynamic (below) model in a typical subject. The solid line represents model predicted values, and solid circles represent measured bevirimat (PA-457) or HIV RNA concentrations.
The pharmacodynamic model is based on a previously published model of Dixit and Perelson (2, 11), modified for evaluating the pharmacodynamic effects of bevirimat in this study. The model has the benefit of being physiological in nature, incorporating viral replication and both infected and uninfected CD4 cell populations. The complete model is summarized graphically in Fig. 3.
In this model, CD4 cells are produced at a constant, zero-order rate from tissues such as the thymus (2). These uninfected cells are either removed from the circulation after normal cell death or become infected with HIV. Infected CD4 cells die at a rate that is more rapid than uninfected CD4 cells, which is required for the eventual immunosuppression associated with the progression of HIV to AIDS. The infected CD4 cells also produce HIV, with these virions eventually going on to infect more uninfected CD4 cells at a particular rate. The HIV is removed from the circulation at a particular rate of clearance which is estimated in the model. The action of bevirimat is to inhibit the production of infectious virus, hence reducing the rate of infection of new CD4 cells, leading to a reduction in plasma viral load upon drug administration and absorption into the bloodstream. This drug effect was modeled as a Hill-type function (further defined below).
The following three differential equations describe the time course for infected and uninfected CD4 cells, HIV, and the effect of bevirimat on viral replication:
 |
(1) |
 |
(2) |
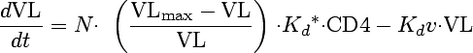 |
(3) |
Each differential equation describes a separate compartment: (equation 1) uninfected CD4 cells (CD4), (equation 2) infected CD4 cells (CD4*), and (equation 3) HIV-1 plasma viral load (VL). The definitions of viral dynamic parameters are provided in Table 2. The model pharmacodynamic parameter estimates are summarized in Table 3. Viral replication (equation 3) required a saturable process, whereas the rate of production approached a maximum at an estimated viral load (VLmax). This was necessary to adequately fit the viral load return towards a baseline equilibrium, during the washout phase of the study.
TABLE 2.
HIV pharmacodynamic model parameters
| Symbol | Parameter definition (units) |
|---|---|
| G | Growth/production of uninfected CD4 cells (ml/h) |
| Kd | Death rate constant of uninfected CD4 cells (1/h) |
| Ki | Infectivity rate constant; the rate that HIV-1 infects CD4 cells (ml/h) |
| Kd* | Death rate constant of infected CD4 cells (1/h) |
| N | Number of new virions produced by an infected CD4 cell |
| VLmax | Maximum rate of viral replication (virions/ml) |
| Kdv | Clearance rate of virions (1/h) |
| INH | Fractional inhibition of viral replication by an antiretroviral |
TABLE 3.
Pharmacodynamic parameters of single-dose bevirimat in HIV-infected patientsa
| Dose (mg) | Kd (1/h) (103) | Ki (1/h) (109) | Kd* (1/h) | Kdv (1/h) (103) | N | EC50 (ng/ml) | H | VLmax (log10 copies/ml) | G (ml/h) |
|---|---|---|---|---|---|---|---|---|---|
| 75 | 0.0031 (146) | 0.14 (168) | 0.013 (126) | 0.008 (228) | 137.8 (127) | 1.19 (94) | 0.35 (68.0) | 5.13 (120) | 320.7 (90.4) |
| 150 | 0.0012 (38) | 0.028 (57) | 0.017 (130) | 0.071 (118) | 126.0 (27) | 0.35 (94) | 0.31 (87) | 4.59 (73) | 1,340.0 (51) |
| 250 | 0.0016 (71) | 0.0349 (141) | 0.007 (129) | 0.024 (73) | 80.20 (69) | 0.55 (113) | 0.42 (101) | 4.46 (84) | 1,856.6 (84) |
| All | 0.0020 (138) | 0.072 (155) | 0.012 (131) | 0.058 (141) | 114.1 (86) | 0.77 (93) | 0.34 (93) | 4.84 (120) | 1,162 (101) |
Data are reported as means with the percent CV in parentheses. N, number of virions; EC50, 50% effective concentration; H, Hill's constant.
The fractional inhibition (INH) was modeled as a function of bevirimat plasma concentrations, according to equation 4, where Cp is plasma concentration, H is Hill's constant, and EC50 is the median effect concentration.
 |
(4) |
The fit of the pharmacodynamic model to the HIV RNA data was excellent, with an overall r2 of 0.87 (Fig. 4). An example fit of the model in a typical subject is shown in Fig. 6. The model describes the effect of bevirimat, linking pharmacokinetics to pharmacodynamics through a multistep process. The pharmacodynamic model parameters illustrated relatively large variability, highlighting the variability in response among HIV patients to therapy. This relatively large degree of variability, with most parameters having a CV greater than 100% (Table 3), has been reported previously in studies with similar models for antiretroviral therapy where patient data have been evaluated (2, 16).
Selection for resistance to bevirimat in vitro, followed by genotyping of the complete Gag and PR coding regions, resulted in the identification of several single-amino-acid substitutions at or near the CA-SP1 cleavage site in Gag that are each sufficient to confer resistance to the drug (7, 17). No mutations have been found in PR or elsewhere in Gag. To determine whether resistance to bevirimat developed in this single-dose study, patient samples were collected prior to dosing, at day 7, at day 10, and at the end of the study (i.e., day 28), and the Gag CA-SP1 cleavage site domain was genotyped. No treatment-related changes were observed in this region during the course of the study for any of the patients.
DISCUSSION
The demonstration of an antiviral effect following a single oral dose of bevirimat validates maturation inhibition as a potential target for antiretroviral therapeutics in humans. The mean maximum observed reduction in viral load for the higher doses approached 0.5 log for both the 150-mg and 250-mg doses. Few prior studies have evaluated antiretroviral activity following single doses; however, studies with tenofovir reported viral load changes of −0.20, −0.33, and +0.20 log10 with single doses of 150 mg, 300 mg, and 600 mg, respectively (8). Clinical studies of dexelvucitabine reported log reductions of −0.45, −0.42, and −0.27 log10 following single 50-mg, 100-mg, and 200-mg doses (15). These data demonstrate the activity of bevirimat and show at least equivalent potency to tenofovir and dexelvucitabine following single oral doses.
The decline in HIV-1 RNA following exposure to effective antiretroviral treatment is biphasic, with the first (alpha) phase representing clearance of virions and loss of productively infected lymphocytes (11, 12). The alpha phase normally begins within 24 h of initiating antiretroviral therapy, continues for 7 to 14 days, and is followed by the slower decay of the beta phase. The rate and extent of viral decay are determined both by drug efficacy and by virus production and clearance rates. Thus, when considered with viral dynamics, the alpha phase can provide an estimate of the relative potency of antiretroviral treatment, and its characterization during abbreviated monotherapy trials allows for selection of a candidate dose(s) for efficacy studies when administered as part of a combination regimen.
The HIV viral dynamic model utilized in these studies has demonstrated its utility to significantly improve our understanding of HIV pathogenesis and antiretroviral pharmacology. There are several assumptions and limitations with this viral dynamic model which should be recognized. The model assumes a population of viral isolates, all of which demonstrate a common susceptibility to the study drug. While other subpopulations of drug-resistant virus may initially be present or develop during the course of therapy, the importance of this assumption appears to be minimal for this short-term single-dose study where the time of exposure was not of sufficient duration to result in the emergence of drug-resistant virus against bevirimat. The model also assumes that CD4 cells are produced at a constant, zero-order rate and that the rate of infected CD4 cell death is greater than for uninfected CD4 cells. In addition, the investigator is typically limited to sampling CD4 cells from the vascular compartment, whereas the majority of CD4 cells exist in lymphoid tissue, continuously migrating into and out of the plasma compartment. The dynamics of CD4 cells, while important in considering longer-term chronic studies, do not change significantly over the course of such short-term studies and thus have little impact on the derived model parameter estimates.
The single-dose pharmacokinetics of bevirimat in this population of HIV-infected patients were similar to those reported in healthy volunteers and were well characterized by a two-compartment oral model. Bevirimat demonstrated rapid oral absorption, linear pharmacokinetics, a long half-life, and slow clearance with low interpatient variability. Based on the pharmacokinetics, this agent appears to be a good candidate for once-daily or even less frequent dosing strategies. The plasma concentrations of bevirimat remained above the in vitro 90% inhibitory concentration for an extended period of time following single doses, which, combined with the long half-life, would be expected to be forgiving from an adherence standpoint, with occasional missed doses having little consequence on overall exposure profiles.
No treatment-related changes in the CA-SP1 region of Gag were found to emerge in any of the patients as detected by population sequencing. This occurred despite the long half-life of bevirimat, as patients were exposed to suboptimal drug concentrations for more than 2 weeks after dosing, thus increasing the potential for resistance development. This lack of treatment-emergent resistance to bevirimat is an important finding, since resistance to the NNRTI nevirapine has been shown to emerge in up to 65% of patients after just a single dose (3, 4, 6). Future studies will determine whether any of the bevirimat resistance changes identified in vitro emerge after more prolonged treatment.
In summary, single oral doses of bevirimat were well tolerated in this population of HIV-infected patients. Due in part to the long half-life, a significant reduction in plasma viral load was detected following a single dose, particularly at the higher dose levels. Additional studies to assess the multiple-dose effectiveness and to further characterize the pharmacokinetics/pharmacodynamics of this compound in HIV infection appear warranted.
Footnotes
Published ahead of print on 16 July 2007.
REFERENCES
- 1.Bartlett, J., H. C. Lane, J. Anderson, A. C. Baker, S. Bozzette, C. Carpenter, M. Delaney, L. D. W. El-Sadr, C. Fletcher, et al. 2005. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. http://AIDSinfo.nih.gov.
- 2.Dixit, N. M., and A. S. Perelson. 2004. Complex patterns of viral load decay under antiretroviral therapy: influence of pharmacokinetics and intracellular delay. J. Theor. Biol 226:95-109. [DOI] [PubMed] [Google Scholar]
- 3.Eshleman, S. H., L. A. Guay, A. Mwatha, S. P. Cunningham, E. R. Brown, P. Musoke, F. Mmiro, and J. B. Jackson. 2004. Comparison of nevirapine (NVP) resistance in Ugandan women 7 days vs. 6-8 weeks after single-dose nvp prophylaxis: HIVNET 012. AIDS Res. Hum. Retrovir. 20:595-599. [DOI] [PubMed] [Google Scholar]
- 4.Eshleman, S. H., and J. B. Jackson. 2002. Nevirapine resistance after single dose prophylaxis. AIDS Rev. 4:59-63. [PubMed] [Google Scholar]
- 5.Hurvich, C. M., and C. L. Tsai. 1989. Regression and time series model selection in small samples. Biometrika 76:297-307. [Google Scholar]
- 6.Johnson, J. A., J. F. Li, L. Morris, N. Martinson, G. Gray, J. McIntyre, and W. Heneine. 2005. Emergence of drug-resistant HIV-1 after intrapartum administration of single-dose nevirapine is substantially underestimated. J. Infect. Dis. 192:16-23. [DOI] [PubMed] [Google Scholar]
- 7.Li, F., R. Goila-Gaur, K. Salzwedel, N. R. Kilgore, M. Reddick, C. Matallana, A. Castillo, D. Zoumplis, D. E. Martin, J. M. Orenstein, G. P. Allaway, E. O. Freed, and C. T. Wild. 2003. PA-457: a potent HIV inhibitor that disrupts core condensation by targeting a late step in Gag processing. Proc. Natl. Acad. Sci. USA 100:13555-13560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Louie, M., C. Hogan, A. Hurley, V. Simon, C. Chung, N. Padte, P. Lamy, J. Flaherty, D. Coakley, M. Di Mascio, A. S. Perelson, and M. Markowitz. 2003. Determining the antiviral activity of tenofovir disoproxil fumarate in treatment-naive chronically HIV-1-infected individuals. AIDS 17:1151-1156. [DOI] [PubMed] [Google Scholar]
- 9.Martin, D. E., R. Blum, J. Wilton, J. Doto, H. Galbraith, G. L. Burgess, P. C. Smith, and C. H. Ballow. 2007. Safety and pharmacokinetics of bevirimat (PA-57), a novel inhibitor of human immunodeficiency virus maturation, in healthy volunteers. Antimicrob. Agents Chemother. 51:3063-3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martin, D. E., C. H. Ballow, R. Blum, J. Doto, C. T. Wild, and G. P. Allaway. 2007. Multiple dose pharmacokinetics and safety of bevirimat, a novel inhibitor of human immunodeficiency virus maturation, in healthy volunteers. Clin. Pharmacokinet. 46:589-598. [DOI] [PubMed] [Google Scholar]
- 11.Perelson, A. S., P. Essunger, Y. Cao, M. Vesanen, A. Hurley, K. Saksela, M. Markowitz, and D. D. Ho. 1997. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature 387:188-191. [DOI] [PubMed] [Google Scholar]
- 12.Perelson, A. S., P. Essunger, and D. D. Ho. 1997. Dynamics of HIV-1 and CD4+ lymphocytes in vivo. AIDS 11(Suppl. A):S17-S24. [PubMed] [Google Scholar]
- 13.Smith, P. F., A. Forrest, C. Rayner, R. Wood, and L. Proulx. 2000. Integrating viral dynamics and pharmacokinetics in a 7-day monotherapy trial of dOTC in HIV-infected, antiretroviral naive patients. Presented at the 13th World AIDS Conference, Durban, South Africa, July 2000.
- 14.Stoddart, C. A., J. Bare, and D. E. Martin. 2004. Potent in vivo antiviral activity of the HIV-1 maturation inhibitor PA103001-01. Presented at the Keystone Symposium on HIV Pathogenesis, Whistler, British Columbia, Canada, 12 to 18 April 2004.
- 15.Stuyver, L. J., T. R. McBrayer, D. Schurmann, I. Kravec, A. Beard, L. Cartee, R. F. Schinazi, A. DeLaRosa, R. L. Murphy, and M. J. Otto. 2004. Potent antiviral effect of reverset in HIV-1 infected adults following a single oral dose. Antivir. Ther. 9:529-536. [PubMed] [Google Scholar]
- 16.Wu, H., Y. Huang, E. P. Acosta, J. G. Park, S. Yu, S. L. Rosenkranz, D. R. Kuritzkes, J. J. Eron, A. S. Perelson, and J. G. Gerber. 2006. Pharmacodynamics of antiretroviral agents in HIV-1 infected patients: using viral dynamic models that incorporate drug susceptibility and adherence. J. Pharmacokinet. Pharmacodyn. 33:399-419. [DOI] [PubMed] [Google Scholar]
- 17.Zhou, J., X. Yuan, D. Dismuke, B. M. Forshey, C. Lundquist, K. H. Lee, C. Aiken, and C. H. Chen. 2004. Small-molecule inhibition of human immunodeficiency virus type 1 replication by specific targeting of the final step of virion maturation. J. Virol. 78:922-929. [DOI] [PMC free article] [PubMed] [Google Scholar]