

Abstract
Human atherosclerotic plaques express the metalloprotease tumor necrosis factor (TNF)-α converting enzyme (TACE/ADAM-17), which cleaves several transmembrane proteins including TNF and its receptors (TNFR-1 and TNFR-2). Plaques also harbor submicron vesicles (microparticles, MPs) released from plasma membranes after cell activation or apoptosis. We sought to examine whether TACE/ADAM17 is present on human plaque MPs and whether these MPs would affect TNF and TNFR-1 cellular shedding. Flow cytometry analysis detected 12,867 ± 2007 TACE/ADAM17+ MPs/mg of plaques isolated from 25 patients undergoing endarterectomy but none in healthy human internal mammary arteries. Plaque MPs harbored mainly mature active TACE/ADAM17 and dose dependently cleaved a pro-TNF mimetic peptide, whereas a preferential TACE/ADAM17 inhibitor (TMI-2) and recombinant TIMP-3 prevented this cleavage. Plaque MPs increased TNF shedding from the human cell line ECV-304 overexpressing TNF (ECV-304TNF), as well as TNFR-1 shedding from activated human umbilical vein endothelial cells or ECV-304TNF cells, without affecting TNF or TNFR-1 synthesis. MPs also activated the shedding of the endothelial protein C receptor from human umbilical vein endothelial cells. All these effects were inhibited by TMI-2. The present study shows that human plaque MPs carry catalytically active TACE/ADAM17 and significantly enhance the cell surface processing of the TACE/ADAM17 substrates TNF, TNFR-1, and endothelial protein C receptor, suggesting that TACE/ADAM17+ MPs could regulate the inflammatory balance in the culprit lesion.
Atherosclerosis is a chronic inflammatory disease of the vessel wall resulting from the interactions between modified lipoproteins, monocytes/macrophages, lymphocytes, and vascular cells.1 The development and the progression of atherosclerotic plaques are associated with apoptotic cell death and accumulation of microparticles (MPs) within the lesion.1,2,3 MPs are submicron plasma membrane vesicles released during cell activation or apoptosis and harbor at their surface transmembrane proteins initially present at the parent cell surface, conferring to MPs a dynamic storage pool of bioactive molecules.4,5,6 MPs have been isolated from human atherosclerotic plaque but are absent in healthy blood vessels.7 Human plaque MPs originate mainly from leukocytes, red blood cells, endothelial cells, and smooth muscle cells.7 They also express a procoagulant activity associated with the presence of phosphatidylserine and tissue factor at their surface, which could lead to thrombus formation at the time of plaque rupture.7,8,9
Inflammatory processes are regulated by the balance between pro- and anti-inflammatory mediators or cytokines. Sheddases also modulate this equilibrium by cleaving transmembrane proteins (cytokines, receptors, adhesion molecules, and so forth) at the cell surface, releasing soluble ectodomains with altered function.10 The typical example is the tumor necrosis factor (TNF)-α converting enzyme (TACE). Initially discovered as the protease that cleaves the 26-kDa proform of TNF (pro-TNF) to yield the TNF soluble form (sTNF),11,12 TACE also cleaves ectodomains of several other transmembrane proteins13 such as TNFR-1 and TNFR-2.14,15 TACE belongs to the ADAM family (ADAM17) and is synthesized as an inactive proform that is further cleaved into an active form by proprotein convertases, such as furin.16,17
We recently reported that TACE/ADAM17 is expressed in both cellular and acellular areas of lesions from apoE−/− mice and in human atherosclerotic plaques.18 We therefore hypothesized that MPs present in the plaque are potential carriers of TACE/ADAM17. MPs were isolated from human atherosclerotic plaques and analyzed for their content in TACE/ADAM17 protein and activity. Results showed that MPs carry TACE/ADAM17, mainly in its mature active form, catalyze in vitro hydrolysis of a mimetic peptide containing the cleavage site of pro-TNF, and activate the shedding of TACE/ADAM17 substrates such as TNF, TNFR-1, and endothelial protein C receptor (EPCR).
Materials and Methods
Isolation of MPs from Human Atherosclerotic Plaque and Human Umbilical Vein Endothelial Cells
MPs were isolated from human atherosclerotic plaques removed from 25 patients undergoing carotid endarterectomy (73 ± 2 years of age; 79% male), as recently reported.7 Plaques were obtained either from symptomatic patients (70% with ischemic attacks and 30% with stroke, n = 10) or from asymptomatic patients (n = 15) with critical asymptomatic stenosis of the carotid artery (>75% narrowing). As control experiments, healthy human internal mammary arteries (n = 3, obtained as surgical waste) were submitted to the same isolation protocol. All patients gave their informed consent to the study, which was approved by our local ethical committee. Surgical samples obtained within 90 minutes after excision were rapidly rinsed in ice-cold sterile phosphate-buffered saline (PBS) solution supplemented with streptomycin and penicillin (100 U/ml each). Atherosclerotic lesions were then mechanically separated from the apparently healthy vessel wall. Plaques were thoroughly minced for 15 minutes into 1-mm3 tissue fragments using fine scissors in a volume of Dulbecco’s modified Eagle’s medium (supplemented with 10 μg/ml polymyxin B, streptomycin, and penicillin, and filtered through a 0.22-μm membrane) corresponding to the respective weight of each lesion. The resulting preparations were centrifuged first at 400 × g (15 minutes) and then at 12,500 × g (5 minutes) to remove cells and cell debris. The resulting supernatants referred to as plaque homogenates were subsequently used for flow cytometry analysis of plaque microparticle cellular origins.7 The remaining plaque homogenate was further centrifuged at 20,500 × g for 150 minutes at 4°C to pellet MPs. The supernatant was discarded, and MP pellets were gently suspended in fresh Dulbecco’s modified Eagle’s medium (1/10 of volume corresponding to the respective weight of each lesion) and were used for in vitro purposes (activity and cell stimulation). Endothelial MPs were obtained from human umbilical vein endothelial cells (HUVECs) (fourth passage) maintained in serum-deprived medium for 72 hours. The medium was first centrifuged at 300 × g for 10 minutes to eliminate cell debris and then at 20,500 × g for 150 minutes at 4°C to pellet MPs that were subsequently resuspended in fresh Dulbecco’s modified Eagle’s medium.
Flow Cytometry Analysis of MPs
All analyses were performed on homogenates prepared from atherosclerotic plaques or normal arteries and were performed on a Coulter EPICS XL flow cytometer (Beckman Coulter, Villepinte, France) as recently reported.7 MP gate was defined as events with a 0.1- to 1-μm diameter and then plotted on a FL/FSC fluorescence dot plot to determinate positively labeled MPs by specific antibodies. MP concentration was assessed by comparison to Flowcount calibrator beads. MPs bearing phosphatidylserine were labeled using fluorescein isothiocyanate (FITC)-conjugated Annexin V (Roche Diagnostics, Meylan, France) in the presence or in the absence of CaCl2 (5 mmol/L). The cellular origin of human plaque MPs was determined as follows. We incubated 10 μl of plaque homogenates with different fluorochrome-labeled antibodies or their corresponding isotype-matched IgG controls at room temperature for 30 minutes. Anti-CD4-phycoerythrin (PE) was provided by BD Biosciences Pharmingen (Le Pont-de-Claise, France); anti-CD41-PE-cyanin5 (PC5), anti-CD66b-FITC, anti-CD144-PE, and anti-CD235a-FITC were obtained from Beckman Coulter; anti-CD14-PE was from Caltag Laboratories (Burlingame, CA). MPs derived from endothelial cells, monocytes/macrophages, lymphocytes, erythrocytes, and granulocytes were identified as CD144+, CD14+, CD4+, CD235a+, and CD66b+, respectively. The presence of intracellular smooth muscle cell actin was assayed after MP fixation in paraformaldehyde (2%) and permeabilization by saponin (0.1%). Anti-smooth muscle cell actin antibodies (rabbit IgG, dilution 1:2; LabVision, Runcorn, UK), or rabbit IgG (as a negative control), were incubated for 1 hour at room temperature. MP samples were washed once in PBS, and Alexa Fluor 555 donkey anti-rabbit IgGs (Invitrogen, Cergy-Pontaise, France) were then added for 30 minutes at room temperature. To investigate the presence of TACE/ADAM17 at the surface of plaque MPs, we incubated 10 μl of homogenate with 5 μl of monoclonal anti-human TACE/ADAM17-PE (clone no. 111633) or its corresponding isotype-matched IgG control (R&D Systems, Lille, France) at room temperature for 20 minutes in the dark. The same anti-TACE-PE antibody (5 μl) was used in co-labeling with the anti-CD66b-FITC (20 μl) and the anti-235a-FITC (20 μl). For the co-labeling experiments with PE-conjugated antibodies (20 μl each of anti-CD4, -CD14, and -CD144), we used the monoclonal antibody anti-human TACE/ADAM17 conjugated to fluorescein (clone no. 111633) (5 μl). All subsequent in vitro experiments were performed using the 20,500 × g-pelleted plaque MPs. In these assays, plaque MPs reached a final concentration of 14,250 ± 4990 Annexin V+ MPs/μl, which corresponded to 11% of the average MP concentration in the plaque.
Fluorogenic Assays of Protease Activity of MPs
Plaque MPs (n = 4) were incubated with the classical pro-TNF mimetic fluorogenic peptide (peptide III; R&D Systems) harboring the consensus sequence A-V (Mca-PLAQA↓V-Dpa-RSSSR-NH2) cleaved by TACE/ADAM17.19 MPs (10 μl) were suspended in 100 μl of the final activity buffer (25 mmol/L Tris/HCl, pH 8.0, containing 2.5 μmol/L ZnCl2). Fluorogenic peptide I (substrate of MMP-1, -2, -7, -8, -9, -12, -13, -14, -15, and -16), peptide II (substrate of MMP-3 and -10) (R&D Systems), and peptide III were diluted in the activity buffer at the final concentration of 10 μmol/L. Recombinant ectodomain of human TACE/ADAM17 (R&D Systems), used as the positive control, or MPs were extemporaneously mixed with the substrate in a final volume of 100 μl at room temperature to initiate the reaction. TACE/ADAM17 inhibitor was premixed with MPs or recombinant TACE/ADAM17 at 4°C for 15 minutes. Mixtures were immediately delivered in a 96-well black plate and read in a microplate fluorescence reader (Chameleon; Hidex, Turku, Finland). For all substrates, fluorescence-related enzymatic cleavage was monitored at 320-nm excitation and 405-nm emission wavelength for 2 to 3 hours. Blank (buffer and MPs and substrate, separately) was subtracted from sample measurements for calculations.
Cell Culture
HUVECs were isolated and cultured as previously described.20 They were used at the third passage for MP stimulation experiments. The human endothelial cell line ECV-304 and ECV-304 cells stably overexpressing TNF (ECV-304TNF) were cultured as described.21 Monocytic mouse cell line homozygous for TACE/ADAM17 mutation, which deletes the Zn2+ binding domain (TACE/ADAM17ΔZn/ΔZn cells), and monocytic mouse TACE/ADAM17 cells expressing active TACE/ADAM17 were kindly provided by Dr. J. Peschon (Amgen Inc., Thousand Oaks, CA).11
Incubation of Cells with MPs
In HUVECs, the constitutive release of TNF or TNFR-1 was less than the detection limit in the present experimental conditions. Thus, HUVECs were first exposed to phorbol ester (PMA; 20 nmol/L, 4 hours), washed with serum-free medium (Dulbecco’s modified Eagle’s medium, 0.2% bovine serum albumin, 0.1 μmol ZnCl2, 1% penicillin/streptomycin, and l-Glu) and incubated for 2 hours in this medium with plaque MPs with or without TMI-2 (1.0 μmol/L) (VF 0.4 ml). ECV-304TNF cells were exposed for 2 hours to plaque-pelleted MPs in the presence or the absence of TMI-2 (1.0 μmol/L) in the above serum-free medium (VF 0.4 ml). Conditioned medium was collected and centrifuged at 20,500 × g to remove the MPs. Cells were lysed with a lysis buffer (PBS, 0.2% Triton X-100, and 1 μg/ml Pefabloc) for subsequent cellular protein assay (enzyme-linked immunosorbent assay and total proteins). In some cases, the 20,500 × g supernatant cleared of the MPs was divided in two parts: one was stored at 4°C, and the other one was centrifuged at 170,000 × g for 16 hours at 4°C (Beckman Optima TLX ultracentrifuge, TLA-100.2 rotor) to sediment exosome-like particles that could be potentially released by cells.22 TNF or TNFR-1 shedding was expressed as the ratio of TNF or TNFR-1 in the culture medium over cellular TNF or TNFR-1, respectively, to take into account variations in cellular protein level.
Western Blotting
MP pellets were lysed with the above lysis buffer containing TMI-2 to prevent the autocleavage of TACE/ADAM17. MP proteins were first submitted to concanavalin A column separation and then submitted to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (12% acrylamide or NuPAGE 10% acrylamide; Invitrogen) followed by immunoblotting as previously described.21 TACE/ADAM17 and ADAM10 were revealed with the rabbit polyclonal anti-human TACE/ADAM17 (R&D Systems) and the rabbit polyclonal anti-human ADAM10 (eBioscience, Montrouge, France), respectively. The potential presence of exosomes in MP preparations was analyzed using the anti-TSG-101 antibody (Sigma, L’Isles d’Abeau Chesnes, France) and the anti-lactadherin antibody.23 The MW was estimated with the BenchMark ladder (Invitrogen).
Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR)
Total RNA from HUVECs was extracted with the RNeasy mini kit from Qiagen (Courtaboeuf, France). Primer pairs for human TNFR-1 and eEF1α and RT-PCR conditions were described previously.24
Protein Assay
Total protein content of cell lysates was assayed using the bicinchoninic acid protein assay kit from Sigma-Aldrich. Levels of human sTNF, sTNFR-1, sICAM-1, and MCP-1, and murine sTNFR-1 were determined by enzyme-linked immunosorbent assay according to the specifications of the supplier (R&D Systems). Soluble endothelial protein C receptor (sEPCR) was assayed by enzyme-linked immunosorbent assay using the specification of the Asserachrom sEPCR kit (Diagnostica Stago, Asnières, France).
Reagents
Culture media and reagents were from Gibco BRL (Invitrogen). Bovine serum albumin and Pefabloc were from Sigma. TACE/ADAM17 inhibitor TMI-2 was kindly donated by Dr. J. Levin (Wyeth Research, Cambridge, MA). Recombinant active human TACE/ADAM17 was from R&D Systems.
Statistics
Results were expressed as mean ± SD or SEM where indicated. Differences of the means between two groups were evaluated by the Mann-Whitney U-test, with P < 0.05 considered as significant.
Results
Presence of the Mature Form of TACE/ADAM17 on MPs Isolated from Human Atherosclerotic Plaques
Flow cytometry analysis of plaque homogenates (Figure 1A) detected the presence of 119,639 ± 26,325 Annexin V+ MPs, and 12,867 ± 2007 TACE/ADAM17+ MPs per mg of plaque (Figure 1B) (mean ± SEM, n = 25). In contrast, isolation of MPs from healthy human internal mammary arteries did not yield detectable levels of TACE/ADAM17+ MPs (n = 3) (Figure 1B). TACE+ MP abundance was not different between asymptomatic and symptomatic plaques (13,487 ± 2529 versus 11,975 ± 3397 TACE+ MPs/mg plaque, respectively; P = 0.48).
Figure 1.

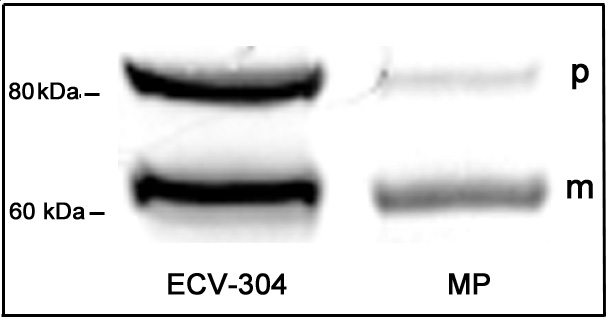
MPs isolated from atherosclerotic human plaque, which contain the mature form of TACE/ADAM17, are of diverse cellular origin and do not contain exosomes: Analysis of TACE/ADAM17 on MPs isolated from human atherosclerotic plaque and human internal mammary arteries. A: Expression of TACE/ADAM17 on MPs from plaque homogenates. This graph is representative of the different plaque preparations. The shaded peak corresponds to negative isotype control. B: Levels of TACE/ADAM17+ MPs in human internal mammary arteries (M.Art., n = 3) and atherosclerotic plaque (plaque, n = 25); values are mean ± SEM. C: Co-labeling of TACE/ADAM17+ MPs with various cellular markers (from left to right: lymphocytes, monocytes, granulocytes, endothelial cells, and erythrocytes) (n = 12). Results are expressed as percentage of total TACE/ADAM17+ MPs (mean ± SEM). D: Immunoblotting of the exosomal marker TSG-101 in the post 20,500 × g pellet (left) and corresponding supernatant further centrifuged at 170,000 × g (right). Because protein profiles were different in both fractions (see Ponceau red staining), two times more MP materials (20 μg) than exosomal-like material were loaded. Representative of three different samples analyzed. E: Immunoblotting of TACE/ADAM17 from two different MP preparations containing 2.8 × 106 and 0.1 × 106 Annexin V+ MPs/μl on the middle and right lanes, respectively. The TACE/ADAM17 of MPs in the middle lane is to illustrate that only a highly MP-enriched plaque allows the detection of TACE proform. The right lane is representative of the five MP preparations tested. mTACE and pTACE indicate the positions of the mature and proform of TACE/ADAM17, respectively, validated by the migration of these forms present in COS-7 cells.
The cellular origin of TACE/ADAM17+ MPs was determined by positive co-labeling of TACE/ADAM17 and cellular markers (n = 12, Figure 1C). TACE/ADAM17+ MPs mostly originated from leukocytes (lymphocytes, granulocytes, and monocytes/macrophages) and also from erythrocytes and endothelial cells. None seems to be of smooth muscle cell origin because TACE/ADAM17+ MPs did not co-label with the smooth muscle cell actin antibody. There was no difference in the cellular origin of TACE/ADAM17+ MPs between symptomatic and asymptomatic plaques (data not shown).
Co-pelleting of plaque MPs with exosomes that sediment at much higher centrifugation speeds than MPs was excluded in the 20,500 × g pellet because there was no significant labeling for the exosomal markers TSG-10125,26 (Figure 1D) or lactadherin (data not shown).22,27,28 In plaque MP pellets, the mature form of TACE/ADAM17 was primarily predominant over the proform. Only a minor band of the proform was detected when a preparation exceptionally rich in MPs was examined (Figure 1E).
The Mature Form of TACE/ADAM17 Present on Plaque MPs Is Active in Vitro
The presence of the mature form of TACE/ADAM17 on MPs prompted us to investigate whether MPs could be catalytically active in vitro. The human recombinant TACE/ADAM17 ectodomain (10 ng/assay), which contains the active catalytic site, time dependently cleaved the pro-TNF mimetic peptide (peptide III). TMI-2, a preferential inhibitor of TACE/ADAM17,29 inhibited this cleavage by 87 and 100% at 5 and 50 nmol/L, respectively (Figure 2A). The natural endogenous TACE/ADAM17 inhibitor TIMP-330 (100 nmol/L) inhibited by 85% the TACE/ADAM17-dependent cleavage of the peptide.
Figure 2.

MPs isolated from human atherosclerotic plaque are active on fluorogenic peptides that are substrates of TACE/ADAM17 or MMPs. Proteolytic activity of recombinant human TACE/ADAM17 and MPs were measured on various fluorogenic substrates. Details of assay conditions are indicated in Materials and Methods. A: Time-dependent cleavage by recombinant TACE/ADAM17 of the fluorogenic peptide III, mimetic of the cleavage zone of pro-TNF (10 ng) in the presence or not of TMI-2 at 5 and 50 nmol/L or TIMP-3 (100 nmol/L), mean ± SD of two separate measurements. B: Time-dependent cleavage by MPs (10 μl) of the fluorogenic peptide III in the presence of various concentrations of TMI-2, and TIMP-3. For the sake of clarity, data are presented only as the dose of 100 nmol/L TIMP-3 because 200 nmol/L gave similar inhibition, and SEM in place of SD to avoid overlapping of error bars. n = 4. C: Dose-dependent effect of MPs [expressed as fold of the maximal amount used (10 μl)] on the cleavage of the fluorogenic peptide III. For clarity of the graph, only two time points are presented. Values are mean ± SD of two separate MP preparations. D: Time-dependent cleavage by MPs of fluorogenic peptide I and peptide II in the presence or not of TMI-2 (1 μmol/L). Values are mean ± SD of four separate MP preparations identical to those used in B.
Plaque MPs pelleted from homogenates cleaved the fluorogenic peptide in a time-dependent manner (Figure 2B). The MP-induced increase in fluorescence was strongly reduced by TMI-2, in a concentration-dependent manner. Inhibition by TMI-2 averaged 63% at 5 nmol/L and was optimal (87%) at 500 nmol/L, after 120 minutes of incubation (Figure 2B). TIMP-3 (100 nmol/L) inhibited the MP-induced increase in fluorescence to the same extent as TMI-2 (5 nmol/L) did. The TACE/ADAM17-dependent hydrolysis of the peptide augmented with the increasing concentrations of MPs (Figure 2C). No TACE/ADAM17 activity could be detected in the supernatant resulting from MP pelleting (45 minutes; 20,500 × g), indicating that the activity is carried by MPs and not by smaller vesicular structures.
We also investigated whether plaque MPs hydrolyze two other fluorogenic peptides that are substrates of several MMPs. Peptide I is cleaved by a large panel of MMPs (MMP-1, -2, -7, -8, -9, -12, -13, -14, -15, and -16), whereas peptide II is preferentially cleaved by MMP-3 and MMP-10. Peptides I and II were not hydrolyzed by recombinant TACE/ADAM17 because the increase in fluorescence after 120 minutes resulting from their cleavage was less than 10% of the initial fluorescence value (data not shown). However, peptides I and II were cleaved by plaque MPs (Figure 2D), but TMI-2 (1 μmol/L) inhibited their cleavage by only 28 and 20%, respectively, indicating that plaque MPs carry other active protease(s)31 in addition to TACE/ADAM17.
We observed that plaque MPs also contain ADAM10 (Supplemental Figure 1, see http://ajp.amjpathol.org), a protease able to cleave in vitro pro-TNF,32 although its physiological relevance as a TNF convertase is considered to be of minor importance.33,34 When testing the cleavage of the pro-TNF mimetic peptide, we observed that the increase in fluorescence induced by recombinant ADAM10 (100 ng) was only 5% of that recorded for recombinant TACE/ADAM17 (100 ng), indicating that, in TACE/ADAM17 assay conditions, ADAM10 activity on pro-TNF was minimal. Furthermore, ADAM10 activity was inhibited by 10 and 40% in the presence of TMI-2 concentrations (5 and 50 nmol/L, respectively) that inhibited TACE/ADAM17 activity by 87 and 100%. We then investigated if plaque MPs were able to stimulate the cleavage of TACE/ADAM17 substrates present on the cell surface.
Effects of TMI-2 on the Release of TACE/ADAM17 Substrates in Cell-Based Conditions
We first examined TMI-2 inhibitory activity on TNF cleavage, the prototypical substrate of TACE/ADAM17, in ECV-304TNF cells that constitutively express TACE/ADAM17 and produce and release TNF.21 ECV-304TNF cells were stimulated for 1 hour by PMA (200 nmol/L), which preferentially activates TACE/ADAM17 over ADAM10.35,36 As shown in Figure 3A, significant inhibition of TNF release occurred for concentrations of TMI-2 at 0.5 μmol/L and reached a plateau at 1.0 μmol/L (63% inhibition). We investigated the effect of TMI-2 also in a murine monocytic cell line deficient in TACE/ADAM17 activity (TACE/ADAM17ΔZn/ΔZn).11 After PMA stimulation, TACE/ADAM17ΔZn/ΔZn cells did not significantly release TNF as expected, but presented a residual significant release of TNFR-1 (Figure 3B), which was much lower than that released by TACE/ADAM17+/+ wild-type cells, as previously described.37 TMI-2 (0.1 to 1.0 μmol/L) dose dependently decreased up to 50% the TNFR-1 release from wild-type cells, without affecting the residual release in TACE/ADAM17ΔZn/ΔZn cells. These data indicate that in these monocytic cells, the TACE/ADAM17-independent TNFR-1 shedding is insensitive to TMI-2, even at 1.0 μmol/L. A concentration of 1.0 μmol/L TMI-2 was selected for further studies.
Figure 3.

Effects of TMI-2 on TNF and TNFR-1 release in cell-based conditions. A: ECV-304TNF cells were preincubated for 10 minutes with TMI-2 at concentrations ranging from 0 to 2 μmol/L and then stimulated with PMA (200 nmol/L) for 1 hour. Culture medium was collected for TNF assay. Values are mean ± SD of two separate experiments each performed in duplicate. B: Murine TACE/ADAM17+/+ and TACE/ADAM17ΔZn/ΔZn monocytic cells were preincubated with TMI-2 at concentrations ranging from 0 to 1.0 μmol/L and then stimulated with PMA (200 nmol/L) for 1 hour. Culture medium was collected for TNFR-1 assay. Values are mean ± SD of two separate experiments each performed in triplicate.
MPs Isolated from Human Atherosclerotic Plaques Stimulate the Release of the TACE/ADAM17 Substrates, TNFR-1 and EPCR, from HUVECs
HUVECs, even after PMA stimulation, did not release detectable amounts of TNF, in agreement with previous studies showing that HUVECs did not release TNF.38 We therefore focused on TNFR-1, which was measurable in the culture medium of PMA-treated HUVECs. Exposure of PMA-treated HUVECs to plaque MPs significantly augmented by 76% the amount of TNFR-1 in the culture medium, without affecting TNFR-1 mRNA expression (Figure 4A). The increased TNFR-1 levels in the culture medium probably did not result from MPs themselves because TNFR-1 was undetectable in plaque MPs. TMI-2 (1.0 μmol/L) significantly reduced by 83 and 70% the amount of TNFR-1 released from unstimulated and MP-stimulated HUVECs, respectively.
Figure 4.

MPs isolated from human atherosclerotic plaque activate the release of TNFR-1, ICAM-1, and EPCR from HUVECs. HUVECs were incubated with MPs as described in Materials and Methods. Release in the culture medium of TNFR-1 (A) (values are mean ± SD, n = 12, each in duplicate), ICAM-1 (B) (values are mean ± SD, n = 3, each in duplicate), and EPCR (C) (values are mean ± SD, n = 8, each in duplicate). Significance of MP and TMI-2 effects was calculated by t-test (Mann-Whitney U-test).
We also investigated MP effects on the release of ICAM-1 and EPCR and on the secretion of MCP-1. MPs significantly increased the amount of ICAM-1 by twofold in the culture medium (Figure 4B), but both basal and MP-induced release of ICAM-1 were unaffected by TMI-2. Plaque MPs significantly augmented the release of EPCR by 62% (Figure 4C). Both basal and MP-stimulated releases of EPCR were inhibited by TMI-2 by 82 and 85%, respectively. MCP-1 secretion was analyzed as a marker of cell activation. Exposure of HUVECs to plaque MPs did not affect the secretion of MCP-1 (control, 43 ± 3; with MPs, 41 ± 7 pg/μg cell proteins; mean ± SD, n = 6; P = 0.7), and this secretion was unaffected by TMI-2 (1.0 μmol/L), both in control and MP-treated cells. These results suggest that MPs did not alter the cell secretory activity within the 2 hours of incubation. Exposure of PMA-treated HUVECs to the recombinant active soluble ectodomain of TACE/ADAM/17 for 2 hours did not alter the amount of TNFR-1 in the culture medium (data not shown).
MPs Isolated from Human Atherosclerotic Plaques Stimulate the Release of the TACE/ADAM17 Substrates, TNF and TNFR-1, from ECV-304TNF Cells
ECV-304 cells did not release significant measurable amounts of TNF, either under basal conditions or in the presence of plaque MPs (data not shown). However, in ECV-304TNF cells,21 plaque MPs significantly increased the release of TNF by 47% (Figure 5A). TMI-2 (1.0 μmol/L) inhibited both basal and MP-stimulated release of TNF by 59 and 54%, respectively. Plaque MP-induced TNF release increased with the amount of MPs (Figure 5B). In the culture medium of ECV-304TNF cells, the amount of TNFR-1 was significantly increased by approximately twofold in the presence of plaque MPs. TMI-2 (1.0 μmol/L) inhibited both basal and MP-stimulated TNFR-1 release by 66 and 70%, respectively (Figure 5C). The addition of the active soluble ectodomain of TACE/ADAM/17 did not enhance TNF release (data not shown).
Figure 5.

MPs isolated from human atherosclerotic plaque activate the release of TNF and TNFR-1 from the human cell line ECV-304TNF. ECV-304TNF cells were incubated with MPs as described in Materials and Methods. A: Release of TNF. Values are mean ± SD (n = 6, each performed in duplicate). Significance of MP and TMI-2 effects was calculated by t-test (Mann-Whitney U-test). B: Dose-dependent effect of MPs (expressed as Annexin V+/μl) on TNF release. C: Release of TNFR-1 measured on the same samples as in A.
MPs Isolated from Human Atherosclerotic Plaques Did Not Induce the Release of Exosome-Associated Full-Length TNFR-1 and TNF from Endothelial Cells
It has been shown that under certain conditions, full-length TNFR-122 and full-length TNF39 can be found on exosomes. We therefore examined if this process might account for the increase in TNFR-1 or TNF in culture medium of HUVECs and ECV-304TNF cells stimulated by plaque MPs. The conditioned medium was sequentially centrifuged to pellet exosome-like vesicles, possibly released during incubation (see Materials and Methods). In HUVECs (Figure 6A), tiny amounts of TNFR-1 sedimented at 170,000 × g, whereas the amounts of TNFR-1 present in the supernatants before and after the 170,000 × g centrifugation were not significantly changed whether MPs were added or not. Similar results were obtained with ECV-304TNF cells releasing TNFR-1 (data not shown) or TNF (Figure 6B). The low amount of TNFR-1 released by HUVECs or ECV-304TNF cells did not allow a reliable detection of the cleaved form by immunoblotting even with enhanced detection. However, the higher amount of TNF released from ECV-304TNF cells exposed to plaque MPs allowed detection of only the cleaved TNF soluble form (Figure 6C).
Figure 6.

Plaque MPs do not induce the release of exosome-associated full-length TNFR-1 and TNF from endothelial cells. HUVECs (A) or ECV-304TNF cells (B) were incubated with or without plaque MPs as indicated in Figures 4 and 5, respectively. The culture medium was centrifuged at 20,500 × g (45 minutes at 4°C) to pellet MPs, and half of the resulting supernatant (Sn 20,500 × g) was further centrifuged at 170,000 × g (16 hours at 4°C) to pellet exosomes and the other half left for the same time at 4°C. The two supernatants (Sn 20,500 × g; Sn 170,000 × g) and the 170,000 × g pellet were assayed for TNFR-1 (expressed as total amount, pg). For both cellular types, at least 98% of TNFR-1 or TNF was recovered in the 170,000 × g supernatant (Sn). Values are mean ± SD; n = 3. C: The culture medium of ECV-304TNF cells exposed to plaque MPs (Sn) contain only the cleaved form of TNF compared with 10 μg of recombinant human TNF (rTNF) on the left lane. Representative of two different samples analyzed.
MPs Devoid of TACE/ADAM17 Did Not Enhance TNFR-1 Release from HUVECs
To examine the contribution of TACE/ADAM17 carried by MPs in the observed effects, we took advantage of MPs emitted by apoptotic HUVECs. These MPs did not express detectable levels of TACE/ADAM17 as judged by Western blot and flow cytometry analyses. PMA-stimulated HUVECs were exposed to apoptotic HUVEC-derived MPs (40,260 Annexin V+ MPs/μl) for 2 hours. These MPs did not enhance significant TNFR-1 release, whereas endothelial ICAM-1 release was concomitantly augmented after HUVEC-derived MP exposure by a factor of 5.3 (P < 0.001) (data not shown).
Discussion
The present study demonstrates that the mature active form of TACE/ADAM17 is present at the surface of MPs isolated from human atherosclerotic lesions and that plaque MPs stimulate the shedding of the TACE/ADAM17 substrates TNF, TNFR-1, and EPCR. The present data also confirm that MPs, which have been previously identified in human plaques, can be isolated from human carotid atherosclerotic lesions but not from healthy arteries.2,7,8 The large preponderance of the mature form of TACE/ADAM17 on MPs raises the question on how MPs are particularly enriched with this form. We recently reported that lipid rafts of THP-1 cell membranes contain specifically the mature form of TACE/ADAM17 and that THP-1-derived MPs are enriched in lipid rafts and mature active form of TACE/ADAM17.40 MPs generated from human blood-derived monocytes are also enriched in lipid rafts.41 Similarly, exosomes in which the mature form of TACE/ADAM17 was found,28 also contain large amounts of lipid rafts.42 Thus, the exclusive sequestration of the mature form of TACE/ADAM17 in lipid rafts may account for the preferential recovery of this form in lipid raft-enriched membrane vesicular structures, herein plaque MPs.
The mature form of TACE/ADAM17 detected on plaque MPs is active as judged by their ability to cleave in vitro a mimetic peptide of the cleavage site of pro-TNF. The pro-TNF cleavage induced by MPs was strongly inhibited by TMI-2, an inhibitor reported to preferentially (500-fold) inhibit TACE/ADAM17 over ADAM10,29 a property confirmed herein, and by TIMP-3, the unique known endogenous TACE/ADAM17 inhibitor.30 Human plaque MPs mainly derive from activated leukocytes and also from erythrocytes.7,8 Interestingly, the present study shows that TACE/ADAM17 labeling is preferentially associated with plaque MPs originating from leukocytes or erythrocytes, but also with MPs of endothelial origin, consistent with previous findings.11,43
Our results demonstrate that plaque MPs stimulate the release of TNF, TNFR-1, and EPCR from endothelial cells. The contribution of the sheddase activity of TACE/ADAM17 carried by plaque MPs to these ectodomain cleavages is supported by several findings. First, TNFR-1 or TNF released by target cells did not pellet at 170,000 × g, excluding a possible release of the full-length molecule associated to exosome-like vesicles.22 Second, TACE/ADAM17-negative-like MPs prepared from apoptotic HUVECs, although not completely assimilated to plaque MPs, did not significantly enhance the shedding of TNFR-1, which supports the role of TACE/ADAM17 carried by plaque MPs in substrate ectodomain cleavage. Third, a global increase in protein synthesis caused by MPs within the 2 hours of incubation might have accounted for higher TNFR-1 or TNF secretion. This was, however, not the case because in HUVECs MPs altered neither TNFR-1 mRNA levels nor MCP-1 secretion, and in ECV-304TNF cells TNF synthesis is under a strong but poorly regulated viral promoter. This is consistent with previous studies examining MP effects on endothelial cell cytokine secretion, for which a significant effect could not be seen before 6 or 12 hours of incubation.44,45 Finally, the contribution of TACE/ADAM17 to MP-induced TNF and TNFR-1 shedding is supported by the TMI-2-dependent inhibition of MP effect on TNF, TNFR-1, and EPCR release. Although TACE/ADAM17 is more efficient in TNF shedding when compared with other ADAMs,33,34 we do not exclude the possibility that these proteases, such as ADAM10, which was detected on plaque MPs, may also contribute to TNF shedding. However, TNFR-1 and EPCR shedding is, to the best of our knowledge, recognized to be processed only by TACE/ADAM17. We also observed that plaque MPs stimulated the shedding of ICAM-1, but neither basal nor MP-induced shedding of ICAM-1 was affected by TMI-2. These findings suggest that TACE/ADAM17 was not involved in ICAM-1 shedding, in agreement with previous studies21,46 but not with another one.47
The cellular mechanisms at the basis of MP-induced TACE/ADAM17 substrate shedding from the cell surface remain to be clarified. These may include i) a direct trans-cleavage, ii) a fusion of MPs with cells and delivery of their active TACE/ADAM17 to the cell plasma membrane subsequently allowing cis-cleavage, and iii) a specific activation of the endothelial sheddase at the cell surface by the vesicle itself through an unknown mechanism. The third hypothesis seems unlikely because there was no enhancement of TNFR-1 release on exposure to TACE/ADAM17-negative-like MPs. Our data also indicated that the recombinant soluble active ectodomain of TACE/ADAM17 is unable to cleave TNFR-1 or TNF at the cell surface. This suggests that the integration of TACE/ADAM17 in a cell-derived structure like MPs, may favor its subsequent delivery at the cell surface through fusion of MPs with membrane and then allowing the cis-cleavage of transmembrane substrates. Indeed, a membrane-to-membrane protein transfer has already been reported after MP interaction with target cells.41,48,49 This process may also account for the drastic enhancement of endothelial sICAM-1 release induced by MPs.
The present data also demonstrate that plaque MPs cause the release of endothelial EPCR, a receptor involved in protein C activation at the cell surface. Higher plasma levels of sEPCR, which are associated with an increased risk of thrombosis, are linked to EPCR gene polymorphisms responsible for increased EPCR ectodomain shedding.50 This results in an elevated procoagulant state at the endothelial surface when compared with the common genotype.51 We show herein that the constitutive and MP-stimulated release of EPCR from HUVECs is strongly inhibited by TMI-2, pointing out TACE/ADAM17 as a likely candidate on plaque MPs to cleave EPCR, consistent with the recent data demonstrating that TACE/ADAM17 cleaves EPCR ectodomain.52 Thus, our data raise the possibility that an enhanced TACE/ADAM17-dependent cleavage of EPCR by MPs could add to their already known prothombogenic effect in human atherosclerotic plaques.8 Taken altogether, these results warrant further study to examine the potential role of TACE/ADAM17+ MPs in atherosclerotic plaque development and rupture.
In conclusion, the present study demonstrates that MPs isolated from human carotid atherosclerotic plaques harbor active TACE/ADAM17 and significantly enhance the shedding of TACE/ADAM17 substrates such as TNF, TNFR-1, and EPCR from endothelial cells. This process could contribute to MP-induced alteration and propagation of inflammatory signals in human atherosclerotic lesions.
Supplementary Material
Footnotes
Address reprint requests to Gilles Nalbone, Ph.D., INSERM U626, Faculté de Médecine Timone, 27 Bd. Jean Moulin, 13385 Marseille Cedex 05. E-mail: gilles.nalbone@univmed.fr.
Supported by the Institut National de la Santé et de la Recherche Médicale (Paris), the European Vascular Genomics Network of Excellence (contract LSHM-CT-2003-503254), the “Université de la Méditerranée” (Marseille), the Nouvelle Société Française d’Athérosclérose (Paris) (to M.C.), and the Ministère de la Recherche et de l’Enseignement Supérieur (Paris) (to A.S.L.).
M.C. and A.S.L. equally contributed to this work.
Supplemental material for this article can be found on http://ajp.amjpathol.org.
References
- Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–1695. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- Kockx MM, De Meyer GRY, Muhring J, Jacob W, Bult H, Herman AG. Apoptosis and related proteins in different stages of human atherosclerotic plaques. Circulation. 1998;97:2307–2315. doi: 10.1161/01.cir.97.23.2307. [DOI] [PubMed] [Google Scholar]
- Mallat Z, Tedgui A. Current perspective on the role of apoptosis in atherothrombotic disease. Circ Res. 2001;88:998–1003. doi: 10.1161/hh1001.090571. [DOI] [PubMed] [Google Scholar]
- Morel O, Toti F, Hugel B, Freyssinet JM. Cellular microparticles: a disseminated storage pool of bioactive vascular effectors. Curr Opin Hematol. 2004;11:156–164. doi: 10.1097/01.moh.0000131441.10020.87. [DOI] [PubMed] [Google Scholar]
- Distler JH, Pisetsky DS, Huber LC, Kalden JR, Gay S, Distler O. Microparticles as regulators of inflammation: novel players of cellular crosstalk in the rheumatic diseases. Arthritis Rheum. 2005;52:3337–3348. doi: 10.1002/art.21350. [DOI] [PubMed] [Google Scholar]
- Boulanger CM, Amabile N, Tedgui A. Circulating microparticles: a potential prognostic marker for atherosclerotic vascular disease. Hypertension. 2006;48:180–186. doi: 10.1161/01.HYP.0000231507.00962.b5. [DOI] [PubMed] [Google Scholar]
- Leroyer AS, Isobe H, Leseche G, Castier Y, Wassef M, Mallat Z, Binder BR, Tedgui A, Boulanger CM. Cellular origins and thrombogenic activity of microparticles isolated from human atherosclerotic plaques. J Am Coll Cardiol. 2007;49:772–777. doi: 10.1016/j.jacc.2006.10.053. [DOI] [PubMed] [Google Scholar]
- Mallat Z, Hugel B, Ohan J, Leseche G, Freyssinet J-M, Tedgui A. Shed membrane microparticles with procoagulant potential in human atherosclerotic plaques: a role for apoptosis in plaque thrombogenicity. Circulation. 1999;99:348–353. doi: 10.1161/01.cir.99.3.348. [DOI] [PubMed] [Google Scholar]
- Bonderman D, Teml A, Jakowitsch J, Adlbrecht C, Gyongyosi M, Sperker W, Lass H, Mosgoeller W, Glogar DH, Probst P, Maurer G, Nemerson Y, Lang IM. Coronary no-reflow is caused by shedding of active tissue factor from dissected atherosclerotic plaque. Blood. 2002;99:2794–2800. doi: 10.1182/blood.v99.8.2794. [DOI] [PubMed] [Google Scholar]
- Garton KJ, Gough PJ, Raines EW. Emerging roles for ectodomain shedding in the regulation of inflammatory responses. J Leukoc Biol. 2006;79:1105–1116. doi: 10.1189/jlb.0106038. [DOI] [PubMed] [Google Scholar]
- Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, Castner BJ, Stocking KL, Reddy P, Srinivasan S, Nelson N, Boiani N, Schooley KA, Gerhart M, Davis R, Fitzner JN, Johnson RS, Paxton RJ, March CJ, Cerretti DP. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature. 1997;385:729–733. doi: 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
- Moss ML, Jin SL, Milla ME, Bickett DM, Burkhart W, Carter HL, Chen WJ, Clay WC, Didsbury JR, Hassler D, Hoffman CR, Kost TA, Lambert MH, Leesnitzer MA, McCauley P, McGeehan G, Mitchell J, Moyer M, Pahel G, Rocque W, Overton LK, Schoenen F, Seaton T, Su JL, Warner J, Willard D, Becherer JD. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature. 1997;385:733–736. doi: 10.1038/385733a0. [DOI] [PubMed] [Google Scholar]
- Smalley DM, Ley K. L-selectin: mechanisms and physiological significance of ectodomain cleavage. J Cell Mol Med. 2005;9:255–266. doi: 10.1111/j.1582-4934.2005.tb00354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy P, Slack JL, Davis R, Cerretti DP, Kozlosky CJ, Blanton RA, Shows D, Peschon JJ, Black RA. Functional analysis of the domain structure of tumor necrosis factor-α converting enzyme. J Biol Chem. 2000;275:14608–14614. doi: 10.1074/jbc.275.19.14608. [DOI] [PubMed] [Google Scholar]
- Solomon KA, Pesti N, Wu G, Newton RC. Cutting edge: a dominant negative form of TNF-α converting enzyme inhibits ProTNF and TNFRII secretion. J Immunol. 1999;163:4105–4108. [PubMed] [Google Scholar]
- Peiretti F, Canault M, Deprez-Beauclair P, Berthet V, Bonardo B, Juhan-Vague I, Nalbone G. Intracellular maturation and transport of tumor necrosis factor alpha converting enzyme. Exp Cell Res. 2003;285:278–285. doi: 10.1016/s0014-4827(03)00052-1. [DOI] [PubMed] [Google Scholar]
- Srour N, Lebel A, McMahon S, Fournier I, Fugere M, Day R, Dubois CM. TACE/ADAM-17 maturation and activation of sheddase activity require proprotein convertase activity. FEBS Lett. 2003;554:275–283. doi: 10.1016/s0014-5793(03)01159-1. [DOI] [PubMed] [Google Scholar]
- Canault M, Peiretti F, Kopp F, Bonardo B, Bonzi M-F, Coudeyre J-C, Alessi M-C, Juhan-Vague I, Nalbone G. The TNF alpha converting enzyme (TACE/ADAM17) is expressed in the atherosclerotic lesions of apolipoprotein E-deficient mice: possible contribution to elevated plasma levels of soluble TNF alpha receptors. Atherosclerosis. 2006;187:82–91. doi: 10.1016/j.atherosclerosis.2005.08.031. [DOI] [PubMed] [Google Scholar]
- Black RA, Doedens JR, Mahimkar R, Johnson R, Guo L, Wallace A, Virca D, Eisenman J, Slack J, Castner B, Sunnarborg SW, Lee DC, Cowling R, Jin G, Charrier K, Peschon JJ, Paxton R. Substrate specificity and inducibility of TACE (tumour necrosis factor α-converting enzyme) revisited: the Ala-Val preference, and induced intrinsic activity. Biochem Soc Symp. 2003;70:39–52. doi: 10.1042/bss0700039. [DOI] [PubMed] [Google Scholar]
- Peiretti F, Alessi MC, Henry M, Anfosso F, Juhan-Vague I, Nalbone G. Intracellular calcium mobilization suppresses the TNF-α-stimulated synthesis of PAI-1 in human endothelial cells. Indications that calcium acts at a translational level. Arterioscler Thromb Vasc Biol. 1997;17:1550–1560. doi: 10.1161/01.atv.17.8.1550. [DOI] [PubMed] [Google Scholar]
- Peiretti F, Canault M, Bernot D, Bonardo B, Deprez-Beauclair P, Juhan-Vague I, Nalbone G. Proteasome inhibition activates the transport and the ectodomain shedding of TNF-α receptors in human endothelial cells. J Cell Sci. 2005;118:1061–1070. doi: 10.1242/jcs.01696. [DOI] [PubMed] [Google Scholar]
- Hawari FI, Rouhani FN, Cui X, Yu Z-X, Buckley C, Kaler M, Levine SJ. Release of full-length 55-kDa TNF receptor 1 in exosome-like vesicles: a mechanism for generation of soluble cytokine receptors. Proc Natl Acad Sci USA. 2004;101:1297–1302. doi: 10.1073/pnas.0307981100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvestre J-S, Thery C, Hamard G, Boddaert J, Aguilar B, Delcayre A, Houbron C, Tamarat R, Blanc-Brude O, Heeneman S, Clergue M, Duriez M, Merval R, Levy B, Tedgui A, Amigorena S, Mallat Z. Lactadherin promotes VEGF-dependent neovascularization. Nat Med. 2005;11:499–506. doi: 10.1038/nm1233. [DOI] [PubMed] [Google Scholar]
- Lopez S, Peiretti F, Bonardo B, Juhan-Vague I, Nalbone G. Tumor necrosis factor alpha up-regulates in an autocrine manner the synthesis of plasminogen activator inhibitor type-1 during induction of monocytic differentiation of human HL-60 leukemia cells. J Biol Chem. 2000;275:3081–3087. doi: 10.1074/jbc.275.5.3081. [DOI] [PubMed] [Google Scholar]
- Stoorvogel W, Kleijmeer MJ, Geuze HJ, Raposo G. The biogenesis and functions of exosomes. Traffic. 2002;3:321–330. doi: 10.1034/j.1600-0854.2002.30502.x. [DOI] [PubMed] [Google Scholar]
- Théry C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol. 2002;2:569–579. doi: 10.1038/nri855. [DOI] [PubMed] [Google Scholar]
- Théry C, Regnault A, Garin J, Wolfers J, Zitvogel L, Ricciardi-Castagnoli P, Raposo G, Amigorena S. Molecular characterization of dendritic cell-derived exosomes: selective accumulation of the heat shock protein hsc73. J Cell Biol. 1999;147:599–610. doi: 10.1083/jcb.147.3.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeck A, Keller S, Riedle S, Sanderson MP, Runz S, Le Naour F, Gutwein P, Ludwig A, Rubinstein E, Altevogt P. A role for exosomes in the constitutive and stimulus-induced ectodomain cleavage of L1 and CD44. Biochem J. 2006;393:609–618. doi: 10.1042/BJ20051013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Hegen M, Xu J, Keith JC, Jin G, Du X, Cummons T, Sheppard BJ, Sun L, Zhu Y, Rao VR, Wang Q, Xu W, Cowling R, Nickerson-Nutter C, Gibbons J, Skotnicki J, Lin L-L, Levin J. Characterization of (2R,3S)-2-([4-(2-butynyloxy)phenyl]sulfonyl amino)-N,3-dihydroxybutanamide, a potent and selective inhibitor of TNF-α converting enzyme. Int Immunopharmacol. 2004;4:1845–1857. doi: 10.1016/j.intimp.2004.08.003. [DOI] [PubMed] [Google Scholar]
- Amour A, Slocombe PM, Webster A, Butler M, Knight CG, Smith BJ, Stephens PE, Shelley C, Hutton M, Knauper V, Docherty AJ, Murphy G. TNF-alpha converting enzyme (TACE) is inhibited by TIMP-3. FEBS Lett. 1998;435:39–44. doi: 10.1016/s0014-5793(98)01031-x. [DOI] [PubMed] [Google Scholar]
- Taraboletti G, D’Ascenzo S, Borsotti P, Giavazzi R, Pavan A, Dolo V. Shedding of the matrix metalloproteinases MMP-2, MMP-9, and MT1-MMP as membrane vesicle-associated components by endothelial cells. Am J Pathol. 2002;160:673–680. doi: 10.1016/S0002-9440(10)64887-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosendahl MS, Ko SC, Long DL, Brewer MT, Rosenzweig B, Hedl E, Anderson L, Pyle SM, Moreland J, Meyers MA, Kohno T, Lyons D, Lichenstein HS. Identification and characterization of a pro-tumor necrosis factor-α-processing enzyme from the ADAM family of zinc metalloproteases. J Biol Chem. 1997;272:24588–24593. doi: 10.1074/jbc.272.39.24588. [DOI] [PubMed] [Google Scholar]
- Killar L, White J, Black R, Peschon J. Adamalysins: a family of metzincins including TNF-α converting enzyme (TACE). Ann NY Acad Sci. 1999;878:442–452. doi: 10.1111/j.1749-6632.1999.tb07701.x. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Saftig P, Hartmann D, Blobel C. Evaluation of the contribution of different ADAMs to tumor necrosis factor α (TNFα) shedding and of the function of the TNFα ectodomain in ensuring selective stimulated shedding by the TNFα convertase (TACE/ADAM17). J Biol Chem. 2004;279:42898–42906. doi: 10.1074/jbc.M403193200. [DOI] [PubMed] [Google Scholar]
- Blobel CP. ADAMS: key components in EGFR signalling and development. Nat Rev Mol Cell Biol. 2005;6:32–43. doi: 10.1038/nrm1548. [DOI] [PubMed] [Google Scholar]
- Ludwig A, Hundhausen C, Lambert MH, Broadway N, Andrews RC, Bickett DM, Leesnitzer MA, Becherer JD. Metalloproteinase inhibitors for the disintegrin-like metalloproteinases ADAM10 and ADAM17 that differentially block constitutive and phorbol ester-inducible shedding of cell surface molecules. Comb Chem High Throughput Screen. 2005;8:161–171. doi: 10.2174/1386207053258488. [DOI] [PubMed] [Google Scholar]
- Peschon JJ, Slack JL, Reddy P, Stocking KL, Sunnarborg SW, Lee DC, Russell WE, Castner BJ, Johnson RS, Fitzner JN, Boyce RW, Nelson N, Kozlosky CJ, Wolfson MF, Rauch CT, Cerretti DP, Paxton RJ, March CJ, Black RA. An essential role for ectodomain shedding in mammalian development. Science. 1998;282:1281–1284. doi: 10.1126/science.282.5392.1281. [DOI] [PubMed] [Google Scholar]
- Imaizumi T, Itaya H, Fujita K, Kudoh D, Kudoh S, Mori K, Fujimoto K, Matsumiya T, Yoshida H, Satoh K. Expression of tumor necrosis factor-α in cultured human endothelial cells stimulated with lipopolysaccharide or interleukin-1α. Arterioscler Thromb Vasc Biol. 2000;20:410–415. doi: 10.1161/01.atv.20.2.410. [DOI] [PubMed] [Google Scholar]
- Zhang H-G, Liu C, Su K, Yu S, Zhang L, Zhang S, Wang J, Cao X, Grizzle W, Kimberly RP. A membrane form of TNF-α presented by exosomes delays T cell activation-induced cell death. J Immunol. 2006;176:7385–7393. doi: 10.4049/jimmunol.176.12.7385. [DOI] [PubMed] [Google Scholar]
- Tellier E, Canault M, Rebsomen L, Bonardo B, Juhan-Vague I, Nalbone G, Peiretti F. The shedding activity of ADAM17 is sequestered in lipid rafts. Exp Cell Res. 2006;312:3969–3980. doi: 10.1016/j.yexcr.2006.08.027. [DOI] [PubMed] [Google Scholar]
- Del Conde I, Shrimpton CN, Thiagarajan P, Lopez JA. Tissue-factor-bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood. 2005;106:1604–1611. doi: 10.1182/blood-2004-03-1095. [DOI] [PubMed] [Google Scholar]
- de Gassart A, Geminard C, Fevrier B, Raposo G, Vidal M. Lipid raft-associated protein sorting in exosomes. Blood. 2003;102:4336–4344. doi: 10.1182/blood-2003-03-0871. [DOI] [PubMed] [Google Scholar]
- Kieseier BC, Pischel H, Neuen-Jacob E, Tourtellotte WW, Hartung HP. ADAM-10 and ADAM-17 in the inflamed human CNS. Glia. 2003;42:398–405. doi: 10.1002/glia.10226. [DOI] [PubMed] [Google Scholar]
- Nomura S, Tandon NN, Nakamura T, Cone J, Fukuhara S, Kambayashi J. High-shear-stress-induced activation of platelets and microparticles enhances expression of cell adhesion molecules in THP-1 and endothelial cells. Atherosclerosis. 2001;158:277–287. doi: 10.1016/s0021-9150(01)00433-6. [DOI] [PubMed] [Google Scholar]
- Mesri M, Altieri DC. Leukocyte microparticles stimulate endothelial cell cytokine release and tissue factor induction in a JNK1 signaling pathway. J Biol Chem. 1999;274:23111–23118. doi: 10.1074/jbc.274.33.23111. [DOI] [PubMed] [Google Scholar]
- Garton KJ, Gough PJ, Philalay J, Wille PT, Blobel CP, Whitehead RH, Dempsey PJ, Raines EW. Stimulated shedding of vascular cell adhesion molecule 1 (VCAM-1) is mediated by tumor necrosis factor-α-converting enzyme (ADAM 17). J Biol Chem. 2003;278:37459–37464. doi: 10.1074/jbc.M305877200. [DOI] [PubMed] [Google Scholar]
- Tsakadze NL, Sithu SD, Sen U, English WR, Murphy G, D’Souza SE. Tumor necrosis factor-α converting enzyme (TACE/ADAM-17) mediates the ectodomain cleavage of intercellular adhesion molecule-1 (ICAM-1). J Biol Chem. 2006;281:3157–3164. doi: 10.1074/jbc.M510797200. [DOI] [PubMed] [Google Scholar]
- Mack M, Kleinschmidt A, Bruhl H, Klier C, Nelson PJ, Cihak J, Plachy J, Stangassinger M, Erfle V, Schlondorff D. Transfer of the chemokine receptor CCR5 between cells by membrane-derived microparticles: a mechanism for cellular human immunodeficiency virus 1 infection. Nat Med. 2000;6:769–775. doi: 10.1038/77498. [DOI] [PubMed] [Google Scholar]
- Mause SF, von Hundelshausen P, Zernecke A, Koenen RR, Weber C. Platelet microparticles: a transcellular delivery system for RANTES-promoting monocyte recruitment on endothelium. Arterioscler Thromb Vasc Biol. 2005;25:1512–1518. doi: 10.1161/01.ATV.0000170133.43608.37. [DOI] [PubMed] [Google Scholar]
- Saposnik B, Reny J-L, Gaussem P, Emmerich J, Aiach M, Gandrille S. A haplotype of the EPCR gene is associated with increased plasma levels of sEPCR and is a candidate risk factor for thrombosis. Blood. 2004;103:1311–1318. doi: 10.1182/blood-2003-07-2520. [DOI] [PubMed] [Google Scholar]
- Qu D, Wang Y, Song Y, Esmon NL, Esmon CT. The Ser219→Gly dimorphism of the endothelial protein C receptor contributes to the higher soluble protein levels observed in individuals with the A3 haplotype. J Thromb Haemost. 2006;4:229–235. doi: 10.1111/j.1538-7836.2005.01676.x. [DOI] [PubMed] [Google Scholar]
- Qu DWY, Esmon NL, Esmon CT. Regulated endothelial protein C receptor shedding is mediated by tumor necrosis factor-α converting enzyme/ADAM17. J Thromb Haemost. 2007;5:395–402. doi: 10.1111/j.1538-7836.2007.02347.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}