

Abstract
A phospholipid flippase activity from the endoplasmic reticulum (ER) of the model organism Saccharomyces cerevisiae has been characterized and functionally reconstituted into proteoliposomes. Analysis of the transbilayer movement of acyl-7-nitrobenz-2-oxa-1,3-diazol-4-yl (acyl-NBD)-labeled phosphatidylcholine in yeast microsomes using a fluorescence stopped-flow back exchange assay revealed a rapid, ATP-independent flip-flop (half-time, <2 min). Proteoliposomes prepared from a Triton X-100 extract of yeast microsomal membranes were also capable of flipping NBD-labeled phospholipid analogues rapidly in an ATP-independent fashion. Flippase activity was sensitive to the protein modification reagents N-ethylmaleimide and diethylpyrocarbonate. Resolution of the Triton X-100 extract by velocity gradient centrifugation resulted in the identification of a ∼4S protein fraction enriched in flippase activity as well as of other fractions where flippase activity was depleted or undetectable. We estimate that flippase activity is due to a protein(s) representing ∼2% (wt/wt) of proteins in the Triton X-100 extract. These results indicate that specific proteins are required to facilitate ATP-independent phospholipid flip-flop in the ER and that their identification is feasible. The architecture of the ER protein translocon suggests that it could account for the flippase activity in the ER. We tested this hypothesis using microsomes prepared from a temperature-sensitive yeast mutant in which the major translocon component, Sec61p, was quantitatively depleted. We found that the protein translocon is not required for transbilayer movement of phospholipids across the ER. Our work defines yeast as a promising model system for future attempts to identify the ER phospholipid flippase and to test and purify candidate flippases.
Glycerophospholipid flip-flop across biogenic membranes such as the endoplasmic reticulum (ER) is an essential feature of biomembrane assembly. Glycerophospholipid synthesis occurs at the cytoplasmic leaflet of the ER membrane (5); for uniform bilayer growth, some of the newly synthesized lipids must be flipped to the luminal leaflet. In addition to glycerophospholipids (referred to simply as “phospholipids” in this paper), complex glycophospholipids have to flip across the ER membrane during the biosynthesis of glycoproteins, including glycosylphosphatidylinositol-anchored proteins; the synthesis of these glycoconjugates requires flipping of glucosaminyl phosphatidylinositol, dolichol-phosphate-mannose, dolichol-phosphate-glucose, and a dolichol pyrophosphoryl oligosaccharide from the cytoplasmic side to the luminal face of the ER (12, 25, 32, 34, 37).
Previous analyses with rat liver microsomes showed that flip-flop of phospholipids occurs very rapidly, with a half-time of seconds to minutes (6, 9, 14, 23, 38). These studies also indicated that transbilayer phospholipid movement is bidirectional, ATP independent, and nonspecific for phospholipid head groups or glycerol stereochemistry. Because the spontaneous movement of phospholipids across protein-free liposomes is orders of magnitude slower, this led to the idea that phospholipid flip-flop is protein mediated and involves one or more ATP-independent phospholipid flippases. Consistent with this notion, transbilayer phospholipid movement in the ER is sensitive to proteases and protein-modifying reagents (6, 9, 14). The successful reconstitution of transport-active proteoliposomes from a protein extract of rat liver microsomes under conditions where protein-free liposomes were inactive provided further evidence for the requirement of specific proteins in the flipping process (1). More-recent studies show that flippase activity can be enriched by fractionation of detergent-solubilized ER proteins (11, 26).
Despite this recent progress, ATP-independent phospholipid flippases in biogenic membranes have yet to be identified. Proteins that have been proposed to act as lipid flippases in the ER include enzymes of lipid metabolism that possess multitransmembrane-spanning structures, the protein translocon complex, which mediates the transmembrane translocation of secretory proteins and the insertion of transmembrane proteins (26, 39); the Rft1 protein, which has been suggested to have an essential role in the translocation of dolichol pyrophosphoryl oligosaccharides from the cytoplasmic to the luminal leaflet of the ER (13); and Rsb1p, a candidate flippase with a predicted ATP-binding motif that appears to be required for the translocation of sphingoid long-chain bases toward the luminal side of the ER (18). However, there is little or no biochemical evidence to document flippase activity of these candidates or to rule it out. Given that basic mechanisms of phospholipid synthesis are common in Saccharomyces cerevisiae and mammalian cells, the tools of yeast genetics and molecular biology offer promising means to identify and dissect molecular components involved in the flipping process. A single report documents the ability to reconstitute flipping of in situ-labeled phosphatidylethanolamine (PE) in proteoliposomes prepared from a detergent extract of yeast microsomes (28). Extending this approach to investigate membrane proteins that might play a role in the transbilayer movement of phospholipids in the ER, we now describe the results of experiments in which we assayed flippase activity in yeast microsomal membranes as well as in proteoliposomes reconstituted from a detergent extract of yeast microsomes. We used 7-nitrobenz-2-oxa-1,3-diazol-4-yl (NBD)-labeled short-chain phospholipid analogues as probes to monitor flip-flop, making use of their ability to be extracted from the membrane by bovine serum albumin (BSA) as well as to be chemically reduced by dithionite, a membrane-impermeant dianion. Our experiments reveal fast flip-flop of NBD-phospholipids (NBD-PLs) in yeast microsomes and demonstrate that flipping is facilitated by a protein(s) that sediments operationally at ∼4S and whose activity is compromised by treatment with the protein modification reagents N-ethylmaleimide (NEM) and diethylpyrocarbonate (DEPC). Fractionation of the detergent extract prior to reconstitution yielded protein mixtures enriched in flippase activity and other fractions devoid of activity, indicating unambiguously that a specific protein (or a specific subset of ER proteins) is required to facilitate flip-flop. We also show clearly that the protein translocon, a promising flippase candidate, is not required for the efficient transbilayer movement of phospholipids across the ER. Our results define yeast as a promising model system for future attempts to identify flippases by isolating and probing candidate proteins.
MATERIALS AND METHODS
Lipids.
Egg phosphatidylcholine (egg PC) and all NBD-PLs were purchased from Avanti Polar Lipids (Alabaster, AL).
Yeast strains and culture conditions.
S. cerevisiae strain sec61 (MATα ura3-52 leu2-3,112 ade2-10 sec61ts), obtained from J. Holthuis (Utrecht University, Utrecht, The Netherlands), was used. Cells were grown at 27°C in liquid YPD medium (1% Bacto yeast extract, 2% Bacto peptone [Difco], 2% glucose). Cells were harvested in the exponential phase, and growth was monitored by A600.
Microsome preparation and characterization.
Microsomes were prepared by differential centrifugation of a yeast homogenate essentially according to the work of Zinser and Daum (42). Briefly, cells were pretreated with 10 mM dithiothreitol in 100 mM Tris sulfate (pH 9.4) for 10 min, incubated with Zymolyase 100T (ICN Biomedicals, Eschwege, Germany) at 5 mg/g (wet weight) of cells in a buffer (20 mM potassium phosphate [pH 7.2], 1.2 M sorbitol) at 30°C for 30 min, harvested, and then lysed in homogenizing buffer (10 mM Tris-HCl [pH 7.4], 0.6 M sorbitol) containing protease inhibitors (1 mg/ml aprotinin, 1 mg/ml leupeptin, 1 mg/ml pepstatin A, 5 mg/ml antipain, 1 mM benzamidine, 1 mM phenylmethylsulfonyl fluoride) using a Dounce homogenizer with a tight-fitting pestle. The cell lysate was centrifuged at 3,000 × gav (average centrifugal force) for 5 min. The pellet was resuspended in homogenizing buffer, homogenized, and centrifuged as described above. Both supernatants were pooled and designated the postnuclear supernatant. Membrane pellets from differential centrifugation (9,000 × gav for 10 min, 20,000 × gav for 30 min, 40,000 × gav for 30 min, 100,000 × gav for 45 to 75 min) were screened for organelle marker proteins by immunoblotting. Pellets from the 100,000 × gav centrifugation enriched in the ER membrane were resuspended in buffer T (10 mM Tris-HCl [pH 7.4], 1 mM EDTA) using a tight fitting Dounce homogenizer and were stored at −80°C. From 10 g (wet weight) of cells, 60 mg of protein was obtained in the final P100 microsome fraction. For isolation of intact microsomes, the final centrifugation (100,000 × gav for 45 to 75 min) was carried out onto a sucrose cushion (60% [wt/wt] in homogenizing buffer) and membrane pellets were resuspended in buffer T supplemented with 100 mM NaCl. Immunoblot analysis was performed using antibodies against Dpm1p (Molecular Probes), Aac2p (H. Rottensteiner, Bochum University, Germany), Vac8p (M. Veit, Free University Berlin, Berlin, Germany), Tlg2p (J. Holthius, Utrecht University, Utrecht, The Netherlands), Sec61p and Sec62p (T. Sommer, Max-Delbrück-Center Berlin, Berlin, Germany), and Wbp1p (35). Protein blots were probed with horseradish peroxidase-conjugated secondary antibodies (Bio-Rad), which were detected using ECL (Amersham). Chemiluminescent bands were quantified using a GS-710 calibrating imaging densitometer (Bio-Rad) with QuantityOne software. The intactness of the microsomes (6 mg of protein/ml) was tested by a protease protection assay using trypsin (0.25 mg/ml). After termination of protease digestion by addition of a trypsin inhibitor (1 mg/ml), samples were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotting.
Preparation and fractionation of the Triton X-100 extract.
P100 microsomal membranes (20 mg/ml protein) were solubilized by diluting with an equal volume of 1.6% (wt/vol) Triton X-100 (Roche Diagnostics) in buffer T. After incubation for 45 min on ice, insoluble material was removed by centrifugation (177,000 × gav, 30 min, 4°C), resulting in a supernatant fraction designated the Triton extract (TE). By this procedure, 10 to 20% of the protein and 90% of the phospholipid were solubilized from the microsomes. For TE fractionation, a linear gradient of 10 to 35% (wt/vol) glycerol in buffer TX (10 mM Tris-HCl [pH 7.4], 1 mM EDTA, 0.8% [wt/vol] Triton X-100) was prepared, loaded with 250 μl TE to a final volume of 4 ml, and centrifuged to equilibrium in a swing-out rotor (165,000 × gav, 20 h, 4°C). Fractions (300 μl) were collected from the top and pooled pairwise, discarding the first one and the very bottom part. The obtained samples I to VI (600 μl each) were desalted using BioGel-P6 spin columns (200 μl sample per 1 ml [bed volume] of resin [Bio-Rad]) to remove glycerol; aliquots of each sample were taken for protein determination and SDS-PAGE analysis before the majority of the material was used for reconstitution and flippase activity measurement. Sedimentation standards (150 μg each of ovalbumin [3.6S], BSA [4.2S], β-amylase [8.9S], and catalase [11S]) were loaded onto a parallel gradient; fractions (300 μl) were harvested from the gradient and analyzed by SDS-PAGE and Coomassie staining to locate the migration positions of the sedimentation standards.
Reconstitution of liposomes and proteoliposomes.
Proteoliposomes were prepared from a mixture of crude or fractionated TE and Triton X-100-solubilized egg PC and NBD-PL as previously described (26). Briefly, a mixture of egg PC (4.5 μmol) and NBD-PL (22.5 nmol or 0.5 mol% of total phospholipid) in chloroform was dried under nitrogen in a glass screw-cap tube and then dissolved in buffer TX. Various amounts of TE or TE-derived fractions were added to generate proteoliposomes with different protein/phospholipid ratios (PPR). The proportion of reconstituted protein correlated linearly with the amount of TE used for the reconstitution (data not shown, but see reference 10). The final phospholipid concentration was 4.5 mM. Protein-free liposomes were prepared similarly by replacing TE with buffer TX. To remove detergent and generate vesicles, 100 mg/ml of SM2 Bio-Beads (Bio-Rad) was added. After 3 to 4 h of rotating at room temperature, the samples were supplemented with an additional 200 mg/ml of Bio-Beads and then transferred to 4°C for a further 14 to 16 h of mixing. The resulting turbid suspensions were withdrawn carefully to avoid collecting any beads and were adjusted to an 8- to 10-fold excess of buffer T. The vesicles were collected by centrifugation (200,000 × gav, 50 min, 4°C) and resuspended at 9 mM phospholipid using a Dounce homogenizer. For some experiments, the vesicle suspension was used directly after removal of the Bio-Beads, without centrifugation and resuspension. Protein recovery in the reconstituted vesicles was ∼40%; phospholipid recovery was 75 to 90%. More than 99.98% of the initial Triton X-100 amount was removed, as determined by extraction with 4 volumes of chloroform-methanol (1/2, vol/vol) and measurement of the absorbance of the supernatant at 275 nm (15).
Protein modification.
The sensitivity of flippase activity to proteolysis was assessed by treating proteoliposomes with proteinase K as described previously (38). Control samples were mock treated. DEPC and NEM treatment of TE was done as described previously (10) except that 200 mM stock solutions or reagent was used (freshly prepared in 10 mM HEPES-NaOH [pH 7.4]-100 mM NaCl) and incubations were carried out for 45 min at room temperature.
Preparation and labeling of liposomes.
Egg PC dissolved in chloroform-methanol (1:1, vol/vol) was mixed with NBD-PLs (1 mol%) and dried under a stream of nitrogen. The dried lipid film containing NBD-PLs was hydrated by vortexing in buffer T supplemented with 100 mM NaCl to give a final lipid concentration of 1 mM. The resulting aqueous phospholipid dispersion was subjected to five freeze-thaw cycles and was then extruded 10 times through two 100-nm-pore-size polycarbonate filters (extruder from Lipex Biomembranes; filters from Costar Nuclepore). This procedure gives liposomes with a symmetrical transbilayer distribution of NBD-PLs. To prepare vesicles with NBD-PLs located exclusively on the outer leaflet (asymmetrically labeled vesicles), liposomes were labeled after preparation.
Stopped-flow analysis of C6-NBD-PC flip-flop.
Microsomes were adjusted to a phospholipid concentration of 1 mM in buffer T supplemented with 100 mM NaCl. Aliquots were incubated at room temperature with 1-palmitoyl-2-C6-NBD-PC (added from stock solutions of 160 μM in buffer T supplemented with 100 mM NaCl) for 30 or 90 min such that the final amount of the NBD-PL was ∼1 mol% of total phospholipid. Fluorescence measurements were performed in quartz cuvettes at 20°C using an Aminco Bowman series 2 spectrofluorometer (SLM Instruments, Rochester, NY) equipped with a stopped-flow accessory (RX 1000; Applied Photophysics, Leatherhead, United Kingdom).
The transfer of C6-NBD-PC from the aqueous phase into the membranes of microsomes was determined by monitoring the increase in fluorescence emission at 540 nm (excitation, 470 nm; slit widths, 4 nm; resolution, 1 s); insertion was essentially complete within 2 min. In order to determine background scattering and efficiency of label extraction from membranes by BSA, emission scans were recorded (wavelength, 490 to 600 nm; excitation, 470 nm; slit widths, 4 nm) and values at 540 nm for two independent experiments were taken for quantification. For stopped-flow measurements, a 100-μl aliquot of labeled vesicles was mixed with an equal volume of fatty acid-free BSA dissolved in the same buffer by simultaneous injection into the chamber of a stopped-flow accessory to give final concentrations of 1% (wt/vol) BSA and 75 μM membrane phospholipid containing 1 mol% of NBD-PL. Fluorescence decay traces were recorded at 540 nm for 500 s (excitation, 470 nm; slit widths, 4 nm; resolution, 1 s).
Kinetic analysis.
The stopped-flow data were evaluated according to a three-compartment model as described previously (20, 23). This kinetic model describes transbilayer movement as well as the transfer of phospholipid analogues between the outer leaflet of the membrane vesicle and BSA. The outward and inward movements of phospholipid analogues are described by the rate constants k+1 and k−1, respectively. The movement of the analogues from microsomal membranes to BSA is characterized by the rate constant k+2 (extraction of the analogues by BSA), and k−2 describes the movement of analogues back from BSA to the vesicle membrane. As previously shown (20, 23), due to the excess of BSA used, the exchange process described by k−2 did not contribute to the kinetics, and the values for this time constant were very small (typically 10−12 s−1), as expected. Furthermore, being aware that the phospholipid analogues used are partially water soluble, one has to consider that analogues partition between membranes and the aqueous compartments, including the vesicular lumen. However, since the analogue concentration is very low with respect to that of microsomal lipids or nonlabeled lipids (reconstitution experiments), the fraction of analogues in the aqueous compartments is negligible. This was verified by measuring the NBD fluorescence associated with the microsomes and supernatant after centrifugation and addition of Triton X-100, showing that essentially all analogues were incorporated into membranes. Therefore, the following model can be applied.
[PLo] and [PLi] are the concentrations of analogue in the outer and inner leaflets of the microsomal membranes, respectively. At the time of BSA addition (t = 0 s), the transmembrane distribution is at steady state, i.e.,
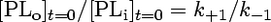 |
(1) |
The concentration of analogue transferred to BSA ([PLtr]) is taken to be zero at the time of addition. The model is represented by the following system of differential equations:
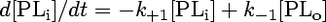 |
(2) |
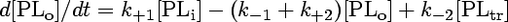 |
(3) |
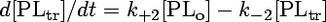 |
(4) |
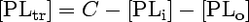 |
(5) |
 |
(6) |
Experimental data were fitted to the model by the Marquardt-Levenberg algorithm using SigmaPlot for Windows (Systat Software, Inc). This algorithm seeks the values of the parameters that minimize the sum of the squared differences between the observed and predicted values of the dependent variable (fluorescence intensity). Three time traces of two independent experiments were fitted in order to derive rate constants.
Analysis of NBD-PL flip-flop by albumin extraction.
To assay flip-flop of 1-myristoyl-2-C6-NBD-PC in reconstituted vesicle preparations by albumin extraction, the fluorescence of NBD-containing liposomes or proteoliposomes was measured at 540 nm (excitation, 470 nm; slit width, 4 nm) before and after BSA treatment for 5 min (final concentration, 2 mg/ml). Fluorescence was measured at 25°C using quartz microcuvettes and an Aminco Bowman series 2 spectrofluorometer (SLM Instruments, Rochester, NY). The accessible pool of fluorescent lipid (Pext) was calculated as [1 − (FBSA/Fbuffer)]/(1 − 0.55) × 100, where FBSA is the plateau value of fluorescence of vesicles after extraction with BSA, Fbuffer is the initial fluorescence of vesicles in buffer without BSA, Pext is the proportion of labeled lipid that is extracted by BSA, and the relative quantum yield of BSA-bound fluorescent lipid is 0.55 compared with a value of 1 for membrane-associated fluorescent lipid (20).
Analysis of NBD-lipid flip-flop by dithionite reduction.
To assay the flip-flop of NBD-PLs (1-myristoyl-2-C6-NBD-PC, 1-acyl-2-C6-NBD-PC, 1-acyl-2-C12-NBD-PC, and PE) in reconstituted vesicle preparations by dithionite reduction (10, 38), the fluorescence of NBD-containing liposomes or proteoliposomes was measured as for BSA extraction, except that instead of BSA, 1 M sodium dithionite (freshly dissolved in 1 M Tris base) was added to a final concentration of 5 mM. For complete reduction of all analogues by dithionite, Triton X-100 was then added to a final concentration of 0.5% (wt/vol). The percentage of NBD-PL that was reduced (Pred) was calculated as (Fbuffer − Fdithionite)/(Fbuffer − F0) × 100, where Fbuffer is the initial fluorescence of vesicles in buffer without dithionite, Fdithionite is the plateau value of fluorescence of vesicles after incubation with dithionite for 5 min, and F0 is the final fluorescence after addition of Triton X-100.
Collisional quenching experiments.
To assess the steady-state distribution of the NBD-PLs across the membranes of liposomes and proteoliposomes, collisional quenching of NBD probes with potassium iodide (KI) was performed according to the method of Lakowicz (21) as follows. NBD-PL-containing liposomes and proteoliposomes were reconstituted using buffer TX supplemented with 0.25 M KCl. Excitation and emission wavelengths were set at 470 and 530 nm, respectively. Fluorescence intensity was measured for samples consisting of 50 μl of vesicles diluted into 1.95 ml of buffer containing 10 mM HEPES-NaOH (pH 7.4), 40 mM Na2S2O3, and the quencher KI (0 to 0.25 M, as indicated); KCl was used to adjust the ionic strength to 0.25 M so that the osmolarity of the solution inside and outside the vesicles was the same. Na2S2O3 was included to prevent production of I2 by keeping the iodide ion reduced; it had no detectable effect on NBD fluorescence. Parallel samples were measured in which KCl was used instead of KI. Data were analyzed according to the modified Stern-Volmer equation (21): Fo/ΔF = (1/fa·K[Q]) + (1/fa) where Fo is the fluorescence intensity in the absence of the quencher, ΔF is the fluorescence intensity in the presence of the quencher at concentration [Q], fa is the fraction of fluorescence which is accessible to the quencher, and K is the Stern-Volmer quenching constant.
Other methods.
Protein content was quantified following trichloroacetic acid precipitation or delipidation according to the procedure of Wessel and Flügge (40) using either modified Lowry reagent (Sigma-Aldrich) or Micro BCA protein assay reagent (Pierce, Rockford, IL) with BSA as a standard. In some experiments, the Kaplan-Pedersen method for protein determination was used (16). The phospholipid content was determined by extracting lipids according to the method of Bligh and Dyer (7) and measuring the amount of phosphorus (33). NBD-PLs were checked for purity and stability during the course of reconstitution experiments by thin-layer chromatography on Silica Gel 60 plates (Merck, Darmstadt, Germany) using chloroform-methanol-water (65/25/4, vol/vol) as the solvent system.
RESULTS AND DISCUSSION
Flip-flop of a fluorescent PC analogue in yeast microsomes.
Yeast microsomes were isolated by differential centrifugation and characterized by immunoblotting using antibodies directed against organelle-specific proteins. Figure 1A shows that the P100 microsome fraction is enriched in ER membranes: it contains the ER marker Dpm1p but is almost entirely depleted of mitochondrial (Aac2p), vacuolar (Vac8p), and Golgi (Tlg2p) markers. The integrity of the ER vesicles in the P100 fraction was determined by testing the susceptibilities of ER membrane protein markers (Wbp1p and Dpm1p) to exogenously added trypsin in the presence or absence of detergent. Wbp1p is a type I ER-resident membrane protein with a large luminal domain and a small cytoplasmic tail (17); in contrast, the bulk of the Dpm1p protein is oriented toward the cytoplasm (29). The luminal domain of Wbp1p remained unaffected when trypsin was added to P100 vesicles, while Dpm1p was rapidly degraded under the same conditions (Fig. 1B). Both proteins were cleaved when trypsin was added to vesicles that had been permeabilized with Triton X-100. These results indicate that ER vesicles in the P100 fraction are sealed and right side out.
FIG. 1.

Characterization of microsomal membrane preparations. (A) Cell homogenates were prepared and subjected to differential centrifugation. Equal protein amounts (5 μg) of membrane pellets obtained at 3,000 × gav (P3), 9,000 × gav (P9), 20,000 × gav (P20), 40,000 × gav (P40), and 100,000 × gav (P100) were analyzed by SDS-PAGE and immunoblotting using antibodies against the marker proteins indicated. (B) The intactness of microsomal vesicles was assayed by protease protection. P100 membranes were incubated with trypsin (0.25 mg/ml) in the absence or presence of 0.5% Triton X-100 (TX-100). At the times indicated, protease digestion was terminated by addition of a trypsin inhibitor (1 mg/ml), and proteins were analyzed by SDS-PAGE and immunoblotting using antibodies against Wbp1p and Dpm1p. The integrity of microsomal membranes is demonstrated by the protection of Wbp1p from proteolysis in the absence of detergent.
To measure phospholipid flip-flop in the P100 vesicles, we used NBD-PLs with a short C6 acyl chain in the sn-2 position. C6-NBD-PLs are readily extracted from membranes by defatted BSA, and since the quantum yield of BSA-bound NBD-PLs is ∼55% of that of membrane-incorporated NBD-PLs (20), extraction is readily monitored by following the decrease in the fluorescence intensity of the sample. In preliminary experiments we prepared asymmetrically labeled liposomes in which C6-NBD-PC was located exclusively in the outer leaflet, as well as symmetrically labeled liposomes that contained C6-NBD-PC in both membrane leaflets. We used a stopped-flow accessory to rapidly mix defatted BSA with the liposome preparations and measured the kinetics of fluorescence decay. When BSA was added in excess to asymmetrically labeled liposomes, the fluorescence rapidly decreased to ∼50% of the initial value (Fig. 2B, trace a), indicating complete extraction of C6-NBD-PC by BSA. For symmetrically labeled liposomes, fluorescence rapidly decreased to 75% of the initial value (Fig. 2B, trace b), consistent with the extraction of ∼50% of the analogues by BSA (C6-NBD-PC molecules residing in the inner leaflet of the liposomes cannot be extracted, since phospholipid flip-flop does not occur in liposomes during the time scale of the experiment [6]). The fluorescence decrease in both cases could be described well by a monoexponential function, consistent with a single rate process, i.e., the essentially irreversible extraction of C6-NBD-PC from the outer leaflet of the vesicles by BSA.
FIG. 2.

Stopped-flow analyses of C6-NBD-PC flip-flop in liposomes and yeast microsomes. (A) Schematic illustration of the assay, showing the transport steps for which rate constants are determined by fitting the fluorescence data to a three-compartment model. k+1 and k−1 correspond to the flop (translocation of lipid from the inner leaflet of the ER membrane bilayer to the BSA-accessible outer leaflet) and the flip (translocation from the outer leaflet to the inner leaflet) of the NBD-PL, respectively. The rate constant of the extraction of NBD-PLs by BSA is represented by k+2; k−2 describes the backward movement of analogues from BSA into the solution or membrane. We note that due to the excess of BSA used, the exchange process described by k−2 did not contribute to the kinetics, and the values for this rate constant were very small (typically 10−12 s−1), as expected. (B) Kinetics of 1-palmitoyl-C6-NBD-PC extraction from liposomes and yeast microsomes by BSA. Labeled liposomes (traces a and b) or microsomes (P100 membranes) (trace c) were mixed with 1% (wt/vol) BSA (final concentration) using a stopped-flow accessory, and the fluorescence decay was recorded. Trace c shows the extraction of NBD-PLs from microsomes. For comparison, the extraction kinetics measured with asymmetrically (trace a) and symmetrically (trace b) labeled liposomes are shown. Trace c (microsomes) was fitted to the model diagramed in panel A by using the Marquardt-Levenberg algorithm (continuous line) (see also Materials and Methods). The following values for the rate constants were obtained by fitting trace c to the model: k+2 (extraction of analogues by BSA), 0.029 s−1; k+1 (outward movement), 0.011 s−1; k−1 (inward movement), 0.007 s−1. The half-times for the outward and inward movements obtained from independent preparations are summarized in Table 1. (C) Residuals from fitting trace c (microsomes) either to the model (A) (left) or to a monoexponential (Monoexp.) decay (right). As evident from the residuals, the model provides a much better fit for trace c than the monoexponential decay. Fluorescence is presented as a percentage of the initial value.
We next labeled P100 vesicles with C6-NBD-PC for 30 min and used stopped-flow kinetic analysis to determine the accessibility of the NBD-PL to BSA extraction. Upon mixing of the labeled P100 vesicles with BSA, the fluorescence intensity dropped to ∼50% of its initial value, indicating complete extraction of C6-NBD-PC by BSA (Fig. 2B, trace c). However, in contrast to that for asymmetrically labeled liposomes, the decay of fluorescence intensity occurred more slowly and displayed biphasic kinetics. We interpret these data as follows. During the 30-min labeling period, C6-NBD-PC equilibrates across the two leaflets of the P100 membrane vesicles as a result of the action of the ER phospholipid flippase. This results in symmetrically labeled vesicles. Upon incubation of the labeled vesicles with BSA, the pool of C6-NBD-PC molecules in the outer leaflet is rapidly extracted; however, C6-NBD-PC molecules initially located in the inner leaflet must first be translocated across the membrane before gaining access to BSA. The rate of transbilayer translocation is lower than the rate of NBD-PL extraction by BSA, as also found for other membranes (20, 23, 38), accounting for the second (slower) phase of fluorescence decay. Although the data in Fig. 2B (trace c) refer to measurements on P100 vesicles labeled for 30 min with C6-NBD-PC, essentially the same results were obtained with vesicles labeled for 90 min. This indicates that the NBD-PLs are already equilibrated between the two leaflets of the microsomal membrane after 30 min of labeling. To determine the half-time of NBD-PL flip-flop in microsomes, the data were fitted to a three-compartment model as diagramed in Fig. 2A (residuals for the fit to the model versus a monoexponential fit are shown in Fig. 2C). The results (Table 1) indicate that C6-NBD-PC is rapidly translocated across yeast microsomal membranes (half-time, <2 min) and that the rate of flipping resembles that seen for C6-NBD-PC and -PE in rat liver microsomes (23, 38). The movement of C6-NBD-PC to the luminal leaflet of the microsomes was slightly slower than that to the cytoplasmic layer. This difference might be related to the insensitivity of the stopped-flow assay or to a slightly nonsymmetric transbilayer distribution of the C6-NBD-PC.
TABLE 1.
Half-times of transbilayer movement of NBD-labeled PC and PE in yeast microsomes and rat liver ER vesicles at 22°Ca
| Organism and NBD-PL | Half-time for:
|
|
|---|---|---|
| Outward movement (s) | Inward movement (s) | |
| Yeast C6-NBD-PC | 103 ± 20 | 176 ± 39 |
| Rat liverb | ||
| C6-NBD-PC | 98 ± 9 | 148 ± 12 |
| C6-NBD-PE | 81 ± 8 | 62 ± 4 |
Experimental traces (see Fig. 2) were fitted according to the model to obtain the rate constants of the outward and inward movements of phospholipid analogues, k+1 and k−1, respectively (see Materials and Methods). Half-times of the outward and inward movements across the ER membrane were derived from k+1 and k−1, respectively, according to the formula τ = (ln2)/k, where τ is the half-time. Data shown are means ± standard deviations for two independent experiments. For each experiment, three single kinetics were averaged and subsequently analyzed.
Data are from Marx et al. (23).
Reconstitution of phospholipid flippase activity in proteoliposomes generated from Triton X-100-solubilized P100 microsomal membranes.
To further characterize the yeast ER phospholipid flippase activity, we reconstituted flippase activity according to previously described procedures (10, 11, 15, 20, 22, 26, 38, 39). P100 microsomal membranes were solubilized in Triton X-100 to generate TE. Different amounts of TE were combined with Triton X-100-solubilized egg PC and trace quantities of NBD-PL (0.5 mol%); the solution was then treated with SM2 Bio-Beads to remove detergent and form proteoliposomes. Liposomes lacking protein were prepared in parallel by the same method except that TE was omitted. We used NBD-PLs with either a C6- or a C12-NBD-labeled fatty acyl chain at the sn-2 position. As reported previously, we found that the majority of vesicles formed by this procedure are unilamellar (data not shown) (15, 22, 26). Liposomes had an average diameter of ∼150 nm, while proteoliposomes displayed a larger average diameter of ∼175 nm. A comparison of the protein profiles of TE versus proteoliposomes by SDS-PAGE and silver staining revealed that microsomal membrane proteins in the TE were represented in the reconstituted vesicles without apparent bias (Fig. 3A).
FIG. 3.

Characterization of reconstituted proteoliposomes. Proteoliposomes were reconstituted from mixtures of Triton X-100-solubilized egg PC and TE; liposomes were prepared similarly except that TE was omitted. (A) Silver-stained SDS-PAGE (12% acrylamide) gel of TE and reconstituted proteoliposomes (P). Molecular mass markers are indicated on the left. Samples of TE (30 μl) and proteoliposomes (prepared from 60 μl TE) were precipitated with trichloroacetic acid and washed with acetone before being dissolved in an SDS-containing sample buffer for PAGE analysis. (B) Collisional quenching of NBD fluorescence with iodide ions to determine the fraction of NBD-PL accessible on the outer leaflets of vesicles. Proteoliposomes (open squares; protein/phospholipid ratio, 4.5 mg/mmol) and liposomes (open circles) were reconstituted from Triton X-100-solubilized mixtures containing C12-NBD-PC. Asymmetrically labeled liposomes (filled circles) were prepared by adding C12-NBD-PC to preformed vesicles. The data are presented as modified Stern-Volmer plots (see Materials and Methods). Fo is the fluorescence intensity of the sample in the absence of quencher, whereas ΔF is the fluorescence intensity at a given iodide ion concentration. The connecting lines were obtained by linear regression. The inverse of the y-intercept represents the fraction of C12-NBD-PC that is accessible to the quencher.
Before proceeding with the flippase assay, we tested whether the NBD-PLs were incorporated symmetrically into the liposomal and proteoliposomal membranes. To do this, we used a collisional quenching approach in which membrane-impermeant iodide ions were used to eliminate the fluorescence contribution of NBD-PLs located in the outer (accessible) leaflets of the vesicles. We note that the collisional quenching approach reports on the pool size of NBD-PLs in the outer leaflets of the vesicles and does not provide information on whether or not NBD-PL flipping occurs in the vesicles. We first tested asymmetrically labeled liposomes generated by addition of NBD-PC after vesicle preparation. Analysis of the quenching data via a modified Stern-Volmer plot (21) (the fraction of NBD-PL accessible to iodide quenching is calculated as the inverse of the y-intercept) indicated that the majority (>80%) of the NBD-PC in these vesicles was accessible to iodide ions, as expected (Fig. 3B). For “symmetrically labeled” liposomes and proteoliposomes generated by our reconstitution procedure, ∼50% and ∼56% of NBD-PC, respectively, was accessible to iodide ions. We conclude that the NBD-PLs are symmetrically distributed across the membranes of the reconstituted vesicles and that this distribution is largely unaffected by the presence of membrane proteins.
Next, C6-NBD-PC-containing proteoliposomes were assayed for flippase activity by using defatted BSA to extract the NBD-PLs from the outer leaflets of the vesicles. On liposomes, BSA depleted ∼50% of the C6-NBD-PC, consistent with a symmetric distribution of the NBD-PL across the liposomal membrane and the absence of lipid flip-flop in protein-free vesicles (Fig. 4A). In similar experiments carried out with proteoliposomes, a larger fraction of C6-NBD-PC was accessible to BSA, suggesting that C6-NBD-PC molecules located in the inner leaflets of proteoliposomes can be translocated to the outer leaflets, where they are extracted by BSA. The accessible proportion of C6-NBD-PC increased as the PPR of the vesicle preparation increased until a plateau maximum was reached at a PPR of ∼10 mg/mmol (determined by fitting the dose-response data with two linear segments as described in reference 11). This can be explained as follows. As the PPR increases, the number of flippase-competent vesicles in the sample increases to a stage where each vesicle in the ensemble is equipped at least with a single flippase. At this point (corresponding to a PPR of ∼10 mg/mmol), all NBD-PLs in the preparation should be accessible to BSA. In practice, more than 90% of the NBD-PL is extracted from proteoliposome samples prepared at a PPR of >10 mg/mmol.
FIG. 4.

Reconstitution of phospholipid flippase activity in proteoliposomes. Vesicles were prepared from Triton X-100-solubilized egg PC, TE, and either 1-C14-2-C6-NBD-PC, 1-acyl-2-C6-NBD-PC, 1-acyl-2-C12-NBD-PC, or PE. Different amounts of TE were used to generate vesicle populations with different PPR (expressed in milligrams per millimole). (A) Plot of the extent of BSA-extractable 1-C14-2-C6-NBD-PC as a function of the PPR. (B) Plot showing the percentage of reduction in fluorescence obtained upon addition of dithionite to the reconstituted vesicle preparations. The data are fitted by a monexponential function that is simplified by deconvolution into two linear segments: a line of positive slope generated by linear regression of data points from the rising section of the graph and a plateau determined by the monoexponential curve fit. The point where the rising linear segment intersects the plateau (PPR, ∼10 mg/mmol) is interpreted as the point where each vesicle contains a single functional flippase. The fatty acid composition at the 1 position of the acyl-NBD-PLs used in panel B is as follows: palmitic acid, 62%; stearic acid, 29%; oleic acid, 5.5%.
As an additional test to establish that proteoliposomes derived from TE and egg PC are capable of flipping NBD-PLs, we used a different assay procedure involving dithionite, a membrane-impermeant dianion that reduces the NBD moiety and destroys its fluorescence. Upon addition of dithionite to NBD-PL-containing liposomes, fluorescence was reduced by ∼50%, whereas for proteoliposomes the accessible proportion of NBD-PLs increased as the PPR of the vesicle preparation increased, irrespective of the NBD-PL used as a transport reporter (Fig. 4B). These data reinforce our conclusion that ER-derived proteoliposomes are competent to flip NBD-PLs.
Both the BSA and dithionite assays displayed the same dependence of transport amplitude on PPR, with an inflection point at a PPR of ∼10 mg/mmol. We note that while the dithionite assay displays a built-in efficiency of ∼70 to 75%, the BSA assay approaches the theoretically predicted range of transport amplitudes (i.e., 50% for liposomes and ∼90% for flippase-equipped proteoliposomes). The reason for this is unclear, but similar results have been reported previously in studies of outward translocation of NBD lipids in membrane vesicles (10, 38) and in assays of outward translocation of natural phospholipids (11). We established that dithionite itself had no unduly minimizing effect on flippase activity, since treatment of microsomes with dithionite prior to solubilization did not alter flippase activity in reconstituted vesicles (data not shown). The PPR at which the reduction amplitude reaches its maximum can be used to infer that flippase constitutes ∼2% by weight of proteins in the TE (see reference 26 for details of the calculation). This is higher than the estimate of flippase abundance in TE from rat liver ER (20), possibly reflecting selective solubilization of the flippase in cold Triton X-100 (2).
Effects of protein modification reagents on flippase activity.
To confirm that a membrane protein(s) is required for NBD-PL flip-flop in reconstituted vesicles, the effects of the protein modification reagents NEM (directed at cysteine residues [3, 24]) and DEPC (directed at histidine residues [27]) were tested. Treatment of TE with these reagents prior to reconstitution of proteoliposomes (prepared with PPR in the range of 2.9 to 4.1 g/mol, providing for vesicles with a maximum of 1 flippase) resulted in ∼60% and ∼40% losses of flippase-active vesicles, respectively (the PPR of inhibitor-treated and mock-treated vesicles were similar). However, when TE was treated with NEM and DEPC simultaneously, the effect was roughly additive: >80% of flippase-active vesicles were inactivated (Fig. 5). These results can be explained by postulating two classes of flippase protein, each defined by its sensitivity to one of the two inhibitors. Alternatively, NEM and DEPC could modify critical residues within the same protein to yield the partial and combined effects that we observe (see reference 10 for further discussion of this issue).
FIG. 5.

Inhibition of flippase activity by treating TE with NEM and DEPC. Aliquots of TE treated with NEM (40 mM), DEPC (40 mM), or a combination of the reagents (40 mM each) were used to generate C12-NBD-PC-containing proteoliposomes with PPR in the range of 2.1 to 4.1 g/mol. Mock-treated TE was used as a control. The vesicles were assayed for flippase activity by using the dithionite method. Data are means and standard deviations for measurements from two independent experiments, each with duplicate samples.
NEM did not affect flippase activity when applied to intact membranes of yeast microsomes or proteoliposomes prepared from yeast microsomes. Although NEM is membrane permeant and should be able to access cysteine residues within and on either side of intact membrane vesicles, it reacts more rapidly (by orders of magnitude) with the thiolate (S−) ion than it does with protonated thiols (SH) (3). Our data therefore suggest that a functionally critical cysteine residue in the flippase is located in a hydrophobic environment, where it is less reactive with NEM (data not shown). We did not test the effect of DEPC on intact vesicles.
We were interested to know whether flippase activity could be reduced by protease-treatment of proteoliposomes. Proteoliposomes containing different amounts of protein (PPR ranging from 2.9 to 7.3 g/mol) were treated with proteinase K and subsequently assayed for their ability to mediate flip-flop of C12-NBD-PC. This treatment resulted in a 42.5% ± 8.5% decrease in the amount of reduced analogues relative to that for mock-treated proteoliposomes. Since the vesicles were prepared with a PPR range where each vesicle has no flippases or at least 1 flippase, this result implies that proteinase K treatment destroys flippase activity in ∼40% of the vesicles while having no effect on the remaining ∼60% of the flippase-containing vesicle population. The ability to eliminate some but not all flippase-containing vesicles by protease treatment requires explanation. One possibility is that at least two distinct flippases with different protease sensitivities might be present in the TE, giving rise to at least two classes of flippase-containing vesicles, as discussed above. An alternative possibility is that the flippase protein is reconstituted symmetrically in the vesicle population, i.e., some vesicles contain the flippase oriented such that its “proteinase K-sensitive segment” is on the outside and accessible to the protease, whereas in other vesicles, the cleavage site is in the vesicle interior. If the flippase functions symmetrically, both topological orientations would yield a flippase-active vesicle, but only the activities of those proteoliposomes containing the flippase oriented with its proteinase K-sensitive segment on the outside would be destroyed by proteinase K treatment (10).
Taken together, these data confirm that transbilayer movement of NBD-PLs requires the participation of membrane proteins and that the flippase protein(s) contains functionally critical cysteine and histidine residues. Similar results have been reported and discussed previously for flippase activity in a TE prepared from rat liver ER vesicles (10).
Role of the ER protein translocon in transbilayer phospholipid movement.
It has been speculated that the protein translocon could act as a phospholipid flippase (26, 39). The translocon is an oligomeric structure with a transverse aqueous channel that opens laterally toward the lipid bilayer (8, 36). Such a molecular architecture could provide a transverse diffusion conduit for phospholipids (39). We used two different approaches to test whether the presence of Sec61p, an essential component of the protein translocon in yeast (41), would contribute significantly to phospholipid flippase activity in the yeast ER. Sec61p was depleted in vivo by using a temperature-sensitive SEC61 mutant. In contrast to wild-type Sec61p, mutant versions of this protein are selectively degraded at 38°C, resulting in reduced cellular Sec61p levels (4). Microsomal membranes were prepared by differential centrifugation of total-cell lysates derived from 38°C-shifted wild-type and sec61ts cells as described in Material and Methods. Depletion of Sec61p in preparations from 38°C-shifted sec61ts cells was verified by SDS-PAGE and immunoblotting (Fig. 6A), showing that membranes from the sec61ts cells contained less than 2% of the Sec61p in wild-type cells, whereas levels of Dpm1p, an unrelated ER protein, were unchanged.
FIG. 6.

Depletion of Sec61p does not affect flippase activity. Wild-type and sec61ts cells were shifted to 38°C prior to preparation of microsomal membranes. (A) Sec61p is specifically depleted from P100 membranes. Membrane proteins of 38°C-shifted wild-type and sec61ts cells were analyzed by SDS-PAGE and immunoblotting using polyclonal antibodies against Sec61p. The blot was reprobed with polyclonal antibodies to the ER membrane protein Dpm1p in order to control for equal loading. (B) Stopped-flow kinetics of extraction of 1-palmitoyl-2-C6-NBD-PC from wild-type (squares) and Sec61p-depleted (circles) microsomal membranes (P100 membranes) by BSA. Labeled microsomal membranes were mixed with 1% (wt/vol) BSA (final concentration) using a stopped-flow accessory, and the fluorescence decay was recorded. Solid lines represent the fit of the experimental data to the three-compartment model diagramed in Fig. 2. The following values for the rate constants were obtained from this fit: for wild-type microsomal membranes, k+1 = 0.011 s−1, k−1 = 0.007 s−1, and k+2 = 0.029 s−1; for Sec61p-depleted microsomal membranes, k+1 = 0.014 s−1, k−1 = 0.009 s−1, and k+2 = 0.053 s−1. (Inset) Residuals for a fit of the data to the model and a monoexponential (Monoexp.) function of the extraction kinetics.
We assayed NBD-PL flip-flop in wild-type and Sec61p-depleted membranes by labeling the membrane vesicles with C6-NBD-PC and using stopped-flow kinetic analysis to determine the accessibility of the fluorescent phospholipid to BSA extraction. As shown in Fig. 6B, a rapid, two-component decay of fluorescence was observed with both wild-type and Sec61p-depleted membranes. The fluorescence decay curve in both cases could not be described as a monoexponential process (see the plots of residuals in the Fig. 6B inset) but fitted well with the kinetic model outlined in Fig. 2A. Although the rate constants (k+1, k−1, k+2) obtained from data fitting were of the same order for both wild-type and Sec61p-depleted membranes, they were slightly higher for Sec61p-depleted membranes than for wild-type membranes. This could be a result of changes in the lipid and protein compositions of the ER membrane during Sec61p depletion.
We next assayed flippase activity in proteoliposomes reconstituted from Triton X-100 extracts of wild-type versus Sec61p-depleted membranes. The protein dependence of NBD-PL flipping was identical in Sec61p-depleted and Sec61p-equipped proteoliposomes (data not shown). We conclude that flippase activity in Sec61p-depleted membranes is comparable to that seen in wild-type membranes. Thus, the protein translocation apparatus is not required for the efficient transbilayer movement of phospholipids across the ER membrane of yeast.
Flippase activity sediments slowly in velocity gradients.
TE prepared from wild-type P100 membranes was fractionated on a linear glycerol velocity gradient, yielding six pools of separated ER membrane proteins (Fig. 7B). SDS-PAGE resolution of the different fractions revealed successful separation of solubilized microsomal membrane proteins, with clear differences in protein profiles between pools I to VI. Reconstitution of these pools into proteoliposomes resulted in vesicles with different flippase activities, as assayed using C12-NBD-PC and the dithionite method. The majority of flippase activity was recovered in pool II, corresponding to proteins that sediment at ∼4S (Fig. 7A). Little or no activity was detected in pools IV, V, and VI, which are enriched in Sec61p (Fig. 7A and D). The latter result reinforces the conclusion from the preceding section that the protein translocon plays no role in phospholipid flip-flop in the ER. Importantly, it also indicates that not all membrane proteins promote rapid phospholipid flip-flop and that distinct proteins are required.
FIG. 7.

Fractionation of Triton X-100 extract on a glycerol gradient. TE was subjected to glycerol gradient centrifugation as described in Materials and Methods. Fourteen fractions were collected from the top; fractions 1 (corresponding to the load) and 14 were discarded, while the remaining fractions were pooled pairwise to yield pools I to VI. Equal volumes per pool were used for reconstitution and the flippase assay, as well as to assay for protein content and to perform SDS PAGE and Western blot analysis. (A) Extent of fluorescence reduction by dithionite in 1-acyl-2-C12-NBD-PC-containing proteoliposomes reconstituted from desalted pools I to VI. Data are means ± ranges from triplicate determinations. (B) Protein content (bars) and fraction densities (dots) within the glycerol gradient. Relative positions and sedimentation values of standards run on a parallel gradient are indicated at the top. (C) Protein band pattern of fraction pools as visualized by 12% SDS-PAGE and silver staining. Sizes of molecular mass standards are given on the left. (D) Immunoblotting to identify the sedimentation behavior of Sec61p.
Conclusions.
We used a combination of fluorescence-based methods to characterize phospholipid flippase activity in microsomes of the model organism Saccharomyces cerevisiae. Using BSA back-extraction in conjunction with stopped-flow kinetic analyses, we demonstrated rapid, ATP-independent flip-flop (half-time, ∼100 s) of NBD-PL analogues in intact P100 microsomal vesicles. We also biochemically reconstituted flippase activity in proteoliposomes generated from a Triton X-100 extract of P100 membranes. Using this approach, we demonstrated that flippase activity requires a membrane protein(s) that is sensitive to proteinase K, NEM, and DEPC and that sediments slowly, as assessed by velocity gradient centrifugation analyses (operational sedimentation coefficient, <4S, consistent with data reported for the flippase from rat liver ER [26] and Bacillus subtilis [15]). Our experiments also allowed us to estimate that the flippase protein represents ∼2% (wt/wt) of membrane proteins in the TE. Using ER membrane protein-containing proteoliposomes such as those described here, we recently succeeded in reconstituting flippase activity in giant unilamellar vesicles (31), allowing us to study the time scale of flippase-mediated transbilayer movement of unlabeled phospholipids. These studies revealed half-times of phospholipid flip-flop on the order of few minutes, similar to those that have been determined here for flip-flop of fluorescently labeled phospholipid analogues.
The reconstitution approach described here and elsewhere (10, 11, 15, 20, 26, 28, 38, 39) provides a basis with good prospects for the identification of the flippase through conventional protein purification. Attempts to do this are under way. Our experiments also present a general method for identifying the flippase by testing promising candidates, in particular, to check whether a protein contributes at all to the number of functional flippase proteins in the TE. Taking advantage of a temperature-sensitive SEC61 yeast mutant, we tested the role of the protein translocon in phospholipid flipping in the ER. Early work predicted that the transmembrane domains of translocating nascent membrane proteins must exit laterally from the channel of the translocon into the lipid phase (8). This idea was confirmed by recent structural analyses of the bacterial translocon that revealed the presence of a gate where the translocon complex could open toward the lipid phase (36). The proximate connection between the lipid-lined lateral channel and the translocon pore could constitute a transverse diffusion conduit for phospholipids, as suggested previously (26, 39). However, depletion of Sec61p had no effect on the flippase activity in native membranes as well as in proteoliposomes. This is in agreement with a previous study showing that depletion of the protein translocon in a bacterial inner membrane system had no effect on the translocation of a dibutyroyl-PC, a water-soluble PC analogue (39). Thus, in both eukaryotes and prokaryotes, the functionally assembled protein-conducting channel is not required for the transbilayer movement of phospholipids. In a similar approach, other candidate flippases, such as members of the major facilitator superfamily (30), could be tested. One such protein, Rft1p, has been suggested to play a role in flipping dolichol pyrophosphoryl oligosaccharide intermediates in the pathway of protein N glycosylation (13); it would be interesting to test whether it has any role in glycerophospholipid flip-flop.
It has been proposed that the mere presence of transmembrane proteins may be sufficient to catalyze rapid flip-flop (19). However, fractionation of detergent-solubilized rat liver ER has previously yielded specific protein pools with increased flip-flop activities (11, 26). Likewise, protein pools with higher specific flippase activities were obtained in this study by fractionation; conversely, other fractions replete with membrane proteins were inactive. In addition, a recent study clearly demonstrated that the presence of membrane proteins in giant unilamellar vesicles per se is not sufficient to facilitate the flip-flop of phospholipids (31). These results reinforce the conclusion that specific proteins are required to facilitate phospholipid flip-flop; the data presented in this paper indicate that the identification of these proteins is feasible.
Acknowledgments
We gratefully acknowledge Ursula Muschick for excellent technical assistance, Gudrun Lutsch for performing electron microscopy, the Maxfield lab at Weill-Cornell for the use of a fluorescence spectrometer, and Thomas Sommer, Joost Holthuis, Hanspeter Rottensteiner, and Michael Veit for generously providing yeast strains or antibodies.
This work was supported in part by the Deutsche Forschungsgemeinschaft (grant He1928-6 to A.H. and Po748-4 to T.P.), the Schering Foundation (to N.A.-B.), and the National Institutes of Health (grant GM071041 to A.K.M.).
Footnotes
Published ahead of print on 6 July 2007.
REFERENCES
- 1.Backer, J. M., and E. A. Dawidowicz. 1987. Reconstitution of a phospholipid flippase from rat liver microsomes. Nature 327:341-343. [DOI] [PubMed] [Google Scholar]
- 2.Bagnat, M., S. Keranen, A. Shevchenko, A. Shevchenko, and K. Simons. 2000. Lipid rafts function in biosynthetic delivery of proteins to the cell surface in yeast. Proc. Natl. Acad. Sci. USA 97:3254-3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bednar, R. A. 1990. Reactivity and pH dependence of thiol conjugation to N-ethylmaleimide: detection of a conformational change in chalcone isomerase. Biochemistry 29:3684-3690. [DOI] [PubMed] [Google Scholar]
- 4.Biederer, T., C. Volkwein, and T. Sommer. 1996. Degradation of subunits of the Sec61p complex, an integral component of the ER membrane, by the ubiquitin-proteasome pathway. EMBO J. 15:2069-2076. [PMC free article] [PubMed] [Google Scholar]
- 5.Bishop, W. R., and R. M. Bell. 1988. Assembly of phospholipids into cellular membranes: biosynthesis, transmembrane movement and intracellular translocation. Annu. Rev. Cell Biol. 4:579-610. [DOI] [PubMed] [Google Scholar]
- 6.Bishop, W. R., and R. M. Bell. 1985. Assembly of the endoplasmic reticulum phospholipid bilayer: the phosphatidylcholine transporter. Cell 42:51-60. [DOI] [PubMed] [Google Scholar]
- 7.Bligh, E. G., and W. J. Dyer. 1959. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37:911-917. [DOI] [PubMed] [Google Scholar]
- 8.Blobel, G., and B. Dobberstein. 1975. Transfer of proteins across membranes. I. Presence of proteolytically processed and unprocessed nascent immunoglobulin light chains on membrane-bound ribosomes of murine myeloma. J. Cell Biol. 67:835-851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buton, X., G. Morrot, P. Fellmann, and M. Seigneuret. 1996. Ultrafast glycerophospholipid-selective transbilayer motion mediated by a protein in the endoplasmic reticulum membrane. J. Biol. Chem. 271:6651-6657. [DOI] [PubMed] [Google Scholar]
- 10.Chang, Q. L., S. N. Gummadi, and A. K. Menon. 2004. Chemical modification identifies two populations of glycerophospholipid flippase in rat liver ER. Biochemistry 43:10710-10718. [DOI] [PubMed] [Google Scholar]
- 11.Gummadi, S. N., and A. K. Menon. 2002. Transbilayer movement of dipalmitoylphosphatidylcholine in proteoliposomes reconstituted from detergent extracts of endoplasmic reticulum. Kinetics of transbilayer transport mediated by a single flippase and identification of protein fractions enriched in flippase activity. J. Biol. Chem. 277:25337-25343. [DOI] [PubMed] [Google Scholar]
- 12.Helenius, J., and M. Aebi. 2002. Transmembrane movement of dolichol linked carbohydrates during N-glycoprotein biosynthesis in the endoplasmic reticulum. Semin. Cell Dev. Biol. 13:171-178. [DOI] [PubMed] [Google Scholar]
- 13.Helenius, J., D. T. Ng, C. L. Marolda, P. Walter, M. A. Valvano, and M. Aebi. 2002. Translocation of lipid-linked oligosaccharides across the ER membrane requires Rft1 protein. Nature 415:447-450. [DOI] [PubMed] [Google Scholar]
- 14.Herrmann, A., A. Zachowski, and P. F. Devaux. 1990. Protein-mediated phospholipid translocation in the endoplasmic reticulum with a low lipid specificity. Biochemistry 29:2023-2027. [DOI] [PubMed] [Google Scholar]
- 15.Hrafnsdóttir, S., and A. K. Menon. 2000. Reconstitution and partial characterization of phospholipid flippase activity from detergent extracts of the Bacillus subtilis cell membrane. J. Bacteriol. 182:4198-4206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaplan, R. S., and P. L. Pedersen. 1989. Sensitive protein assay in presence of high levels of lipid. Methods Enzymol. 172:393-399. [DOI] [PubMed] [Google Scholar]
- 17.Kelleher, D. J., and R. Gilmore. 2006. An evolving view of the eukaryotic oligosaccharyltransferase. Glycobiology 16:47R-62R. [DOI] [PubMed] [Google Scholar]
- 18.Kihara, A., and Y. Igarashi. 2002. Identification and characterization of a Saccharomyces cerevisiae gene, RSB1, involved in sphingoid long-chain base release. J. Biol. Chem. 277:30048-30054. [DOI] [PubMed] [Google Scholar]
- 19.Kol, M. A., A. I. de Kroon, J. A. Killian, and B. de Kruijff. 2004. Transbilayer movement of phospholipids in biogenic membranes. Biochemistry 43:2673-2681. [DOI] [PubMed] [Google Scholar]
- 20.Kubelt, J., A. K. Menon, P. Muller, and A. Herrmann. 2002. Transbilayer movement of fluorescent phospholipid analogues in the cytoplasmic membrane of Escherichia coli. Biochemistry 41:5605-5612. [DOI] [PubMed] [Google Scholar]
- 21.Lakowicz, J. R. 1983. Principles of fluorescence spectroscopy. Plenum Press, New York, NY.
- 22.Lévy, D., A. Bluzat, M. Seigneuret, and J. L. Rigaud. 1990. A systematic study of liposome and proteoliposome reconstitution involving Bio-Bead-mediated Triton X-100 removal. Biochim. Biophys. Acta 1025:179-190. [DOI] [PubMed] [Google Scholar]
- 23.Marx, U., G. Lassmann, H. G. Holzhutter, D. Wustner, P. Muller, A. Hohlig, J. Kubelt, and A. Herrmann. 2000. Rapid flip-flop of phospholipids in endoplasmic reticulum membranes studied by a stopped-flow approach. Biophys. J. 78:2628-2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Means, G., and R. Feeney. 1971. Chemical modification of proteins. Holden-Day, San Francisco, CA.
- 25.Menon, A. K. 1995. Flippases. Trends Cell Biol. 5:355-360. [DOI] [PubMed] [Google Scholar]
- 26.Menon, A. K., W. E. Watkins III, and S. Hrafnsdottir. 2000. Specific proteins are required to translocate phosphatidylcholine bidirectionally across the endoplasmic reticulum. Curr. Biol 10:241-252. [DOI] [PubMed] [Google Scholar]
- 27.Miles, E. W. 1977. Modification of histidyl residues in proteins by diethylpyrocarbonate. Methods Enzymol. 47:431-442. [DOI] [PubMed] [Google Scholar]
- 28.Nicolson, T., and P. Mayinger. 2000. Reconstitution of yeast microsomal lipid flip-flop using endogenous aminophospholipids. FEBS Lett. 476:277-281. [DOI] [PubMed] [Google Scholar]
- 29.Orlean, P., C. Albright, and P. W. Robbins. 1988. Cloning and sequencing of the yeast gene for dolichol phosphate mannose synthase, an essential protein. J. Biol. Chem. 263:17499-17507. [PubMed] [Google Scholar]
- 30.Pao, S. S., I. T. Paulsen, and M. H. Saier, Jr. 1998. Major facilitator superfamily. Microbiol. Mol. Biol Rev. 62:1-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Papadopulos, A., S. Vehring, I. Lopez-Montero, L. Kutschenko, M. Stockl, P. F. Devaux, M. Kozlov, T. Pomorski, and A. Herrmann. 2007. Flippase activity detected with unlabeled lipids by shape changes of giant unilamellar vesicles. J. Biol. Chem. 282:15559-15568. [DOI] [PubMed] [Google Scholar]
- 32.Pomorski, T., and A. K. Menon. 2006. Lipid flippases and their biological functions. Cell. Mol. Life Sci. 63:2908-2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rouser, G., S. Fleischer, and A. Yamamoto. 1970. Two dimensional thin layer chromatographic separation of polar lipids and determination of phospholipids by phosphorus analysis of spots. Lipids 5:494-496. [DOI] [PubMed] [Google Scholar]
- 34.Schenk, B., F. Fernandez, and C. J. Waechter. 2001. The ins(ide) and out(side) of dolichyl phosphate biosynthesis and recycling in the endoplasmic reticulum. Glycobiology 11:61R-70R. [DOI] [PubMed] [Google Scholar]
- 35.te Heesen, S., R. Rauhut, R. Aebersold, J. Abelson, M. Aebi, and M. W. Clark. 1991. An essential 45 kDa yeast transmembrane protein reacts with anti-nuclear pore antibodies: purification of the protein, immunolocalization and cloning of the gene. Eur. J. Cell Biol. 56:8-18. [PubMed] [Google Scholar]
- 36.Van den Berg, B., W. M. Clemons, Jr., I. Collinson, Y. Modis, E. Hartmann, S. C. Harrison, and T. A. Rapoport. 2004. X-ray structure of a protein-conducting channel. Nature 427:36-44. [DOI] [PubMed] [Google Scholar]
- 37.Vishwakarma, R. A., and A. K. Menon. 2005. Flip-flop of glycosylphosphatidylinositols (GPI's) across the ER. Chem. Commun. (Cambridge) 2005:453-455. [DOI] [PubMed] [Google Scholar]
- 38.Vishwakarma, R. A., S. Vehring, A. Mehta, A. Sinha, T. Pomorski, A. Herrmann, and A. K. Menon. 2005. New fluorescent probes reveal that flippase-mediated flip-flop of phosphatidylinositol across the endoplasmic reticulum membrane does not depend on the stereochemistry of the lipid. Org. Biomol. Chem. 3:1275-1283. [DOI] [PubMed] [Google Scholar]
- 39.Watkins, W. E., and A. K. Menon. 2002. Reconstitution of phospholipid flippase activity from E. coli inner membrane: a test of the protein translocon as a candidate flippase. Biol. Chem. 383:1435-1440. [DOI] [PubMed] [Google Scholar]
- 40.Wessel, D., and U. I. Flügge. 1984. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem. 138:141-143. [DOI] [PubMed] [Google Scholar]
- 41.Wilkinson, B. M., M. Regnacq, and C. J. Stirling. 1997. Protein translocation across the membrane of the endoplasmic reticulum. J. Membr. Biol. 155:189-197. [DOI] [PubMed] [Google Scholar]
- 42.Zinser, E., and G. Daum. 1995. Isolation and biochemical characterization of organelles from the yeast, Saccharomyces cerevisiae. Yeast 11:493-536. [DOI] [PubMed] [Google Scholar]