

Abstract
The cytochrome P450 (P450) superfamily metabolizes many endogenous signaling molecules and drugs. P450 enzymes are regulated by post-translational mechanisms in vivo, which hinders their functional characterization by conventional genomic or proteomic methods. Here, we describe a chemical proteomic strategy to profile P450 activities directly in living systems. Derivatization of a mechanism-based inhibitor with a “clickable” handle provided an activity-based probe that labels multiple P450s both in proteomic extracts and in vivo. This probe was used to record alterations in liver P450 activities triggered by chemical agents, including inducers of P450 expression and direct P450 inhibitors. The chemical proteomic strategy described herein thus offers a versatile method to monitor P450 activities and small molecule interactions in any biological system and, through doing so, should facilitate the functional characterization of this large and diverse enzyme class.
The mammalian cytochrome P450 (P450) membrane-bound monooxygenase enzyme family metabolizes a large number of xenobiotics, drugs, and endogenous signaling molecules [1]. The human genome encodes 57 distinct P450 enzymes [2], and, despite sharing a conserved structural core with a reactive heme group at the active site [3], these enzymes are remarkably diverse, exhibiting as little as 16% sequence identity. Multiple P450 enzymes participate in drug metabolism, and several demonstrate broad substrate specificity [1]. Five human P450 enzymes in particular contribute to the metabolism of 90% of current clinical drugs, with P450 3A4 having a role in more than 60% [2]. Other P450 enzymes are involved in the biosynthesis and/or degradation of bioactive sterols, fatty acids, and eicosanoids. Some of these enzymes, such as aromatase (P450 19A1), which is involved in the biosynthesis of estrogen, have emerged as important drug targets for the treatment of hormone-sensitive cancers [4]. Finally, a significant portion of the P450 family remains essentially unannotated, with 20-30% of human P450s lacking known substrates [2].
The study of P450 drug metabolism has become a critical part of the pharmaceutical development process. In vitro testing of P450 metabolism, inhibition, and induction is typically used to model P450-drug interactions in vivo. Challenges in predicting from In vitro assays the impact of drugs on P450 expression and/or activity in vivo are well-documented [5-12]. In vitro systems, such as liver microsomes, hepatocyte cultures, and recombinant P450 expression systems, do not account for the variable distribution and uptake of drugs in living animals, nor do they report accurately on the dynamic regulation of P450 function in vivo. Indeed, the activity of P450 enzymes is regulated by multiple factors in vivo, including membrane composition and localization, protein binding partners (e.g., Dap1/PGRMC1) [13], post-translational modification [14-16], and endogenous concentrations of cofactors (e.g., NAPDH) and regulatory enzymes (e.g., NADPH-P450 reductases) [8]. Finally, the mechanisms of drug interactions with P450 enzymes may differ In vitro and in vivo, as in the case of the H2 receptor antagonist cimetidine, which acts as a reversible inhibitor In vitro, but appears to irreversibly inactivate P450s in vivo [17-20].
Due to the large size of the P450 family and the methodological complications associated with predicting their activity and drug interactions using In vitro assays, there has been great interest in developing methods to assess P450 function in living systems. To date, these efforts have largely been restricted to the co-administration of drugs with P450 substrates, followed by evaluation of excreted products to estimate drug effects on P450 activity. However, given the broad substrate selectivity of many P450 enzymes, extrapolating from indirect substrate assays the identity of specifically affected P450 enzymes remains difficult [8-12]. Here, we report an alternative strategy that utilizes chemical probes to covalently label P450 enzymes in an activity-based manner. Specifically, we convert a broad-spectrum, mechanism-based P450 inhibitor, 2-ethynylnaphthalene (2EN), to an activity-based protein profiling (ABPP) probe by derivatization with a versatile “click chemistry (CC)” handle that enables the selective tagging, detection, enrichment, and identification of P450 enzymes in any biological system. The 2EN-activity based probe (2EN-ABP) was found to label several P450 enzymes in rodent liver in an NADPH-dependent manner, and proved capable of monitoring both drug induction and inhibition of these enzymes in vivo.
RESULTS
Design and synthesis of an activity-based probe for P450 enzymes
Methods to characterize P450s in proteomes have been intensely pursued; however to date, these approaches have been restricted to measurements of P450 expression rather than activity [21]. An ideal functional proteomics probe for the P450 superfamily would react with many members of this enzyme class in a purely activity-based manner. With this goal in mind, we selected the mechanism-based inhibitor 2-ethynylnapthalene (2EN) as the reactive group for a P450-directed activity-based probe (Figure 1A). The inactivation of P450 enzymes by aryl acetylenes, such as 2EN, has been well characterized [22-25]. Briefly, the naphthalene acts as a hydrophobic binding group to direct the inhibitor toward P450 active sites, wherein enzyme-catalyzed oxidation of the conjugated 2-acetylene generates a highly reactive ketene (Figure 1A). The ketene intermediate can then acylate nucleophilic residues within the P450 active site or N-alkylate the bound heme molecule to generate covalent enzyme-inhibitor adducts. Although only the former reaction product may be stable enough to follow by ABPP methods, we anticipated that a certain level of “non-productive” heme alkylation could be tolerated, as this process would only be expected to impact the sensitivity (not fidelity) of probe-P450 reactions. Based on these mechanisms, we envisaged that a 2EN-derived probe would exhibit high selectivity for labeling catalytically active, but not inactive P450 enzymes. 2EN has also been shown to inhibit multiple sub-classes of P450s, including the 1, 2, and 3 families, and thus offered the potential for broad proteomic coverage of the P450 superfamily [22-28].
Figure 1.

Reactivity of 2-ethynylnaphthalene (2EN) agents with cytochrome P450 enzymes. (A) Mechanism of inhibition of P450 enzymes by 2EN, where the aryl alkyne group of 2EN is first metabolized to an electrophilic ketene that then reacts with amino acid residues in the P450 active site. (B) Conversion of 2EN into an ABP for P450 enzymes. Addition of an alkyl (unconjugated) acetylene group to 2EN provides a handle for click chemistry (CC) conjugation to azide-modified rhodamine and/or biotin reporter groups (red ball).
Original strategies for ABPP employed chemical probes that contained large reporter tags, such as biotin or fluorophores, to permit the detection and enrichment of labeled enzymes [29-31]; however, these bulky tags can, in certain cases, negatively impact probe-protein interactions and hinder probe uptake and distribution in living systems. Since our goal was to create an activity-based probe that could target P450 enzymes in vivo, we elected to modify 2EN with an alkyl (unconjugated) acetylene group (Figure 1B) to enable coupling of probe-modified enzymes to reporter tags after the proteome labeling step via the Cu(I)-catalyzed azide-alkyne [3 + 2] cycloaddition reaction [click chemistry (CC)] [32-34]. The resulting agent is referred to hereafter as 2EN-activity-based probe, or 2EN-ABP (Figure 1B).
Evidence that 2EN-ABP labels P450s in an activity-dependent manner
The catalytic cycle of P450 enzymes requires the transfer of two electrons from cytochrome P450 reductase [3]. These electrons are formally donated by NAPDH; therefore, the catalytic activity of P450s is NADPH-dependent. To test whether 2EN-ABP labels P450s in an activity-dependent manner, this probe was added to mouse liver microsomes, a proteomic fraction that contains several P450s, in the presence or absence of NADPH. Following a 1 h incubation, proteomes were treated with an azide-conjugated rhodamine tag under CC conditions and resolved by SDS-PAGE. Ingel fluorescence scanning identified an intensely labeled set of protein bands in the molecular mass range of 48-55 kDa selectively in the NADPH-treated proteome (Figure 2A). Labeling of these proteins was dependent on the concentration of 2EN-ABP, with signals saturating at approximately 20 μM probe (Figure 2B). The 48-55 kDa proteins were not detected in soluble liver proteome (Figure 2C), nor were they observed at high levels in other tissues, including kidney, lung, testes, heart, and brain (Figure 2D). However, NADPH-dependent labeling was observed in the small intestine upon increase of the gel scan intensity (Figure 2E). The collective features displayed by the 48-55 kDa targets of 2EN-ABP, including NADPH-dependent labeling, molecular mass, and enhanced expression in liver microsomes (with lower levels also observed in small intestine), suggested that they could represent P450s from the 1, 2, and/or 3 families. To directly test this premise, we next pursued the molecular characterization of these proteins.
Figure 2.

ABPP of P450 enzymes with 2EN-ABP. (A) Treatment of mouse liver microsomal proteome (1 mg protein/mL) with 2EN-ABP (10 μM) in the presence or absence of NADPH (1 mM) resulted in selective NADPH-dependent labeling of a set of 48-55 kDa proteins (asterisk). Probe targets were detected by CC with a rhodamine-azide tag, SDS-PAGE analysis, and in-gel fluorescence scanning (fluorescent gel shown in grayscale). (B) Concentration-dependence of 2EN-ABP labeling of 48-55 kDa liver microsomal proteins. (C) Selective 2EN-ABP labeling of 48-55 kDa proteins in liver microsomal proteomes compared to cytosolic proteomes. (D) Enhanced 2EN-ABP labeling of 48-55 kDa proteins in liver microsomal proteomes compared to other tissue microsomal proteomes. (E) Low levels of NADPH-dependent labeling for 48-55 kDa proteins were also observed at higher gel scanning intensity in small intestine. (F) NADPH-dependent labeling by 2EN-ABP (10 μM) of human P450 1a2 recombinantly expressed in COS-7 cells. Probe labeling was blocked by treatment with the P450 1a2 inhibitor α-naphthoflavone (αNF) (20 μM).
Liver microsomal proteome was treated with 2EN-ABP in the presence or absence of NADPH for 1 h, after which the samples were reacted with a trifunctional azido-biotin-rhodamine tag [33] under CC conditions. 2EN-ABP-labeled proteins were enriched using streptavidin-conjugated beads, separated by SDS-PAGE, and detected by in-gel fluorescence scanning. Strong fluorescent signals were observed in the 48-55 kDa region of the gel in the sample containing NADPH, but not in the sample lacking NAPDH (Supplemental Figure 1). This region of the gel was excised for both samples, subjected to in-gel trypsin digestion, and the resulting peptides analyzed by multidimensional LC-MS [35-36]. Proteins were identified and quantified using the SEQUEST search algorithm and spectral counting [36], respectively. Five independent experiments were performed, and specific targets of 2EN-ABP were identified by filtering data for proteins that exhibited: 1) at least five spectral counts in each NADPH(+) sample, 2) at least a three-fold increase in average spectral counts in NADPH(+) versus NAPDH(-) samples; and 3) a p value of ≤ 0.01 for average spectral count differences in NADPH(+) versus NAPDH(-) samples.
Based on these criteria, several specific targets of 2EN-ABP were identified, all of which represented members of the P450 superfamily: 1a2, 3a11, 2c29, and 2d9/2d10 (Table 1). In the case of 2d9 and 2d10, a significant number of shared peptides were detected (Supplemental Figure 2), which precluded a confident decision on whether one or both of these enzymes was targeted by 2EN-ABP. To confirm that P450s were legitimate targets of 2EN-ABP, we recombinantly expressed P450 1a2 in COS-7 cells. A strongly labeled, NADPH-dependent target of 2EN-ABP was detected in P450 1a2-transfected, but not mock-transfected COS-7 cells (Figure 2F).
Table 1.
P450 enzyme activities labeled by 2EN-ABP in mouse liver microsomes. Data represent the average ± standard error of five independent experiments.
| Tryptic peptide spectral counts |
||||
|---|---|---|---|---|
|
in vitro labeling |
in vivo labeling | |||
| Protein | IPI number | NADPH (+) | NADPH (-) | |
| P450 1a2 | IPI00128287 | 9 ± 1 | 0 ± 0 | 28 ± 6* |
| P450 3a11 | IPI00134504 | 9 ± 1 | 0.6 ± 0.5 | 9 ± 2 |
| P450 2c29 | IPI00134503 | 13 ± 2 | 2 ± 1 | 21 ± 5 |
| P450 2d9** | IPI00116572 | 7 ± 1 | 1.4 ± 0.4 | 4 ± 2 |
| P450 2d10** | IPI00323908 | 10 ± 1 | 3.0 ± 1 | 7 ± 2 |
p = 0.02 for in vivo versus in vitro [NADPH (+)] labeled samples (planned comparison).
P450 2d9 and 2d10 shared ∼3-4 peptides in common per NADPH (+) samples, which precluded confident assignment of whether one or both of these enzymes was labeled by 2EN-ABP. See Supplemental Figure 2 for a list of peptides identified for each P450 enzyme.
Profiling P450 induction and drug interactions In vitro with 2EN-ABP
Several drugs can induce, inhibit, or induce and inhibit the expression and activity of P450 enzymes. For example, β-naphthoflavone (βNF) and dexamethasone (DEX) are known to induce the mouse P450 1a and 3a subfamilies, respectively [37]. To test whether 2EN-ABP could detect changes in P450 activity induced by drugs, we treated mice with βNF (40 mg/kg), DEX (80 mg/kg), or vehicle daily by intraperitoneal (i.p.) injection for three days, and, on the fourth day, livers were harvested and microsomal proteomes prepared. Labeling of proteomes with 2EN-ABP, followed by CC reaction with a rhodamine-azide tag, SDS-PAGE, and in-gel fluorescence scanning revealed a striking elevation of multiple P450 activities in βNF-treated mice (Figure 3A, arrowheads). In contrast, DEX treatment did not significantly alter the 2EN-ABP labeling profiles as judged by SDS-PAGE analysis (Figure 3A). These findings were confirmed by quantification of results from six independent experiments (Supplemental Figure 3).
Figure 3.
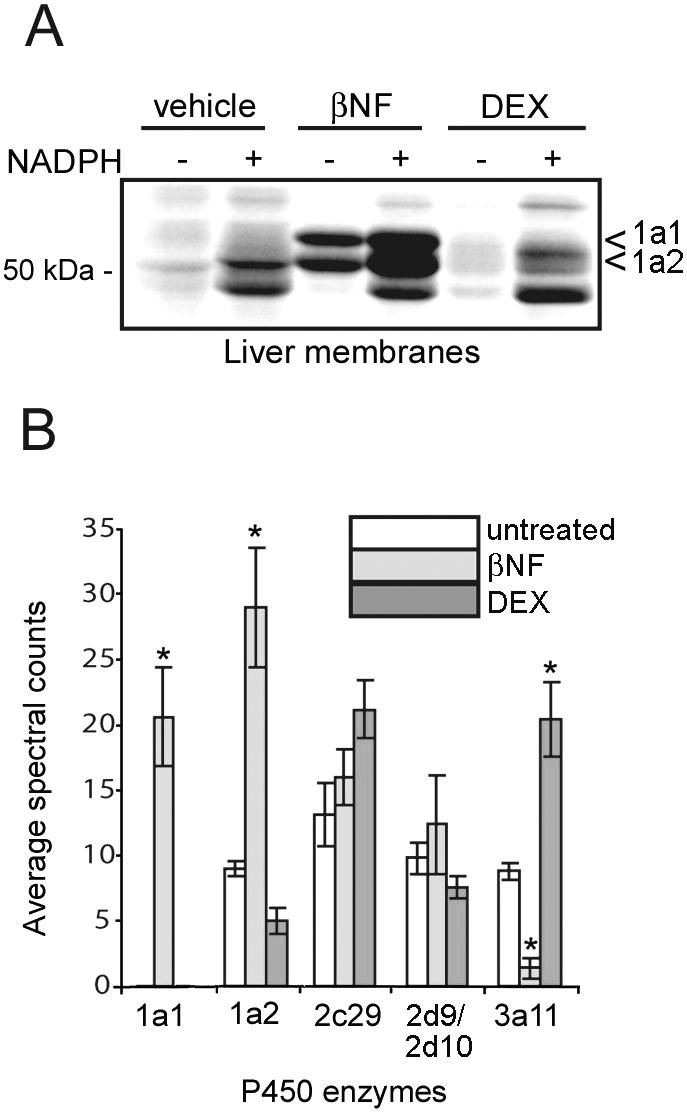
Monitoring drug induction of P450 activities with 2EN-ABP. (A) Liver proteomes from mice treated with β-naphthoflavone (βNF) (40 mg/kg, 3 days) showed augmented 2EN-ABP labeling of two 51-55 kDa (arrowheads) proteins compared to vehicle controls. Liver proteomes from dexamethasone (DEX)-treated mice did show significant changes in 2EN-ABP labeling profiles compared to vehicle or βNF -treated mice as judged by SDS-PAGE. (B) LC-MS analysis of 2EN-ABP-labeled enzymes from liver proteomes of untreated mice or mice treated with βNF and DEX. Data are reported as average spectral counts (± standard error) for five independent experiments per group. *p < 0.01 for βNF- or DEX-induced samples relative to other groups. The signals for 2d9 and 2d10 are shown together because these enzymes shared too many peptides in common to permit their individual assignment.
We next used LC-MS methods to identify the targets of 2EN-ABP in liver proteomes from βNF- and DEX-treated mice and compared their activity signals to those observed in untreated mice. βNF treatment induced a dramatic increase in the activities of P450 1a1 and 1a2 (Figure 3B). The induction of P450 1a1 was particularly striking, as this enzyme was virtually undetectable in livers from untreated or DEX-treated mice (< 5 spectral counts per sample). A comparison of these LC-MS measurements to the P450 activity signals observed by SDS-PAGE permitted us to assign the βNF-induced 52 and 55 kDa targets as P450s 1a2 and 1a1, respectively (Figure 3A). LC-MS analysis of 2EN-ABP targets in DEX-treated mice revealed a selective elevation in the activity of P450 3a11 compared to untreated or βNF-treated mice (Figure 3B). We assume that the inability to detect similar DEX-induced elevations in P450 3a11 activity by SDS-PAGE was due to the presence of additional 2EN-ABP-labeled P450s that co-migrated with P450 3a11. These results thus serve to underscore the enhanced resolution afforded by LC-MS-based methods for ABPP, which is especially relevant for enzyme classes like P450s that possess many members of similar molecular mass. Interestingly, LC-MS data also revealed that P450 3a11 activity was significantly reduced in βNF-treated mice compared to untreated animals (Figure 3B), indicating that the same drug can have opposing effects on the activity of different members of the P450 family.
To test whether 2EN-ABP could also report on the direct binding of drugs to P450 active sites, membrane proteomes from P450 1a2-transfected COS7 cells were incubated with the reversible P450 1a2 inhibitor, α-naphthoflavone (αNF, 20 μM), for 10 min prior to addition of 2EN-ABP (10 μM). αNF completely blocked labeling of P450 1a2 by 2EN-ABP (Figure 2F). Equivalent results were obtained using liver microsomal proteomes from βNF-induced mice, where 2EN-ABP labeling of endogenous P450 1a2 was disrupted by treatment with αNF (Figure 4A). An IC50 value of 2.0 ± 0.3 μM was calculated for αNF inhibition of 2EN-ABP labeling of P450 1a2 in liver proteomes (Figure 4B). Notably, this IC50 value matched closely the IC50 value reported for αNF inhibition of mouse P450 1a2 using standard substrate assays (0.8 μM) [38].
Figure 4.
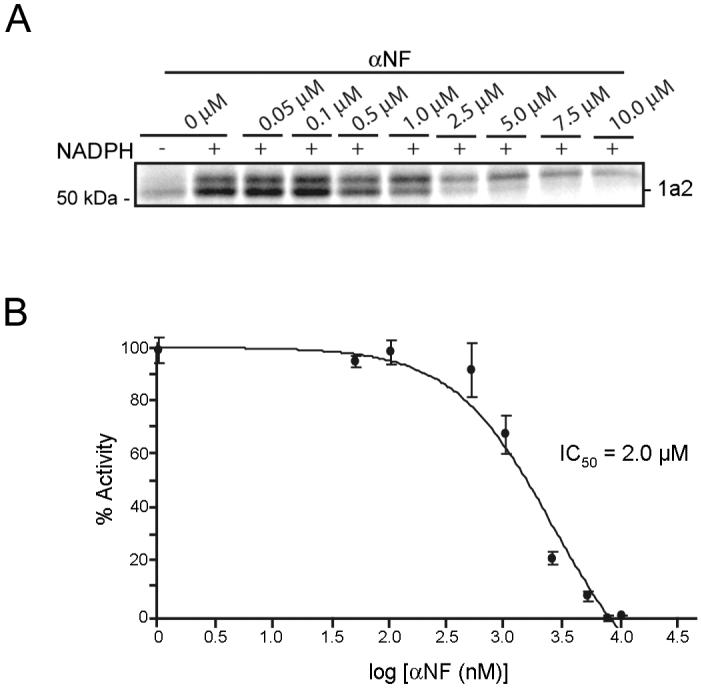
Monitoring inhibition of P450 activities with 2EN-ABP. (A) Inhibition of 2EN-ABP labeling of P450 1a2 by αNF. Treatment of liver microsomal proteome with increasing concentrations of αNF (0-10 μM) resulted in concentration-dependent blockade of 2EN-ABP labeling of the 52 kDa band designated as P450 1a2. Labeling conditions: 2EN-ABP (2.5 μM) proteome (0.1 mg/mL), 6 min. (B) IC50 curve for inhibition of 2EN-labeling of P450 1a2 by αNF. The calculated IC50 value of 2.0 ± 0.3 μM for αNF matched closely previous literature values (0.8 μM40). Data represent the average ± standard error of four independent experiments per group.
Collectively, these data demonstrate that 2EN-ABP can accurately monitor changes in P450 activity in native proteomes caused by either indirect induction or direct inhibition by drugs. We next considered whether 2EN-ABP could provide similar data on P450 activities in vivo.
Profiling P450-drug interactions in vivo with 2EN-ABP
We next investigated whether 2EN-ABP could label P450 activities in vivo by administering this probe to adult male mice over a dose range of 5-20 mg/kg (i.p.). Livers were harvested 30 minutes after probe treatment and processed for detection of labeled enzymes as described above. Clear dose-dependent labeling of P450s was observed by SDS-PAGE analysis (Figure 5A). These in vivo-labeled proteins were identified by LC-MS analysis as the major P450 enzymes observed previously with In vitro preparations of mouse liver (Table 1). At the 20 mg/kg dose of 2EN-ABP, the signals for most P450s were on par with or greater than those observed in liver microsomes treated with 2EN-ABP In vitro (Table 1), indicating that in vivo profiling of P450 activities can be accomplished without a significant loss in sensitivity. The signals for P450 1a2, in particular, were significantly higher for in vivo-labeled samples, perhaps indicating that this enzyme is more active in the context of living animals.
Figure 5.
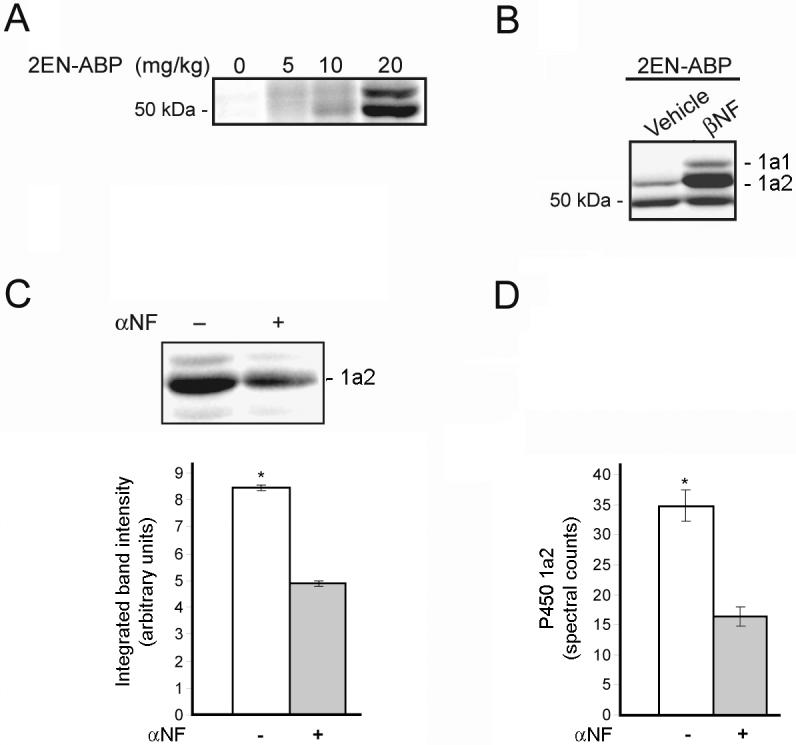
Profiling P450 activities in vivo with 2EN-ABP. (A) Labeling of P450 enzymes by 2EN-ABP in mice. Mice were treated with the indicated doses of 2EN-ABP for 30 minutes and then sacrificed and their liver proteomes analyzed for P450 labeling using CC methods. Dose-dependent labeling was observed of P450 enzymes by 2EN-ABP. (B) Following treatment with βNF (40 mg/kg; i.p., once-per-day dosing for 3 days), mice were administered 2EN-ABP (20 mg/kg, i.p.) and, after 30 min, sacrificed and their liver proteomes analyzed for P450 labeling. Elevated labeling of protein bands corresponding to P450s 1a1 and 1a2 was observed in βNF-treated mice. (C and D) Blockade of 2EN-ABP labeling of P450 1a2 in vivo by pre-treatment with αNF (80 mg/kg), as judged by in-gel fluorescence scanning (C) or LC-MS/MS analysis and spectral counting (D). Mice pre-treated with βNF were used in this study to augment P450 1a2 signals. *p < 0.01 for αNF-treated versus vehicle controls (planned comparison). Data represent the average ± standard error of four independent experiments per group.
Can in vivo profiling with 2EN-ABP detect changes in the activity of P450s caused by drug treatment? To answer this question, we first compared the in vivo labeling profiles of mice treated with βNF or vehicle. A dramatic increase in the in vivo labeling intensities of P450s 1a1 and 1a2 was observed in βNF-treated mice (Figure 5B), analogous to the P450 activity patterns observed previously for liver proteomes treated In vitro with 2EN-ABP (Figure 3A). The significance of the βNF-induced changes in the in vivo activities of P450 1a1 and 1a2 was confirmed by quantification of six independent experiments (Supplemental Figure 3). We next tested whether 2EN-ABP could detect direct P450-drug interactions in vivo by treating βNF-induced mice with the P450 1a2 inhibitor αNF (80 mg/kg, i.p.) or vehicle followed 30 min later by 2EN-ABP (20 mg/kg, i.p.). Mice were sacrificed 30 min later and liver tissues harvested and processed to detect P450 activities. A significant reduction in the labeling intensity of P450 1a2 was observed in mice treated with αNF (Figure 5C). A similar reduction in P450 1a2 signals was observed by LC-MS/MS analysis of avidin-enriched samples from vehicle- versus _NF-treated mice (Figure 5D). These results indicate that 2EN-ABP can be used to monitor the direct binding of drugs to P450 active sites in living animals.
DISCUSSION
ABPP technologies have facilitated the functional proteomic analysis of several enzyme classes [29-31]. The recent advent of tag-free methods for ABPP has provided avenues for profiling enzyme activities directly in living systems [29,31-33]. Building on these precedents, we have reported herein the first activity-based probe for the cytochrome P450 superfamily of enzymes. This agent, designated 2EN-ABP, satisfies all of the major criteria expected for a bona fide functional proteomics probe for the P450 enzyme class: 1) specific labeling of active, but not inactive P450s, 2) targeting of several members of the P450 family, and 3) minimal cross-reactivity with other proteins. Although it is unlikely that 2EN-ABP will serve as a universal probe for all P450s due to differences in their substrate selectivity, the general principles applied in this study, which exploit conserved features of the P450 mechanism, should prove suitable for the design of ABPs for other members of this enzyme class. Indeed, the creation of a library of structurally diverse “clickable” P450 probes might prove particularly useful for mapping the small molecule-binding preferences of less well-characterized members of this enzyme class. On this note, we should qualify that our original probe 2EN-ABP could conceivably react with P450s at either of its two alkyne groups (even though literature precedent would suggest preferred reactivity with the aryl alkyne, at least with regards to P450 1a1 and 1a2 [25]). Reaction at either site would still leave a free alkyne for subsequent click chemistry, which has been shown to be compatible with either alkyl or aryl alkynes [39].
We have demonstrated a broad range of potential applications for 2EN-ABP. First, this probe can be used to inventory P450 activities in any biological sample, including cells, tissues, and living animals. We anticipate that the 2EN-ABP will prove of value for comparatively profiling P450 activities in diseases such as cancer, where these enzymes play important roles in the metabolism of signaling hormones and chemotherapeutic agents [40-42]. We have also shown that key aspects of P450-drug interactions, including enzyme induction and inhibition, can be discerned with 2EN-ABP both In vitro and in vivo. Notably, 2EN-ABP was able to detect the inhibition of P450 1a2 in mice by αNF, an inhibitor of only moderate potency with mouse P450 1a2 (IC50 value of ∼ 2.0 μM). These data indicate that even relatively modest affinity P450-drug interactions can be detected in vivo using 2EN-ABP.
The ability to monitor drug effects on P450 activities in living systems, where the various dynamic processes that regulate P450 function are maintained, may help researchers make informed decisions about the suitability of drug candidates at early preclinical stages of development. Historically, the pharmaceutical industry has relied on in vitro data and modeling to predict drug interactions with P450s in vivo [7-11]. Though many time-tested pharmaceuticals have been developed using this standard approach, complications from drug-drug interactions still present a serious risk that results in hospitalization for millions worldwide per year [3,43]. This problem is likely to escalate in the future as our population ages and polytherapy becomes more common. P450 subfamilies 1, 2, and 3 play important roles in both human and mouse xenobiotic metabolism [2]. Our data indicate that 2EN-ABP targets several key members of these P450 subfamilies. Human P450 1A2 is inducible by cigarette smoking and oxidizes procarcinogens, and is suggested to participate in the metabolism of approximately 10-15% of marketed drugs [44,45]. P450 3a11 is considered to be the mouse congenator of human P450 3A4, which is involved in the metabolism of greater than 60% of marketed drugs [45]. P450 2d9 is a male-specific enzyme in mice that may be related to human 2D6, an important P450 for drug metabolism, including the therapeutics minaprine and indinivar [46]. These findings collectively indicate that 2EN-ABP is primed to report on P450-drug interactions of relevance to the development of wide range of pharmaceuticals.
One limitation of our chemical proteomic approach for in vivo profiling of P450 activities is that its application is restricted to cell and animal model systems. Although rodents are routinely used as in vivo models for P450 functional analysis and drug metabolism studies, their P450 repertoires are quite distinct from that of humans (e.g., mice contain 102 putatively functional P450 enzymes compared to only 57 for humans) [47,48]. These differences create complications in making direct comparisons between the function of P450s in rodents and humans. Nonetheless, significant research efforts have been devoted to determining functional orthologues for rodent and human P450s, and it does appear that correlations can be drawn between the 1a, 2d and 3a subfamilies across these species. In contrast, other sub-families, such as 2c, are more challenging to interpret [47,48]. To overcome this drawback, additional animal models, such as rabbits, dogs, and primates, that are known to possess closer correlates of human P450s could be used for in vivo ABPP experiments [47]. The continued creation of transgenic mouse models that express human P450 enzymes should also prove useful [49-52].
In summary, we envision that P450-directed ABPP probes will serve as valuable research tools for the functional characterization of this large and diverse enzyme family. In addition to their application for the proteomic profiling of P450 activities and small-molecule interactions, the compatibility of these probes with living systems opens up future possibilities for their incorporation into cell-based screens [53] to assess the functional contribution of P450 enzymes in wide range of biological processes.
Significance
Cytochrome P450 enzymes represent a large and diverse protein family in mammals that play key roles in the metabolism of endogenous signaling molecules, xenobiotics, and drugs. As our understanding of P450 biology has increased, it has become apparent that these enzymes can be regulated by a number of post-translational events in vivo, including availability of catalytic cofactors, post-translational modifications, and protein-protein interactions. These layers of biological complexity emphasize the need for advanced proteomic tools to monitor the functional state of P450s in native biological systems. Herein, we have described the design, synthesis, and characterization of the first class of chemical proteomic probes for profiling P450 activities. A broad-spectrum, mechanism-based P450 inhibitor, 2-ethynylnaphthalene, was converted into an activity-based probe by derivatization with a versatile click chemistry handle that enables the selective tagging, detection, enrichment, and identification of P450 enzymes in any biological system. The 2EN-activity based probe (2EN-ABP) was found to label several P450 enzymes in rodent liver in an NADPH-dependent manner, and proved capable of monitoring both drug induction and inhibition of these enzymes in vivo. We anticipate that 2EN-ABP and related chemical proteomic probes will serve as valuable research tools for the functional characterization of P450 activities and small-molecule interactions in a wide range of biological systems.
EXPERIMENTAL PROCEDURES
Synthesis of 2EN-ABP
See Supplemental Materials.
Preparation of mouse tissue proteomes
Livers were harvested from male C57Bl/6J mice and processed according to prior methods with some minor changes [54]. Mice were anesthetized with a CO2/O2 mixture, and euthanized with CO2. Livers were harvested, rinsed with ice-cold 1.5% KCl, finely diced with a razor blade, and dounce homogenized in 250 mM sucrose (∼5 mL per liver) in PBS buffer. EDTA was not included in the buffer due to incompatibility with subsequent CC reactions. Liver homogenates were then treated with a tissue tearor for five pulses. High-speed centrifugation was used to remove the heavy membrane components and ultimately isolate microsomes: 10,000 × g (25 min, pellet = heavy membrane fraction) to obtain the S9 fraction, 100,000 × g (90 min, pellet = microsome fraction). Microsomes were resuspended in 250 mM sucrose in PBS buffer with minimal dounce homogenization. All samples were stored at -80 °C until use. Samples were subjected to no more than two freeze-thaw cycles.
Recombinant expression of P450 enzymes in COS-7 cells
COS-7 cells were grown to 80% confluency, and transfections were carried out with a human P450 1a2 cDNA (Open Biosystems) according to previously described methods [55]. Transfected cells were then harvested, sonicated, dounce homogenized in PBS buffer, and centrifuged at 100,000 × g (60 min, pellet = membrane fraction). The membrane fraction was resuspended in PBS buffer with minimal sonication.
In vitro labeling of P450 enzymes in cell and tissue proteomes with 2EN-ABP
Proteomes (50 μL of 1.0 mg/mL protein in PBS) were treated with 2EN-ABP (10 μM - unless otherwise noted, 0.5 μL of a stock solution in DMSO) in the presence or absence of NADPH (1 mM final concentration, 0.5 _L of a stock solution in PBS). Samples were incubated at 37 °C for 45 min. For drug induction studies, male C57Bl/6J mice were treated with dexamethasone or β-naphthoflavone (βNF) (80 or 40 mg/kg, i.p., respectively) in vehicle (18:1:1 saline:emulphor:ethanol) once daily for 3 days. On the fourth day livers were collected and analyzed as described above.
Following incubation with 2EN-ABP, proteomes were treated with an azide-rhodamine reporter group (0.5 μL of a 6 mM stock solution in DMSO) followed by 1 mM TCEP (25× stock in water) and 100 μM ligand (17× stock in DMSO:t-butanol (1:4)). The samples were vortexed and then 1 mM CuSO4 (50× stock in water) was added. Samples were vortexed again and left at room temperature in the dark for 1 hr at which time 2× SDS-PAGE loading buffer (reducing) was added to each reaction. The samples were then heated at 90 °C for 8 min. The samples were then loaded onto gels (30 μL per well).
Inhibition of 2EN-ABP labeling of P450 1a2 by αNF
Liver microsomal proteomes (50 μL of 0.1 mg/mL protein in PBS) from βNF-induced mice were treated with αNF (0-10 μM, 0.5 μL of a stock solution in DMSO for each concentration) and NADPH was added to the samples (0.5 mM final concentration, 0.5 μL of a stock solution in PBS). Samples were incubated at 37 °C for 10 min, after which 2EN-ABP was added (2.5 μM, 0.5 μL of a stock solution in DMSO), and incubations proceeded at 37 °C for an additional 6 min prior to analysis.
In vivo labeling of P450 enzymes in mice with 2EN-ABP
Male C57Bl/6J mice were administered 2EN-ABP (20 mg/kg, i.p. - unless otherwise noted) in a vehicle of 18:1:1 saline:emulphor:ethanol at a volume of 10 μL/g weight. Control mice were injected with vehicle alone (listed as 0 mg/kg). After 30 min the mice were anesthetized with a CO2/O2 mixture, and euthanized with CO2. Livers were harvested as described above. For inhibition studies, αNF (80 mg/kg in above vehicle) was administered by i.p. injection to βNF-induced mice on the fourth day. After 30 min, mice were administered 2EN-ABP. Following an additional 30 min, the mice were euthanized and liver tissue harvested and analyzed as described above.
Supplementary Material
ACKNOWLEDGMENTS
We thank H. Hoover for assistance with cell culture experiments and the Cravatt laboratory for helpful discussions and critical reading of the manuscript. This work was supported by the National Institutes of Health (CA087660), the California Breast Cancer Research Program (A.T.W.), and the Skaggs Institute for Chemical Biology.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Denisov IL, Makris TM, Sligar SG, Schlichting I. Structure and chemistry of cytochrome P450. Chem. Rev. 2005;105:2253–2277. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- 2.Guengerich FP, Wu Z-L, Bartleson CJ. Function of human cytochrome P450s: Characterization of the orphans. Biochem. Biophys. Res. Commun. 2005;338:465–469. doi: 10.1016/j.bbrc.2005.08.079. [DOI] [PubMed] [Google Scholar]
- 3.Meunier B, de Visser SP, Shaik S. Mechanism of oxidation reactions catalyzed by cytochrome P450 enzymes. Chem. Rev. 2004;104:3947–3980. doi: 10.1021/cr020443g. [DOI] [PubMed] [Google Scholar]
- 4.Brueggemeier RW, Hackett JC, Diaz-Cruz ES. Aromatase inhibitors in the treatment of breast cancer. Endocr. Rev. 2005;26:331–345. doi: 10.1210/er.2004-0015. [DOI] [PubMed] [Google Scholar]
- 5.Wienkers LC, Heath TG. Predicting in vivo drug interactions from In vitro drug discovery data. Nature Rev. Drug. Disc. 2005;4:825–833. doi: 10.1038/nrd1851. [DOI] [PubMed] [Google Scholar]
- 6.Bjornsson TD, et al. The conduct of in vitro and in vivo drug-drug interaction studies: a pharmaceutical research and manufacturers of America (PhRMA) perspective. Drug Metab. Dispos. 2003;31:815–832. doi: 10.1124/dmd.31.7.815. [DOI] [PubMed] [Google Scholar]
- 7.Lin JH, Lu AYH. Inhibition and induction of cytochrome P450 and the clinical implications. Clin. Pharmacokinet. 1998;35:361–390. doi: 10.2165/00003088-199835050-00003. [DOI] [PubMed] [Google Scholar]
- 8.Guengerich FP. Cytochrome P450s and other enzymes in drug metabolism and toxicity. The AAPS Journal. 2006;8:E101–E111. doi: 10.1208/aapsj080112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rodrigues AD, Rushmore TH. Cytochrome P450 pharmacogenetics in drug development: In vitro studies and clinical consequences. Curr. Drug Metab. 2002;3:289–309. doi: 10.2174/1389200023337522. [DOI] [PubMed] [Google Scholar]
- 10.Bachmann KA. Inhibition constants, inhibitor concentrations and the prediction of inhibitory drug-drug interactions: pitfalls, progress, and promise. Curr. Drug Metab. 2006;7:1–14. doi: 10.2174/138920006774832541. [DOI] [PubMed] [Google Scholar]
- 11.Marathe PH, Rodrigues AD. In vivo animal models for investigating potential CYP3A- and Pgp- mediated drug-drug interactions. Curr. Drug. Metab. 2006;7:687–704. doi: 10.2174/138920006778520598. [DOI] [PubMed] [Google Scholar]
- 12.Shou M. Prediction of pharmacokinetics and drug-drug interactions from In vitro metabolism data. Curr. Opin. Drug. Disc. Devt. 2005;8:66–77. [PubMed] [Google Scholar]
- 13.Hughes AL, Powell DW, Bard M, Eckstein J, Barbuch R, Link AJ, Espenshade PJ. Dap1/PGRMC1 binds and regulates cytochrome P450 enzymes. Cell Metab. 2007;5:143–149. doi: 10.1016/j.cmet.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 14.Aguiar M, Masse R, Gibbs BF. Regulation of cytochrome P450 by posttranslational modification. Drug Metab. Rev. 2005;37:379–404. doi: 10.1081/dmr-46136. [DOI] [PubMed] [Google Scholar]
- 15.Oesch-Bartlomowicz B, Oesch F. Phosphorylation of cytochrome P450 isoenzymes in intact hepatocytes and its importance for their function in metabolic processes. Arch. Toxicol. 1990;64:257–261. doi: 10.1007/BF01972984. [DOI] [PubMed] [Google Scholar]
- 16.Venkatakrishnan K, et al. Comparison between cytochrome P450 (CYP) content and relative activity approaches to scaling from cDNA-expressed CYPs to human liver microsomes: ratio of accessory proteins as sources of discrepancies between the approaches. Drug Metab. Dispos. 2000;28:1493–1504. [PubMed] [Google Scholar]
- 17.Lin JH. Pharmacokinetic and pharmacodynamic properties of histamine H2-receptor antagonists: relationship between intrinsic potency and effective plasma concentrations. Pharmacokinet. 1991;20:218–236. doi: 10.2165/00003088-199120030-00004. [DOI] [PubMed] [Google Scholar]
- 18.Knodell RG, et al. Differential inhibition of individual human liver cytochromes P450 by cimetidine. Gastroenterology. 1991;101:1680–1691. doi: 10.1016/0016-5085(91)90408-d. [DOI] [PubMed] [Google Scholar]
- 19.Chang T, et al. Selective inhibition of rat hepatic microsomal P450: I. Effect of the in vivo administration of cimetidine. J. Pharmacol. Exp. Ther. 1991;260:1441–1449. [PubMed] [Google Scholar]
- 20.Chang T, et al. Selective inhibition of rat hepatic microsomal P450: II. Effect of the In vitro administration of cimetidine. J. Pharmacol. Exp. Ther. 1991;260:1450–1455. [PubMed] [Google Scholar]
- 21.Lane CS, Wang Y, Betts R, Griffiths WJ, Patterson LH.Comparative cytochrome P450 proteomics in the livers of immune-deficient mice using 18O stable isotope labeling Mol. Cell. Prot In press 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kent UM, Jushchyshyn MI, Hollenberg PF. Mechanism-based inactivators as probes of cytochrome P450 structure and function. Curr. Drug Metab. 2001;2:215–243. doi: 10.2174/1389200013338478. [DOI] [PubMed] [Google Scholar]
- 23.Foroozesh M, Primrose G, Guo Z, Bell LC, Alworth WL, Guengerich F. P. Aryl acetylenes as mechanism-based inhibitors of cytochrome P450-dependent monooxygenase enzymes. Chem. Res. Toxicol. 1997;10:91–102. doi: 10.1021/tx960064g. [DOI] [PubMed] [Google Scholar]
- 24.Roberts ES, Hopkins NE, Foroozesh M, Alworth WL, Halpert JR, Hollenberg PF. Inactivation of cytochrome P450S 2B1, 2B4, 2B6, and 2B11 by arylalkynes. Drug Met. Dis. 1997;25:1242–1248. [PubMed] [Google Scholar]
- 25.Ortiz de Montellano PR, Mico BA. Branchpoint for heme alkylation and metabolite formation in the oxidation of aryl acetylenes by cytochrome P450. J. Biol. Chem. 1985;260:3330–3336. [PubMed] [Google Scholar]
- 26.Hammons GJ, Alworth WL, Hopkins NE, Guengrich FP, Kadlubar FD. 2-Ethynylnaphthalene as a mechanism-based inactivator of the cytochrome P-450 catalyzed N-oxidation of 2-naphthylamine. Chem. Res. Toxicol. 1989;2:367–374. doi: 10.1021/tx00012a003. [DOI] [PubMed] [Google Scholar]
- 27.Yun C-H, Hammons GJ, Jones G, Martin MV, Hopkins NE, Alworth WL, Guengerich FP. Modification of cytochrome P450 1A2 enzymes by the mechanism-based inactivator 2-ethynylnaphthalene and the photoaffinity label 4-azidobiphenyl. Biochem. 1992;31:10556–10563. doi: 10.1021/bi00158a019. [DOI] [PubMed] [Google Scholar]
- 28.Beebe LE, Roberts ES, Fornwald LW, Hollenberg PF, Alworth WL. Mechanism-based inhibition of P4502b-10 by selected arylalkynes. Biochem. Pharmacol. 1996;52:1507–1513. doi: 10.1016/s0006-2952(96)00525-4. [DOI] [PubMed] [Google Scholar]
- 29.Evans MJ, Cravatt BF. Mechanism-based profiling of enzyme families. Chem. Rev. 2006;106:3279–3301. doi: 10.1021/cr050288g. [DOI] [PubMed] [Google Scholar]
- 30.Jessani N, Cravatt BF. The development and application of methods for activity-based protein profiling. Curr. Opin. Chem. Biol. 2004;8:54–59. doi: 10.1016/j.cbpa.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 31.Sadaghiani AM, Verhelst SH, Bogyo M. Tagging and detection strategies for activity-based proteomics. Curr. Opin. Chem. Biol. 2007;11:20–28. doi: 10.1016/j.cbpa.2006.11.030. [DOI] [PubMed] [Google Scholar]
- 32.Speers AE, Adam GC, Cravatt BF. Activity-based protein profiling in vivo using a copper(I)-catalyzed azide-alkyne [3+2] cycloaddition. J. Am. Chem. Soc. 2003;125:4686–4687. doi: 10.1021/ja034490h. [DOI] [PubMed] [Google Scholar]
- 33.Speers AE, Cravatt BF. Profiling enzyme activities in vivo using click chemistry methods. Chem. Biol. 2004;11:535–546. doi: 10.1016/j.chembiol.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 34.Kolb HC, Finn MG, Sharpless KB. Click chemistry: diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 35.Washburn MP, Wolters D, Yates JR. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 2001;19:242–247. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 36.Jessani N, et al. A streamlined platform for high-content functional proteomics of primary human specimens. Nat. Methods. 2005;2:691–697. doi: 10.1038/nmeth778. [DOI] [PubMed] [Google Scholar]
- 37.Zhang Q-Y, Dunbar D, Kaminsky LS. Characterization of mouse small intestinal cytochrome P450 expression. Drug Metab. Dispos. 2003;31:1346–1351. doi: 10.1124/dmd.31.11.1346. [DOI] [PubMed] [Google Scholar]
- 38.Helsby NA, Chipman JK, Gescher A, Kerr D. Inhibition of mouse and human CYP 1A- and 2E1-dependent substrate metabolism by the isoflavonoids genistein and equol. Food Chem. Toxicol. 1998;36:375–382. doi: 10.1016/s0278-6915(97)00171-3. [DOI] [PubMed] [Google Scholar]
- 39.Lee B-Y, Park SR, Jeon HB, Kim KS. A new solvent system for the efficient synthesis of 1,2,3-triazoles. Tetrahedron Lett. 2006;47:5105–5109. [Google Scholar]
- 40.Agundez JAG. Cytochrome P450 gene polymorphism and cancer. Curr. Drug Metab. 2004;5:211–224. doi: 10.2174/1389200043335621. [DOI] [PubMed] [Google Scholar]
- 41.Patterson LH, Murray GI. Tumour cytochrome P450 and drug activation. Curr. Pharm. Design. 2002;8:1335–1347. doi: 10.2174/1381612023394502. [DOI] [PubMed] [Google Scholar]
- 42.McFadyen MCE, Melvin WT, Murray GI. Cytochrome P450 enzymes: novel options for cancer therapeutics. Mol. Cancer Ther. 2004;3:363–371. [PubMed] [Google Scholar]
- 43.Poirier A, Funk C, Lavé T, Noé J. New strategies to address drug-drug interactions involving OATPs. Curr. Opin. Drug Disc. Devt. 2007;10:74–83. [PubMed] [Google Scholar]
- 44.Nishikawa A, Mori Y, Lee IS, Tanaka T, Hirose M. Cigarette smoking, metabolic activation and carcinogenesis. Curr. Drug. Metab. 2004;5:363–373. doi: 10.2174/1389200043335441. [DOI] [PubMed] [Google Scholar]
- 45.Guengerich FP. Human cytochrome P450 enzymes. In: Ortiz de Montellano PR, editor. Cytochrome P450. Kluwer Academic/Plenum Publishers; New York: 2005. pp. 377–530. [Google Scholar]
- 46.Rendic S. Summary of information on human CYP enzymes: human P450 metabolism data. Drug. Metab. Rev. 2002;34:83–448. doi: 10.1081/dmr-120001392. [DOI] [PubMed] [Google Scholar]
- 47.Guengerich FP. Comparisons of catalytic selectivity of cytochrome P450 subfamily enzymes from different species. Chem. Bio. Interact. 1997;106:161–182. doi: 10.1016/s0009-2797(97)00068-9. [DOI] [PubMed] [Google Scholar]
- 48.Nelson DR, Zeldin DC, Hoffman SMG, Maltais LJ, Wain HM, Nebert DW. Comparison of cytochrome P450 (CYP) genes from the mouse and human genomes, including nomenclature recommendations for genes, pseudogenes and alternative-splice variants. Pharmacogenetics. 2004;14:1–18. doi: 10.1097/00008571-200401000-00001. [DOI] [PubMed] [Google Scholar]
- 49.Gonzalez FJ, Kimura S. Study of P450 function using gene knockout and transgenic mice. Arch. Biochem. Biophys. 2003;409:153–158. doi: 10.1016/s0003-9861(02)00364-8. [DOI] [PubMed] [Google Scholar]
- 50.Gonzalez FJ, Yu A-M. Cytochrome P450 and xenobiotic receptor humanized mice. Annu. Rev. Pharmacol. Toxicol. 2006;46:41–64. doi: 10.1146/annurev.pharmtox.45.120403.100007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Katoh M, et al. Expression of human cytochromes P450 in chimeric mice with humanized liver. Drug Metab. Dispos. 2004;32:1402–1410. doi: 10.1124/dmd.104.001347. [DOI] [PubMed] [Google Scholar]
- 52.Katoh M, et al. In vivo induction of human cytochrome P450 3A4 by rifabutin in chimeric mice with humanized liver. Xenobiotica. 2005;35:863–875. doi: 10.1080/00498250500296231. [DOI] [PubMed] [Google Scholar]
- 53.Evans MJ, Saghatelian A, Sorensen EJ, Cravatt BF. Target discovery in small-molecule cell-based screens in situ proteome reactivity profiling. Nat. Biotechnol. 2005;23:1303–1307. doi: 10.1038/nbt1149. [DOI] [PubMed] [Google Scholar]
- 54.Guengerich FP. Analysis and characterization of enzymes and nucleic acids. In: Hayes AW, editor. Principles and Methods of Toxicology. Raven Press, Ltd.; New York: 1994. pp. 1259–1313. [Google Scholar]
- 55.Clark BJ, Waterman M. Heterologous expression of mammalian P450 in COS cells. Methods Enzymol. 1991;206:100–108. doi: 10.1016/0076-6879(91)06081-d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.