

Abstract
Troponin I isoforms play a key role in determining myofilament Ca2+ sensitivity in cardiac muscle. The goal here was to identify domain clusters and residues that confer troponin I isoform-specific myofilament Ca2+ and pH sensitivities of contraction. Key domains/residues that contribute to troponin I isoform-specific Ca2+ and pH sensitivity were studied using gene transfer of a slow skeletal troponin I (ssTnI) template, with targeted cardiac troponin I (cTnI) residue substitutions. Substitutions in ssTnI with cognate cTnI residues R125Q, H132A, and V134E, studied both independently and together (ssTnIQAE), resulted in efficient stoichiometric replacement of endogenous myofilament cTnI in adult cardiac myocytes. In permeabilized myocytes, the pCa50 of tension ([Ca2+] required for half maximal force), and the acidosis-induced rightward shiftof pCa50 were converted to the cTnI phenotype in myocytes expressing ssTnIQAE or ssTnIH132A, and there was no functionally additive effect of ssTnIQAE versus ssTnIH132A. Interestingly, only the acidosis-induced shift in Ca2+ sensitivity was comparable to cTnI in myocytes expressing ssTnIV134E, while ssTnIR125Q fully retained the ssTnI phenotype. An additional ssTnIN141H substitution, which lies within the same structural region of TnI as V134, produced a shift in myofilament Ca2+ sensitivity comparable to cTnI at physiological pH, while the acidic pH response was similar to the effect of wild-type ssTnI. Analysis of sarcomere shortening in intact adult cardiac myocytes was consistent with the force measurements. Targeted substitutions in the carboxyl portion of TnI produced residue-specific influences on myofilament Ca2+ and pH sensitivity of force and give new molecular insights into the TnI isoform-dependence of myofilament function.
Keywords: troponin I, heart, myocyte, Ca2+ sensitivity, contractility
INTRODUCTION
Regulation of myofilament force production by intracellular Ca2+ is critical for rhythmic pressure generation in the heart. The sarcomeric troponin complex is essential for transducing the intracellular Ca2+ signal into force production. Within the heterotrimeric troponin complex, troponin I (TnI) acts as a molecular switch by toggling from actin to the Ca2+ binding troponin C (TnC) subunit as intracellular Ca2+ levels increase. This switch is a key step needed for thin filament activation and strong cross-bridge attachment in the intact myofilament (1). Recent crystal structure and NMR analyses of the troponin (Tn) complex provide a framework for understanding the location of major structural domains within the Tn complex (2;3). However, the function of these domains within the Tn complex remains less clear, especially in the context of the contracting myocyte.
Troponin I isoform expression is a critical determinant of myofilament Ca2+ and pH sensitivity of force in cardiac muscle. The slow skeletal TnI (ssTnI) isoform is expressed during fetal cardiac development (4) and an irreversible perinatal transition to the adult cardiac isoform (cTnI) develops in most rodents (5–7). This transition also occurs in large mammals, but takes place earlier during fetal development (8–10), and during the first year after birth in human hearts (11;12). Gene transfer studies show that expression and thin filament incorporation of the fetal ssTnI isoform confers increased Ca2+ sensitivity and reduced sensitivity to acidic pH compared to the adult cTnI isoform in permeabilized myocytes (13). Work with TnI chimeras derived from the ssTnI and cTnI isoforms (Fig. 1A) demonstrate that the carboxyl half of these TnI isoforms contributes significantly to both myofilament Ca2+ and pH sensitivity of force generation (14–16). The TnI isoform-specific differences in Ca2+ sensitivity and resistance to acidosis play an important role in determining myocardial systolic and diastolic performance.
FIGURE 1.

A. Illustration of amino acid substitutions within cardiac (blue) and slow skeletal troponin I (red). B. The core domain helices (2) are identified below the linear diagram of the troponin I isoforms, and a model of the core domain is shown below the linear diagram for each troponin I to demonstrate the location of these mutations within the defined 3-dimensional structure of TnI. The R125Q mutation lies within the H3 helical domain, while the H132A (cTnIA164H), V134E, and N141H in ssTnI are located in the H4 helix defined by Takeda et al. (2). The ssTnIQAE mutant contains all 3 of these substitutions, and ssTnIR92T, K95A, and K98T were used to construct additional substitution mutants for ssTnIQAETAT. The last 3 substitutions are all located within the H2 helical domain. The cTnIT144P and ssTnIP112T substitution mutants are located within the inhibitory peptide (IP) region of TnI.
The strategy in the present study was to determine whether specific amino acid clusters and/or single residues in the carboxyl portion of TnI contribute to the isoform-specific effects on myofilament Ca2+ and pH sensitivity of tension, and significantly influence relaxation performance at the single myocyte level. Based on previous results with TnI isoform chimeras (14;15), experiments here primarily focused on amino acid differences located in the carboxyl portion of TnI, and residing in the inhibitory peptide (IP), as well as the H3 and H4 amphiphilic α-helices described in cardiac and skeletal muscle structural studies (Fig. 1A,B) (2;17). Substitutions in the IP region, H3 and H4 helices were considered particularly important because they contain the primary and secondary actin and TnC binding domains (18) and the pH sensitivity region previously defined in functional studies (19). An additional group of substitutions in the H2 helix, which is located within the amino half of TnI were also examined. We postulated that charge and/or histidine residue differences in these regions contribute to isoform-specific influences of troponin I on contractile function. Cardiac TnI residues were substituted into the analogous residue in ssTnI using the ssTnI backbone as a template, and expressed in adult rat cardiac myocytes using viral-mediated gene transfer. This approach results in the stoichiometric replacement of the majority of endogenous cTnI with substitution mutants (13;20). The ssTnI backbone was used for ease of detecting the modified TnI, and to determine if modified ssTnI retains the gain in function observed with ssTnI (13) or functions more like cTnI in adult cardiac myocytes. Results demonstrate that specific charge and histidine differences contribute to isoform-specific functional differences between cTnI and ssTnI. These amino acid substitutions result in a complete or partial influence on TnI isoform function, thus defining a new high resolution functional map of TnI residues critical for modifying the molecular switch function of troponin.
METHODS
Mutagenesis strategy
Wild-type rat slow skeletal TnI (ssTnI) and cardiac TnI (cTnI) cDNA were used as described previously (13–15). Figure 1A shows the location of each residue mutated to the cognate residue found in the other isoform (cTnI or ssTnI). For ssTnI, mutants included ssTnIR125Q/H132A/V134E (ssTnIQAE), ssTnIR92T/K95A/K98T/R125Q/H132A/V134E (ssTnIQAETAT), ssTnIR125Q, ssTnIH132A, ssTnIV134E, ssTnIP112T, and ssTnIN141H. The cTnI mutant constructs under investigation included cTnIFLAG, and cTnIT144PFLAG. The FLAG-tag epitope was added to cTnI constructs to distinguish between expression of endogenous and mutant cTnI. These constructs plus the ssTnIP112T were prepared as described earlier (21;22). Primers for construction of mutants and details of construct preparation are described in the on-line supplement.
Constructs were made using a pGEM-3Z vector and the Stratagene QuikChange site-directed mutagenesis kit (15). Primers were then extended using Pfu DNA polymerase. Methylated parental DNA was subsequently digested with Dpn I and DNA containing each mutation was transformed into competent bacterial cells. The presence of each mutant was verified by DNA sequencing of the entire coding sequence.
Generation of adenoviral vectors
Recombinant adenovirus (Ad) vectors were constructed by cotransfection of pCA4TnI cDNA with pJM17 by calcium phosphate into a HEK 293 cell line. Replication deficient, recombinant adenovirus containing wild-type rat cTnI, wild-type ssTnI, or mutant ssTnI cDNAs, a CMV promoter, and SV 40 poly-adenylation signal were obtained following homologous recombination (23).
Myocyte isolation
Adult rat cardiac myocytes were isolated as described earlier(13;22). Myocytes were plated on laminin coated coverslips in DMEM supplemented with 50 U/ml penicillin, 50 μg/ml streptomycin (pen/strep), and 5% serum for 2 hr prior to replacing the media with serum free DMEM containing recombinant adenovirus vector for 1 hr, with regular replacement of media over 6 days in culture. Controls in all subsequent experiments are defined as non-adenovirus treated myocytes.
Analysis of protein expression
Control and viral-treated adult rat cardiac myocytes were maintained in culture for 3–6 days and then scraped from coverslips into sample buffer(13). Coverslips were briefly permeabilized in 0.1% triton X-100 prior to collection in sample buffer for experiments comparing expression in permeabilized versus intact myocytes. Proteins are then separated by gel electrophoresis and transblotted onto PVDF membrane for 2000 V-h with immunodetection carried out as described earlier(13) using a 1:1000 dilution of MAB 1691 (Chemicon Inc., Temecula, CA), a monoclonal antibody recognizing all TnI isoforms.
Indirect immunodetection of myofilament incorporation of mutant troponin I
Dual monoclonal antibodies were used to determine whether the substitution mutant TnI proteins replaced endogenous cTnI within the myofilaments of single cardiac myocytes. These studies were carried out 5–6 days after gene transfer, when there is an absence of cTnI-specific immunostaining in the majority of myocytes, and non-isoform-specific TnI immunostaining then detects ssTnI incorporation. To detect ssTnI expression, the primary monoclonal antibodies (mAbs) used were MAB1691 and the cTnI-specific TI-1 mAb, which does not recognize ssTnI (4;13). MAB1691 binding was detected with fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse Ab and TI-1 was detected with a Texas-Red (TR)-conjugated secondary goat anti-mouse Ab. High resolution images were obtained with a Noran OZ laser scanning confocal microscope. Projection images of z-line scans were assembled for image analysis.
Measurement of isometric force and myofilament Ca2+ sensitivity in permeabilized myocytes
Complete descriptions of the experimental chamber, attachment procedure, solutions, cell permeabilization, and tension measurement on mounted single cardiac myocytes have been reported earlier (24;25). Briefly, the concentration of each metal, ligand, and metal-ligand complex in the relaxing and activating solutions was calculated using previously published stability constants(26). Relaxing and activating solutions used for experiments contained 7 mM EGTA, 20 mM imidazole, 1 mM free Mg2+, 14.5 mM creatine phosphate, and 4 mM MgATP, with sufficient KCl to yield a total ionic strength of 180 mM. Solution pH was adjusted to 7.00 or 6.20 with KOH/HCl. The relaxing solution had a pCa (−log [Ca2+]) of 9.0, and maximal activation was achieved with a pCa of 4.0.
Prior to force measurements, cardiac myocytes attached to a force transducer (403A: Aurora Scientific, Canada) and a high performance moving coil galvanometer (Aurora Scientific) at a sarcomere length set to 2.1 μm were briefly permeabilized with 0.2% Triton X-100. The experimental temperature was set at 15°C to allow comparison with earlier work (13–15) and to optimize viability and minimize sarcomere length nonuniformity.
Force measurements are made over a range of pCa values. At each pCa, the preparation was rapidly (<0.5 ms) slackened after peak steady isometric tension developed to obtain the baseline and then immersed in relaxing solution. Total tension is the difference between peak steady tension measurement and baseline tension after the slack step. Active tension was obtained by subtracting resting tension in relaxing solution at pCa 9.0 from total tension at each pCa. Submaximal activations were bracketed by maximal activations at pCa4.0. Curve fits were performed using the Marquardt-Levenberg nonlinear least squares fitting algorithm for the Hill equation: P= [Ca2+]nH/(KnH+[Ca2+]nH), where P is tension as a fraction of maximum tension. The midpoint of the tension-pCa relationship, termed the pCa50 (Ca2+ required for half-maximal activation) and Hill coefficient (nH) are measures of myofilament Ca2+ sensitivity and cooperativity, respectively.
Myocyte stimulation and sarcomere shortening measurements in single cardiac myocytes
Myocytes used for shortening assays are transferred to a plexiglas stimulation chamber consisting of 8 wells containing platinum electrodes mounted to the sides of each well and a glass bottom (22;27). Myocytes are electrically stimulated (2.5 ms pulse, 0.2 Hz) in media 199 supplemented with pen/strep (M199), 10 mM HEPES, 0.2 mg/ml bovine serum albumin, and 10 mM glutathione (M199+), and media is replaced every 12 hrs (28). The voltage is set so that ~20% of myocytes are stimulated on each coverslip. Four days after gene transfer, individual coverslips are transferred to a temperature-controlled chamber mounted on a Nikon microscope stage, and the chamber is filled with M199+. A video-based detection system (Ionoptix, Milton, MA) is used to detect sarcomere shortening in intact myocytes. The cell chamber temperature is maintained at 37°C. Experiments are recorded using SarcLen software, and an average of 10 twitches per myocyte is collected under basal conditions at 0.2 Hz. Signal averaged data is analyzed to determine resting sarcomere length (μm), sarcomere length shortening (μm), time to peak shortening (TTP), time to 25, 50, and 75% relaxation (TTR50, and TTR75, respectively), and maximum normalized shortening and relaxation velocities (+dl/dtmax, −dl/dtmax, respectively).
Statistical Analysis
Values are expressed as mean±SEM. Grouped comparisons are analyzed using a Student’s t-test or a one-way analysis of variance. Statistical differences detected by the 1-way ANOVA were compared with a Newman-Keuls or Turkey’s multiple comparison test with p<0.05 considered significantly different.
RESULTS
Our experimental strategy targeted both single and clustered amino acid substitutions in TnI with the goal of defining a functional map of TnI isoform-dependent myofilament Ca2+ and pH sensitivities of tension. We hypothesized that charge differences and/or histidine substitutions within the secondary actin/TnC binding regions in the carboxyl portion of TnI (Fig. 1) provide an isoform-specific influence on Ca2+ and/or pH sensitivity. Myofilament replacement of endogenous cTnI with each modified ssTnI was examined by Western blot analysis and immunohistochemistry, along with parallel functional studies of Ca2+-dependent isometric force production and sarcomere shortening in electrically paced single myocytes.
The first studies focused on charge differences between ssTnI and cTnI in the H2, H3 and H4 α-helical domains defined for cTnI (2) and skeletal TnI (17). Isoform differences of interest included a R125Q charge difference within the H3 domain, a H132A substitution in the H4 region, and a V134E in ssTnI. Additional charge substitutions were located in the upstream H2 helix and included ssTnI R92T, K95A and K98T. These differences were used to construct ssTnIQAE and ssTnIQAETAT (Fig. 1), which are named for the underlined residue substitutions described above. A reduced net negative charge was predicted to make ssTnI substitution mutants function more like cTnI in the myofilaments of adult cardiac myocytes.
Expression studies
Expression of ssTnIQAE, ssTnIQAETAT, and the individual substitutions, ssTnIR125Q, ssTnIH132A, and ssTnIV134E each resulted in high level (>80%) stoichiometric replacement with endogenous cTnI (Fig. 2A,C, Table 1). There were no differences in expression between intact versus permeabilized myocytes (Fig. 2B,C), and there were no significant changes in thin filament stoichiometry for any of the substitutions (Table 2). The striated pattern of immunostaining verified myofilament incorporation of each substitution mutant (Fig. 3).
FIGURE 2.

Expression and myofilament incorporation of ssTnI and the substitutions constructed within ssTnI. A. Representative Western blot analysis of cardiac (cTnI) and slow skeletal TnI (ssTnI) expression in myocytes 6 days after gene transfer of wildtype ssTnI, ssTnIR125Q, ssTnIH132A, ssTnIV134E, or ssTnIQAE and in non-infected controls (control). Expression of the thin filament proteins tropomyosin (Tm) and troponin T (TnT), and a silver stained (Ag stain) portioned of the gel are also included here. Composite results for relative expression of TnI, TnT and Tm are shown in Table 2. B. Representative expression before (intact; I) and after (permeabilized; P) detergent permeabilization of non-infected control myocytes and myocytes expressing ssTnI substitution mutants shown in A. These results demonstrate TnI expressed after gene transfer is incorporated into the myofilament and does not accumulate in the cytosol. Composite results for expression of substitution mutants in intact and permeabilized myocytes are shown in Table 1. C. Representative Western blot analyses of ssTnIQAETAT expression in intact (I) and detergent-permeabilized (P) myocytes 6 days after gene transfer. Labels shown below the figure indicate source of muscle (soleus), control myocytes or myocytes after gene transfer with adenoviral vector (AdssTnIQAETAT).
Table 1.
Comparison of expression in intact and detergent permeabilized myocytes. Expression in intact versus permeabilized myocyte preparations were compared by an unpaired Student’s t-test with p<0.05 (*) considered significantly different.
| ssTnI/Total TnI(Intact myocytes) | n | ssTnI/Total TnI(Permeabilized myocytes | n | |
|---|---|---|---|---|
| cTnI | 0.0 | 7 | 0.0 | 7 |
| ssTnI | 88.3±4.1 | 12 | 80.2±5.7 | 6 |
| ssTnIR125Q | 85.8±3.2 | 17 | 81.4±3.5 | 5 |
| ssTnIH132A | 80.0±4.1 | 9 | 74.3±2.9 | 4 |
| ssTnIV134E | 82.0±3.6 | 9 | 83.0±1.9 | 3 |
| ssTnIQAE | 91.3±2.2 | 14 | 85.4±3.1 | 6 |
| ssTnIQAETAT | 91.6±2.9 | 7 | 89.4±6.3 | 4 |
| ssTnIN141H | 93.9±4.4 | 5 | 92.5±4.6 | 4 |
Table 2.
Expression of TnI, TnT and Tm relative to a silver stained portion of the gel for each ssTnI construct expressed in adult myocytes 6 days after gene transfer. Expression was set to 1.0 for all non-infected control myocytes, and then expression in myocytes after gene transfer was normalized to the values in the non-infected control myocytes. There were no significant differences (p>0.05) in expression compared to control using a 1-way ANOVA and post-hoc Dunnett’s multiple comparison test.
| Control | TnI/Ag | n | TnT/Ag | n | Tm/Ag | n |
|---|---|---|---|---|---|---|
| 1 | 16 | 1 | 15 | 1 | 16 | |
| cTnI | 1.13±0.18 | 6 | 1.38±0.27 | 5 | 0.95±0.11 | 6 |
| ssTnI | 0.91±0.13 | 11 | 1.29±0.22 | 9 | 1.00±0.11 | 11 |
| ssTnIR125Q | 1.34±0.28 | 16 | 1.00±0.10 | 14 | 0.90±0.06 | 16 |
| ssTnIH132A | 1.20±0.11 | 7 | 1.12±0.09 | 6 | 0.97±0.05 | 7 |
| ssTnIV134E | 1.27±0.13 | 7 | 1.05±0.09 | 6 | 0.94±0.11 | 7 |
| ssTnIQAE | 1.29±0.21 | 13 | 1.18±0.11 | 12 | 1.05±0.11 | 13 |
| ssTnIQAETAT | 1.56±0.27 | 7 | 1.01±0.13 | 6 | 0.99±0.08 | 7 |
| ssTnIN141H | 1.24±0.49 | 5 | 1.11±0.23 | 5 | 0.80±0.15 | 5 |
Figure 3.
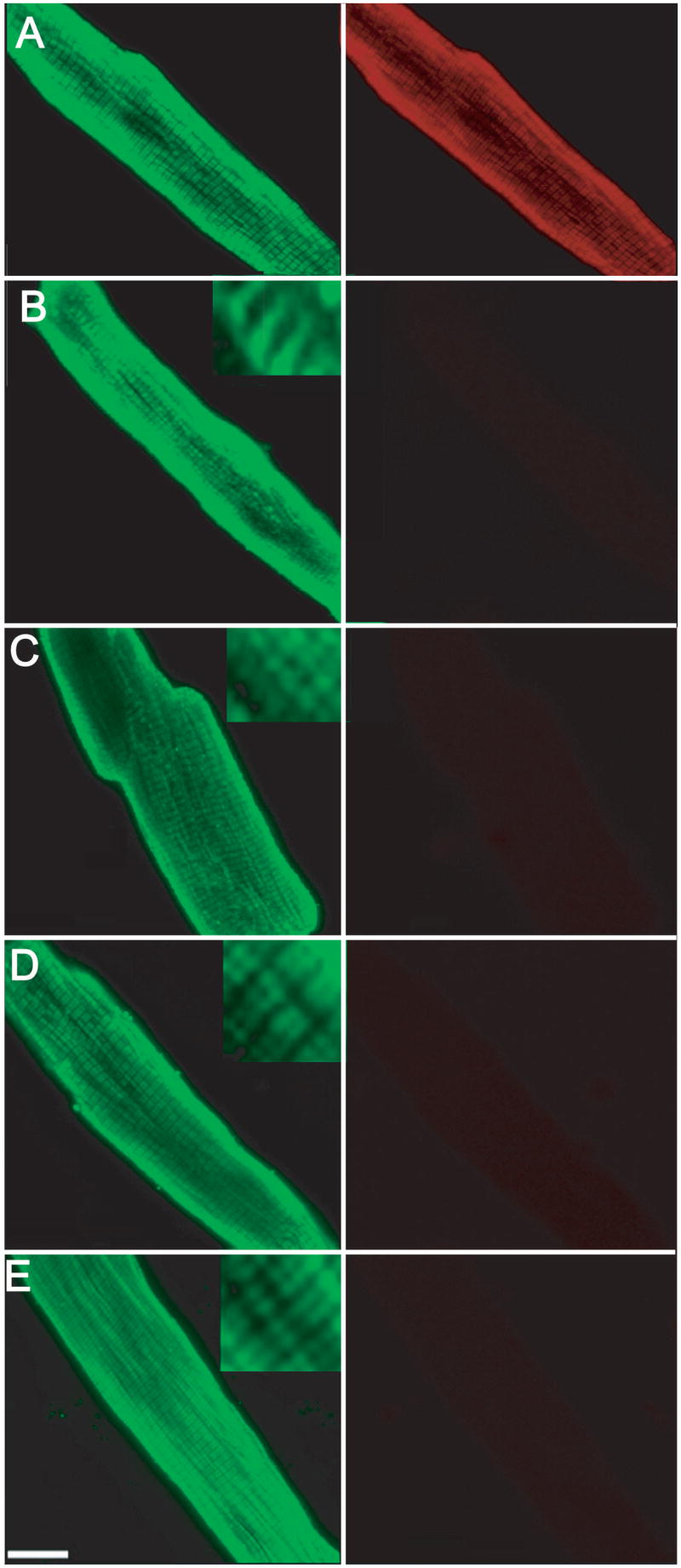
Representative immunofluorescence projection images of myocytes expressing ssTnI substitution mutants 6 days after gene transfer. Myocytes were immunostained with MAB1691 (left panel), which recognizes all troponin I isoforms and a FITC-conjugated secondary antibody, while cTnI-specific staining was detected with TI-1 antibody (right panel) and a Texas-Red-conjugated secondary antibody. Immunostaining of cTnI (TI-1 Ab) disappears in myocytes expressing ssTnI and ssTnI substitution mutants (right panels of B–E). The striated pattern of TnI staining using the non-isoform-specific MAB1691 indicates the ssTnI mutants are incorporated into the myofilaments. A. Control myocytes, B. ssTnIR125Q, C. ssTnIH132A, D. ssTnIV134E, and E ssTnIQAE. A similar pattern of staining was obtained in myocytes expressing ssTnIQAETAT (results not shown).
Isometric force studies with ssTnIH132A
We recently demonstrated that substitution of rat ssTnIH132 into cTnIA164 to form cTnIA164H produced a gain-of-function for myofilament Ca2+ and pH sensitivity of force after gene transfer into rat myocytes and in transgenic mice (20). This substitution is located within the linker between H3 and H4 in structural studies (2). The cTnIA164H substitution proved to be protective against acidosis- and ischemia-related systolic and diastolic dysfunction in the transgenic mouse hearts. A starting place for the current studies was to determine whether the converse ssTnI substitution, ssTnIH132A, produced a phenotype analogous to cTnI. As predicted, a rightward shift in myofilament Ca2+ sensitivity at both pH 7.0 and 6.2 that was comparable to cTnI was observed in myocytes expressing ssTnIH132A (Fig. 4).
FIGURE 4.

Results showing pCa50 values (Ca2+ required for half maximal activation of tension) at pH 7.0 (left panel), 6.20 (middle panel), and the delta shift in pCa50 between pH 7.0 and 6.20 (right panel) in detergent permeabilized control myocytes (n=8), and myocytes expressing ssTnI (n=8), and ssTnIH132A (n=6) 5–6 days after gene transfer. Values are expressed as mean±SEM and the statistical comparisons shown in each panel demonstrate that the Ca2+ sensitivity at each pH was significantly different in myocytes expressing ssTnI versus controls (*<0.05 vs control by ANOVA). In myocytes expressing ssTnIH132A, the Ca2+ sensitivity at each pH also was significantly changed compared to ssTnI (p<0.001 vs ssTnI by ANOVA) and was comparable to results obtained in myocytes expressing cTnI.
Function of clustered ssTnI substitutions
Efforts then focused on the potential influence of other residues that lie within proximity of H132 to modify or add to the effects produced by ssTnIH132A. In functional studies on myocytes expressing ssTnIQAE and ssTnIQAETAT, the Ca2+ sensitivity determined from the pCa50 of tension at pH 7.0 and 6.2 showed conversion to the cTnI phenotype, and a significant shift in function compared to wildtype ssTnI (Fig. 5). The desensitization to Ca2+ when pH was decreased from 7.0 to 6.2 (ΔpCa50) also was significantly greater in myocytes expressing ssTnIQAE or ssTnIQAETAT than in ssTnI-expressing myocytes (Fig. 5C, right panel). Maximal force and the Hill coefficient in myocytes expressing these substitution clusters were not different from control or wildtype ssTnI-expressing myocytes at pH 7.0 and 6.2, respectively (Table 3). These results are direct evidence that the residues located within the H3 and H4 helices, namely the ssTnI R125, H132, and V134 contribute substantially to isoform-specific differences in myofilament Ca2+ and pH sensitivity of tension. The R92, K95, and K98 residues located within the α-helical H2, TnT binding domain (ssTnI residues 58–103) did not appear to further influence myofilament Ca2+ and pH sensitivity to a significant extent (Fig. 5).
FIGURE 5.

Representative normalized tension/pCa relationship at pH 7.0 (closed circles) and 6.2 (open circles) in control myocytes (A) and in myocytes expressing ssTnIQAE (B), and composite results for the pCa50 of isometric tension (C) at pH 7.0 (left panel), 6.20 (middle panel), and the delta shift in pCa50 between pH 7.0 and 6.20 (right panel) in detergent permeabilized myocytes expressing cTnI (n=7), ssTnI (n=8), ssTnIQAE (n=6), and ssTnIQAETAT (n=4) 5–6 days after gene transfer. These results demonstrate that the pCa50s in myocytes expressing the ssTnIQAE and ssTnIQAETAT substitution mutants were comparable to myocytes expressing cTnI, and were significantly less sensitive to Ca2+ and acidic pH than myocytes expressing ssTnI. Values are expressed as mean ± SEM and results were compared by a 1-way ANOVA and Tukey’s multiple comparison test, with *p<0.05 versus control and **p<0.05 for substitution mutants versus ssTnI.
Table 3.
Hill coefficient (nH) and maximum force at pH 7.0 (Po, kN/m2) and 6.2 (P6.2/Po) for myocytes expressing cTnI, ssTnI and substitution mutants. Results were compared by a 1-way analysis of variance with p<0.05 considered significantly different. There were no significant differences between groups with this statistical analysis.
| Substitution | nH7.0 | Po7.0 | P6.2/Po | nH6.2 |
|---|---|---|---|---|
| Control | 2.26±0.36 (8) | 14.2±1.8 (8) | 0.52±0.05 (8) | 2.41±0.18 (8) |
| CTnI | 1.75±0.18 (7) | 8.1±1.1 (7) | 0.59±0.04 (7) | 1.84±0.32 (7) |
| ssTnIQAE | 1.97±0.38 (6) | 11.8±2.7 (6) | 0.51±0.05 (6) | 3.08±0.96 (6) |
| ssTnIQAETAT | 2.51±0.71 (4) | 15.3±3.7 (4) | 0.60±0.09 (4) | 2.97±0.31 (4) |
| ssTnIR125Q | 1.67±0.12 (5) | 14.5±1.7 (5) | 0.53±0.04 (5) | 3.59±1.51 (5) |
| ssTnIH132A | 2.14±0.33 (6) | 19.9±3.4 (6) | 0.54±0.04 (6) | 2.58±0.20 (6) |
| ssTnIV134E | 1.59±0.10 (6) | 14.4±1.9 (6) | 0.63±0.04 (6) | 2.29±0.16 (6) |
| ssTnIN141H | 1.50±0.10 (5) | 17.4±2.7 (5) | 0.58±0.01 (5) | 1.37±0.16 (5) |
| SsTnI | 1.43±0.14 (8) | 14.2±4.5 (8) | 0.58±0.05 (8) | 4.20±1.16 (8) |
Additional single charge substitutions in troponin I
The next set of studies focused on single substitutions within ssTnI to further map functionally important amino acid residues in the TnI structure. The ssTnIR125Q substitution is located within the H3 helical region and regulatory head domain defined earlier in structural studies of cTnI(2;17) (Fig. 1). Van der Waals contacts are predicted between this region of TnI and the hydrophobic cleft of the amino-terminal TnC regulatory Ca2+ binding domain in the presence of Ca2+. Thus, this H3 region is expected to act as a molecular switch that binds to TnC in response to Ca2+, and may independently influence TnI isoform function. Representative tension-pCa curves demonstrated that ssTnIR125Q retained the ability of ssTnI to increase pCa50 as compared to cTnI-expressing myocytes at both pH 7.0 and 6.2 (Fig. 6A,B), and the pH-induced rightward shift remained comparable to ssTnI-expressing myocytes (Fig. 6C, right panel). Maximal isometric tension (Po), ratio of maximal tension at pH 6.20/Po (P/Po), and Hill coefficient (Hill n) also were not significantly different in myocytes expressing ssTnIR125Q versus ssTnI at pH 7.0 or 6.20 (Table 3). Thus, the absence of functional alterations in ssTnIR125Q versus ssTnI, indicates that charge differences per se do not contribute to isoform-specific differences in myofilament Ca2+ sensitivity, and instead depend on specific residues within TnI.
FIGURE 6.

Representative normalized tension/pCa relationship at pH 7.0 (closed circles) and 6.2 (open circles) in myocytes expressing ssTnI (A) and ssTnIR125Q (B) 5–6 days after gene transfer, and comparison of ssTnI and ssTnI single substitution mutants on pCa50 values (C) at pH 7.0 (left panel) and 6.2 (middle panel), as well as the delta shift in pCa50 between the 2 pH values (right panel). The effect of ssTnIR125Q (n=5), and ssTnIV134E (n=6) on the pCa50 of isometric tension are shown for pH 7.0 (left panel), pH 6.2 (middle panel), and the delta shift in pCa50 between the 2 pH values (right panel). The response in myocytes expressing ssTnIR125Q was comparable to ssTnI (n=8), and the expression of ssTnIV134E produced an intermediate response between ssTnI and cTnI, in which Ca2+ sensitivity was similar to ssTnI at pH 7.0 and the response to acidic pH was more comparable to cTnI. Values are expressed as mean ± SEM and results were compared by a 1-way ANOVA and Tukey’s multiple comparison test, with *p<0.05 versus control and **p<0.05 for substitution mutants versus ssTnI.
Single substitution within the H4 helix
In addition to ssTnIH132A, the ssTnIV134E substitution is the other substitution located at the beginning of the H4 helix. Expression of ssTnV134E produced an intermediate phenotype, with pCa50 values comparable to ssTnI at pH 7.0, but a hybrid of the ssTnI-cTnI phenotype at pH6.20 (Fig. 6C). As a result, the ΔpCa50 was not different from cTnI-expressing myocytes (Fig. 6C, right panel).
The remainder of the H4 region is highly conserved across isoforms, with the exception of a histidine residue in cTnI that is substituted with asparagines in the slow and fast isoforms. Thus, the ssTnIN141H substitution was studied to determine whether this histidine residue within cTnI in the H4 region influences myofilament Ca2+ and pH sensitivity of tension. Introduction of this histidine to ssTnI produced a pCa50 at pH 7.0 that was comparable to cTnI, yet the rightward shift in pCa50 caused by the change in pH from 7.0 to 6.2 was comparable to ssTnI (Fig. 7C). These results suggest that while H132 in ssTnI is a key residue determining Ca2+ sensitivity at pH 7.0 and 6.2, the V134E and N141H substitutions within this H4 region provide partial effects in terms of modulating myofilament Ca2+ at physiological or acidic pH, respectively. These studies also provide evidence that specific substitutions within this H4 helix influence the inhibitory properties of TnI in the myofilament.
FIGURE 7.

Expression of ssTnIN141H 6 days after gene transfer (A, B) and influence on myofilament Ca2+ sensitivity of force at pH 7.0 and 6.2, 5–6 days after gene transfer (C). A. Representative expression of ssTnIN141H, and comparison to non-infected control myocytes and myocytes treated with adenoviral vectors for cTnI and ssTnI 5–6 days after gene transfer. Similar results were obtained in intact and permeabilized myocytes 6 days after gene transfer (results not shown). B. Immunhistochemical staining of troponin I showing a striated pattern of TnI expression (left panel), and loss of cTnI expression as ssTnIN141H replaces endogenous TnI (right panel). C. The pCa50 at pH 7.0 (left panel), 6.2 (middle panel), and the shift in pCa50 between pH 7.0 and 6.2 (right panel) for myofilament tension generation in myocytes expressing ssTnIN141H (n = 5) compared to cTnI- (control n = 8; cTnI n = 5) and ssTnI (n = 8) -expressing myocytes. Values are expressed as mean ± SEM and results were compared by a 1-way ANOVA and Tukey’s multiple comparison test, with *p<0.05 versus control and **p<0.05 for substitution mutant versus ssTnI. The pCa50 in myocytes expressing ssTnIN141H is very similar to cTnI at pH 7.0, but the response to acidic pH more closely resembles the response observed in ssTnI-expressing myocytes.
Sarcomere length detection of shortening and re-lengthening was examined in intact myocytes to determine whether the shifts in Ca2+ sensitivity of tension at pH 7.0 in myocytes expressing H132A, V134E or N141H would produce significant changes in myocyte shortening or relaxation at physiological temperatures. Baseline sarcomere length and the normalized rate of contraction (+dl/dtmax) were comparable among groups (Fig. 8). Relaxation, measured by −dl/dtmax, TTR50 and TTR75 in myocytes expressing ssTnIH132A and ssTnIN141H, was significantly accelerated compared to ssTnI expressing myocytes. Myocytes expressing ssTnIV134E and ssTnI showed comparable relaxation values. These same measures of relaxation were comparable to controls in myocytes expressing ssTnIH132A, and to a partial extent in myocytes expressing ssTnIN141H (Fig. 8). However, the amplitude of sarcomere shortening in myocytes expressing ssTnIH132A and ssTnIV134E was significantly decreased compared to ssTnI-expressing myocytes. In contrast, myocytes expressing ssTnIN141H demonstrated sarcomere shortening values that were comparable to ssTnI.
FIGURE 8.

Analysis of sarcomere shortening in myocytes expressing ssTnIH132A, ssTnIV134E, and ssTnIN141H compared to control and ssTnI-expressing myocytes 4 days after gene transfer. Results are expressed as mean ± SEM for baseline sarcomere length (Baseline SL, μm), sarcomere shortening (μm), normalized +dl/dtmax (s−1), normalized −dl/dtmax (s−1), and time from peak to 50% and 75% relaxation (TTR50 and TTR75, respectively in ms). Recordings were made from 14 myocytes in 7 different hearts, with *p<0.05 considered significantly different for wild-type and substituted ssTnI versus cTnI-expressing control (C) and **p<0.05 for substitution mutants versus ssTnI.
Inhibitory peptide domain substitutions
The final studies were on the reciprocal substitutions, ssTnIP112T and cTnIT144P located within the inhibitory peptide region of ssTnI and cTnI. Biochemical studies point to the 12 amino acid IP domain as necessary to inhibit actomyosin interactions in the absence of Ca2+, and therefore, we examined whether this single amino acid difference between isoforms contributes to isoform-specific differences in Ca2+ and pH sensitivity of isometric tension output. Here, there were no significant changes in Ca2+-activated isometric tension in myocytes expressing either mutant. Both the pCa50 (Fig. 9B) and peak force at pH 7.0 and 6.2 were comparable to results obtained with myocytes expressing the analogous TnI isoform (mean±SEM Po at pH 7.0 in kN/m2; ssTnIP112T=17.6±2.1, n=7; cTnIT144PFLAG = 21.7±2.8, n=6; P6.2/Po7.0 ssTnIP112T = 0.56±0.04, cTnIT144PFLAG = 0.47±0.04; p>0.05 vs control shown in Table 3 with ANOVA). Expression of cTnIT144P also did not detectably alter sarcomere shortening in intact myocytes (27), in agreement with the present findings. However, this residue is important due to the phosphorylation of T144 in cTnI by protein kinase C (27).
FIGURE 9.

Expression and myofilament Ca2+ sensitivity of isometric force in permeabilized adult rat myocytes expressing cTnIT144PFLAG and ssTnIP112T 5–6 days after gene transfer. Expression of these substitution mutants can be compared to cTnI in non-infected control and AdcTnI-treated myocytes in Figs. 2 and 7. A. Expression of TnI in control myocytes and after gene transfer of cTnI, cTnIFLAG, ssTnIP112T and cTnIT144PFLAG 5 days after gene transfer. B. The pCa50 at pH 7.0 and 6.20 in myocytes expressing cTnIT144PFLAG (n = 6) and ssTnIP112T (n=7) 5–6 days after gene transfer compared to myocytes expressing cTnI (n=8) or ssTnI (n=8). Values are expressed as mean ± SEM and results were compared by a 1-way ANOVA and Tukey’s multiple comparison test, with *p<0.05 versus control.
DISCUSSION
Our results with genetically engineered TnIs demonstrate that critical amino acid differences in and around the TnI H4 helix contribute to isoform-specific effects on myofilament force under physiological and acidic pH. The ssTnIH132 residue is a key isoform determinant of Ca2+- and pH- sensitivity of myofilament tension, which translates into significantly decreased peak shortening and accelerated relaxation compared to ssTnI in the intact myocyte. Interestingly, ssTnIV134E and ssTnIN141H each partially substitute the isoform-specific effects on myofilament Ca2+ sensitivity of tension. The ssTnIV134E substitution primarily influences the pH-dependent shift in Ca2+ sensitivity of tension, while ssTnIN141H decreases the pCa50 of tension at physiological pH. Substitutions within the IP region, ssTnIP112T and cTnIT144P, and the H3 helix, ssTnIR125Q play no significant role in conferring TnI isoform-specific influences on basal myofilament Ca2+ sensitivity of tension. Collectively, these results provide a new high resolution functional map of the regulatory domain of TnI in the context of the intact myocyte.
Integration of TnI structure and function
The present results, together with earlier studies (14;15;20) significantly expand our understanding of the role played by TnI within the intact myofilament. Recent structural studies of the troponin core identified the amphiphilic H1 region (rat cTnI residues 44–80; rat ssTnI residues 15–51), H2 helix (cTnI residues 91–136; ssTnI residues 59–104), H3 helix (cTnI residues 151–160; ssTnI residues 119–128), a flexible, pivoting linker region (cTnI residues 161–164; ssTnI residues 129–132) and H4 helix (cTnI residues 165–189; ssTnI residues 133–157), plus a structural inhibitory peptide domain (cTnI residues 138–149; ssTnI residues 106–117) and a mobile domain at the carboxyl end of TnI (2;29). Analogous structural regions were also identified in fast skeletal TnI (fsTnI) (17), which is highly homologous with ssTnI (30). These findings suggest the structural domains are common to all three troponin I isoforms: cTnI, ssTnI and fsTnI. Functional studies with TnI chimeras identified an isoform-dependent pH sensitivity domain in the carboxyl-half of TnI, and separate Ca2+ sensitivity domains within the amino- and carboxyl- half of the ssTnI and cTnI isoforms (19). The substitutions in the H2, IP, H3 and H4 helices studied here are located within the carboxyl region identified in these earlier functional studies on TnI chimeras(14;15). Earlier biochemical studies on TnI fragments also mapped out the inhibitory peptide region (31–33) and secondary actin and TnC binding domains in the carboxyl half of TnI (18;34;34). Based on these biochemical studies, Pearlstone and colleagues (35) predicted that Q157, A164 and E166 in cardiac TnI may be key residues involved in conferring myofilament pH sensitivity.
Myofilament and myocyte function with ssTnIH132A replacement
The present results demonstrate that the ssTnIH132A substitution located in the pivoting flexible linker between helices H3 and H4 (2;29) produces a dominant isoform-specific functional influence on Ca2+ sensitivity of myofilament tension. The reciprocal effects of ssTnIH132A and cTnIA164H on Ca2+ sensitivity of force in rat myocytes (Fig. 4, (20)) and transgenic mice(20) also resulted in altered myocyte re-lengthening. Similar effects on relaxation in intact myocytes were observed after gene transfer of cTnIA164H into rat myocytes (results not shown). These findings suggest that ssTnIH132 and cTnIA164 play an important role in the release of TnI from actin and/or its interaction with TnC, as Ca2+ binds to troponin C.
The influence of rat ssTnIH132A observed here is in contrast to the biochemical evaluation of the reciprocal human cTnIA162H substitution (rat cTnIA164H). The cTnIA162H reduced myofilament pH sensitivity, but lacked a significant influence on Ca2+ sensitivity at pH 7.0 when measuring actomyosin ATPase activity (36). Fast skeletal TnIH130A (fsTnIH130A), which is equivalent to ssTnIH132A, produced an intermediate change in pH sensitivity in actomyosin ATPase studies. The lack of effect produced by these substitutions on actomyosin ATPase at physiological pH suggests that this biochemical approach may not be sufficiently sensitive for translating the influence of these mutants on force generation and/or myocyte shortening. Our results provide strong evidence that this single amino acid difference (ssTnIH132A, cTnIA164H) is capable of conferring TnI isoform specificity for Ca2+ and pH sensitivity in both the intact myofilament lattice and electrically stimulated myocytes.
The decrease in peak shortening observed in myocytes expressing ssTnIH132A compared to ssTnI (Fig. 8) also is an isoform-specific effect. In earlier work, ssTnI significantly enhanced peak shortening compared to cTnI in myocytes from transgenic mice(37). This increase was independent of peak Ca2+ and the velocity of unloaded shortening. Although decreased peak tension could lead to decreased peak shortening, there were no significant influences of ssTnIH132A on maximal isometric tension (Table 3). Thus, the rightward shift in pCa50 at physiological pH is predicted to be responsible for the decrease in peak shortening in myocytes expressing ssTnIH132A versus ssTnI.
Analysis of H4 helix substitutions
Although results with cTnIA164H, and ssTnIH132A indicate a single amino acid can confer TnI isoform specificity, it remained unclear whether other substitutions in the carboxyl portion of TnI were functionally redundant or able to confer isoform-specific effects on myofilament Ca2+ and/or pH sensitivity. In work with human cTnIQ155R/H162A/E164V (cTnIRAV; rat equivalent is cTnIQ157R/A164H/E166V), Ca2+ sensitivity of actomyosin ATPase increased relative to cTnI at 7.0 and 6.5 and behaved more like ssTnI. The cTnIQ155R/E164V (cTnIRV; rat cTnIQ157R/E166V) substitution did not show these increases in Ca2+ sensitivity (36). Based on these findings, the charge substitution in cTnIE164V (ssTnIV134E) was not predicted to influence Ca2+ or pH sensitivity. In contrast, our results demonstrate that ssTnI residues 134 and 141 are able to confer partial isoform-specific influences on contractile function. The ssTnIV134E substitution produced isoform-specific shifts in the acidosis response (Fig. 6C), while ssTnIN141H caused a cTnI isoform-specific shift in myofilament Ca2+ sensitivity at physiological pH (Fig. 7) relative to ssTnI. Thus, V134E and N141H each have unique and non-complementary functional effects. The contributions of V134E and H132A are not additive based on results with ssTnIQAE, and instead provide further evidence for redundancy within TnI isoforms. Both ssTnIV134E and ssTnIN141H are located within the protruded alpha helical H4 domain (18). Future investigations into the differential influence of the reciprocal cTnI substitutions on myocyte and cardiac function may be important for ultimately understanding the detailed mechanism of the H4 region within TnI.
The mechanism responsible for the effects of ssTnIV134E and ssTnIN141H on peak shortening in intact myocytes is not yet known. The ssTnIV134E substitution does not influence Ca2+ sensitivity at physiological pH, which suggests that effects on myofilament Ca2+ sensitivity are unrelated to this response in intact myocytes. These substitutions may influence the velocity of unloaded shortening, although ssTnI in transgenic mouse hearts did not significantly influence shortening velocity (37). In the future, shortening velocity and cross-bridge-mediated activation studies with these substitutions are needed to determine the mechanism responsible for the effects on peak shortening.
Results obtained here demonstrate there are multiple residues capable of conferring all or some portion of TnI isoform specificity. The ssTnIH132A substitution has a dominant effect, while ssTnIV134E and ssTnIN141H alone provide partially redundant effects. The gain-in-function observed in transgenic mouse hearts expressing cTnIA164H, which is the reciprocal of the ssTnIH132A substitution, protects the heart against ischemia and acidosis and improves myocyte function in failing hearts(20). We speculate that organ-level expression of the reciprocal cTnI mutation of ssTnIN141H and ssTnIV134E (e.g. cTnIH173N, cTnIE166V) would respond with a decrease in Ca2+ sensitivity and pH sensitivity, respectively. These later substitutions may help in identifying whether the change in Ca2+ sensitivity, pH sensitivity or both are needed for the protective effect of cTnIA164H on transgenic hearts. Therapy targeted to the amino acids that confer isoform specificity may one day help to restore myocardial function in patients with heart failure.
Acknowledgments
We gratefully acknowledge technical assistance provided by Andrea Borton, Stan Forfa, and Dustin Robinson, and helpful comments by Todd Herron on earlier versions of this manuscript. This study was supported by National Institutes of Health grants HL67254 (MVW) and HL59301 (JMM). Immunohistochemical analysis of myofilament incorporation utilized the Morphology and Image Analysis Core of the Michigan Diabetes Research and Training Center funded by NIH5P60 DK20572 from the National Institute of Diabetes & Digestive & Kidney Diseases.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gordon AM, Regnier M, Homsher E. Skeletal and cardiac muscle contractile activation: tropomyosin “rocks and rolls”. News Physiol Sci. 2001;16:49–55. [PubMed] [Google Scholar]
- 2.Takeda S, Yamashita A, Maeda K, Maeda Y. Structure of the core domain of human cardiac troponin in the Ca(2+)-saturated form. Nature. 2003;424(6944):35–41. doi: 10.1038/nature01780. [DOI] [PubMed] [Google Scholar]
- 3.Pirani A, Vinogradova MV, Curmi PM, King WA, Fletterick RJ, Craig R, et al. An atomic model of the thin filament in the relaxed and Ca2+-activated states. J Mol Biol. 2006;357(3):707–717. doi: 10.1016/j.jmb.2005.12.050. [DOI] [PubMed] [Google Scholar]
- 4.Saggin L, Gorza L, Ausoni S, Schiaffino S. Troponin I switching in the developing heart. J Biol Chem. 1989;264:16299–16302. [PubMed] [Google Scholar]
- 5.Reiser PJ, Westfall MV, Schiaffino S, Solaro RJ. Tension production and thin-filament protein isoforms in developing rat myocardium. Am J Physiol. 1994;267(4 Pt 2):H1589–H1596. doi: 10.1152/ajpheart.1994.267.4.H1589. [DOI] [PubMed] [Google Scholar]
- 6.Schiaffino S, Gorza L, Ausoni S. Troponin isoform switching in the developing heart and its functional consequences. Trends Cardiovasc Med. 1993;3:12–17. doi: 10.1016/1050-1738(93)90022-X. [DOI] [PubMed] [Google Scholar]
- 7.VanBuren P, Okada Y. Thin filament remodeling in failing myocardium. Heart Fail Rev. 2005;10(3):199–209. doi: 10.1007/s10741-005-5250-8. [DOI] [PubMed] [Google Scholar]
- 8.Solaro RJ, Kumar P, Blanchard EM, Martin AF. Differential effects of pH on calcium activation of myofilaments of adult and perinatal dog hearts. Evidence for developmental differences in thin filament regulation. Circ Res. 1986;58(5):721–729. doi: 10.1161/01.res.58.5.721. [DOI] [PubMed] [Google Scholar]
- 9.Gao L, Kennedy JM, Solaro RJ. Differential expression of TnI and TnT isoforms in rabbit heart during the perinatal period and during cardiovascular stress. J Mol Cell Cardiol. 1995;27(1):541–550. doi: 10.1016/s0022-2828(08)80049-1. [DOI] [PubMed] [Google Scholar]
- 10.Kruger M, Kohl T, Linke WA. Developmental changes in passive stiffness and myofilament Ca2+ sensitivity due to titin and troponin-I isoform switching are not critically triggered by birth. Am J Physiol Heart Circ Physiol. 2006;291(2):H496–H506. doi: 10.1152/ajpheart.00114.2006. [DOI] [PubMed] [Google Scholar]
- 11.Hunkeler NM, Kullman J, Murphy AM. Troponin I isoform expression in human heart. Circ Res. 1991;69(5):1409–1414. doi: 10.1161/01.res.69.5.1409. [DOI] [PubMed] [Google Scholar]
- 12.Bhavsar PK, Dhoot GK, Cumming DV, Butler-Browne GS, Yacoub MH, Barton PJ. Developmental expression of troponin I isoforms in fetal human heart. FEBS Lett. 1991;292(1–2):5–8. doi: 10.1016/0014-5793(91)80820-s. [DOI] [PubMed] [Google Scholar]
- 13.Westfall MV, Rust EM, Metzger JM. Slow skeletal troponin I gene transfer, expression, and myofilament incorporation enhances adult cardiac myocyte contractile function. Proc Natl Acad Sci U S A. 1997;94(10):5444–5449. doi: 10.1073/pnas.94.10.5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Westfall MV, Albayya FP, Turner II, Metzger JM. Chimera analysis of troponin I domains that influence Ca(2+)-activated myofilament tension in adult cardiac myocytes. Circ Res. 2000;86(4):470–477. doi: 10.1161/01.res.86.4.470. [DOI] [PubMed] [Google Scholar]
- 15.Westfall MV, Albayya FP, Metzger JM. Functional analysis of troponin I regulatory domains in the intact myofilament of adult single cardiac myocytes. J Biol Chem. 1999;274(32):22508–22516. doi: 10.1074/jbc.274.32.22508. [DOI] [PubMed] [Google Scholar]
- 16.Metzger JM, Westfall MV. Covalent and noncovalent modification of thin filament action: the essential role of troponin in cardiac muscle regulation. Circ Res. 2004;94(2):146–158. doi: 10.1161/01.RES.0000110083.17024.60. [DOI] [PubMed] [Google Scholar]
- 17.Vinogradova MV, Stone DB, Malanina GG, Karatzaferi C, Cooke R, Mendelson RA, et al. Ca(2+)-regulated structural changes in troponin. Proc Natl Acad Sci U S A. 2005;102(14):5038–5043. doi: 10.1073/pnas.0408882102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tripet B, Van Eyk JE, Hodges RS. Mapping of a second actin-tropomyosin and a second troponin C binding site within the C terminus of troponin I, and their importance in the Ca2+-dependent regulation of muscle contraction. J Mol Biol. 1997;271(5):728–750. doi: 10.1006/jmbi.1997.1200. [DOI] [PubMed] [Google Scholar]
- 19.Westfall MV, Metzger JM. Troponin I isoforms and chimeras: tuning the molecular switch of cardiac contraction. News Physiol Sci. 2001;16:278–281. doi: 10.1152/physiologyonline.2001.16.6.278. [DOI] [PubMed] [Google Scholar]
- 20.Day SM, Westfall MV, Fomicheva EV, Hoyer K, Yasuda S, La Cross NC, et al. Histidine button engineered into cardiac troponin I protects the ischemic and failing heart. Nat Med. 2006;12(2):181–189. doi: 10.1038/nm1346. [DOI] [PubMed] [Google Scholar]
- 21.Michele DE, Albayya FP, Metzger JM. Thin filament protein dynamics in fully differentiated adult cardiac myocytes: toward a model of sarcomere maintenance. J Cell Biol. 1999;145(7):1483–1495. doi: 10.1083/jcb.145.7.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Westfall MV, Borton AR. Role of troponin I phosphorylation in protein kinase C-mediated enhanced contractile performance of rat myocytes. J Biol Chem. 2003;278:33694–33700. doi: 10.1074/jbc.M305404200. [DOI] [PubMed] [Google Scholar]
- 23.Westfall MV, Rust EM, Albayya F, Metzger JM. Adenovirus-mediated myofilament gene transfer into adult cardiac myocytes. Methods Cell Biol. 1997;52:307–322. [PubMed] [Google Scholar]
- 24.Metzger JM. Effects of troponin C isoforms on pH sensitivity of contraction in mammalian fast and slow skeletal muscle fibres. J Physiol (Lond) 1996;492 ( Pt 1):163–172. doi: 10.1113/jphysiol.1996.sp021298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Metzger JM, Wahr PA, Michele DE, Albayya F, Westfall MV. Effects of myosin heavy chain isoform switching on Ca2+-activated tension development in single adult cardiac myocytes. Circ Res. 1999;84(11):1310–1317. doi: 10.1161/01.res.84.11.1310. [DOI] [PubMed] [Google Scholar]
- 26.Godt RE, Lindley BD. Influence of temperature upon contractile activation and isometric force production in mechanically skinned muscle fibers of the frog. J Gen Physiol. 1982;80(2):279–297. doi: 10.1085/jgp.80.2.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Westfall MV, Lee AM, Robinson DA. Differential contribution of troponin I phosphorylation sites to the endothelin modulated contractile response. J Biol Chem. 2005;280:41324–41331. doi: 10.1074/jbc.M506043200. [DOI] [PubMed] [Google Scholar]
- 28.Coutu P, Metzger JM. Optimal range for parvalbumin as relaxing agent in adult cardiac myocytes: gene transfer and mathematical modeling. Biophys J. 2002;82(5):2565–2579. doi: 10.1016/S0006-3495(02)75599-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murakami K, Yumoto F, Ohki SY, Yasunaga T, Tanokura M, Wakabayashi T. Structural basis for Ca2+-regulated muscle relaxation at interaction sites of troponin with actin and tropomyosin. J Mol Biol. 2005;352(1):178–201. doi: 10.1016/j.jmb.2005.06.067. [DOI] [PubMed] [Google Scholar]
- 30.Hastings KE, Koppe RI, Marmor E, Bader D, Shimada Y, Toyota N. Structure and developmental expression of troponin I isoforms. cDNA clone analysis of avian cardiac troponin I mRNA. J Biol Chem. 1991;266(29):19659–19665. [PubMed] [Google Scholar]
- 31.Syska H, Wilkinson JM, Grand RJ, Perry SV. The relationship between biological activity and primary structure of troponin I from white skeletal muscle of the rabbit. Biochem J. 1976;153(2):375–387. doi: 10.1042/bj1530375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Talbot JA, Hodges RS. Comparative studies on the inhibitory region of selected species of troponin-I. The use of synthetic peptide analogs to probe structure-function relationships. J Biol Chem. 1981;256(23):12374–12378. [PubMed] [Google Scholar]
- 33.Van Eyk JE, Strauss JD, Hodges RS, Ruegg JC. A synthetic peptide mimics troponin I function in the calcium-dependent regulation of muscle contraction. FEBS Lett. 1993;323(3):223–228. doi: 10.1016/0014-5793(93)81344-y. [DOI] [PubMed] [Google Scholar]
- 34.Van Eyk JE, Thomas LT, Tripet B, Wiesner RJ, Pearlstone JR, Farah CS, et al. Distinct regions of troponin I regulate Ca2+-dependent activation and Ca2+ sensitivity of the acto-S1-TM ATPase activity of the thin filament. J Biol Chem. 1997;272(16):10529–10537. doi: 10.1074/jbc.272.16.10529. [DOI] [PubMed] [Google Scholar]
- 35.Pearlstone JR, Sykes BD, Smillie LB. Interactions of structural C and regulatory N domains of troponin C with repeated sequence motifs in troponin I. Biochemistry. 1997;36(24):7601–7606. doi: 10.1021/bi970200w. [DOI] [PubMed] [Google Scholar]
- 36.Dargis R, Pearlstone JR, Barrette-Ng I, Edwards H, Smillie LB. Single mutation (A162H) in human cardiac troponin I corrects acid pH sensitivity of Ca2+-regulated actomyosin S1 ATPase. J Biol Chem. 2002;277(38):34662–34665. doi: 10.1074/jbc.C200419200. [DOI] [PubMed] [Google Scholar]
- 37.Fentzke RC, Buck SH, Patel JR, Lin H, Wolska BM, Stojanovic MO, et al. Impaired cardiomyocyte relaxation and diastolic function in transgenic mice expressing slow skeletal troponin I in the heart. J Physiol (Lond) 1999;517 ( Pt 1):143–157. doi: 10.1111/j.1469-7793.1999.0143z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]