

Abstract
Background and aims
The formation of cholesterol gallstones is a complex process involving contributions from genes and environmental factors. Although gallbladder inflammation is believed to be common during cholelithogenesis, the role of immunological factors is unknown.
Methods
The role of adaptive immunity in cholesterol cholelithogenesis was analyzed utilizing immunocompetent Helicobacter spp. infected and uninfected BALB/c and congenic immunodeficient Rag2−/− (Rag) mice. Lymphocyte transfer studies were performed to determine which cellular subset was responsible for cholesterol gallstone formation. Also, gallbladder inflammation was quantified to determine the nature of the inflammatory response associated with cholelilithogenesis.
Results
When fed a lithogenic diet for eight-weeks, wild-type mice developed significantly more cholesterol gallstones (27–80% prevalence) than Rag mice (~5%, P<0.05). Helicobacter spp. infected BALB/cJ mice displayed mild, but statistically significant increases in cholesterol gallstone prevalence compared to uninfected mice (81% vs. 39%; P<0.05). Transfer of splenocytes, or T-lymphocytes to Rag2−/− mice increased stone prevalence markedly (26% and 40% respectively, P<0.05) whereas transfer of B-cells was not appreciably cholelithogenic (13%). The adaptive immune response increased the expression ofgallbladder Muc genes and accumulation of mucin gel. In addition, T-cells and cholesterol monohydrate crystals induced proinflammatory gene expression in the gallbladder which likely contributes to gallbladder dysfunction.
Conclusions
These studies indicate that T-cells are critical in murine cholesterol cholelithogenesis. Furthermore, cholesterol monohydrate crystals induce expression of proinflammatory cytokines in a T-cell dependent fashion. Acquired immunity and inflammation are likely to be crucial factors in cholesterol gallstone pathogenesis rather then merely the result of cholelithogenesis.
Introduction
Cholesterol gallstones are composed predominantly of cholesterol monohydrate crystals within a mucin glycoprotein scaffolding 1. They form following nucleation and phase separation of cholesterol monohydrate crystals from supersaturated bile 1. Despite more than a half-century of basic and clinical research, there is currently no satisfactory, non-surgical treatment for definitive prevention or management of gallstone patients. As a consequence, the disease continues to be a serious economic burden with an annual cost in the United States approaching 10 billion dollars 1.
Most commonly, cholesterol gallstones result from the interaction of multiple genes and environmental factors 1. A large twin study proposed that, in symptomatic gallstone patients, genes contribute 25% to the phenotype, shared environmental factors 13%, and unique environmental factors 62% 2. Regardless of the predisposing causes, cholesterol gallstones occur when excess hepatically-secreted cholesterol in bile phase separates as unilamellar vesicles which eventually fuse to form multilamellar vesicles (liquid crystals)1, 3. Cholesterol gallstone formation proceeds when these liquid crystals nucleate cholesterol monohydrate crystals in an inflamed and hypomotile gallbladder 1, 3–5. Biliary proteins, in particular, mucin glycoproteins promote cholesterol nucleation and precipitation 6–9. In addition, a variety of other proteins, including all classes of biliary immunoglobulins, may have pronucleating properties 10–12.
In vivo studies analyzing the pathogenesis of cholesterol gallstones rely predominantly on inbred mouse models 1, 13. These models allow investigators to analyze genetic differences in cholelithogenesis under well-defined conditions. However, despite these controlled environments, different cholesterol gallstone prevalence rates are often noted in inbred mice under seemingly identical conditions 13. We demonstrated recently that several murine enterohepatic bacterial pathogens, specifically Helicobacter hepaticus, H. bilis, and coinfection with H. hepaticus and H. rodentium increased cholesterol gallstone formation in C57L/J mice by as much as 70%. These organisms likely contributed to the susceptibility differences noted historically with this model 13. Enterohepatic Helicobacter spp. are enzootic in mouse colonies worldwide, and colonize the distal intestine and bile canaliculi 14. Interestingly, the human gastric pathogen H. pylori, and two urease-negative enterohepatic helicobacters are not able to promote cholesterol gallstone formation in this model 13, 15, 16. Aside from the gallstone phenotype, the most notable findings in mice infected with cholelithogenic Helicobacter spp. is an increase in gallbladder weight (a surrogate marker for gallbladder stasis), mucin gel accumulation, inflammation, and hyperplasia of the gallbladder epithelium 13. However, Helicobacter spp. infection does not alter the absolute or relative compositions of biliary lipids in hepatic bile supporting the notion that the target cholelithogenic organ for their effect is not the liver 13.
Helicobacter spp. generally cause disease by inducing a T helper Type 1 (Th1) mediated pro-inflammatory immune response 17, 18. In many cases, disease is mediated by the response of the adaptive immune system. Most notably, without an adaptive immune response, H. pylori fails to produce gastritis 19. When adaptive immunity is restored by splenocyte transfer, gastritis ensues. In the case of H. pylori this effect is mediated by T-cells 19, 20.
We hypothesized that adaptive immunity might be important in cholesterol gallstone formation. Further, we hypothesized that mechanistically, gallstone promotion could occur in one of two ways: i) Either by increased production of pronucleating immunoglobulins; or ii) T-cell mediated production of pro-inflammatory cytokines altering mucin production (or other unidentified pronucleating proteins) and promoting gallbladder inflammation and dysfunction. To test these competing hypotheses, we compared wild-type BALB/c mice and congenic BALB/c mice lacking the Rag2 gene and therefore lacking functional T and B-lymphocytes.
Methods
Animal maintenance and husbandry
Male mice were housed in a facility accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care (AAALAC) and all protocols were approved by the institutional animal care and use committee. Animals were provided with rodent chow (Purina Mills, St. Louis MO) and water ad libitum and housed in micro-isolator cages under specific pathogen free (SPF) conditions (free of known bacterial, including Helicobacter spp., viral and parasitic pathogens). Mice were fasted for approximately 12 hours prior to being euthanized with a CO2 overdose followed by necropsy.
Helicobacter spp. infection
Helicobacter hepaticus 3B1 and H. rodentium ATCC 700285 were grown on blood agar plates (Remel; Lenexa, KS) under microaerobic conditions at 37°C. Bacterial colonies were removed from plates and suspended in Brucella broth (Becton, Dickinson and Company, Franklin Lakes, NJ) to a final optical density of 0.6–2.0 at 660nm. Mice were infected as previously described at 4–6 weeks of age via gavage and redosed at 4-week intervals with H. hepaticus 13, 15.
Bile analysis, tissue harvest, and histopathological analysis
Gallbladders were removed intact and weighed. Bile was removed from full gallbladders and analyzed for mucin gel accumulation and biliary lipid phenotype by direct and polarized light microscopy. Mucin gel was scored visually by microscopy on a scale of 0–5 (0=0% mucin, 1=20% of the gallbladder filled with mucin, 2= 40% of the gallbladder filled with mucin, 3=60% of the gallbladder filled with mucin, 4=80% of the gallbladder filled with mucin and 5=100% of the gallbladder filled with mucin) 13, 15, 21. Empty gallbladders were immediately flash-frozen in liquid N2 and stored at −80°C for transcription analysis or were fixed in 10% neutral-buffered formalin. Formalin fixed gallbladders were routinely processed and stained with hematoxylin and eosin. Gallbladder sections were analyzed by a comparative pathologist (ABR) blinded as to sample identity.
BALB/cJ and Rag mouse studies
Male BALB/cJ mice were purchased from The Jackson Laboratory (Bar Harbor, ME; referred to as Jackson) and male C.129S6(B6)-Rag2tm1FwaN12 (Rag mice, BALB/c background) were bred at MIT following acquisition from Taconic Farms, Inc. (Germantown, NY; referred to as Taconic). Mice were further divided into Helicobacter spp. infected and uninfected groups (n=15–18 mice per group). Infected groups were dosed with both H. hepaticus and H. rodenitum by oral gavage as described elsewhere 13. Mice were fed standard rodent diet until 8–10 weeks of age, at which time they were converted to the lithogenic diet (containing 0.5% cholic acid, 1% cholesterol and 15% dairy derived triglyceride). Mice were fed the lithogenic diet for 8 weeks and were then euthanized with CO2 and analyzed for biliary phenotype as described 13, 15.
BALB/cAnNTac, Rag and Nude mice and Adoptive Transfer Studies
Male BALB/cAnNTac, and C.Cg/AnNTac-Foxn1nu N9 (nude mice) were purchased from Taconic. Male Rag mice for these studies were either acquired directly from Taconic or were bred at MIT from mice originally obtained from Taconic. Adoptive transfer of splenocytes, T-cells or B-cells was performed when Rag mice were approximately 8–9 weeks of age (approximately one-week prior to initiation of feeding the lithogenic diet). Seven mouse groups in total were included in these studies: Rag mice (n=36), Rag mice receiving splenocytes (n=27), nude mice (n=12), Rag mice receiving T-cells (n=15), Rag mice receiving B-cells (n=15), and wild-type mice (n=15). Mice were fed a standard rodent diet (Purina Mills, St. Louis MO) until 9–10 weeks of age at which time they were converted to the lithogenic diet. Mice were fed the lithogenic diet for 8 weeks and were then humanely euthanized and the biliary phenotype analyzed 13, 15, 21.
Splenocyte transfer
Splenocytes prepared from BALB/cAnNTac donors were adoptively transferred into Rag recipients one week prior to the initiation of lithogenic diet feeding. Spleens were aseptically removed from euthanized donor mice and single cell suspensions from the pooled spleens were prepared in RPMI-1640 medium supplemented with 10% fetal bovine serum. After osmotic lysis of red blood cells, splenocytes were washed and examined for viability with Trypan blue exclusion. Cell number was quantified by an automated cell-counter and validated by counting with a hemocytometer. Then, the cell concentration was adjusted such that each mouse received 2×106 viable splenocytes intraperitoneally in 0.2 ml serum-free Hanks balanced salt solution (HBSS pH 7.2).
T-cell transfer
For T-cell transfer studies, splenocytes were prepared initially as described above. Enrichment utilized Dynal® Mouse T Cell Negative Isolation Kit (Invitrogen Corporation; Carlsbad, CA) per the manufacturer’s recommendations. Briefly, the splenocytes were incubated with a cocktail of magnetized-beads coupled to antibodies that bind non-T-cell populations. The non-T cell fraction i.e. bead-bound was then separated from the T-cell enriched fraction by placing the sample on a magnetic column. This initial purification provides a highly purified T-cell population. Following this negative selection strategy, a second flow cytometry-based approach was used to further eliminate contaminating B-cells. The T-cell enriched fraction was incubated with Cy-labeled anti-B220 antibody (Pharmingen, BD Biosciences, San Diego, CA) and subjected to cell-sorting to “gate out” B cells stained with the antibody. An aliquot of the collected cells was stained with fluorescein isothiocyanate (FITC) labeled pan-T-cell specific αβ+ TCR-specific antibody (Cedarlane, Burlington, NC) confirming a T cell population of >95% purity. Lastly, these highly purified T cells were injected retroorbitally with 2×106/0.2 ml HBSS into recipient mice anesthetized with isoflurane.
B-cell transfer
For B-cell transfer studies, splenocytes were first prepared as described elsewhere in this text. Initial enrichment utilized Dynal® Mouse B Cell Negative Isolation Kit (Invitrogen Corporation; Carlsbad, CA) per the manufacturer’s instructions. Briefly, the splenocytes were incubated with a cocktail of magnetized-bead coupled antibodies that bind non-B-cell populations, and the bead-bound non-B cell fraction was retained on a magnetic column as described. Further, to remove any residual T cells, the B-cell enriched fraction was incubated with FITC-labeled pan-T-cell specific αβ+ TCR-specific antibody (Cedarlane, Burlington, NC). During sorting, the FITC-labeled T cells were eliminated and B cells were isolated. An aliquot of the sorted cells was labeled with anti-B220 antibody (Pharmingen, BD Biosciences, San Diego, CA) and shown to contain >95% pure B-cell population on re-analysis. Subsequently, these highly purified B cells with 2×106/0.2 ml HBSS were injected retroorbitally into recipient mice anesthetized with isoflurane.
mRNA and cDNA preparation
The mouse gallbladder is small and as a result contains minute concentrations of RNA. To maximize RNA yield, the RNeasy Micro Kit (Qiagen Corporation; Valencia, CA), a kit designed to maximize RNA yield from small tissue samples was utilized per the manufacturer’s recommendations. Following mRNA isolation, cDNA was prepared utilizing SuperScript™ III First-Strand Synthesis System (Invitrogen Corporation; Carlsbad, CA) according to the manufacturer’s recommendation for Muc gene analysis. For cytokine gene expression analysis the ReactionReady™ First Strand cDNA Synthesis Kit (Superarry Bioscience; Frederick, MD) was utilized for cDNA preparation.
Muc gene expression analysis
To determine mucin (Muc) gene expression, four Fam-labeled murine mucin gene (Muc 1, Muc 3, Muc 4, and Muc 5ac) primer-probe sets were acquired (Applied Biosystems; Foster City, CA). Quantitative PCR (QPCR) was performed utilizing the ABI-Prism 7700, and Muc gene expression levels were normalized to Gapdh expression; differences between groups (n= ≥10 per group) were analyzed via the ΔΔ CT method utilizing Rag mice as the comparative delta value.
Inflammatory gene expression analysis
Large scale transcriptional screening of genes involved in immunity and inflammation was performed utilizing the RT² Profiler™ PCR Array Mouse Inflammatory Cytokines & Receptors (Superarray Bioscience; Frederick, MD). This kit is a SYBR green 96 well based quantitative PCR assay which includes 84 immune and inflammatory based genes, 5 housekeeping genes (Gusb, Hprt1, Hspcb, Gapdh, Actb) and appropriate controls. Expression of each gene of interest is normalized to the housekeeping genes. Groups (n=5–6 per group) are compared by the ΔΔ CT method for each individual gene utilizing Rag mice that developed cholesterol monohydrate crystals as the baseline delta (expression of 1). Additionally, relative expression of three common, well characterized Th1 cytokine genes (Il-1β, Ifn-γ, and Tnf-α) and three common, well characterized Th2 cytokine genes (Il-4, Il-10, and Il-13) were analyzed to determine the relative Th1/Th2 balance of each group.
Statistical analyses
All statistical analyses were performed utilizing GraphPad Prism (Graphpad, Inc; San Diego, CA). Cholesterol monohydrate, sandy stone and cholesterol gallstone formation were compared by Fisher’s exact test. Non-parametric data were analyzed by the Mann-Whitney Test. Gene expression analysis was performed utilizing one-way analysis of variance and individual data pairs were analyzed by the unpaired t-test. Data were considered statistically significant if the P values were < 0.05.
Results
Biliary phenotype of BALB/cJ mice compared to Rag mice
To determine the role of adaptive immunity in cholesterol gallstone formation, BALB/cJ (The Jackson Laboratories, Bar Harbor, ME; Jax) mice were compared to T and B-cell deficient Rag mice (C.129S6(B6)-Rag2tm1FwaN12) on a BALB/c background. Regardless of Helicobacter spp. infection status BALB/cJ mice display significant increases in mucin gel accumulation, and increased prevalence of cholesterol monohydrate crystals, sandy stones and true cholesterol gallstones when compared to Rag mice (P<0.05; Fig. 1A–F). Infected BALB/cJ mice displayed a significant increase in cholesterol gallstone prevalence when compared to uninfected BABL/cJ mice (P<0.05). Gallbladders from uninfected and infected BALB/cJ mice were infiltrated with modest numbers of eosinophils and lymphocytes, and developed mild mucosal hyperplasia compared to Rag mice that displayed little or no gallbladder inflammation and lacked hyperplasia (Fig. 1 G–H). There were no significant changes in the gallbladder weight of mice regardless of immune or infection status (data not shown). These data strongly suggest that adaptive immunity is important in cholesterol gallstone formation. Further, because only the most end-point phenotype (cholesterol gallstones) differs between infected and uninfected BALB/cJ mice it appears that infection in this strain is not necessary and merely increases the kinetics of cholesterol gallstone formation.
Figure 1.
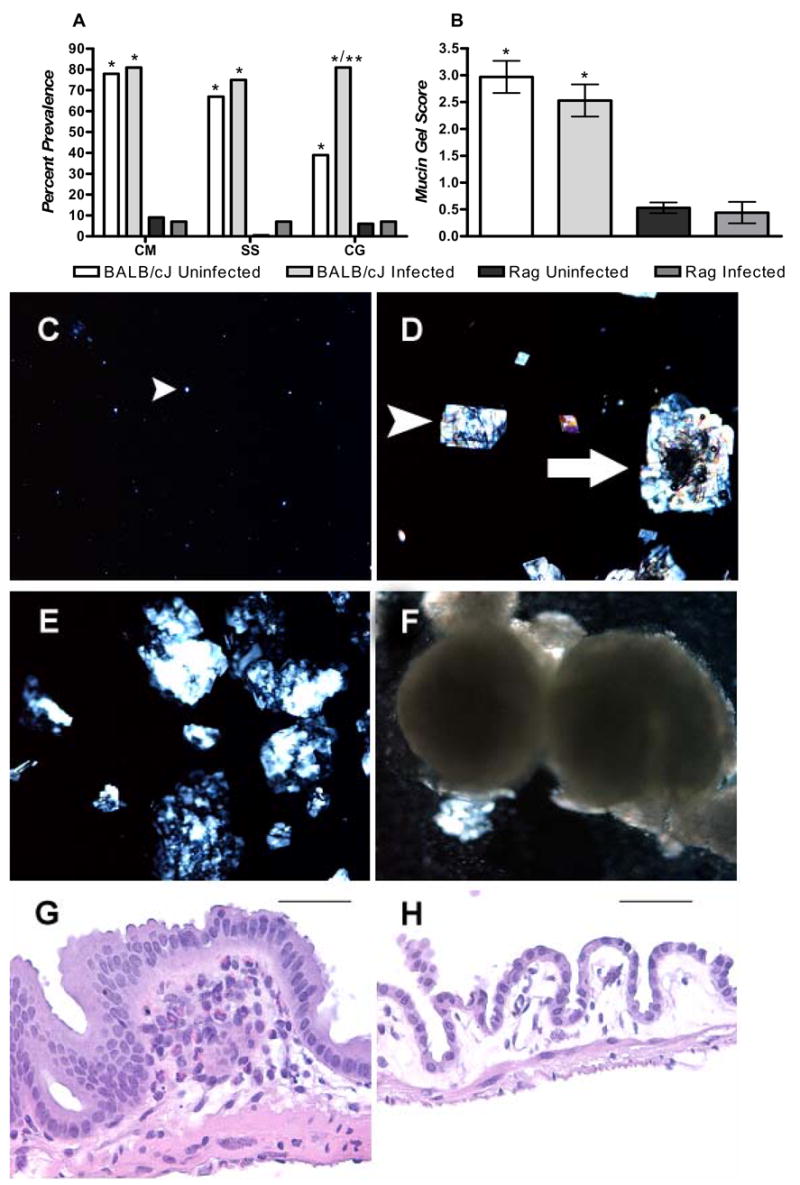
(A) Data represent percent prevalence of cholesterol monohydrate crystals (CM), sandy stones (SS) cholesterol gallstones (CG) and (B) mucin gel score (0–5) in male BALB/cJ and Rag mice (n=15–18/group). All groups of mice developed lithogenic bile as evidenced by liquid crystals (C) or further progression (D–E). Regardless of infection status BALB/cJ mice formed cholesterol monohydrate crystals (A, D), sandy stones (A, D white arrow, E), and cholesterol gallstones (A, F) significantly more frequently then Rag mice and accumulated significantly more mucin gel (B) then Rag mice (*: P<0.05 compared to Rag mice). Infected BALB/cJ differed from uninfected BALB/cJ mice only in the final stage (true cholesterol gallstones) of cholesterol gallstone pathogenesis (**: P<0.05 compared to uninfected BALB/cJ mice). Gallbladder tissue of BALB/c mice (G) was infiltrated by a mixed eosinophilic/lymphocytic inflammatory cell population. Additionally, these tissues were typically thickened with mild mucosal hyperplasia. Rag mice (H) seldom displayed inflammatory infiltrates in the gallbladder and mucosa of Rag mice generally consisted of cuboidal epithelium that was rarely hyperplastic. G,H: Bars= 60 αM
Biliary phenotype of Rag mice following splenocyte transfer
To determine whether transfer of wild-type lymphocytes could restore cholesterol gallstone susceptibility, splenocytes were harvested from BALB/cAnNTac (WT) mice and transferred to their Rag counterparts. To eliminate any genetic effects on phenotype we utilized a genetically homogeneous group from Taconic Farms Inc. (Germantown, NY; Taconic). Additionally, nude mice (C.Cg/AnNTac-Foxn1nuN9) on a BALB/c background were analyzed to determine if B-cells, in the absence of T-cells, are key players in cholelithogenesis.
Both WT and Rag mice receiving splenocytes (splenocyte transfer, ST) exhibit a statistically higher prevalence of gallstones when compared to Rag mice (P<0.05, Fig. 2A). The ST group also significantly increased cholesterol monohydrate crystal formation compared to Rag mice (P<0.05, Fig. 2A). WT, nude and ST mice all display significant increases in mucin-gel accumulation when compared to Rag mice (P<0.05, Fig. 2B). Interestingly, WT BALB/c mice from Taconic (BALB/cAnNTac) express a lower prevalence of cholesterol monohydrate crystals, sandy stones and true cholesterol gallstones when compared to JAX BALB/c (BALB/cJ) mice (Fig. 1A and 2A). These data indicate that restoration of the cholesterol gallstone phenotype occurs with transfer of functional lymphocytes. Furthermore, nude mice, which lack circulating T-cells, are not capable of completely inducing the gallstone phenotype, although they do promote mucin gel accumulation and moderate, non-significant increases in cholesterol monohydrate crystal and gallstone formation. Finally, cholelithogenesis was substrain dependent in the BALB/c mouse with mice of Jackson origin being more susceptible to cholesterol gallstone formation than mice of Taconic origin.
Figure 2.

(A) Data represent percent prevalence of cholesterol monohydrate crystals, sandy stones and cholesterol gallstones (B) and mucin gel score in Helicobacter spp. infected male BALB/cAnNTac (WT, n=15), Rag mice (n=36), Rag mice receiving splenocytes (ST, n=27), and nude mice (n=12). All mice developed liquid crystals (not shown). Mice receiving splenocytes formed cholesterol monohydrate crystals, sandy stones, and cholesterol gallstones significantly more frequently then Rag mice (*: P<0.05 when compared to Rag mice). WT mice displayed increased cholesterol gallstone prevalence when compared to Rag mice (P<0.05). (B) WT, ST, and nude mice all significantly increased mucin gel accumulation when compared to Rag mice as indicated by the mucin gel score (P<0.05). As shown in A, BALB/cAnNTac mice exhibited a lower prevalence of cholesterol monohydrate crystals, sandy stones and cholesterol gallstones when compared to BALB/cJ mice (see also Fig 1A).
Mucin gene expression analysis
Mucin gene expression patterns differ in WT mice and ST mice (P<0.05, Fig. 3A–D). Nude mice display increased expression of Muc1 compared to ST mice (Fig. 3A). ST mice upregulate Muc3 when compared to all other groups (P<0.05, Figure 3B), Muc4 when compared to WT mice (P<0.05, Fig. 3C), and Muc5ac when compared to either nude or Rag mice (P<0.05, Fig. 3D). Moreover, WT mice display increased expression of Muc5ac compared to Rag mice (P=0.06, Fig. 3D). These data demonstrate that expression of mucin genes is dependent upon the immune status of the mice examined. Interestingly, both ST and WT mice display increases in Muc5ac which was demonstrated recently to be under inflammatory regulation in the gallbladder 22.
Figure 3.

(A–D) Data represent normalized expression of Muc1 (A), Muc3 (B), Muc4 (C), and Muc5ac (D) from the gallbladders of previously described (Fig 2) experimental groups (n ≥ 10 gallbladder analyzed/group). Gene expression is normalized to Gapdh expression and groups are compared by the ΔΔ CT method utilizing Rag mice as the baseline expression level. ST mice demonstrated significant increases (*: P<0.05) in Muc3 (B) compared to all other groups, Muc4 compared to wild-type mice, (C) and Muc5ac compared to either nude or Rag mice (D). Wild-type mice increased expression of Muc5ac to nearly significant levels (+: P=0.06) compared to Rag mice (D). Nude mice demonstrated a significant increase in Muc1 compared to ST mice (A).
Biliary phenotype of Rag mice adoptively transferred with T or B-cells
To determine whether T or B-cells alone are critical effectors in cholesterol gallstone pathogenesis, individual cell types were harvested, purified and transferred to Rag mice. Rag mice receiving T-cells (T-cell) form cholesterol monohydrate crystals significantly more frequently compared to Rag mice or mice receiving B-cells (B-cell) (P<0.05, Fig. 4A). Moreover, they demonstrate a significant increase in cholesterol gallstone prevalence rate compared to Rag mice (P<0.05, Fig. 4A). Both B and T-cell reconstituted mice increase mucin gel accumulation compared to Rag mice (P<0.05, Fig. 4B). These data indicate that although both B and T-cells induce mucin gel accumulation, only T-cells increase cholesterol gallstone prevalence.
Figure 4.

(A) Data represent percent prevalence of cholesterol monohydrate crystals, sandy stones and cholesterol gallstones (B) and mucin gel score in male Helicobacter spp. infected Rag mice receiving T-cells only (T-cell, n=15), Rag mice receiving B-cells only (B-cell, n=15), or Rag mice receiving no cells (n=36). All mice developed liquid crystals (not displayed). T-cell mice demonstrated an increased prevalence of cholesterol monohydrate crystals, sandy stones, and cholesterol gallstones when compared to Rag mice (* P<0.05) (A). Further, these mice had an increased prevalence of cholesterol monohydrate crystals when compared to B-cell mice (**: P<0.05) (A). Both T-cell and B-cell mice significantly accumulated mucin gel when compared to Rag mice (B) (*: P<0.05).
Cytokine and inflammatory gene product expression analysis
Others have described the ability of cholesterol monohydrate crystals to induce inflammation and dysfunction in otherwise healthy gallbladders 23, 24. To determine if T-cell mediated cholesterol gallstone formation could be due, at least in part, to their ability to promote local cytokine production following solid cholesterol phase separation cytokine expression was analyzed in gallbladder tissue. Mice were divided into four groups based upon lymphocyte status (T-cell positive or Rag) and biliary phenotype (formation of liquid crystals or progressing beyond the liquid crystalline stage). The following four groups were analyzed: 1. Rag mice developing cholesterol monohydrate crystals whether free or agglomerated as stones (Rag CM); 2. Rag mice developing only liquid crystals (Rag LC); 3. T-cell mice developing cholesterol monohydrate crystals (T-cell CM); 4. T-cell mice developing only liquid crystals (T-cell LC).
T-cell CM mice increase consistently expression of genes indicative of a Th1 inflammatory response (Fig. 5). In contrast, T-cell LC mice displayed a dampened Th1 response (Fig. 5). Furthermore, in the absence of T-cells (Rag mice), solid cholesterol crystals did not promote a Th1 inflammatory response (Fig. 5). Essentially, Rag CM mice responded to solid cholesterol crystals in the opposite manner to mice reconstituted with T-cells (Fig. 5). These global changes are readily demonstrated by comparing the mean expression of three well-described Th1 cytokines (Il1β, Ifn-γ. Tnf-α) with three well defined Th2 cytokines (Il4, Il10, Il13). Th1 cytokines are highly expressed in T-cell CM mice compared to all other groups (P<0.05, Fig. 5N). In contrast, common Th2 cytokines are not expressed differentially nor are they expressed to any large degree in any group (Fig. 5O). Additionally, Tgf-β, which was shown previously to be expressed during cholesterol gallstone-induced gallbladder damage and fibrosis 25 is highly expressed in T-cell CM mice (Table 1). Finally, the CD40 ligand, which is specifically expressed on T-cells is markedly upregulated in both groups of mice receiving T-cells (Table 1). These data demonstrate that these mice received functional T-cells and that a population of these T-cells migrated to the gallbladder.
Figure 5.

(A–M) Data represent the gallbladder expression of genes frequently involved in a Th1 immune response. Expression values are normalized to five control genes included in the assay and compared to Rag CM mice by the ΔΔCT method and logarithmically converted and expressed in relation to Rag CM mice (see Methods). All groups are male mice infected with Helicobacter spp. (n=5–6 gallbladders analyzed/group). (N) Common Th1 cytokines (Il1-β, Ifn- γ, and Tnf-α) and (O) Th2 cytokines (Il-4, Il-10, Il-13) were analyzed by taking the mean CT value of each gene group relative to the five control genes, logarithmically converting this value and multiplying by 104 for ease of display (see Methods). All graphs depict mean expression +/− standard error of the mean (calculated prior to logarithmic conversion). Significant changes are indicated as *:P<0.05 when compared to bracketed group, #:P<0.05 when compared to all Rag mice (Rag CM + Rag LC), Δ:P<0.05 when compared to all other groups. T-cell mice that developed cholesterol monohydrate crystals increase Th1 cytokines significantly compared to all other groups analyzed. For all groups analyzed Th1 cytokines are expressed 10 fold or greater compared to Th2 cytokines (N–O).
Table 1.
Fold changes of gallbladder inflammatory gene expression relative to Rag CM mice
| Gene NameA | Rag LC | T-cell LC | T-cell CM |
|---|---|---|---|
| Th1 genes | |||
| Casp1 | 6.3 (3.9–9.5)B | 2.3 (1.4–3.5) | 9.9 (6.7–13.8)* |
| Ccl1 | 1.3 (0.4–4.1) | 0.9 (0.4–1.9) | 32.9 (7.3–147.9)† |
| Ccl5 | 23.3 (13.8–38.9)‡ | 8.0 (4.4–14.4) | 26.4 (16.8–40.9)‡ |
| Ccr5 | 3.8 (2.5–5.7)‡ | 1.1 (0.5–2.2) | 5.2 (3.8–7.1)‡ |
| Cxcl 5 | 0.6 (0.4–0.8) | 1.0 (0.5–1.8) | 19.0 (5.5–65.7)† |
| Cxcl9 | 27.6 (8.8–86.6) | 4.4 (1.1–18.0) | 40.3 (22.3–72.8)‡ |
| Cxcl10 | 6.2 (3.5–11.1) | 1.0 (0.4–2.6) | 14.9 (7.8–28.2)‡,* |
| Cxcl11 | 29.8 (15.8–56.2)‡ | 15.4 (7.3–32.6) | 133.9 (63.9–280.7)‡ |
| Cxcr 3 | 9.6 (6.0–15.6)‡ | 2.7 (0.9–8.3) | 24.2 (8.4–69.2)‡ |
| Ifn-γ | 30.7 (15.4–57.9)‡ | 3.3 (0.8–13.5) | 125.4 (55.8–266.1)‡,* |
| Il1β | 3.1 (2.3–4.0)‡ | 1.1 (0.7–1.7) | 6.7 (4–11.3) ‡,* |
| IL8 rb | 2.7 (0.6–12.8) | 1.5 (0.4–5.1) | 43.0 (20.7–89.4)‡,*,† |
| Tnf-α | 6.7 (3.2–13.8) | 2.0 (1.1–3.7) | 31.0 (14.4–66.8) ‡,*,† |
| Th2 genes | |||
| Ccl22 | 4.3 (3.4–5.5)‡ | 0.5 (0.2–1.4) | 11.7 (4.3–31.6) |
| Ccr3 | 8.3 (5.7–12.1)‡ | 2.0 (1.1–3.7) | 7.2 (4.7–11.1)‡ |
| Il10ra | 5.2 (3.8–6.9)‡ | 2.8 (2.1–3.8) | 6.8 (4.5–10.3)‡ |
| Mixed function Genes | |||
| Ccl2 | 19.5 (12.1–31.4)‡ | 1.9 (0.6–5.4) | 16.0 (8.6–29.7)‡ |
| Ccl8 | 25.2 (16–40) | 10.2 (4.7–22.0) | 43.0 (22.1–83.7)‡ |
| Ccl9 | 7.9 (4.5–14.1)‡ | 1.3 (0.7–2.6) | 3.5 (1.8–6.9) |
| Ccr1 | 2.1 (1.5–2.8)* | 0.6 (0.4–0.8) | 2.2 (1.4–3.4) |
| Ccr2 | 11.8 (7.8–17.7)‡ | 2.5 (1.3–4.9) | 12.8 (9.2–17.9)‡,* |
| Ccr4 | 7.7 (2.0–29.0) | 15.9 (7.7–32.8) | 226.8 (85.2–603.4)‡,† |
| Cxcl 4 | 0.9 (0.7–1.0) | 0.45 (0.3–0.8) | 2.3 (1.5–3.6)* |
| Il2rg | 3.7 (2.4–5.7)‡ | 1.1 (0.6–2.1) | 5.5 (3.6–8.4)‡ |
| Il 15 | 7.9 (5.4–11.5)‡ | 3.3 (2.5–4.3) | 10.9 (4.3–27.9) |
| IL16 | 8.7 (6.7–11.1)‡ | 5.1 (3.8–6.9) | 8.7 (5.2–14.4)‡ |
| Ltb | 5.8 (4.2–8.0)‡ | 2.0 (1.2–3.2) | 5.2 (3.5–7.7)‡ |
| Tgfβ1 | 1.6 (1.3–2.0) | 0.7 (0.5–1.0) | 2.1 (1.7–2.5)* |
| Innate immune genes | |||
| Crp | 2.5 (2.3–2.8)‡,*,**,¥ | 1.5 (1.3–1.7) | 0.7 (0.5–1.1) |
| Undefined genes | |||
| Il1f6 | 2.0 (0.8–5.0) | 4.1 (2.9–5.8) | 73.4 (28.2–191.2)‡,+,† |
| Il1r2 | 4.7 (2.6–8.4) | 0.7 (0.2–1.9) | 22.1 (7.8–62.9)‡,*,† |
| IL17b | 32.4 (19.2–54.9) ‡,* | 1.3 (0.4–4.0) | 44.0 (14.6–132.3)‡ |
| T-cell expressed genes | |||
| Tnfsf5 (CD40L) | 0.52 (0.2–1.3) | 21.4 (8.3–55.0)‡,+,† | 40.8 (16.1–103.5) ‡,+,† |
Only genes demonstrating significant changes are displayed: Abcf1, Bcl6, Blr1, C3, Ccl3, Ccl4, Ccl6, Ccl7, Ccl12, Ccl17, Ccl19, Ccl20, Ccl22, Ccl24, Ccl25, Ccr6, Ccr7, Ccr8, Ccr9, Cxcl1, Cxcl12, Cxcl13, Cxcl15, Gpr2, Il1a, Il1f8, Il1r1, Il2rb, Il3, Il4, Il10, Il10ra, Il10rb, Il11, Il2rb, Il13, IL13ra1, Il5ra, Il6ra, Il6st, Il18, Il20, Itgam, Itgb2, Lta, Mif, Scye1, Spp1, Tnfrsf1a, Tnfrsf1b, Tollip, and Xcr1 did not demonstrate any significant changes between any of the groups and are not displayed.
Data represent fold changes and ranges compared to Rag mice developing cholesterol monohydrate crystals (expression of 1 for each gene). Ranges are based on plus or minus standard error of the mean prior to logarithmic conversion All groups represent male mice infected with Helicobacter spp. (n=5–6/group).
P<0.05 vs. T-cell LC mice
: P<0.05 vs. Rag mice (combined)
: P<0.05 vs. Rag CM mice
P<0.05 vs. T-cell CM mice
P<0.05 vs. Rag LC mice
P<0.05 vs. T-cell mice (combined)
Discussion
This study provides the first detailed description linking adaptive immunity to cholesterol gallstone pathogenesis. The data indicates that T-cells exert a profound effect in promoting cholesterol gallstone formation. Additionally, we demonstrated that T-cells and solid cholesterol crystals induce a potent, Th1-mediated, local inflammatory response in the gallbladder. In the absence of T-cells, solid cholesterol crystals, as inferred from the cytokine data, down-regulate inflammatory genes. This downregulation likely prevents subsequent cytokine-mediated damage and ensuing gallbladder mucosal and muscle defects.
The potential potent influence of inflammation and the immune system on cholesterol gallstone formation was first evidenced by Lee and coworkers in a diet-induced prairie dog model of cholesterol gallstones 26. When treated with high-dose acetylsalicylic acid (ASA), these rodents maintained supersaturated bile but did not nucleate solid cholesterol monohydrate crystals nor form cholesterol gallstones 26. However, ASA has a wide range of in vivo activities and the exact mechanism whereby it prevented cholesterol gallstone formation was never elucidated subsequently 27. Additional publications attempting to link the immune system to cholesterol gallstone formation have suggested the possible importance of immunoglobulins in promoting solid cholesterol crystal nucleation 10–12, 28, 29. In vitro studies indicate that pro-inflammatory products influence mucin production in cultured gallbladder epithelial cells 30, 31. Moreover, in several previous investigations, attempts were made to examine the in vivo influence of inflammation on cholesterol gallstone pathogenesis 32, 33. These investigators invariably concluded that gallbladder inflammation and immunoglobulin production occurred when mice were fed a lithogenic diet 32, 33 but both gallstone susceptible (C57L) and resistant (AKR) murine strains studied display marked differences in their ability to supersaturate hepatic and gallbladder bile 32, 33. Therefore, the confounding influence of biliary cholesterol supersaturation and phase separation could not be eliminated 32, 33. The current murine study eliminates this variable by utilizing congenic mice capable of identical biliary cholesterol supersaturation levels but differing only in their capacity for adaptive immunity.
In the present study, B-cells and immunoglobulins were not necessary to achieve the high cholesterol gallstone prevalence rates observed. Nude mice displayed a greater quantity of cholesterol monohydrate crystals and cholesterol gallstones when compared with Rag mice or mice receiving B-cells. These differences are likely due to the presence of intraepithelial T-cells in nude mice 34, 35. In contrast, Rag mice lack any functional T and B-cells and therefore are a robust model in which to study the individual effects of T and B cells on cholelithogenesis.
Mice that received B-cells only significantly accumulated gallbladder mucin gel. Therefore, it seems likely that mucin gene expression and mucin gel accumulation are not the only mechanisms whereby T-cells promote cholelithogenesis. Instead, based upon the inflammatory gene expression data we propose that T-cell-derived cytokine-mediated damage of the gallbladder wall promotes gallbladder mucosal and muscle dysfunction. Although the data did not demonstrate motility defects directly, we hypothesize that cytokine-mediated damage will produce similar contractile deficits to those induced by free radical damage of the gallbladder wall 36, 37.
A plausible mechanism for the observations noted in the current study is that mucin gel accumulation promotes cholesterol monohydrate crystal formation; however, it is the subsequent cytokine response to accumulated cholesterol monohydrate crystals that determines T-cell mediated progression. Without T-cells and Th1 inflammation, cholesterol monohydrate crystals are most likely removed via normal gallbladder emptying even in the presence of mucin gel. Our data from Rag mice that developed cholesterol monohydrate crystals illustrate that these mice down-regulate Th1 cytokines, chemokines and receptors and as a result, the cholesterol monohydrate crystals do not progress to cholesterol gallstone formation. Interestingly, mice adoptively transferred with T-cells and developing only liquid crystals, displayed less pronounced cytokine expression than Rag mice with liquid crystals. We speculate that these mice may represent a population with greater percentages of regulatory T-cells which would prevent T-effector mediated cytokine damage as is the case in animal models of inflammatory bowel disease 38.
The data in the current study parallel several important findings in the pathogenesis of Helicobacter pylori gastritis. In the absence of T-cells, H. pylori-mediated gastritis is markedly attenuated 19. However, when T-cells are transferred into the animal host, gastritis ensues and occurs via a Th1-mediated immune response 19, 20. Interestingly, others have putatively identified H. pylori in the gallbladders of patients with cholesterol gallstones and speculated that the organism may promote cholesterol gallstones 39–41. Unfortunately, most of these studies based their phylogenetic classification on limited biochemical tests and morphological criteria 39, sequencing small DNA fragments, or PCR results 39–41 all of which can inadvertently identify enterohepatic Helicobacter spp. (and other similar organisms) as H. pylori 14, 42, 43. Several critical factors argue that H. pylori is not a primary pathogen of the biliary tree. Specifically, a polymicrobial flora was found in many of the gallbladders analyzed indicating altered biliary defenses 39. Also H. pylori is inhibited in vitro 44, 45 and in vivo 15 by bile salts which would likely prevent it from colonizing a healthy gallbladder. Finally in a prospective mouse study, we established that H. pylori did not promote cholesterol gallstone formation 15. We speculate that if H. pylori infection plays any role in cholesterol gallstone formation in humans, it could do so indirectly through promotion of chronic inflammation and upregulation of systemic proinflammatory cytokines rather than the ability of the organism to colonize the hepatobiliary tree.
In one of our earlier studies13 we demonstrated that without enteroheptic Helicobacter spp. infection C57L/J mice rarely form cholesterol gallstones. In the current study, helicobacter infection is unnecessary to produce a high prevalence of cholesterol gallstones in BALB/cJ mice. Uninfected mice developed cholesterol monohydrate crystals and sandy stones to levels virtually identical to infected mice. Because, sandy stones, which are the immediate precursors of cholesterol gallstones, would rapidly become cholesterol gallstones, Helicobacter spp. infection appears to only mildly alter the kinetics of cholesterol gallstone formation in BALB/cJ mice.
We were surprised to find that BALBc/J (Jackson) and BALB/cAnNTac (Taconic) mice exhibited markedly different cholesterol gallstone prevalence rates (Fig. 1 and 2). These differences could be due to either genetic or environmental influences. Although genetic drift (the cumulative effect of spontaneous mutations over time) cannot be discounted entirely, a lack of genetic contamination of BALB/c mice was demonstrated previously and therefore seems unlikely 46. Alternatively, environmental i.e. microbial factors could play an important role. Specifically, husbandry practices differ between Taconic and Jax. Taconic rederives mice in a germ-free state and then colonizes them with altered Schaedler flora (ASF) containing eight defined gastrointestinal bacteria 47–49. These mice are then maintained within a specific pathogen free (SPF) barrier, where they may become colonized with other non-pathogenic microbes. In contrast, Jax does not rederive their mice into a germ-free state nor do they colonize the mice with ASF. Jax mice are maintained in a SPF environment and possess a spontaneously acquired, formally undefined gut microflora 48. Indeed microbial differences were confirmed in a recent analysis of Jax mice, which demonstrated that they possessed a diverse population of intestinal microbes 50. Interestingly, Jax mice were found to be free of lactobacilli which account for 2 of the 8 bacteria in ASF 48, 49. Some Lactobacilli protect against Th1-mediated disease and diminish proinflammatory cytokine production 51, 52. It is intriguing to hypothesize that these organisms may also afford protection against cholesterol gallstone formation. In support of this hypothesis, is the observation that a variety of inbred strains of Taconic mice were resistant to experimental infection with Giardia lamblia whereas isogenic Jax mice were susceptible. When these mice were co-housed, Giardia spp. resistance was transferred to the Jax mice indicating that resistance was readily transmissible 48. Further evidence for the role of indigenous microbes was demonstrated in studies using BALB/cAnNCr (from the National Cancer Institute, Belthesda, MD) mice which are genetically most closely related to BALB/cAnNTac mice (both are of the BALB/cAnN lineage). Despite their genetic similarity, BALB/cAnNCr developed cholesterol gallstones with 100% prevalence rate, whereas 27% of BALB/cAnNTac mice developed gallstones under similar dietary conditions 23. BALB/cAnNCr mice behave phenotypically more like the genetically less related BALB/cJ strain (80% gallstone prevalence).
In summary, T-cell function appears critical in the pathogenesis of murine cholesterol gallstones. We propose a biological model whereby the presence of T-cells and cholesterol monohydrate crystals elicits gallbladder inflammation, which in turn promotes tissue damage and defects in gallbladder function, as well as further production of pronucleating agents. Furthermore, we hypothesize that infectious agents and other Th1-associated inflammatory responses are likely to contribute to cholesterol gallstone formation in mice as well as in humans. For example in humans, hepatitis C virus infection and Crohn’s disease, both chronic Th1-inducing diseases, correlate strongly with increased prevalence of cholesterol gallstones 53–56. In our study we describe a hitherto unappreciated new dimension in cholesterol gallstone pathogenesis which may prove crucial in our understanding of the disease in humans. Defining the importance of the adaptive immune system in cholesterol gallstone formation may change not only the way we study cholesterol gallstone pathogenesis and the disease, but could alter the paradigms whereby the disease is treated and possibly prevented. The findings in the present study are reminiscent of other diseases in humans (ie. gastric cancer, diabetes mellitus, atherosclerosis) which were once thought to represent merely disturbances in biochemistry, biophysical-chemistry and metabolism, but can now be clearly identified as polyfactorial conditions with strong immunologic components 57–59.
Acknowledgments
The authors wish to thanks K. Clapp and J. Miyamae for technical support. This work was supported by R01-CA67529, P30-ES02109, and T32-RR07036 to JGF and R37-DK36588 and R01-DK73687 to MCC.
Footnotes
No conflict of interest exists for any of the authors
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Paigen B, Carey MC. Gallstones. In: King RA, Rotter JI, Motulsky AG, editors. Genetic Basis of Common Diseases. 2. New York: Oxford University Press, Inc; 2002. pp. 298–335. [Google Scholar]
- 2.Katsika D, Grjibovski A, Einarsson C, Lammert F, Lichtenstein P, Marschall HU. Genetic and environmental influences on symptomatic gallstone disease: a Swedish study of 43,141 twin pairs. Hepatology. 2005;41:1138–43. doi: 10.1002/hep.20654. [DOI] [PubMed] [Google Scholar]
- 3.Carey MC, LaMont JT. Cholesterol gallstone formation. 1. Physical-chemistry of bile and biliary lipid secretion. Prog Liver Dis. 1992;10:139–63. [PubMed] [Google Scholar]
- 4.Carey MC. Pathogenesis of gallstones. Recenti Prog Med. 1992;83:379–91. [PubMed] [Google Scholar]
- 5.Hofmann AF. Pathogenesis of cholesterol gallstones. J Clin Gastroenterol. 1988;10(Suppl 2):S1–11. [PubMed] [Google Scholar]
- 6.Afdhal NH, Niu N, Gantz D, Small DM, Smith BF. Bovine gallbladder mucin accelerates cholesterol monohydrate crystal growth in model bile. Gastroenterology. 1993;104:1515–23. doi: 10.1016/0016-5085(93)90364-i. [DOI] [PubMed] [Google Scholar]
- 7.Levy PF, Smith BF, LaMont JT. Human gallbladder mucin accelerates nucleation of cholesterol in artificial bile. Gastroenterology. 1984;87:270–5. [PubMed] [Google Scholar]
- 8.Smith BF. Gallbladder mucin as a pronucleating agent for cholesterol monohydrate crystals in bile. Hepatology. 1990;12:183S–186S. discussion 186S–188S. [PubMed] [Google Scholar]
- 9.Yamasaki T, Chijiiwa K, Endo M. Isolation of mucin from human hepatic bile and its induced effects on precipitation of cholesterol and calcium carbonate in vitro. Dig Dis Sci. 1993;38:909–15. doi: 10.1007/BF01295919. [DOI] [PubMed] [Google Scholar]
- 10.Harvey PR, Upadhya GA. A rapid, simple high capacity cholesterol crystal growth assay. J Lipid Res. 1995;36:2054–8. [PubMed] [Google Scholar]
- 11.Harvey PR, Upadhya GA, Strasberg SM. Immunoglobulins as nucleating proteins in the gallbladder bile of patients with cholesterol gallstones. J Biol Chem. 1991;266:13996–4003. [PubMed] [Google Scholar]
- 12.Hattori Y, Tazuma S, Yamashita G, Kajiyama G. Influence of cholesterol crystallization effector proteins on vesicle fusion in supersaturated model bile. J Gastroenterol Hepatol. 1999;14:669–74. doi: 10.1046/j.1440-1746.1999.01933.x. [DOI] [PubMed] [Google Scholar]
- 13.Maurer KJ, Ihrig MM, Rogers AB, Ng V, Bouchard G, Leonard MR, Carey MC, Fox JG. Identification of cholelithogenic enterohepatic helicobacter species and their role in murine cholesterol gallstone formation. Gastroenterology. 2005;128:1023–33. doi: 10.1053/j.gastro.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 14.Fox JG. The non-H pylori helicobacters: their expanding role in gastrointestinal and systemic diseases. Gut. 2002;50:273–83. doi: 10.1136/gut.50.2.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maurer KJ, Rogers AB, Ge Z, Wiese AJ, Carey MC, Fox JG. Helicobacter pylori and cholesterol gallstone formation in C57L/J mice: a prospective study. Am J Physiol Gastrointest Liver Physiol. 2006;290:G175–82. doi: 10.1152/ajpgi.00272.2005. [DOI] [PubMed] [Google Scholar]
- 16.Belzer C, Kusters JG, Kuipers EJ, van Vliet AH. Urease induced calcium precipitation by Helicobacter species may initiate gallstone formation. Gut. 2006;55:1678–9. doi: 10.1136/gut.2006.098319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Whary MT, Morgan TJ, Dangler CA, Gaudes KJ, Taylor NS, Fox JG. Chronic active hepatitis induced by Helicobacter hepaticus in the A/JCr mouse is associated with a Th1 cell-mediated immune response. Infect Immun. 1998;66:3142–8. doi: 10.1128/iai.66.7.3142-3148.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fox JG, Beck P, Dangler CA, Whary MT, Wang TC, Shi HN, Nagler-Anderson C. Concurrent enteric helminth infection modulates inflammation and gastric immune responses and reduces helicobacter-induced gastric atrophy. Nat Med. 2000;6:536–42. doi: 10.1038/75015. [DOI] [PubMed] [Google Scholar]
- 19.Eaton KA, Ringler SR, Danon SJ. Murine splenocytes induce severe gastritis and delayed-type hypersensitivity and suppress bacterial colonization in Helicobacter pylori-infected SCID mice. Infect Immun. 1999;67:4594–602. doi: 10.1128/iai.67.9.4594-4602.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eaton KA, Mefford M, Thevenot T. The role of T cell subsets and cytokines in the pathogenesis of Helicobacter pylori gastritis in mice. J Immunol. 2001;166:7456–61. doi: 10.4049/jimmunol.166.12.7456. [DOI] [PubMed] [Google Scholar]
- 21.Wang DQ, Paigen B, Carey MC. Phenotypic characterization of Lith genes that determine susceptibility to cholesterol cholelithiasis in inbred mice: physical-chemistry of gallbladder bile. J Lipid Res. 1997;38:1395–411. [PubMed] [Google Scholar]
- 22.Finzi L, Barbu V, Burgel PR, Mergey M, Kirkwood KS, Wick EC, Scoazec JY, Peschaud F, Paye F, Nadel JA, Housset C. MUC5AC, a gel-forming mucin accumulating in gallstone disease, is overproduced via an epidermal growth factor receptor pathway in the human gallbladder. Am J Pathol. 2006;169:2031–41. doi: 10.2353/ajpath.2006.060146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rege RV, Prystowsky JB. Inflammation and a thickened mucus layer in mice with cholesterol gallstones. J Surg Res. 1998;74:81–5. doi: 10.1006/jsre.1997.5213. [DOI] [PubMed] [Google Scholar]
- 24.Jennings LJ, Xu QW, Firth TA, Nelson MT, Mawe GM. Cholesterol inhibits spontaneous action potentials and calcium currents in guinea pig gallbladder smooth muscle. Am J Physiol. 1999;277:G1017–26. doi: 10.1152/ajpgi.1999.277.5.G1017. [DOI] [PubMed] [Google Scholar]
- 25.Koninger J, di Mola FF, Di Sebastiano P, Gardini A, Brigstock DR, Innocenti P, Buchler MW, Friess H. Transforming growth factor-beta pathway is activated in cholecystolithiasis. Langenbecks Arch Surg. 2005;390:21–8. doi: 10.1007/s00423-004-0517-4. [DOI] [PubMed] [Google Scholar]
- 26.Lee SP, Carey MC, LaMont JT. Aspirin prevention of cholesterol gallstone formation in prairie dogs. Science. 1981;211:1429–31. doi: 10.1126/science.7466399. [DOI] [PubMed] [Google Scholar]
- 27.Gilroy DW. New insights into the anti-inflammatory actions of aspirin-induction of nitric oxide through the generation of epi-lipoxins. Mem Inst Oswaldo Cruz. 2005;100(Suppl 1):49–54. doi: 10.1590/s0074-02762005000900009. [DOI] [PubMed] [Google Scholar]
- 28.Miquel JF, Nunez L, Rigotti A, Amigo L, Brandan E, Nervi F. Isolation and partial characterization of cholesterol pronucleating hydrophobic glycoproteins associated to native biliary vesicles. FEBS Lett. 1993;318:45–9. doi: 10.1016/0014-5793(93)81324-s. [DOI] [PubMed] [Google Scholar]
- 29.Upadhya GA, Harvey PR, Strasberg SM. Effect of human biliary immunoglobulins on the nucleation of cholesterol. J Biol Chem. 1993;268:5193–200. [PubMed] [Google Scholar]
- 30.Rhodes M, Allen A, Lennard TW. Mucus glycoprotein biosynthesis in the human gall bladder: inhibition by aspirin. Gut. 1992;33:1109–12. doi: 10.1136/gut.33.8.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim HJ, Lee SK, Kim MH, Seo DW, Min YI. Cyclooxygenase-2 mediates mucin secretion from epithelial cells of lipopolysaccharide-treated canine gallbladder. Dig Dis Sci. 2003;48:726–32. doi: 10.1023/a:1022832608466. [DOI] [PubMed] [Google Scholar]
- 32.van Erpecum KJ, Wang DQ, Lammert F, Paigen B, Groen AK, Carey MC. Phenotypic characterization of Lith genes that determine susceptibility to cholesterol cholelithiasis in inbred mice: soluble pronucleating proteins in gallbladder and hepatic biles. J Hepatol. 2001;35:444–51. doi: 10.1016/s0168-8278(01)00173-8. [DOI] [PubMed] [Google Scholar]
- 33.van Erpecum KJ, Wang DQ, Moschetta A, Ferri D, Svelto M, Portincasa P, Hendrickx JJ, Schipper M, Calamita G. Gallbladder histopathology during murine gallstone formation: relation to motility and concentrating function. J Lipid Res. 2006;47:32–41. doi: 10.1194/jlr.M500180-JLR200. [DOI] [PubMed] [Google Scholar]
- 34.Emoto Y, Emoto M, Miyamoto M, Yoshizawa I, Kaufmann SH. Functionally active CD8alphabeta+ TCRgammadelta intestinal intraepithelial lymphocytes in athymic nu/nu mice. Int Immunol. 2004;16:111–7. doi: 10.1093/intimm/dxh008. [DOI] [PubMed] [Google Scholar]
- 35.Guy-Grand D, Azogui O, Celli S, Darche S, Nussenzweig MC, Kourilsky P, Vassalli P. Extrathymic T cell lymphopoiesis: ontogeny and contribution to gut intraepithelial lymphocytes in athymic and euthymic mice. J Exp Med. 2003;197:333–41. doi: 10.1084/jem.20021639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xiao ZL, Andrada MJ, Biancani P, Behar J. Reactive oxygen species (H(2)O(2)): effects on the gallbladder muscle of guinea pigs. Am J Physiol Gastrointest Liver Physiol. 2002;282:G300–6. doi: 10.1152/ajpgi.00241.2001. [DOI] [PubMed] [Google Scholar]
- 37.Xiao ZL, Rho AK, Biancani P, Behar J. Effects of bile acids on the muscle functions of guinea pig gallbladder. Am J Physiol Gastrointest Liver Physiol. 2002;283:G87–94. doi: 10.1152/ajpgi.00536.2001. [DOI] [PubMed] [Google Scholar]
- 38.Izcue A, Coombes JL, Powrie F. Regulatory T cells suppress systemic and mucosal immune activation to control intestinal inflammation. Immunol Rev. 2006;212:256–71. doi: 10.1111/j.0105-2896.2006.00423.x. [DOI] [PubMed] [Google Scholar]
- 39.Abayli B, Colakoglu S, Serin M, Erdogan S, Isiksal YF, Tuncer I, Koksal F, Demiryurek H. Helicobacter pylori in the etiology of cholesterol gallstones. J Clin Gastroenterol. 2005;39:134–7. [PubMed] [Google Scholar]
- 40.Bulajic M, Maisonneuve P, Schneider-Brachert W, Muller P, Reischl U, Stimec B, Lehn N, Lowenfels AB, Lohr M. Helicobacter pylori and the risk of benign and malignant biliary tract disease. Cancer. 2002;95:1946–53. doi: 10.1002/cncr.10893. [DOI] [PubMed] [Google Scholar]
- 41.Silva CP, Pereira-Lima JC, Oliveira AG, Guerra JB, Marques DL, Sarmanho L, Cabral MM, Queiroz DM. Association of the presence of Helicobacter in gallbladder tissue with cholelithiasis and cholecystitis. J Clin Microbiol. 2003;41:5615–8. doi: 10.1128/JCM.41.12.5615-5618.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fox JG. Other helicobacters involved in human diseases. Acta Gastroenterol Belg. 2002;65:24–32. [PubMed] [Google Scholar]
- 43.O’Rourke JL, Solnick JV, Neilan BA, Seidel K, Hayter R, Hansen LM, Lee A. Description of ‘Candidatus Helicobacter heilmannii’ based on DNA sequence analysis of 16S rRNA and urease genes. Int J Syst Evol Microbiol. 2004;54:2203–11. doi: 10.1099/ijs.0.63117-0. [DOI] [PubMed] [Google Scholar]
- 44.Mathai E, Arora A, Cafferkey M, Keane CT, O’Morain C. The effect of bile acids on the growth and adherence of Helicobacter pylori. Aliment Pharmacol Ther. 1991;5:653–8. doi: 10.1111/j.1365-2036.1991.tb00533.x. [DOI] [PubMed] [Google Scholar]
- 45.Hanninen ML. Sensitivity of Helicobacter pylori to different bile salts. Eur J Clin Microbiol Infect Dis. 1991;10:515–8. doi: 10.1007/BF01963941. [DOI] [PubMed] [Google Scholar]
- 46.Hilgers J, van Nie R, Ivanyi D, Hilkens J, Michalides R, de Moes J, Poort-Keesom R, Kroezen V, von Deimling O, Kominami R, et al. Genetic differences in BALB/c sublines. Curr Top Microbiol Immunol. 1985;122:19–30. doi: 10.1007/978-3-642-70740-7_3. [DOI] [PubMed] [Google Scholar]
- 47.Dewhirst FE, Chien CC, Paster BJ, Ericson RL, Orcutt RP, Schauer DB, Fox JG. Phylogeny of the defined murine microbiota: altered Schaedler flora. Appl Environ Microbiol. 1999;65:3287–92. doi: 10.1128/aem.65.8.3287-3292.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Singer SM, Nash TE. The role of normal flora in Giardia lamblia infections in mice. J Infect Dis. 2000;181:1510–2. doi: 10.1086/315409. [DOI] [PubMed] [Google Scholar]
- 49.Sarma-Rupavtarm RB, Ge Z, Schauer DB, Fox JG, Polz MF. Spatial distribution and stability of the eight microbial species of the altered schaedler flora in the mouse gastrointestinal tract. Appl Environ Microbiol. 2004;70:2791–800. doi: 10.1128/AEM.70.5.2791-2800.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kuehl CJ, Wood HD, Marsh TL, Schmidt TM, Young VB. Colonization of the cecal mucosa by Helicobacter hepaticus impacts the diversity of the indigenous microbiota. Infect Immun. 2005;73:6952–61. doi: 10.1128/IAI.73.10.6852-6961.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pena JA, Rogers AB, Ge Z, Ng V, Li SY, Fox JG, Versalovic J. Probiotic Lactobacillus spp. diminish Helicobacter hepaticus-induced inflammatory bowel disease in interleukin-10-deficient mice. Infect Immun. 2005;73:912–20. doi: 10.1128/IAI.73.2.912-920.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pena JA, Versalovic J. Lactobacillus rhamnosus GG decreases TNF-alpha production in lipopolysaccharide-activated murine macrophages by a contact-independent mechanism. Cell Microbiol. 2003;5:277–85. doi: 10.1046/j.1462-5822.2003.t01-1-00275.x. [DOI] [PubMed] [Google Scholar]
- 53.Bini EJ, McGready J. Prevalence of gallbladder disease among persons with hepatitis C virus infection in the United States. Hepatology. 2005;41:1029–36. doi: 10.1002/hep.20647. [DOI] [PubMed] [Google Scholar]
- 54.Chang TS, Lo SK, Shyr HY, Fang JT, Lee WC, Tai DI, Sheen IS, Lin DY, Chu CM, Liaw YF. Hepatitis C virus infection facilitates gallstone formation. J Gastroenterol Hepatol. 2005;20:1416–21. doi: 10.1111/j.1440-1746.2005.03915.x. [DOI] [PubMed] [Google Scholar]
- 55.Fraquelli M, Losco A, Visentin S, Cesana BM, Pometta R, Colli A, Conte D. Gallstone disease and related risk factors in patients with Crohn’s disease: analysis of 330 consecutive cases. Arch Intern Med. 2001;161:2201–4. doi: 10.1001/archinte.161.18.2201. [DOI] [PubMed] [Google Scholar]
- 56.Hutchinson R, Tyrrell PN, Kumar D, Dunn JA, Li JK, Allan RN. Pathogenesis of gall stones in Crohn’s disease: an alternative explanation. Gut. 1994;35:94–7. doi: 10.1136/gut.35.1.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fox JG, Wang TC. Inflammation, atrophy, and gastric cancer. J Clin Invest. 2007;117:60–9. doi: 10.1172/JCI30111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Reiss AB, Glass AD. Atherosclerosis: immune and inflammatory aspects. J Investig Med. 2006;54:123–31. doi: 10.2310/6650.2006.05051. [DOI] [PubMed] [Google Scholar]
- 59.Zozulinska D, Wierusz-Wysocka B. Type 2 diabetes mellitus as inflammatory disease. Diabetes Res Clin Pract. 2006 [Google Scholar]