

Abstract
Glucose stimulates both insulin secretion and hydrolysis of arachidonic acid (AA) esterified in membrane phospholipids of pancreatic islet β-cells, and these processes are amplified by muscarinic agonists. Here we demonstrate that nonesterified AA regulates the biophysical activity of the pancreatic islet β-cell-delayed rectifier channel, Kv2.1. Recordings of Kv2.1 currents from INS-1 insulinoma cells incubated with AA (5 μM) and subjected to graded degrees of depolarization exhibit a significantly shorter time-to-peak current interval than do control cells. AA causes a rapid decay and reduced peak conductance of delayed rectifier currents from INS-1 cells and from primary β-cells isolated from mouse, rat, and human pancreatic islets. Stimulating mouse islets with AA results in a significant increase in the frequency of glucose-induced [Ca2+] oscillations, which is an expected effect of Kv2.1 channel blockade. Stimulation with concentrations of glucose and carbachol that accelerate hydrolysis of endogenous AA from islet phosphoplipids also results in accelerated Kv2.1 inactivation and a shorter time-to-peak current interval. Group VIA phospholipase A2 (iPLA2β) hydrolyzes β-cell membrane phospholipids to release nonesterified fatty acids, including AA, and inhibiting iPLA2β prevents the muscarinic agonist-induced accelerated Kv2.1 inactivation. Furthermore, glucose and carbachol do not significantly affect Kv2.1 inactivation in β-cells from iPLA2β−/− mice. Stably transfected INS-1 cells that overexpress iPLA2β hydrolyze phospholipids more rapidly than control INS-1 cells and also exhibit an increase in the inactivation rate of the delayed rectifier currents. These results suggest that Kv2.1 currents could be dynamically modulated in the pancreatic islet β-cell by phospholipase-catalyzed hydrolysis of membrane phospholipids to yield non-esterified fatty acids, such as AA, that facilitate Ca2+ entry and insulin secretion.
Glucose metabolism within the pancreatic islet β-cell generates a multitude of signals that regulate insulin secretion. Changes in the relative concentrations of the metabolites ATP and ADP cause β-cell depolarization via closure of ATP-sensitive potassium channels (KATP),3 which results in activation of voltage-operated Ca2+ channels, Ca2+ influx, and insulin secretion (1). The extent of β-cell depolarization and insulin release are regulated in part by the activation of repolarizing ion channels, including the voltage-gated potassium channel, Kv2.1 (2–4). One mechanism employed by pancreatic β-cells to regulate the biophysical activity of the ion channels involved in insulin release involves hydrolysis of membrane phospholipids to yield mediators that include inositol triphosphates and free fatty acids (5–8).
Pharmacologic, biochemical, and genetic evidence suggests that glucose-stimulated hydrolysis of esterified arachidonic acid from β-cell membrane phospholipids is required for physiological insulin secretion (7–25). Pancreatic islet β-cells contain high levels of arachidonic acid compared with other tissues (7, 9–12 24, 25), and about two-thirds of islet β-cell glycerophospholipids contain arachidonic acid as the sn-2 substituent (23–25). Islet phospholipase A2 (PLA2) enzyme(s) activated by stimulation with glucose and/or by G-protein signaling mechanisms catalyze hydrolysis of arachidonic acid from β-cell membrane phospholipids and cause levels of free arachidonic acid to rise by up to 3-fold to the mid-micromolar range (7, 13, 14, 27, 28). Such concentrations of arachidonic acid amplify both the rise in β-cell [Ca2+] and insulin secretion induced by glucose (7, 15–17, 23, 25, 27, 28).
Pharmacologic inhibition or molecular biologic suppression of expression of the pancreatic islet β-cell Group VIA phospholipase A2 (iPLA2β) enzyme (24, 29, 30) results in attenuation of insulin secretory responses induced by glucose and other secretagogues (18–22, 24). The products of iPLA2β action on its phospholipid substrates include a nonesterified fatty acid, such as AA, and a 2-lysphophosplipid, such as lysophosphatidylcholine (LPC), and β-cell levels of both products correlate with iPLA2β expression level (21, 22, 31, 32). Modulation of iPLA2β activity and the rate of secretagogue-induced hydrolysis of arachidonic acid from β-cell membrane phospholipids might thus represent a tunable signal to amplify or attenuate islet insulin secretion in various (patho)physiologic settings (19, 24).
The products of iPLA2β action, including physiologically attainable concentrations of nonesterified AA, amplify secretagogue-induced rises in β-cell [Ca2+] (7, 17, 18, 27, 33), and several mechanisms have been proposed to explain these effects. These include facilitating Ca2+ entry (7, 17), perhaps by effects of AA on voltage-operated Ca2+ channels (34), effects of AA and LPC on KATP (35), effects of AA 12-lipoxygenase product(s) on an electrogenic plasma membrane Na+/K+-ATPase (36), and effects of LPC (37) on plasma membrane store-operated cation channels (38). Arachidonic acid modulates the activity of a number of ion channels, including KATP channels and delayed rectifier K+ channels (29–41). The effects of AA on β-cell intracellular [Ca2+] dynamics are likely to reflect the combined actions on such ion channels, possibly in concert with effects on G-protein-coupled receptor 40 signaling.
In this study, we investigated the effects of arachidonic acid and iPLA2β on the Kv2.1-delayed rectifier channel of the pancreatic β-cell and on islet [Ca2+] dynamics. We find that incubating islet β-cells with exogenous arachidonic acid results in significant reduction of total Kv activity and increased glucose-stimulated islet [Ca2+] oscillations. Concentrations of glucose and carbachol that stimulate islet PLA2 activity (13, 14, 18) are also demonstrated here to reduce Kv currents and thereby decrease repolarization. We observe similar reductions in Kv currents with a genetically modified INS-1 insulinoma cell line that overexpresses iPLA2β. As far as we are aware, genetically modified cell lines with stably altered expression of iPLA2β have not heretofore been used to examine the role of the enzyme in modulating the activity of specific β-cell ion channels involved in regulating [Ca2+] dynamics.
EXPERIMENTAL PROCEDURES
Isolation and Culture of Mouse, Rat, and Human Islets of Langerhans, Insulinoma Cells, and HEK Cells
Islets were isolated from pancreata of 1- to 5-month-old C57BL/J6 wild-type mice (Jackson Laboratories); of iPLA2β−/− mice, which were generated as described elsewhere (24); or of rats using collagenase digestion and Ficoll gradients as described previously (2). Human cadaveric islets were a generous gift from Dr. M. Garfinkel, University of Chicago. Islets were dissociated in 0.005% trypsin, placed on glass coverslips, and cultured for 16 h in RPMI 1640 medium supplemented with 10% fetal calf serum, concentrations of glucose specified in the figure legends, 100 IU/ml penicillin, and 100 μg/ml streptomycin. INS-1 cells and HEK cells expressing Kv2.1-GFP (a generous gift from Michael Tamkun (42)) were cultured for 16 h in RPMI 1640 medium containing 11.1 mM glucose and supplemented with 10% fetal calf serum, 100 IU/ml penicillin, and 100 μg/ml streptomycin. Cells and islets were maintained in a humidified incubator at 37 °C under an atmosphere of 95% air/5% CO2.
Electrophysiological Recordings
Voltage-activated currents were recorded using whole-cell ruptured patch clamps with an Axopatch 200B amplifier and pCLAMP software (Molecular Devices, Sunnyvale, CA). Patch electrodes (2–4 micro-ohms) were loaded with intracellular solution containing (in mmol/liter) KCl, 140; MgCl2·6H2O, 1; EGTA, 10; Hepes, 10; MgATP, 5; pH 7.25 with KOH. Cells were perifused with a Krebs-Ringer buffer (KRB) containing (in mmol/liter) NaCl, 119; CaCl2 · [(H2O)6], 2; KCl, 4.7; Hepes, 10; MgSO4, 1.2; KH2PO4, 1.2; glucose, 14.4; adjusted to pH 7.3 with NaOH. When indicated, cells were incubated with arachidonic acid (Sigma) in ethanol (<0.3%). Ethanol (0.03%) treatment of cells alone had no effect on their KV currents. When indicated, cells were pretreated with a bromenol lactone (BEL) iPLA2β inhibitor (43) obtained from Sigma and/or treated with carbachol (Sigma) in KRB with 20 mM glucose. Cells were also pretreated with the S or R enantomers of BEL obtained from Cayman Chemical Co. (Ann Arbor, MI) in 20 mM glucose and then treated with carbachol. S-BEL inactivates iPLA2β but not iPLA2γ, and R-BEL inactivates iPLA2γ but not iPLA2β (71). The inactivation curves were fitted to the waveform with a single exponential function using Excel software and plotted with the standard error of the mean as a dashed line.
Confocal Immunofluorescence Microscopy
An inverted microscope (Olympus, Tokyo, Japan) with a dual rotating-disk confocal scanner (CSU10; Yokogawa Electric, Tokyo, Japan) was used for confocal imaging of HEK cells expressing KV2.1-GFP. The cells were illuminated at 488 nm, and emitted light was filtered through a 530/30 nm filter (Omega Filters, Brattleboro, VT).
Measurement of Cytoplasmic [Ca2+]
Mouse islets were incubated (20 min, 37 °C) in KRB supplemented with 5 μmol/liter of Fura-2 acetoxymethyl ester (Molecular Probes, Eugene, OR). Fluorescence imaging was performed using a Nikon Eclipse TE2000-U microscope equipped with an epifluorescence illuminator and a 20× fluorescence objective (Fryer Company Inc., Huntley, IL) and MetaFluor software (Molecular Devices, Sunnyvale, CA). Cells were perifused at 37 °C at a flow of 1 ml/min with appropriate KRB-based solutions that contained glucose concentrations specified in the figures with or without the indicated concentrations of arachidonic acid or tolbutamide. The ratios of emitted fluorescence intensities at excitation wavelengths of 340 and 380 nm (F340/F380) were determined every 5 s with background subtraction.
RESULTS
Effects of Arachidonic Acid on β-Cell Kv2.1 Channel Activity
Stimulation of pancreatic islet β-cells with glucose induces both the secretion of insulin and hydrolysis of arachidonic acid from membrane phospholipids, and the latter event is thought to facilitate Ca2+ entry and thereby to amplify insulin secretion (7, 15, 17, 18, 27, 33, 36, 44). One mechanism whereby arachidonic acid might affect glucose-induced Ca2+ entry into β-cells is by modulating activity of delayed rectifier K+ channels that limit glucose-induced depolarization of the β-cell plasma membrane (2–4, 40–42, 45–51), and we therefore examined the effects of arachidonic acid on β-cell Kv2.1 channel activity.
As illustrated in Fig. 1A, whole cell recordings from rat INS-1 insulinoma cells show a typical Kv2.1-like delayed rectifier current when stimulated with depolarizing voltage steps. Maximal INS-1 Kv current amplitude at a depolarization step of 60 mV was significantly reduced by 33.4% upon incubation with 5 μM arachidonic acid (n = 7). INS-1 Kv currents also exhibited significantly accelerated current decline from their respective peak current amplitudes (Fig. 1, C and D) upon incubation with 5 μM arachidonic acid. Arachidonic acid also significantly shortened by a mean of 8 ms the (“time-to-peak”) interval required to achieve peak INS-1 Kv current amplitude during a 30 mV depolarization (Fig. 2C). These effects of arachidonic acid are not use-dependent because Kv currents elicited after a 5-min incubation of cells with 5 μM arachidonic acid when held at −80 mV show effects similar those observed (20) with cells subjected to depolarizing steps during the 5 min period of incubation with arachidonate (Fig. 2B). There were also no significant changes in Kv current inactivation with continued depolarizing steps applied after a 5 min preincubation with 5 μM arachidonic acid. The effects of arachidonate on Kv2.1 were rapid and occurred within 45 s.
FIGURE 1. Arachidonic acid negatively regulates β-cell Kv2.1 channel activity.
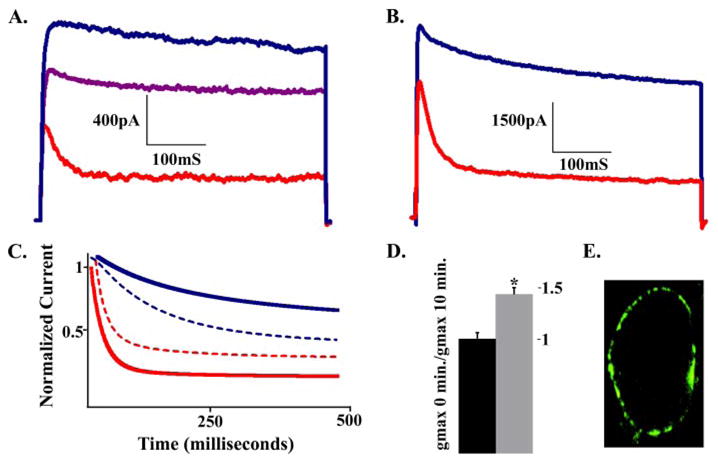
A, Kv currents from an INS-1 cell recorded at +60 mV before (blue trace) or after (red trace) 10 min of incubation with 5 μM arachidonic acid or 15 min (purple trace) after washout of arachidonic acid. B, Kv currents recorded from a HEK cell expressing Kv2.1 at +60 mV before (blue trace) and 10 min after (red trace) incubation with 10 μM arachidonic acid. C, decay curves of Kv current traces from INS-1 cells at +60 mV before (blue line) or after (red line) incubation with 5 μM arachidonic acid. Dashed lines represent standard errors of the mean (n = 7). D, Gmax changes before and after INS-1 incubation with vehicle (black bar) or arachidonic acid (gray bar); displayed values are the mean ± S.E. (n = 7, p < 0.05). E, immunoflouroscent confocal-microscropic localization of a Kv2.1-GFP construct expressed in HEK cells.
FIGURE 2. Arachidonic acid affects β-cell-delayed rectifier currents in a use-independent manner.
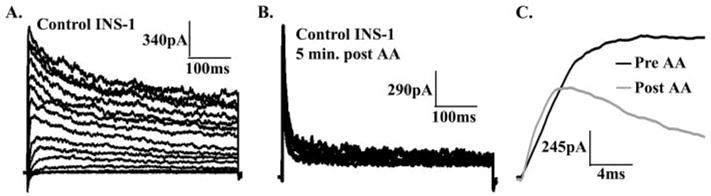
A, control INS-1 cell-delayed rectifier currents elicited by depolarization for 500 ms in 10-mV steps over the range from −70 to +80 mV. B, current recordings from the same INS-1 cell in response to depolarization for 500 ms in 10 mV increments over the range from +30 to +80 mV after 5 min of incubation with 5 μM AA. C, INS-1 KV current traces recorded +30 mV before (black line) or 10 min after (gray line) incubation with 5 μM arachidonic acid.
To examine further effects of arachidonic acid on the activity of the Kv2.1 channel, which is the predominant delayed rectifier of β-cells (4, 46), HEK cells expressing a Kv2.1-GFP construct (42) were studied (Fig. 1E). HEK cells alone have no Kv-like currents whereas HEK cells expressing Kv2.1-GFP show Kv currents similar to INS-1 cells. Like the INS-1 cells Kv2.1-GFP expressing HEK cells developed significantly reduced Kv current amplitudes with increases in their tau’s of inactivation when incubated with arachidonic acid (Fig. 1E). The rate of Kv2.1 activation is not accelerated as significantly in HEK cells as in INS-1 cells. These results demonstrate that arachidonic acid has direct effects on Kv2.1 current amplitude and inactivation kinetics.
Arachidonic Acid Regulates Delayed Rectifier Currents in Both Rodent and Human Pancreatic Islet β-Cells
Because arachidonic acid affects the activity of Kv2.1 channels expressed in immortalized cell lines, we examined the possibility that similar effects would be observed with Kv currents in rodent and human primary β-cells. When incubated with 5 μM arachidonate, both mouse and rat primary β-cells exhibited decreased Kv-like current amplitudes, increased rates of current inactivation, and slightly shorter time-to-peak current intervals than did cells incubated without arachidonic acid (Fig. 3, A and B). Human β-cells also displayed significantly reduced Kv-like current amplitudes and time-to-peak intervals when incubated with arachidonic acid, although the human Kv-like currents exhibited only a slight acceleration in decay rates upon incubation with arachidonic acid (Fig. 3C). These results suggest that arachidonic acid can modulate the activity of the pancreatic islet β-cell repolarizing delayed rectifier Kv currents that are activated upon depolarization of the β-cell plasma membrane.
FIGURE 3. Arachidonic acid affects delayed rectifier currents of primary β-cells prepared from rodent and human pancreatic islets.
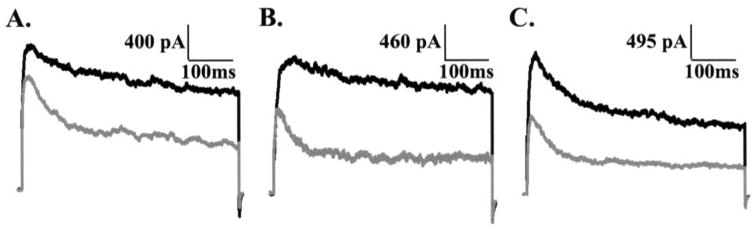
Delayed rectifier currents recorded during a 500 ms depolarization step at +60 mV with primary β-cells isolated from rat (A), mouse (B), or human (C) islets before (black traces) or 10 min after (gray traces) incubation with 5 μM arachidonic acid.
Arachidonic Acid Increases [Ca2+] Fluctuations of Rodent Islets of Langerhans
Because insulin secretion kinetics closely parallel islet β-cell [Ca2+] fluctuations (1) and Kv2.1 participates in regulating β-cell [Ca2+] (2), effects of arachidonate on β-cell [Ca2+] were examined. Stimulation of mouse islets with 14.4 mM glucose induced typical [Ca2+] oscillations, and their frequency was minimally increased upon incubation with 5 μM arachidonic acid and returned to basal frequency after washout (Fig. 4A). Incubating islets with 10 μM arachidonic acid induced a significant increase in [Ca2+] oscillation frequency that returned to prestimulation levels after washout (Fig. 4B). Because arachidonic acid has also been found to modulate β-cell KATP channel activity as well as the sodium potassium ATPase (Na+/K+-ATPase; Refs. 36 and 39), we next examined effects of AA on islet [Ca2+] fluctuations when KATP activity was blocked with tolbutamide alone or in combination with ouabain to block Na+/K+-ATPase activity. When islets were induced to undergo [Ca2+] oscillations by stimulation with glucose, adding 250 μM tolbutamide or 50 μM ouabain resulted in an immediate and persistent rise in [Ca2+] (Fig. 4C and supplemental Fig. 1). Subsequent addition of 10 μM AA induced fast oscillations in [Ca2+] (10 out of 15 islets, supplemental Fig. 1), which is an effect typical of Kv2.1 channel inhibition (47). These results suggest that AA modulates the activity of ion channels in addition to the KATP channel or the Na+/K+-ATPase that affect β-cell [Ca2+], such as Kv2.1.
FIGURE 4. Arachidonic acid affects glucose-induced [Ca2+] oscillations in mouse pancreatic islets.
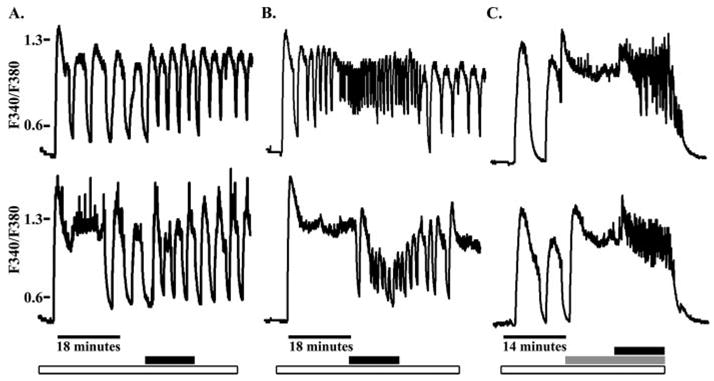
A, representative calcium traces recorded with mouse islets loaded with Fura-2 and incubated with with 14.4 mM glucose (white bar) in the presence (black bar) or absence of 5 μM arachidonic acid. B and C, calcium traces recorded with mouse islets incubated with 14.4 mM glucose alone (white bar) or in combination with 250 μM tolbutamide (gray bar) and 10 μM arachidonic acid (black bar). The intervals in the recordings with no bars represent calcium from islets incubated with 2 mM glucose and no tolbutamide or arachidonic acid.
The Muscarinic Receptor Agonist Carbachol Modulates Kv2.1 Currents in β-Cells
The muscarinic agonist carbachol amplifies both arachidonic acid hydrolysis from pancreatic islet β-cell membrane phospholipids and insulin secretion (14, 18, 44), and we examined the effects of carbachol on β-cell Kv currents. As illustrated in Fig. 5, B and C, Kv currents recorded from INS-1 cells simulated with carbachol (500 μM) displayed a significantly shorter time-to-peak current interval and an increased rate of inactivation compared with currents recorded before stimulation. This suggests that muscarinic receptor occupancy modulates β-cell Kv channel activity. The previously suggested possibility that a PLA2 participates in muscarinic agonist effects on β-cells (14, 18) is consistent with our observation that pretreatment of INS-1 cells with the iPLA2β inhibitor BEL (10 μM; Ref. 43) resulted in a significant reduction in the carbachol-induced increase in the rate of Kv inactivation (Fig. 5, D and F), although the BEL-treated cells still exhibited a slight reduction in the time-to-peak Kv current interval upon stimulation with carbachol (Fig. 5D). Pretreatment of cells with the iPLA2γ-selective (71) enantiomer of BEL (R-BEL), which has little effect on iPLA2β activity, did not influence carbachol-induced inactivatition of INS-1 Kv currents (n = 7; supplemental Fig. 2, A and C), whereas the iPLA2β-selective enantiomer (S-BEL) (71) still prevented Cch induced Kv inactivation (n = 6; supplemental Fig. 2, B and D).
FIGURE 5. Effects of the muscarinic agonist carbachol on INS-1 cell-delayed rectifier currents.
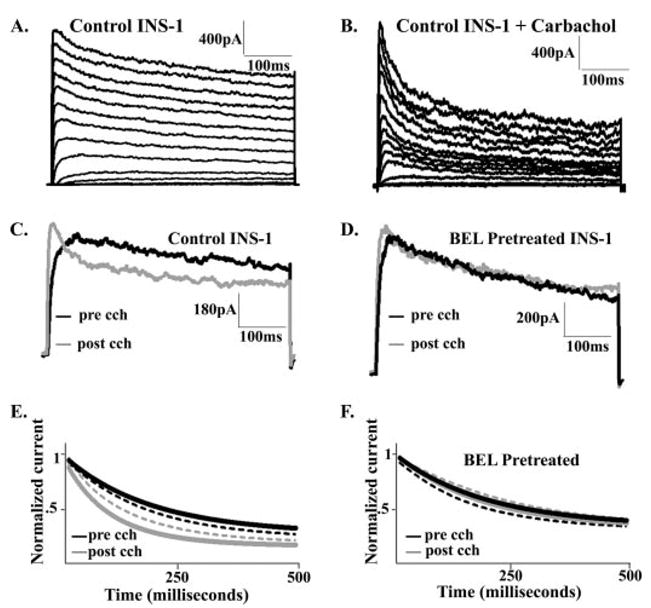
A, traces recorded from an INS-1 cell subjected to 500-ms depolarization in 10-mV increments from −70 to +80 10 mV. B, traces recorded from the same INS-1 cell as in A 10 min after incubation with carba-chol (500 μM). C, traces from an INS-1 cell subjected to a 500 ms depolarization step from −80 to −5 mV. D, traces from an INS-1 cell pretreated with 10 μM BEL and then subjected to a 500-ms step from −80 to −5 mV before (black trace) and 10 min after incubation with 500 μM carbachol (gray bar). E, decay curves of Kv current traces from an INS-1 cell subjected to 500-ms depolarization at +60 mV before (black line) and after (gray line) incubation with 500 μM carbachol. Dashed lines represent standard errors of the mean (n = 7). F, Kv current decay curves from INS-1 cells that had been pretreated with BEL as in D and then subjected to 500-ms depolarization at +60 mV before (black line) and after (gray line) incubation with 500 μM carbachol. Dashed lines represent standard errors of the mean (n = 6).
To examine the physiological effects of iPLA2β on β-cell Kv currents, studies were performed with islet β-cells isolated from iPLA2β-null mice, which were prepared as described else-where (24). Because phospholipase C is also activated by Cch, these experiments utilized intracellular calcium buffering to prevent phosoholipase C activation. Carbachol and glucose were applied to control and iPLA2β−/− mouse pancreatic islet β-cells, and Kv currents were then recorded (20). The control β-cell Kv currents exhibit a significant left shift in their time to peak response to 20 mM glucose, and this response is maintained upon addition of carbachol (500 μM; Fig. 6A, inset). A left shift in Kv activation in response to glucose is also observed with iPLA2β−/− β-cells (Fig. 6B, inset). Interestingly, 20 mM glucose causes a significant increase in inactivation in the control β-cells, and this effect is significantly augmented upon addition of carbachol (500 μM; Fig. 6, A and C, thin black line and thick gray lines, respectively). The iPLA2β−/− β-cells, however, failed to exhibit significant changes in Kv inactivation with glucose (20 mM) in the presence or absence of carbachol (500 μM; Fig. 6, B and D, thin black line and thick gray lines, respectively). These results suggest that muscarinic receptor occupancy increases β-cell Kv current inactivation by iPLA2β-regulated mechanism(s), and this results in a shorter time to peak Kv current response.
FIGURE 6. Delayed rectifier currents from iPLA2β−/− and control mouse β-cells.
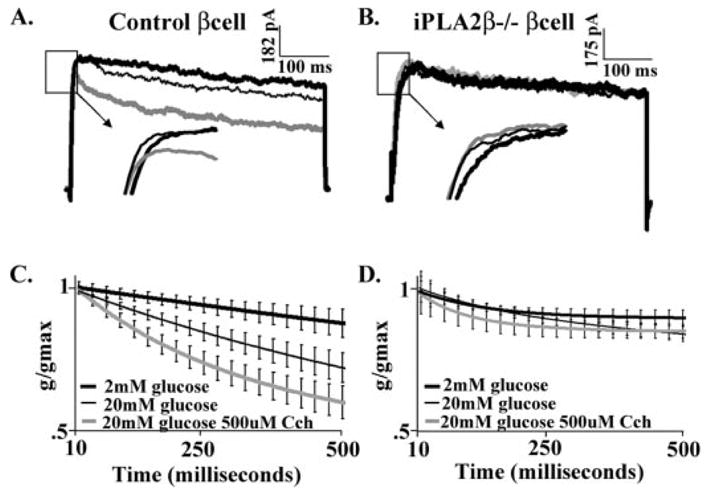
A, Kv currents recorded from a control mouse β-cells treated with 2 mM glucose (thick black line), 20 mM glucose (thin black line), and 20 mM glucose and 500 μM carbachol (thick gray line) during a 500-ms step from −80 mV to −5 mV. B, Kv currents recorded from iPLA2β null mouse β-cell treated with 2 mM glucose (thick black line), 20 mM glucose (thin black line), and 20 mM glucose and 500 μM carbachol (thick gray line) during a 500-ms step from −80 mV to −5 mV. Insets of A and B depict the expanded current traces from their respective boxed areas. C and D, Kv current decay curves from control (C; n = 8) and iPLA2β null (D; n = 7) β-cells cells treated with 2 mM glucose (thick black line), 20 mM glucose (thin black line), and 20 mM glucose and 500 μM carbachol (thick gray line), ± S.E. black bars.
Phospholipase A2 Regulates Delayed Rectifier Current Activity
iPLA2β is expressed by islet β-cells from rats (29), humans (47), and mice (48), and by several insulinoma cell lines (31, 38, 39). Upon activation, iPLA2β hydrolyzes the sn-2 substituent from phospholipids substrates, such as phosphatidylcholine, to yield a free fatty acid and a lysophospholipid, such as LPC (44, 54, 55). Because arachidonate is the most abundant sn-2 substituent in β-cell membrane phospholipids (23, 25, 26), activation of iPLA2β upon stimulation of islets with glucose results in liberation of arachidonate as the preponderant free fatty acid (7). Stably transfected INS-1 cells that overexpress iPLA2β (INS1-iPLA2β) exhibit amplified insulin secretory responses to glucose and other secretatogues (21, 56). We therefore compared Kv activity in control INS-1 cells and those that overexpress iPLA2β (“INS-1-iPLA2β cells”), both treated with 20 mM glucose. As illustrated in Fig. 7, Kv currents from INS-1-iPLA2β cells exhibit accelerated activation rates and a significant increase in decay rates, although the peak Kv-like current amplitudes do not differ significantly between the two cell lines. These results demonstrate that iPLA2β expression level in β-cell lines correlates with their Kv-like channel activity.
FIGURE 7. Delayed rectifier currents from INS-1 cells that express different levels of iPLA2.
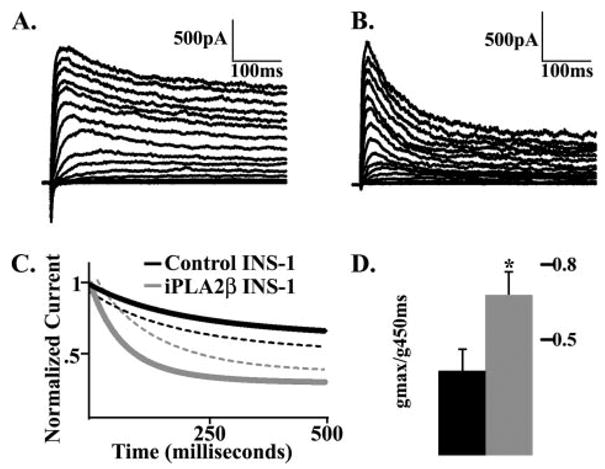
β. A, delayed rectifier currents recorded from control INS-1 cells subjected to 500 ms of depolarization at −70 to +80 mV in 10-mV increments. B, delayed rectifier currents recorded from stably transfected INS-1 cells that overexpress iPLA2β and treated as in A. C, Kv current decay curves recorded from control INS-1 cells (black trace) or from stably transfected INS-1 cells that overexpress PLA2β (gray trace) subjected to a 500-ms depolarization from −80 to +60 mV. Dashed lines represent standard errors of the mean (n = 6). D, change in Gmax observed with control INS-1 cells (gray bar) or with stably transfected cells that overexpress iPLA2β (black bar) subjected to a 500-ms depolarization step at +60 mV. Mean values (±S.E., n = 6) are displayed (p < 0.05).
DISCUSSION
Concentrations of glucose that stimulate insulin secretion also induce phospholipid hydrolysis in pancreatic islet β-cells, and the resultant accumulation of phospholipid-derived mediators including AA is thought to amplify insulin secretion (11–20, 23–25, 27, 28, 31–34, 36, 57, 58). The results presented here demonstrate that AA modulates Kv2.1-delayed rectifier K+ channel currents in β-cells, and these channels are thought to modulate glucose-induced fluctuations in β-cell [Ca2+] and insulin secretion (2–4, 40–42, 45–51). Whole cell patch clamp recordings of delayed rectifier currents elicited by application of graded degrees of depolarization to β-cells incubated with AA display a shorter time-to-peak current interval, a decrease in peak current amplitude, and accelerated inactivation compared with control cells. The iPLA2β expressed in pancreatic islet β-cells hydrolyzes AA esterified in membrane phospholipids (18–22, 24, 31, 32, 36), and we find that overexpression of iPLA2β in β-cells mimics the effects of exogenous AA on Kv-like currents. Furthermore, β-cells cells that do not express iPLA2β or β-cells treated with pharmacological inhibitors of iPLA2β have attenuated muscarinic agonist-induced Kv channel inactivation. These findings suggest that iPLA2β might participate in dynamic regulation of the repolarizing effects of Kv2.1 channel activation in β-cells.
Arachidonic acid causes other slowly inactivating delayed rectifier K+ channels, such as Kv3.4, to resemble fast inactivating A-type K+ currents (41). The phospholipid content of esterified AA is higher in pancreatic islets than in other tissues (23, 25, 26, 29), and arachidonate is hydrolyzed from phospholipids upon stimulation of islets with secretagogues (13, 14, 27, 31, 36, 44) and accumulates to micromolar concentrations (7, 27, 28) that are sufficient to modulate delayed rectifier Kv channel activity (41). We find that incubating rodent and human pancreatic islet β-cells with low micromolar concentrations of AA results in accelerated inactivation of their delayed rectifier Kv-like currents (Figs. 1 and 2). Kv2.1 is the primary β-cell-delayed rectifier potassium channel (4, 46), and β-cells incubated with AA secrete more insulin when stimulated than do cells incubated in the absence of AA (7). This response mimics the effects of Kv2.1 blockade under similar conditions (28, 29). These results suggest that AA might modulate the duration of stimulus-induced depolarization in β-cells through reduction in the repolarizing function of Kv2.1.
The kinetics of insulin secretion from islets closely parallels β-cell [Ca2+] fluctuations (1), and Kv2.1 participates in regulating [Ca2+] in β-cells stimulated with glucose (2). Secretagogue-induced hydrolysis of AA from β-cell phospholipids (7, 13, 14, 27, 28) is also thought to facilitate Ca2+ entry (15–17, 18, 33) and thereby to amplify insulin secretion (44). The frequency of glucose-induced [Ca2+] oscillations in islets increases when Kv2.1 channels are inhibited (47). This results in a reduction in the rate of β-cell repolarization, increased duration of stimulated depolarization, and amplification of insulin secretion (47). Similarly, incubating islets with AA results in an increased frequency of glucose-induced [Ca2+] fluctuations (Fig. 4B). KATP channels are partially inhibited by AA (39), and this could contribute to the increase in [Ca2+] fluctuations when islets are incubated with AA (39). AA causes increased islet [Ca2+] fluctuations even when KATP channels are inhibited with tolbutamide, however, which supports our findings that modulation of Kv2.1 activity could also participate in effects of AA on β-cell [Ca2+] fluctuations.
Arachidonate is the most abundant sn-2 substituent of islet phospholipids from rodents and humans (23, 25, 26) and accumulates to greater levels than do other fatty acids in secretagogue-stimulated islets (7, 27). The iPLA2β enzyme is expressed by islets from rats, mice, and humans (20, 24–26) and by several insulinoma cell lines (31, 52, 53), and iPLA2β catalyzes the hydrolysis of the sn-2 ester bond of phospholipids to yield a free fatty acid, such as arachidonic acid, and a 2-lysophospholipid, such as LPC (54, 55). Secretagogue-stimulated hydrolysis of AA from β-cell phospholipids is suppressed by the iPLA2β inhibitor BEL (18, 52, 53), and overexpression of iPLA2β in insulinoma cells causes a rise in PLA2 product levels and is associated with amplification of insulin secretory responses (21, 56). Changes in local levels of nonesterified AA within the phospholipid bilayer could affect the activities of proteins in membranes. AA facilitates Ca2+ influx into β-cells (17, 18, 33) and has been suggested to influence ion movement through pumps and channels (34–36). Our observations suggest that Kv2.1 might represent one such channel. The PLA2 product AA blocks the activity of these channels, and overexpression of iPLA2β in insulinoma cells is associated with amplification of insulin secretion and suppression of Kv2.1 channel activity, which is consistent with previous observations that blockade of Kv2.1 activity by other means also results in increased insulin secretory responses (48, 49).
The muscarinic agonist carbachol amplifies both glucose-induced insulin secretion and eicosanoid release that reflects hydrolysis of arachidonate from islet membrane phospholipids (14, 18, 44), and the iPLA2β inhibitor BEL blunts both responses (18). Carbachol also induces accumulation of inositol trisphosphates (18) and diacylglycerol (14) in islets by a BEL-insensitive mechanism that probably involves coupling of the muscarinic receptor with a G-protein that activates a phospholipase C. Accumulation of diacylglycerol in phospholipid bilayers facilitates PLA2 activation (63, 64) and phospholipids hydrolysis to yield free fatty acids, such as arachidonic acid or linoleic acid (50, 51), near Kv2.1 channels might affect channel activity. We observe that carbachol shortens the depolarization-induced time-to-peak Kv2.1 current and accelerates current inactivation in INS-1 cells and primary mouse β-cells; the latter effect is prevented by the iPLA2β inhibitor BEL or in cells from genetically modified mice that do not express iPLA2β. These findings suggest that iPLA2β catalyzes formation of product(s) that negatively regulate the repolarizing effects of β-cell Kv2.1 channels.
Kv1.1 is a delayed rectifier K channel related to Kv2.1 that is expressed at high levels in the central nervous system, and its activity is affected in a manner similar to that reported here by the iPLA2β inhibitor BEL (65). This raises the possibility that regulation of delayed rectifier, voltage-gated K channels by iPLA2β, or products of its action might be a phenomenon that is not confined to β-cells. Kv1.1 channel activity (66) and subcellular location (67) are also affected by free fatty acids, and it is possible that colocalization or direct interaction of iPLA2β and voltage-gated K channels, such as Kv1.1 and Kv2.1, occur.
β-Cell iPLA2β subcellular redistribution is known to occur upon stimulation (21), and iPLA2β undergoes some established interactions with other signaling proteins (68). Interactions with additional proteins are suspected (69), and iPLA2β has an ankyrin-repeat domain (29) and a Smad-4-like protein interaction domain (30) that might mediate interaction with a variety of signaling partners or the assembly of multimeric protein aggregates (70) that allow local, direct delivery of the products of iPLA2β action to effector molecules regulated by them. In β-cells, such molecules could include ion channels like Kv2.1.
The evolution of type II diabetes is associated with changes in blood free fatty acids that have been linked to insulin resistance, including a rise in concentrations of AA and a fall in linoleic acid levels (59–61). Prolonged exposure of β-cells to AA stimulates their proliferation and amplifies secretagogue-induced insulin release (62), both of which also occur during the evolution of type II diabetes. Similarly, overexpression of iPLA2β in insulinoma cell lines is associated with increased proliferation rates and amplified secretory responses (21, 56), and suppression of iPLA2β expression is associated with reduced proliferation rates and insulin secretion (22). The progressive rise in insulin resistance during the evolution of type II diabetes requires development of a compensatory hypersecretion of insulin to maintain glucose homeostasis, and a rise in AA levels might stimulate such a response. The deterioration of glucose tolerance induced in mice fed a high fat diet is exacerbated in iPLA2β-null mice, and pancreatic islets isolated from these mice also fail to develop the exaggerated insulin secretory responses observed with islets isolated from wild-type mice (20). This suggests that iPLA2β participates in β-cell compensatory responses to increasing insulin resistance, and our results suggest that modulation of Kv2.1 channel activity by fatty acids could be one of the steps involved in such β-cell compensation.
In conclusion, our results indicate that AA negatively regulates the activity of the delayed rectifier potassium channel Kv2.1 in β-cells. Reducing Kv current prolongs stimulus-induced depolarization of the β-cell plasma membrane and thereby amplifies insulin secretion (46, 48). This could account for the observations that overexpression of iPLA2β in insulinoma cells is associated with both amplified insulin secretion (21) and reduced Kv2.1 channel activity (Fig. 5) and for the facts that pharmacologic suppression of iPLA2β activity is associated with reduced insulin secretory responses (19) and increased Kv2.1 channel activity (Fig. 6). This series of observations suggests that modulation of β-cell Kv2.1 channel activity by product(s) of iPLA2β action might be an important signaling event in the regulation of insulin secretion and in β-cell compensatory responses to the progression of insulin resistance.
Supplementary Material
Acknowledgments
We appreciate the expert technical assistance of Molly Dodge and Felipe Mendez and helpful discussions with Andrey Kuznetsov, James Lopez, Leonid Fridlyand, and Natalia Tamarina.
Footnotes
This work was supported in part by National Institutes of Health Grants DK44840, DK48494, DK63493, and DK20595; United States Public Health Service Grants R37-DK34388, P60-DK20579, and P30-DK56341; the Diabetes Research and Training Center at the University of Chicago; and the Blum-Kovler Foundation.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1 and 2.
The abbreviations used are: KATP, ATP-sensitive potassium channels; Kv2.1, voltage-dependent delayed rectifier potassium channel; AA, arachidonic acid; iPLA2β, Group VIA Phospholipase A2; INS-1, rat insulinoma cell line; HEK, human embryonic kidney cell line; LPC, lysophosphatidylcholine; GFP, green fluorescent protein; BEL, bromenol lactone; KRB, Krebs-Ringer buffer.
References
- 1.Ashcroft FM, Proks P, Smith PA, Ammala C, Bokvist K, Rorsman PJ. J Cell Biochem. 1994;55:54 – 65. doi: 10.1002/jcb.240550007. [DOI] [PubMed] [Google Scholar]
- 2.Philipson LH, Rosenberg MP, Kuznetsov A, Lancaster ME, Worley JF, 3rd, Roe MW, Dukes ID. J Biol Chem. 1994;269:27787–27790. [PubMed] [Google Scholar]
- 3.Dukes ID, Philipson LH. Diabetes. 1996;45:845–853. doi: 10.2337/diab.45.7.845. [DOI] [PubMed] [Google Scholar]
- 4.Roe MW, Worley JF, III, Mittal AA, Kuznetsov A, DasGupta S, Mertz RJ, Witherspoon SM, III, Blair N, Lancaster ME, McIntyre MS, Shehee WR, Dukes ID, Philipson LH. J Biol Chem. 1996;271:32241–32246. doi: 10.1074/jbc.271.50.32241. [DOI] [PubMed] [Google Scholar]
- 5.Laychock SG. Diabetes. 1983;32:6–13. doi: 10.2337/diab.32.1.6. [DOI] [PubMed] [Google Scholar]
- 6.Morgan NG, Montague W. Biochem J. 1985;226:571–576. doi: 10.1042/bj2260571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wolf BA, Pasquale SM, Turk J. Biochemistry. 1991;30:6372–6379. doi: 10.1021/bi00240a004. [DOI] [PubMed] [Google Scholar]
- 8.Tamarina NA, Kuznetsov A, Rhodes CJ, Bindokas VP, Philipson LH. Diabetes. 2005;54:3073–3081. doi: 10.2337/diabetes.54.11.3073. [DOI] [PubMed] [Google Scholar]
- 9.Turk J, Colca JR, Kotagal N, McDaniel ML. Biochim Biophys Acta. 1984;794:110–124. doi: 10.1016/0005-2760(84)90304-7. [DOI] [PubMed] [Google Scholar]
- 10.Turk J, Wolf BA, Comens PG, Colca J, Jakschik B, McDaniel ML. Biochim Biophys Acta. 1985;835:1–17. doi: 10.1016/0005-2760(85)90023-2. [DOI] [PubMed] [Google Scholar]
- 11.Dixon JF, Hokin LE. J Biol Chem. 1984;259:14418–14425. [PubMed] [Google Scholar]
- 12.Chaudhry A, Laychock SG, Rubin RP. J Biol Chem. 1987;262:17426–17431. [PubMed] [Google Scholar]
- 13.Konrad RJ, Jolly YC, Major C, Wolf BA. Biochim Biophys Acta. 1992;1135:215–220. doi: 10.1016/0167-4889(92)90139-3. [DOI] [PubMed] [Google Scholar]
- 14.Konrad RJ, Jolly YC, Major C, Wolf BA. Biochem J. 1992;287:283–290. doi: 10.1042/bj2870283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Turk J, Wolf BA, McDaniel ML. Prog Lipid Res. 1987;26:125–181. doi: 10.1016/0163-7827(87)90010-5. [DOI] [PubMed] [Google Scholar]
- 16.Turk J, Hughes JH, Easom RA, Wolf BA, Scharp DW, Lacy PE, McDaniel ML. Diabetes. 1988;37:992–996. doi: 10.2337/diab.37.7.992. [DOI] [PubMed] [Google Scholar]
- 17.Ramanadham S, Gross R, Turk J. Biochem Biophys Res Commun. 1992;184:647–653. doi: 10.1016/0006-291x(92)90638-2. [DOI] [PubMed] [Google Scholar]
- 18.Ramanadham S, Gross RW, Han X, Turk J. Biochemistry. 1993;32:337–346. doi: 10.1021/bi00052a042. [DOI] [PubMed] [Google Scholar]
- 19.Song K, Zhang X, Zhao C, Ang NT, Ma ZA. Mol Endocrinol. 2005;19:504–515. doi: 10.1210/me.2004-0169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bao S, Song H, Wohltmann M, Ramanadham S, Jin W, Bohrer A, Turk J. J Biol Chem. 2006;281:20958–20973. doi: 10.1074/jbc.M600075200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma Z, Ramanadham S, Wohltmann M, Bohrer A, Hsu FF, Turk J. J Biol Chem. 2001;276:13198–13208. doi: 10.1074/jbc.M010423200. [DOI] [PubMed] [Google Scholar]
- 22.Bao S, Bohrer A, Ramanadham S, Jin W, Zhang S, Turk J. J Biol Chem. 2006;281:87–98. doi: 10.1074/jbc.M509105200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ramanadham S, Bohrer A, Mueller M, Jett P, Gross R, Turk J. Biochemistry. 1993;32:5339–5351. doi: 10.1021/bi00071a009. [DOI] [PubMed] [Google Scholar]
- 24.Ramanadham S, Song H, Hsu FF, Zhang S, Crankshaw M, Grant GA, Newgard CB, Bao S, Ma Z, Turk J. Biochemistry. 2003;42:13929–13940. doi: 10.1021/bi034843p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramanadham S, Bohrer A, Gross RW, Turk J. Biochemistry. 1993;32:13499–13509. doi: 10.1021/bi00212a015. [DOI] [PubMed] [Google Scholar]
- 26.Ramanadham S, Hsu FF, Bohrer A, Nowatzke W, Ma Z, Turk J. Biochemistry. 1998;37:4533–4567. doi: 10.1021/bi9722507. [DOI] [PubMed] [Google Scholar]
- 27.Wolf B, Turk J, Sherman W, McDaniel ML. J Biol Chem. 1986;261:3501–3510. [PubMed] [Google Scholar]
- 28.Turk J, Wolf BA, McDaniel MLJ. Biochem J. 1986;237:259–263. doi: 10.1042/bj2370259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma Z, Ramanadham S, Kempe K, Chi XS, Ladenson J, Turk J. J Biol Chem. 1997;272:11118–11127. [PubMed] [Google Scholar]
- 30.Ma Z, Wang X, Nowatzke W, Ramanadham S, Turk J. J Biol Chem. 1999;274:9607–9616. doi: 10.1074/jbc.274.14.9607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramanadham S, Hsu FF, Bohrer A, Ma Z, Turk J. J Biol Chem. 1999;274:13915–13927. doi: 10.1074/jbc.274.20.13915. [DOI] [PubMed] [Google Scholar]
- 32.Ma Z, Bohrer A, Wohltmann M, Ramanadham S, Hsu FF, Turk J. Lipids. 2001;36:689–700. doi: 10.1007/s11745-001-0774-9. [DOI] [PubMed] [Google Scholar]
- 33.Metz SA, Draznin B, Sussman KE, Leitner JW. Biochem Biophys Res Commun. 1987;142:251–258. doi: 10.1016/0006-291x(87)90478-5. [DOI] [PubMed] [Google Scholar]
- 34.Vacher P, McKebzie J, Dufy F. Am J Physiol. 1989;257:E203–E211. doi: 10.1152/ajpendo.1989.257.2.E203. [DOI] [PubMed] [Google Scholar]
- 35.Eddlestone GT. Am J Physiol. 1995;268:C181–C190. doi: 10.1152/ajpcell.1995.268.1.C181. [DOI] [PubMed] [Google Scholar]
- 36.Owada S, Larsson O, Arkhammar P, Katz AI, Chibalin AV, Berggren PO, Bertorello AM. J Biol Chem. 1999;274:2000–2008. doi: 10.1074/jbc.274.4.2000. [DOI] [PubMed] [Google Scholar]
- 37.Smani T, Zakharov SI, Csutora P, Leno E, Trepakova ES, Bolotina VM. Nat Cell Biol. 2004;6:113–120. doi: 10.1038/ncb1089. [DOI] [PubMed] [Google Scholar]
- 38.Roe MW, Worley JF, Qian F, Tamarina N, Mittal AA, Drayluk F, Blair NT, Mertz RJ, Philipison LH, Dukes ID. J Biol Chem. 1998;273:10402–10410. doi: 10.1074/jbc.273.17.10402. [DOI] [PubMed] [Google Scholar]
- 39.Juhl K, Efanov AM, Olsen HL, Gromada J. Biochem Biophys Res Commun. 2003;310:274–279. doi: 10.1016/j.bbrc.2003.09.018. [DOI] [PubMed] [Google Scholar]
- 40.McGee R, Jr, Sansom MS, Usherwood PN. J Membr Biol. 1988;102:21–34. doi: 10.1007/BF01875350. [DOI] [PubMed] [Google Scholar]
- 41.Oliver D, Lien CC, Soom M, Baukrowitz T, Jonas P, Fakler B. Science. 2004;304:265–270. doi: 10.1126/science.1094113. [DOI] [PubMed] [Google Scholar]
- 42.O’Connell KM, Tamkun MM. J Cell Sci. 2005;118:2155–2166. doi: 10.1242/jcs.02348. [DOI] [PubMed] [Google Scholar]
- 43.Song H, Ramanadham S, Bao S, Hsu FF, Turk J. Biochemistry. 2006;45:1061–1073. doi: 10.1021/bi052065q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Turk J, Gross RW, Ramanadham S. Diabetes. 1993;42:367–374. doi: 10.2337/diab.42.3.367. [DOI] [PubMed] [Google Scholar]
- 45.Dukes ID, Cleemann L, Morad M. J Pharmacol Exp Ther. 1990;254:560–569. [PubMed] [Google Scholar]
- 46.MacDonald PE, Ha XF, Wang J, Smukler SR, Sun AM, Gaisano HY, Salapatek AM, Backx PH, Wheeler MB. Mol Endocrinol. 2001;15:1423–1435. doi: 10.1210/mend.15.8.0685. [DOI] [PubMed] [Google Scholar]
- 47.Tamarina NA, Kuznetsov A, Fridlyand LE, Philipson LH. Am J Physiol. 2005;289:E578–E585. doi: 10.1152/ajpendo.00054.2005. [DOI] [PubMed] [Google Scholar]
- 48.MacDonald PE, Sewing S, Wang J, Joseph JW, Smukler SR, Sakellaropoulos G, Wang J, Saleh MC, Chan CB, Tsushima RG, Salapatek AM, Wheeler MB. J Biol Chem. 2002;277:44938 – 44945. doi: 10.1074/jbc.M205532200. [DOI] [PubMed] [Google Scholar]
- 49.El-Kholy W, Macdonald PE, Lin JH, Wang J, Fox JM, Light PE, Wang Q, Tsushima RG, Wheeler MB. FASEB J. 2003;17:720–722. doi: 10.1096/fj.02-0802fje. [DOI] [PubMed] [Google Scholar]
- 50.McKay MC, Worley JF., III Am J Physiol. 2001;281:C1277–C1284. doi: 10.1152/ajpcell.2001.281.4.C1277. [DOI] [PubMed] [Google Scholar]
- 51.Feng DD, Luo Z, Roh SG, Hernandez M, Tawadros N, Keating DJ, Chen C. Endocrinology. 2006;147:674–682. doi: 10.1210/en.2005-0225. [DOI] [PubMed] [Google Scholar]
- 52.Newgard CB, McGarry JD. Annu Rev Biochem. 1995;64:689–719. doi: 10.1146/annurev.bi.64.070195.003353. [DOI] [PubMed] [Google Scholar]
- 53.Ramanadham S, Wolf M, Jett PA, Gross RW, Turk J. Biochemistry. 1994;33:7442–7452. doi: 10.1021/bi00189a052. [DOI] [PubMed] [Google Scholar]
- 54.Ramanadham S, Wolf M, Li B, Bohrer A, Turk J. Biochim Biophys Acta. 1997;1344:153–164. doi: 10.1016/s0005-2760(96)00139-7. [DOI] [PubMed] [Google Scholar]
- 55.Ma Z, Turk J. Prog Nucleic Acid Res Mol Biol. 2001;67:1–33. doi: 10.1016/s0079-6603(01)67023-5. [DOI] [PubMed] [Google Scholar]
- 56.Turk J, Ramanadham S. Can J Physiol Pharmacol. 2004;82:824–832. doi: 10.1139/y04-064. [DOI] [PubMed] [Google Scholar]
- 57.Turk J, Mueller M, Bohrer A, Ramanadham S. Biochim Biophys Acta. 1992;1125:280–291. doi: 10.1016/0005-2760(92)90057-3. [DOI] [PubMed] [Google Scholar]
- 58.Turk J, Bao S, Ramanadham S. In: Commentaries on Perspectives in Diabetes. Robertson RP, editor. Vol. 2. American Diabetes Association; Alexandria, VA: 2006. pp. 10–20. [Google Scholar]
- 59.Salomaa V, Ahola I, Tuomilehto J, Aro A, Pietinen P, Korhonen HJ, Penttila I. Metabolism. 1990;39:1285–1291. doi: 10.1016/0026-0495(90)90185-f. [DOI] [PubMed] [Google Scholar]
- 60.Pelikanova T, Kohout M, Valek J, Base J, Stefka Z. Metabolism. 1991;40:175–180. doi: 10.1016/0026-0495(91)90170-2. [DOI] [PubMed] [Google Scholar]
- 61.Pelikanova T, Kazdova L, Chvojkova S, Base J. Metabolism. 2001;50:1472–1478. doi: 10.1053/meta.2001.27195. [DOI] [PubMed] [Google Scholar]
- 62.Dixon G, Nolan J, McClenaghan NH, Flatt PR, Newsholme P. Clin Sci Lond. 2004;106:191–199. doi: 10.1042/CS20030261. [DOI] [PubMed] [Google Scholar]
- 63.Prentki M, Matschinsky FM. Physiol Rev. 1987;67:1185–1248. doi: 10.1152/physrev.1987.67.4.1185. [DOI] [PubMed] [Google Scholar]
- 64.Hsu FF, Ma Z, Wohltmann M, Bohrer A, Ramanadham S, Turk J. J Biol Chem. 2000;275:16579–16589. doi: 10.1074/jbc.M908342199. [DOI] [PubMed] [Google Scholar]
- 65.Gubitosi-Klug RA, Yu SP, Choi DW, Gross RW. J Biol Chem. 1995;270:2885–2888. doi: 10.1074/jbc.270.7.2885. [DOI] [PubMed] [Google Scholar]
- 66.Gubitosi-Klug RA, Gross RW. J Biol Chem. 1996;271:32519–32522. doi: 10.1074/jbc.271.51.32519. [DOI] [PubMed] [Google Scholar]
- 67.Gubitosi-Klug RA, Mancuso DJ, Gross RW. Proc Natl Acad Sci U S A. 2005;102:5964–5968. doi: 10.1073/pnas.0501999102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang Z, Ramanadham S, Ma Z, Bao S, Mancuso DJ, Gross RW, Turk J. J Biol Chem. 2005;280:6840–6849. doi: 10.1074/jbc.M405287200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ramanadham S, Wolf M, Ma Z, Li B, Wang J, Gross RW, Turk J. Biochemistry. 1996;35:5464–5471. doi: 10.1021/bi952652j. [DOI] [PubMed] [Google Scholar]
- 70.McConnachie G, Langeberg LK, Scott JD. Trends Mol Med. 2006;12:317–323. doi: 10.1016/j.molmed.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 71.Jenkins CM, Han X, Mancuso DJ, Gross RW. J Biol Chem. 2002;277:32807–32814. doi: 10.1074/jbc.M202568200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.