

Abstract
The regulation of innate immune responses to pathogens occurs through the interaction of Toll-like receptors (TLRs) with pathogen-associated molecular patterns and the activation of several signaling pathways whose contribution to the overall innate immune response to pathogens is poorly understood. We demonstrate a mechanism of control of murine macrophage responses mediated by TLR1/2 heterodimers through c-Jun N-terminal kinase 1 (JNK1) activity. JNK controls tumor necrosis factor alpha production and TLR-mediated macrophage responses to Borrelia burgdorferi, the causative agent of Lyme disease, and the TLR1/TLR2-specific agonist PAM3CSK4. JNK1, but not JNK2, activity regulates the expression of the tlr1 gene in the macrophage cell line RAW264.7, as well as in primary CD11b+ cells. We also show that the proximal promoter region of the human tlr1 gene contains an AP-1 binding site that is subjected to regulation by the kinase and binds two complexes that involve the JNK substrates c-Jun, JunD, and ATF-2. These results demonstrate that JNK1 regulates the response to TLR1/2 ligands and suggest a positive feedback loop that may serve to increase the innate immune response to the spirochete.
Toll-like receptors (TLRs) play critical roles during the initiation of innate immunity and the development of specific cell-mediated immune responses (1). The members of the TLR family recognize specific components conserved among microorganisms (1), such as triacylated antigens of Borrelia burgdorferi, the causative agent of Lyme disease (2). In humans, the levels of expression of TLR1 have been involved in their capacity to respond to lipidated OspA (2), the antigen used for the FDA-approved vaccine against Lyme borreliosis that has been retired from the market. Similarly, the capacity of B. burgdorferi to induce proinflammatory cytokine production is associated with the interaction of lipoproteins of the spirochete with TLR1/TLR2 complexes (2, 16). The comparison of disease in patients with polymorphic forms of different TLRs has demonstrated their importance in the progression of asthma and atherosclerosis (13, 25). TLR expression is regulated by their interaction with specific ligands and by cytokines. Thus, during leprosy, TLR1 expression is upregulated by gamma interferon (IFN-γ) (20), while B. burgdorferi lipoproteins increase the expression of TLR1 and TLR2 and induce the downregulation of TLR5 (7). Lipopolysaccharide has also been shown to induce increased expression of TLRs and accessory molecules, including MyD88 and MD-2 (24, 29). The mechanism by which this regulation occurs is not completely understood. The transcription factors AP-1, Ets, and PU.1 have been involved in the regulation of murine TLR4 expression (28, 34).
The interaction of TLRs with specific ligands results in the activation of several signaling pathways. The activation of the transcription factor NF-κB in response to TLR stimulation results in the expression of chemotactic factors, proinflammatory cytokines, and adhesion molecules in several cell types, such as macrophages, fibroblasts, and synovial cells (19). TLR engagement also results in the activation of the mitogen-activated protein p38 and c-Jun N-terminal kinases (JNK) (19). These pathways are involved in various physiological processes. The JNK pathway regulates apoptosis, development, cell transformation, T-cell activation and differentiation, and cytokine production (10, 18, 31, 33, 35, 38, 39). Three isoforms of JNK have been identified: two ubiquitously expressed isoforms, JNK1 and JNK2, and the tissue-specific isoform JNK3 (9). JNK phosphorylates the transcription factor c-Jun and increases AP-1 transcriptional activity. Other substrates include JunD, ATF-2, ATF-a, Elk-1, Sap-1, and NFATc3 (9).
In this study, we show that JNK1 controls macrophage responses to B. burgdorferi by regulating the expression of the tlr1 gene. Our results clarify the role of this kinase in the innate immune response to B. burgdorferi.
MATERIALS AND METHODS
Cells.
JNK1-deficient (10) and JNK2-deficient (41) C57BL/6 (B6) mice were used to purify splenic macrophages by positive selection, using a biotinylated antibody (Ab) against CD11b (BD Pharmingen, La Jolla, CA). The purity of the cells, as determined by flow cytometry, was >85%. The macrophage cell line RAW264.7 (ATCC, Manassas, VA) was grown in RPMI medium (Sigma, St. Louis, MO) supplemented with 10% fetal calf serum.
All procedures that involved animals were in accordance with institutional guidelines for animal care at UNC Charlotte and the University of Massachusetts at Amherst.
Plasmids, small interfering RNA (siRNA), and transfections.
Plasmids containing a mutant (dominant-negative) version of human JNK1 (dnJNK), constructed by replacement of Thr183 and Tyr185 by Ala and Phe, respectively (27), or the luciferase gene downstream of 2× AP-1 (27) or 5× NF-κB (Stratagene, La Jolla, CA) response elements were used. The plasmids were transfected into 3 × 106 to 5 × 106 RAW264.7 cells using DEAE-Dextran (Promega, Madison, WI) according to the manufacturer's protocol. The plasmid pBluescript SK(−) (SK) was used as a negative control in transfections with the plasmid containing dnJNK.
siRNA targeting jnk1 mRNA (Ambion, Austin, TX) was used to transfect 5 × 105 RAW264.7 cells using the siPort Amine transfection agent (Ambion) following the manufacturer's instructions. Control siRNA (Ambion) containing a random mixture of oligonucleotides was used in parallel. After 48 h, the cells were assessed for jnk1 mRNA by reverse transcriptase (RT) PCR (Table 1) and stimulated as described below.
TABLE 1.
Primers used in the study
| Gene/target | Sequencea | Purpose |
|---|---|---|
| tlr1 | 5′-CGC AAA CCT TAC CAG AGT G-3′ | RT-PCR |
| 5′-GAC TGG CGT ATG CCA AAC TA-3′ | ||
| tlr1 | 5′-CTG GAG TCT GTT GTA GGA C-3′ | Real-time PCR |
| 5′-GAC TGG CGT ATG CCA AAC TA-3′ | ||
| tlr2 | 5′-AAG TGA AGA GTC AGG TGA TGG ATG TCG-3′ | RT-PCR |
| 5′-GCA GAA TCA ATA CAA TAG AGG GAG ACG C-3′ | Real-time PCR | |
| jnk1 | 5′-TGT GGA ATC AAG CAC CTT CAC TCT GCT G-3′ | RT-PCR |
| 5′-GCA AAC CAT TTC TCC CAT AAT GCA CCC-3′ | ||
| gadph | 5′-CCA TCA CCA TCT TCC AGG AGC GAG-3′ | RT-PCR |
| 5′-CAC AGT CTT CTG GGT GGC AGT GAT-3′ | ||
| tlr1 promoter | 5′-AAG AGC TCC TGA GGT AAG GGG AAA CAG AG-3′ | PCR |
| 5′-AAC CCG GGA AGA AAT TCA AGC ACT TCC TTG-3′ | ||
| tlr1 promoter | 5′-AAT CAA CTT GTC AAA AAA GAC GCA TCC ATC CTG TAA CCA GCA CA-3′ | Site-directed mutagenesis |
| 5′-TGT GCT GGT TAC AGG ATG GAT GCG TCT TTT TTG ACA AGT TGA TT-3′ | ||
| tlr1-AP-1 (−230, −236) | 5′-TAG TAA ACT GAC TGT AGT GA-3′ | EMSA |
| tlr1-AP-1 (−471, −480) | 5′-GCA CAT GAA TGA TCT TCC CT-3′ | EMSA |
| tlr1-AP-1 (−502, −508) | 5′-AAA GAC GTG ATT AAC ATC CAT-3′ | EMSA |
| AP-1 consensus | 5′-CGC TTG ATG ACT CAG CCG GAA-3′ | EMSA |
Underlining represents the consensus sequence.
Stimulations.
Low-passage B. burgdorferi N40 lysates were obtained from mid-log phase cultures by sonication. The protein concentration was determined by the Bradford method (Bio-Rad, Hercules, CA). The TRL1/TLR2 agonist N-palmitoyl-S-[2,3-bis(palmitoyloxy)-(2RS)-propyl]-Cys-[S]-Ser-[S]-Lys4 trihydrochloride (PAM3-CSK4) was purchased from Invivogen (San Diego, CA). In vitro stimulations were performed using 10 μg/ml of B. burgdorferi lysate or 1 μg/ml of PAM3-CSK4.
The phosphorylation of STAT-1 in response to IFN-γ stimulation of RAW264.7 cells was determined by stimulation with 100 ng/ml of recombinant murine IFN-γ (R&D Systems, Minneapolis, MN) for 30 min. Phospho-Tyr701 and total STAT1 (Cell Signaling, Danvers, MA) were then detected by immunoblotting.
JNK activity determination.
Five million RAW264.7 or primary CD11b+ cells were incubated with 10 μg/ml of a B. burgdorferi lysate for the indicated times. The cells were washed and lysed, and JNK activity was determined using the JNK activity assay kit (Cell Signaling) according to the manufacturer's protocol. Briefly, JNK was immunoprecipitated from the stimulated cell extracts with a c-Jun fusion protein bound to agarose. The immunoprecipitate was incubated in kinase buffer for 30 min at 30°C. Phospho-c-Jun was then detected by immunoblotting using a rabbit phospho-c-Jun polyclonal Ab.
Cytokine ELISA.
The levels of tumor necrosis factor alpha (TNF-α) produced by B. burgdorferi-stimulated primary CD11b+ and RAW264.7 cells were determined by capture enzyme-linked immunosorbent assay (ELISA), as described previously (3).
Abs and Western blotting.
Anti-IκBα (Santa Cruz Biotechnology), anti-TLR1, and anti-TLR2 (Invivogen) Abs were used. Immunoblotting was performed with stimulated and unstimulated cell extracts, using standard protocols.
RT-PCR.
Total RNA was isolated from splenic purified CD11b+ and RAW264.7 cells using the TRIzol reagent (Invitrogen, Carlsbad, CA) according to the instructions of the manufacturer. PCR was performed to determine the expression of tlr1, tlr2, and the glyceraldehyde-3-phosphate dehydrogenase gene (gapdh) using 95°C denaturation, 55°C (tlr1 and tlr2) or 60°C (gapdh) annealing, and 72°C extension temperatures with the primers listed in Table 1.
Real-time RT-PCR to quantify tlr1 and tlr2 gene expression in dnJNK-transfected cells was performed using the primers listed in Table 1 and 2 μl of cDNA in a final volume of 20 μl. The reaction mixture contained SYBR green (Quanta Biosciences, Gaithersburg, MD) and ROX (as a reference dye). The reaction was performed at an annealing temperature of 55°C. Relative expression of the gene was determined by amplifying β-actin (Applied Biosystems, Foster City, CA) and was referred to control (SK)-transfected cells, according to the following formula:
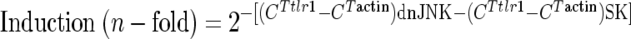 |
where CT represents the threshold cycle for each gene.
Construction of tlr1-luc and tlr1(mut)-luc plasmids.
The proximal DNA fragment corresponding to 1 kb of the human tlr1 gene promoter region was subcloned upstream of the promoterless firefly luciferase (luc) gene in the vector pGL3-basic (Promega). The 1-kb promoter fragment was generated by PCR (Table 1) using genomic DNA isolated from HeLa cells as a template. The fragment was cloned into the pBAD-TOPO (Invitrogen) vector and subcloned using the SmaI and SacI restriction sites into the pGL3 vector (pGL3-tlr1-luc). The construct was sequenced across both junctions to confirm the nucleotide sequence and the predicted orientation. Empty pGL3 was used as a control.
The pGL3-tlr1(mut)-luc plasmid, a derivative of pGL3-tlr1-luc with the AP-1-binding site deleted (nucleotides −502 to −508 relative to the transcription start site), was constructed by using the QuikChange II XL site-directed mutagenesis kit (Stratagene) according to the manufacturer's instructions. The primers used for the deletion of the 7 bp are listed in Table 1. The mutations were confirmed by sequencing.
Nuclear extracts and electromobility shift assay (EMSA).
Mini nuclear extracts were obtained as described previously (26) from 106 RAW264.7 cells unstimulated or stimulated with 10 μg/ml of a B. burgdorferi lysate for 16 h. Binding reactions were performed using 2 μg of nuclear protein in the presence of a specific 32P end-labeled double-stranded oligonucleotide as described previously (26) in the absence or presence of unlabeled oligonucleotides. The oligonucleotides used in this study are listed in Table 1. Supershift/competition assays were performed using 1 μl of anti-c-Jun (N), c-Fos (4), JunD (329), ATF-2 (C-19), and CREB (H-74) Abs (Santa Cruz Biotechnology).
Statistical analyses.
Data are presented as the means and standard errors of at least three independent experiments or means and standard deviations for experiments performed in triplicate. The means of independent experiments were compared with two-way analysis of variance, followed by Bonferroni posttests, using the software Prism version 4.0. The means were considered statistically different when P was <0.05.
RESULTS
JNK activity regulates TNF-α production in response to B. burgdorferi.
The role played by the JNK pathway in macrophage responses to pathogens mediated by TLRs is poorly understood. To address whether the interaction between B. burgdorferi antigens and macrophages results in the activation of JNK, we first stimulated RAW264.7 cells with a B. burgdorferi lysate. JNK activity was evident after 40 min of stimulation and decayed after 80 min (Fig. 1A). The activation of JNK was also detected in primary splenic macrophages (CD11b+ cells) from B6 mice stimulated with a B. burgdorferi lysate (Fig. 1A).
FIG. 1.

JNK activity regulates TNF-α production in response to B. burgdorferi. (A) RAW264.7 (top) and primary CD11b+ (bottom) cells were stimulated with a B. burgdorferi lysate for the indicated times (top) or 40 min (bottom) prior to assaying JNK activity in vitro with c-Jun as a substrate. Phospho-c-Jun was then detected by immunoblotting. The results are representative of three experiments performed. (B) RAW264.7 cells were transfected with a plasmid containing dnJNK1 or a plasmid control (SK) and stimulated with a B. burgdorferi lysate (Bb) or PAM3-CSK4 for 16 h. The TNF-α levels in the stimulation supernatants were then quantified by ELISA. The results shown are the average plus standard error (SE) of three independent experiments. *, P < 0.001 and P < 0.01 for B. burgdorferi and PAM3-CSK4 stimulation, respectively. (C) RAW264 cells were stimulated with 10 μg/ml of a B. burgdorferi lysate (Bb) or 1 μg/ml of PAM3-CSK4 in the presence of increasing concentrations of the JNK inhibitor SP100625. TNF-α was quantified in the stimulation supernatants after 16 h. The results are the average plus SE of three independent experiments.
The involvement of JNK activity in macrophage responses to TLR ligands was then assessed. RAW264.7 cells were transfected with a dominant-negative form of the kinase (dnJNK) (27) and stimulated with a B. burgdorferi lysate or the TLR1/TLR2 agonist PAM3-CSK4, and the supernatants were evaluated for TNF-α production. The repression of JNK activity resulted in a significant reduction of TNF-α production compared to control transfected cells (P < 0.001 for B. burgdorferi and P < 0.01 for PAM3-CSK4 stimulation) (Fig. 1B). Similar results were obtained in macrophages isolated from the spleens of B6 mice (data not shown) and RAW264.7 cells stimulated with a B. burgdorferi extract or PAM3-CSK4 in the presence of increasing concentrations of the JNK inhibitor SP600125 (5) (Fig. 1C). The reduced production of TNF-α was not the result of SP600125-induced cell death, as determined by the analysis of the cells by trypan blue exclusion (data not shown). These results indicated that TLR1/TLR2-mediated TNF-α production in macrophages involves JNK activity.
TLR-mediated responses are dependent on JNK activity.
We further substantiated the contribution of JNK activity to TLR-induced signaling events in macrophages by analyzing the activation of JNK-dependent and independent transcription factors. JNK activity results in the phosphorylation of c-Jun and the formation of AP-1 complexes (9). As expected, the repression of JNK activity during stimulation with B. burgdorferi resulted in decreased AP-1 transcriptional activity (P < 0.001) (Fig. 2A). As a control, we also analyzed the activation of JNK-independent signaling pathways in response to B. burgdorferi antigens. Surprisingly, the repression of JNK activity also resulted in reduced NF-κB transcriptional activity (P < 0.05) (Fig. 2B). No reports have associated JNK activity and the activation of NF-κB. Since the repression of JNK activity during stimulation with TLR ligands results in lower TNF-α production, and this cytokine also activates NF-κB (36), we analyzed IκBα degradation in the cells that had been transfected with the dnJNK plasmid at times when the production of the cytokine was not evident (data not shown). The stimulation of RAW264.7 cells transfected with the dnJNK plasmid also resulted in decreased degradation of IκBα (Fig. 2C), suggesting that the inhibition of JNK results in decreased activation of NF-κB. In order to assess whether the repression of JNK in macrophages results in both TLR-dependent and -independent cell responses, we determined the effect of JNK repression in response to IFN-γ-induced signals. The phosphorylation of STAT1 induced by IFN-γ was not affected by the presence of the dnJNK-containing plasmid in RAW264.7 cells (Fig. 2D), indicating that JNK affects TLR-mediated responses, but not the TLR-independent, IFN-γ-mediated response, in macrophages.
FIG. 2.

TLR1/2-mediated responses are dependent on JNK activity. (A) RAW264.7 cells were cotransfected with a plasmid containing the luciferase gene under the influence of AP-1 (A) or NF-κB (B) response elements plus a plasmid containing the dnJNK form or a plasmid control (SK). The cells were then stimulated with a B. burgdorferi lysate (Bb) for 16 h, and luciferase activity was assayed. The results shown are the average ± standard error of four and three independent experiments, respectively. *, P < 0.001 (AP-1-luc) and P < 0.05 (NF-κB-luc). (C) RAW264.7 cells were transfected with a plasmid containing the dnJNK form or a plasmid control (SK) and stimulated with a B. burgdorferi lysate for the indicated times. The cells were then lysed and assessed for IκBα content by immunoblotting (left). Band quantitation was performed with the Java-based software ImageJ (right). The experiment shown is representative of three performed. (D) RAW264.7 cells were transfected with a plasmid containing the dnJNK form or a plasmid control (SK) and stimulated with recombinant murine IFN-γ for 30 min. Phospho-STAT1 and total STAT-1 levels were determined by immunoblotting. The results shown are representative of two experiments performed with similar results.
JNK1 regulates the expression of the tlr1 gene.
Our results suggested that TLR-mediated responses in macrophages are dependent on JNK activity. These data could be explained by a direct effect of JNK activity on the regulation of the expression of the components involved in the responses, including TLR1 and TLR2, or signaling intermediates that lead to the activation of downstream signaling pathways. To test whether JNK regulates the expression of the components involved in TLR-mediated responses, we analyzed tlr1 and tlr2 mRNA levels in macrophages with repressed or absent JNK activity. RAW264.7 cells transfected with dnJNK showed lower expression levels of tlr1, with no effect on tlr2 (Fig. 3A). These results correlated with lower TLR1 protein levels than in control transfected cells, while TLR2 protein levels were not affected by the dnJNK plasmid (Fig. 3B). The reduction of TLR1 surface protein expression induced by the repression of JNK activity was also observed by flow cytometry in THP-1 cells transfected with dnJNK (data not shown).
FIG. 3.

JNK1 regulates the expression of the tlr1 gene. (A) (Left) RAW264.7 cells were transfected with a plasmid containing a dnJNK form or a plasmid control (SK), and total RNA was extracted and assessed by RT-PCR (left) or real-time RT-PCR (right) for the expression of tlr1 and tlr2 mRNAs. Equal input of RNA (left) was assessed by the amplification of the housekeeping gene gapdh. (B) The cells were also used to determine the levels of TLR1 and TLR2 by Western blotting. Protein input was assessed by immunobloting with an anti-actin Ab. (C) CD11b+ cells were purified from B6 (WT) and JNK1- and JNK2-deficient mice. RNA was extracted and subjected to RT-PCR for the expression of tlr1 and tlr2. RNA input was assessed by the amplification of gapdh mRNA. (D) RAW264.7 cells were transfected with JNK1 siRNA or a control (siNC) and used to extract RNA and to determine the mRNA levels of jnk1, tlr1, and tlr2. Equal input was determined by amplification of gapdh mRNA. (E) Purified CD11b+ cells from B6 (WT) and JNK1- and JNK2-deficient mice were unstimulated (Unst.) or stimulated with a B. burgdorferi lysate (Bb). The levels of TNF-α were determined in the stimulation supernatants after 16 h. (F) dnJNK1-transfected RAW264.7 cells were stimulated with lipopolysaccharide (LPS), poly(dI-dC), and flagellin, and TNF-α was detected 16 h later. (G) RAW264.7 cells were transfected with siRNA oligonucleotides specific for JNK1 or control oligonucleotides (siNC). Forty-eight hours later, the cells were stimulated for 16 h with a B. burgdorferi lysate (Bb) or PAM3-CSK4 and assessed for TNF-α by ELISA. The results represent the average plus standard error (SE) of three independent experiments. *, P < 0.05. (H) RAW264.7 cells were cotransfected with a plasmid encoding TLR1 under the influence of a constitutively expressed promoter (UNO) or a control plasmid (GL3) plus a plasmid containing the dnJNK form or the empty plasmid (SK). All cells were transfected with the same amount of plasmid. The cells were then stimulated with a B. burgdorferi lysate (Bb) or PAM3CysK4. TNF-α levels in the supernatants were determined 16 h after stimulation. The results shown represent the average plus SE of three independent experiments. *, P < 0.05 compared to control-, UNO plus control-, or UNO plus dnJNK-transfected cells stimulated with B. burgdorferi and PAM3-CSK4.
To further demonstrate the regulation of tlr1 gene expression by JNK, we analyzed the levels of expression of both tlr1 and tlr2 genes in primary macrophages from JNK1- and JNK2-deficient mice. While tlr2 mRNA was not affected by the lack of either JNK1 or JNK2, tlr1 mRNA was reduced in JNK1-deficient macrophages (Fig. 3C). Moreover, the silencing of JNK1-encoding mRNA in RAW264.7 cells resulted in lower tlr1 gene expression than in controls (Fig. 3D), suggesting that JNK1 activity specifically regulates the expression of TLR1.
To assess the contribution of the JNK isoforms to TNF-α production in response to B. burgdorferi, we stimulated JNK1- and JNK2-deficient CD11b+ cells with a B. burgdorferi lysate. Both JNK1- and JNK2-deficient macrophages produced lower levels of TNF-α than wild-type macrophages, although the differences were not statistically significant (Fig. 3E). Correspondingly, JNK1 siRNA-transfected RAW264.7 cells produced lower levels of TNF-α in response to a B. burgdorferi lysate and PAM3-CSK4 (P < 0.05 for both stimuli) (Fig. 3G), indicating that JNK1 and JNK2 regulate the production of TNF-α through different mechanisms and that it involves the regulation of tlr1 gene expression by JNK1.
To determine whether JNK1 regulates TLR1/2-mediated responses solely through the control of TLR1 expression, we transfected RAW264.7 cells with a plasmid that drove the constitutive expression of the receptor. The ectopic expression of TLR1 prevented the inhibition of TNF-α production in the presence of the dnJNK plasmid in response to B. burgdorferi antigens and PAM3-CSK4 (Fig. 3H), confirming that JNK1 controls TLR1/2-mediated signals in macrophages by regulating the expression of TLR1.
JNK1 regulates tlr1 promoter activity.
To further demonstrate that JNK activity regulates tlr1 gene transcription, we cloned the proximal 1-kb promoter region of the tlr1 gene (Fig. 4) upstream of a promoterless luciferase gene. The transfection of RAW264.7 cells with this construct resulted in basal levels of promoter activity that were significantly enhanced by the stimulation of the cells (P < 0.05) (Fig. 5A). The increased luciferase expression correlated with augmented TLR1 protein levels in RAW264.7 cells that had been stimulated with a B. burgdorferi lysate (Fig. 5A). The cotransfection of RAW264.7 cells with this plasmid and the dnJNK-containing plasmid resulted in the repression of promoter activity (P < 0.001 for all three conditions) (Fig. 5A), confirming that JNK is involved in the regulation of TLR1 expression.
FIG. 4.

Structure of the human tlr1 gene. The 1-kb proximal promoter and the gene encoding TLR1 are shown. The proximal promoter sequence is shown in the magnified box, with the three putative AP-1 binding sites highlighted. The sequence corresponds to nucleotides 5956071 to 5955050 of contig NT016297 (NCBI/NIH).
FIG. 5.

JNK1 regulates tlr1 promoter activity. (A) (Top) RAW264.7 cells were cotransfected with a plasmid containing the luciferase gene downstream of the 1-kb proximal tlr1 promoter and the dnJNK plasmid or a plasmid control (SK) and stimulated with a B. burgdorferi lysate (Bb) or PAM3-CSK4 or left unstimulated (Unst.). Luciferase activity (AU, arbitrary units) was measured after 16 h of stimulation. The data presented correspond to the average plus standard deviation of triplicate determinations of one of four experiments performed. *, P < 0.05. (Bottom) Western blot of RAW264.7 cells stimulated with 10 μg/ml of a B. burgdorferi lysate for 16 h using anti-TLR1 Ab. Equal protein input was assessed with anti-Actin Ab. (B) EMSA showing nuclear extract binding to oligonucleotides (oligo) containing putative AP-1 binding sites in the proximal 1-kb tlr1 promoter. The reactions were performed in the absence or presence of an excess (100×) of unlabeled (cold) oligonucleotides. (C) EMSA of nuclear extracts of RAW264.7 cells unstimulated and stimulated with a B. burgdorferi lysate for 30 min. The reactions were performed in the absence or the presence of Abs against c-Jun, JunD, c-Fos, CREB, and ATF-2 and in the absence or presence of unlabeled (cold) oligonucleotides corresponding to the tlr1 promoter or the AP-1 consensus binding sequence. The arrows indicate the constitutive (top) and B. burgdorferi-enhanced (bottom) complexes formed. (D) EMSA of tlr1-derived and consensus AP-1 binding oligonucleotides with nuclear extracts from unstimulated or B. burgdorferi-stimulated RAW264.7 cells. (E) RAW264.7 cells were cotransfected with plasmids containing tlr1- or tlr1Δ(−502,−508) [tlr1(mut)]-luciferase and a plasmid containing dnJNK or a plasmid control (SK). After 48 h, the cells were stimulated with a B. burgdorferi lysate (Bb) or PAM3-CSK4 or left unstimulated (Unst.). Luciferase activity was measured 16 h poststimulation. The results shown are the average plus standard error of three independent experiments. *, P < 0.05.
Three putative AP-1 binding sites that potentially could serve as AP-1 binding sites in the tlr1 promoter were identified (Fig. 4). Double-stranded oligonucleotides that spanned these regions were used in EMSAs (Table 1). Only the oligonucleotide based on the sequence found at −502 to −508 showed binding of nuclear extract preparations of unstimulated (Fig. 5B) and B. burgdorferi-stimulated (not shown) RAW264.7 cells. We observed two binding complexes at this site. The lower complex increased in B. burgdorferi-stimulated RAW264.7 compared to unstimulated cells (Fig. 5C) and was competed when the binding reactions contained anti-c-Jun and anti-c-Fos Abs (Fig. 5C). This complex was competed by unlabeled double-stranded oligonucleotides corresponding to the AP-1 consensus binding site (Fig. 5C) and coincided with the complex obtained with a consensus AP-1 binding site oligonucleotide (Fig. 5D). The presence of ATF-2 and JunD Abs competed the formation of both complexes, while Abs to CREB did not result in competition (Fig. 5C). The upper band observed with anti-CREB was not consistently seen in other experiments performed. These results suggested that this region serves as a docking area for two complexes that contain ATF-2 plus JunD and c-Jun plus c-Fos. These data also suggested that the upper complex is involved in the constitutive expression of the tlr1 gene while the lower complex is formed in response to stimulation with B. burgdorferi antigens. Since ATF-2 and JunD are substrates of JNK (12), this interpretation is in agreement with the control of basal levels of tlr1 gene expression by JNK. Overall, these data demonstrated the presence of functional AP-1 binding sites in the proximal promoter region of the tlr1 promoter.
The contribution of the −502 to −508 binding site to the regulation of tlr1 gene expression mediated by JNK was tested with a deletion mutant of the tlr1-luc construct. The deletion resulted in decreased promoter activity that was not enhanced by stimulation with a B. burgdorferi lysate or PAM3-CSK4 (P < 0.05 for both stimuli) (Fig. 5E). Furthermore, compared to the wild-type promoter, the mutated form was not affected by the coexpression of dnJNK (Fig. 5E). Overall, these data demonstrate that JNK1 activity regulates the expression of TLR1 in macrophages.
DISCUSSION
The interaction of pathogen-associated molecular patterns with the germ line-encoded TLRs is a fundamental feature of the immune response associated with infection. It is becoming evident that these interactions in innate immune cells have profound consequences for the ability of the body to mount efficient or detrimental responses to pathogens. Thus, deficiencies in TLR1, TLR2, or the adaptor protein MyD88 lead to increased B. burgdorferi burdens in mice (2, 4, 6, 21, 40), underscoring the importance of these receptors for the efficient control of infection. Moreover, the severity of pathology associated with infection with Mycobacterium leprae is associated with the host's capacity to respond to mycobacterial antigens and is also related to TLR expression (20). In addition, TLR ligands are being considered as potential adjuvants that could help boost the response to vaccines compared to the currently limited choices in humans (17, 32). TLR-mediated signals are also required for the successful vaccination of individuals against infectious diseases. For example, the deficient expression of TLR1 in patients hyporesponsive to lipidated outer surface protein A of B. burgdorferi is associated with decreased responses to the vaccine against the spirochete (2). It is therefore imperative to understand the control of these responses, including how the expression of TLRs is regulated in innate immune cells.
Our results demonstrate that JNK activation upon engagement of TLR1/2 complexes with specific ligands initiates a positive feedback cascade that increases the expression of TLR1. They also show that in our model the ectopic expression of the receptor restores the capacity of macrophages to respond to B. burgdorferi and PAM3-CSK4, indicating that in our system, JNK1 activity contributes exclusively to TLR1 expression. Experiments with splenic macrophages derived from deficient mice demonstrated that JNK1, but not JNK2, activity is involved in the control of TLR1 expression, while neither JNK1 nor JNK2 regulates the expression of TLR2 in RAW264.7 cells or primary macrophages. These results correlate with previous observations in the macrophage cell line RAW264.7 that showed no implication of JNK in the expression of the tlr2 gene (22). Furthermore, our results show that JNK2 also contributes to the induction of TNF-α in response to B. burgdorferi antigens and the TLR1/2 agonist PAM3-CSK4 through a mechanism that does not involve the regulation of TLR1. In correlation, the induction of TNF-α by the TLR4, TLR9, and TLR5 agonists lipopolysaccharide, poly(dI-dC), and flagellin, respectively, was lowered in cells transfected with the plasmid containing dnJNK (Fig. 3F). These data therefore imply nonoverlapping functions of the two kinases that have also been demonstrated in T cells and other cell types (8, 10, 30, 41) and that have been associated with differential binding of both isoforms with their substrates (11).
Several reports have shown the regulation of TLR expression by different stimuli, including B. burgdorferi stimulation and cytokines (7, 20, 24). Similarly, corticotropin-releasing factor and the urocortins regulate the expression of TLR4 through the activation of the transcription factors PU.1 and AP-1 (34), although the exact mechanisms of regulation have not been described. The regulation of TLR expression may be stimulus dependent, since B. burgdorferi does not affect the expression of the tlr4 gene (reference 7 and data not shown). Similarly, the regulation of tlr2 gene expression by NF-κB has been documented (22-24), and NF-κB binding sites have been described in the tlr2 promoter (23, 37). However, our results indicate that despite lower NF-κB activation induced by TLR ligands in the absence of JNK activity due to reduced expression levels of TLR1, the levels of TLR2 remained unchanged.
The interaction of B. burgdorferi antigens with macrophages potentially occurs through different TLRs: TLR1/TLR2 dimers respond to triacylated lipoproteins, such as lipidated OspA (2), while TLR5 and TLR9 probably contribute to the response of bacterial lysates through their interaction with flagellin and hypomethylated CpG motifs, respectively (14, 15). In turn, these interactions may be physiologically relevant, due to bacterial death in vivo. Our results strongly suggest, however, that the predominant response under our experimental conditions occurred through TLR1/TLR2, since (i) the response was mimicked by the use of the TLR1/TLR2 agonist PAM3-CSK4 and (ii) the ectopic expression of TLR1 in JNK-repressed RAW264.7 cells completely restored the responses to both B. burgdorferi extracts and PAM3-CSK4. In light of our results, we propose that the interactions of spirochetal antigens with TLRs result in the activation of JNK. JNK substrates contribute to the upregulation of the tlr1 gene, which in turn increases the response of macrophages to TLR1/TLR2 ligands. Thus, we propose that JNK1 contributes to the response of macrophages to B. burgdorferi by regulating the expression of TLR1.
Several signaling pathways are activated as a result of the ligation of TLRs with specific ligands. The specific contribution of each pathway to phagocytic responses is still unclear. During infection with B. burgdorferi, p38 mitogen-activated protein kinase controls inflammation (3). Our results indicate that the different signal pathways activated as a result of the interactions between TLRs and their ligands are not redundant, although they may all be needed for a full response to infectious agents. Since macrophage responses to B. burgdorferi are largely dependent on TLR-mediated interaction with ligands present in the bacterium, our results can provide the basis for a full understanding of the immune response to this prevalent infectious agent.
Acknowledgments
This work was supported by NIH grant AR048265 to J.A.
Editor: F. C. Fang
Footnotes
Published ahead of print on 30 July 2007.
REFERENCES
- 1.Akira, S., S. Uematsu, and O. Takeuchi. 2006. Pathogen recognition and innate immunity. Cell 124:783-801. [DOI] [PubMed] [Google Scholar]
- 2.Alexopoulo, L., T. Thomas, M. Schnare, Y. Lobet, J. Anguita, R. T. Schoen, R. Medzhitov, E. Fikrig, and R. A. Flavell. 2002. Hyporesponsiveness to vaccination with Borrelia burgdorferi OspA in humans and in TLR1- and TLR2-deficient mice. Nat. Med. 8:878-884. [DOI] [PubMed] [Google Scholar]
- 3.Anguita, J., S. W. Barthold, R. Persinski, M. N. Hedrick, C. A. Huy, R. J. Davis, R. A. Flavell, and E. Fikrig. 2002. Murine Lyme arthritis development mediated by p38 mitogen-activated protein kinase activity. J. Immunol. 168:6352-6357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Behera, A. K., E. Hildebrand, R. T. Bronson, G. Perides, S. Uematsu, S. Akira, and L. T. Hu. 2006. MyD88 deficiency results in tissue-specific changes in cytokine induction and inflammation in interleukin-18-independent mice infected with Borrelia burgdorferi. Infect. Immun. 74:1462-1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bennett, B. L., D. T. Sasaki, B. W. Murray, E. C. O'Leary, S. T. Sakata, W. Xu, J. C. Leisten, A. Motiwala, S. Pierce, Y. Satoh, S. S. Bhagwat, A. M. Manning, and D. W. Anderson. 2001. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. USA 98:13681-13686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bolz, D. D., R. S. Sundsbak, Y. Ma, S. Akira, C. J. Kirschning, J. F. Zachary, J. H. Weis, and J. J. Weis. 2004. MyD88 plays a unique role in host defense but not arthritis development in Lyme disease. J. Immunol. 173:2003-2010. [DOI] [PubMed] [Google Scholar]
- 7.Cabral, E. S., H. Gelderblom, R. L. Hornung, P. J. Munson, R. Martin, and A. R. Marques. 2006. Borrelia burgdorferi lipoprotein-mediated TLR2 stimulation causes the down-regulation of TLR5 in human monocytes. J. Infect. Dis. 193:849-859. [DOI] [PubMed] [Google Scholar]
- 8.Conze, D., T. Krahl, N. Kennedy, L. Weiss, J. Lumsden, P. Hess, R. A. Flavell, G. Le Gros, R. J. Davis, and M. Rincon. 2002. c-Jun NH2-terminal kinase (JNK)1 and JNK2 have distinct roles in CD8+ T cell activation. J. Exp. Med. 195:811-823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dong, C., R. J. Davis, and R. A. Flavell. 2002. MAP kinases in the immune response. Annu. Rev. Immunol. 20:55-72. [DOI] [PubMed] [Google Scholar]
- 10.Dong, C., D. D. Yang, M. Wysk, A. J. Whitmarsh, R. J. Davis, and R. A. Flavell. 1998. Defective T cell differentiation in the absence of Jnk1. Science 282:2092-2095. [DOI] [PubMed] [Google Scholar]
- 11.Gupta, S., T. Barrett, A. J. Whitmarsh, J. Cavanagh, H. K. Sluss, B. Derijard, and R. J. Davis. 1996. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 15:2760-2770. [PMC free article] [PubMed] [Google Scholar]
- 12.Gupta, S., D. Campbell, B. Derijard, and R. J. Davis. 1995. Transcription factor ATF2 regulation by the JNK signal transduction pathway. Science 267:389-393. [DOI] [PubMed] [Google Scholar]
- 13.Hajishengallis, G., A. Sharma, M. W. Russell, and R. J. Genco. 2002. Interactions of oral pathogens with toll-like receptors: possible role in atherosclerosis. Ann. Periodontol. 7:72-78. [DOI] [PubMed] [Google Scholar]
- 14.Hayashi, F., K. D. Smith, A. Ozinsky, T. R. Hawn, E. C. Yi, D. R. Goodlett, J. K. Eng, S. Akira, D. M. Underhill, and A. Aderem. 2001. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 410:1099-1103. [DOI] [PubMed] [Google Scholar]
- 15.Hemmi, H., O. Takeuchi, T. Kawai, T. Kaisho, S. Sato, H. Sanjo, M. Matsumoto, K. Hoshino, H. Wagner, K. Takeda, and S. Akira. 2000. A Toll-like receptor recognizes bacterial DNA. Nature 408:740-745. [DOI] [PubMed] [Google Scholar]
- 16.Hirschfeld, M., C. J. Kirschning, R. Schwandner, H. Wesche, J. H. Weis, R. M. Wooten, and J. J. Weis. 1999. Cutting edge: inflammatory signaling by Borrelia burgdorferi lipoproteins is mediated by toll-like receptor 2. J. Immunol. 163:2382-2386. [PubMed] [Google Scholar]
- 17.Jiang, Z. H., and R. R. Koganty. 2003. Synthetic vaccines: the role of adjuvants in immune targeting. Curr. Med. Chem. 10:1423-1439. [DOI] [PubMed] [Google Scholar]
- 18.Johnson, N. L., A. M. Gardner, K. M. Diener, C. A. Lange-Carter, J. Gleavy, M. B. Jarpe, A. Minden, M. Karin, L. I. Zon, and G. L. Johnson. 1996. Signal transduction pathways regulated by mitogen-activated/extracellular response kinase kinase kinase induce cell death. J. Biol. Chem. 271:3229-3237. [DOI] [PubMed] [Google Scholar]
- 19.Kawai, T., and S. Akira. 2006. TLR signaling. Cell Death Differ. 13:816-825. [DOI] [PubMed] [Google Scholar]
- 20.Krutzik, S. R., M. T. Ochoa, P. A. Sieling, S. Uematsu, Y. W. Ng, A. Legaspi, P. T. Liu, S. T. Cole, P. J. Godowski, Y. Maeda, E. N. Sarno, M. V. Norgard, P. J. Brennan, S. Akira, T. H. Rea, and R. L. Modlin. 2003. Activation and regulation of Toll-like receptors 2 and 1 in human leprosy. Nat. Med. 9:525-532. [DOI] [PubMed] [Google Scholar]
- 21.Liu, N., R. R. Montgomery, S. W. Barthold, and L. K. Bockenstedt. 2004. Myeloid differentiation antigen 88 deficiency impairs pathogen clearance but does not alter inflammation in Borrelia burgdorferi-infected mice. Infect. Immun. 72:3195-3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matsuguchi, T., T. Musikacharoen, T. Ogawa, and Y. Yoshikai. 2000. Gene expressions of Toll-like receptor 2, but not Toll-like receptor 4, is induced by LPS and inflammatory cytokines in mouse macrophages. J. Immunol. 165:5767-5772. [DOI] [PubMed] [Google Scholar]
- 23.Musikacharoen, T., T. Matsuguchi, T. Kikuchi, and Y. Yoshikai. 2001. NF-kappa B and STAT5 play important roles in the regulation of mouse Toll-like receptor 2 gene expression. J. Immunol. 166:4516-4524. [DOI] [PubMed] [Google Scholar]
- 24.Oshikawa, K., and Y. Sugiyama. 2003. Gene expression of Toll-like receptors and associated molecules induced by inflammatory stimuli in the primary alveolar macrophage. Biochem. Biophys. Res. Commun. 305:649-655. [DOI] [PubMed] [Google Scholar]
- 25.Redecke, V., H. Hacker, S. K. Datta, A. Fermin, P. M. Pitha, D. H. Broide, and E. Raz. 2004. Cutting edge: activation of Toll-like receptor 2 induces a Th2 immune response and promotes experimental asthma. J. Immunol. 172:2739-2743. [DOI] [PubMed] [Google Scholar]
- 26.Rincón, M., and R. A. Flavell. 1994. AP-1 transcriptional activity requires both T-cell receptor-mediated and co-stimulatory signals in primary T lymphocytes. EMBO J. 13:4370-4381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rincón, M., A. Whitmarsh, D. D. Yang, L. Weiss, B. Derijard, P. Jayaraj, R. J. Davis, and R. A. Flavell. 1998. The JNK pathway regulates the in vivo deletion of immature CD4+CD8+ thymocytes. J. Exp. Med. 188:1817-1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roger, T., I. Miconnet, A. L. Schiesser, H. Kai, K. Miyake, and T. Calandra. 2005. Critical role for Ets, AP-1 and GATA-like transcription factors in regulating mouse Toll-like receptor 4 (Tlr4) gene expression. Biochem. J. 387:355-365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Romics, L., Jr., A. Dolganiuc, K. Kodys, Y. Drechsler, S. Oak, A. Velayudham, P. Mandrekar, and G. Szabo. 2004. Selective priming to Toll-like receptor 4 (TLR4), not TLR2, ligands by P. acnes involves up-regulation of MD-2 in mice. Hepatology 40:555-564. [DOI] [PubMed] [Google Scholar]
- 30.Sabapathy, K., K. Hochedlinger, S. Y. Nam, A. Bauer, M. Karin, and E. F. Wagner. 2004. Distinct roles for JNK1 and JNK2 in regulating JNK activity and c-Jun-dependent cell proliferation. Mol. Cell 15:713-725. [DOI] [PubMed] [Google Scholar]
- 31.Sato, K., H. Nagayama, K. Tadokoro, T. Juji, and T. A. Takahashi. 1999. Extracellular signal-regulated kinase, stress-activated protein kinase/c-Jun N-terminal kinase, and p38mapk are involved in IL-10-mediated selective repression of TNF-alpha-induced activation and maturation of human peripheral blood monocyte-derived dendritic cells. J. Immunol. 162:3865-3872. [PubMed] [Google Scholar]
- 32.Seya, T., T. Akazawa, T. Tsujita, and M. Matsumoto. 2006. Role of Toll-like receptors in adjuvant-augmented immune therapies. Evid Based Complement Alternat Med. 3:31-38, 133-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sluss, H. K., and R. J. Davis. 1997. Embryonic morphogenesis signaling pathway mediated by JNK targets the transcription factor JUN and the TGF-beta homologue decapentaplegic. J. Cell Biochem. 67:1-12. [PubMed] [Google Scholar]
- 34.Tsatsanis, C., A. Androulidaki, T. Alissafi, I. Charalampopoulos, E. Dermitzaki, T. Roger, A. Gravanis, and A. N. Margioris. 2006. Corticotropin-releasing factor and the urocortins induce the expression of TLR4 in macrophages via activation of the transcription factors PU.1 and AP-1. J. Immunol. 176:1869-1877. [DOI] [PubMed] [Google Scholar]
- 35.Verheij, M., R. Bose, X. H. Lin, B. Yao, W. D. Jarvis, S. Grant, M. J. Birrer, E. Szabo, L. I. Zon, J. M. Kyriakis, A. Haimovitz-Friedman, Z. Fuks, and R. N. Kolesnick. 1996. Requirement for ceramide-initiated SAPK/JNK signalling in stress-induced apoptosis. Nature 380:75-79. [DOI] [PubMed] [Google Scholar]
- 36.Vermeulen, L., G. De Wilde, P. Van Damme, W. Vanden Berghe, and G. Haegeman. 2003. Transcriptional activation of the NF-κB p65 subunit by mitogen- and stress-activated protein kinase-1 (MSK1). EMBO J. 22:1313-1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang, T., W. P. Lafuse, and B. S. Zwilling. 2001. NFκB and Sp1 elements are necessary for maximal transcription of toll-like receptor 2 induced by Mycobacterium avium. J. Immunol. 167:6924-6932. [DOI] [PubMed] [Google Scholar]
- 38.Weston, C. R., A. Wong, J. P. Hall, M. E. Goad, R. A. Flavell, and R. J. Davis. 2004. The c-Jun NH2-terminal kinase is essential for epidermal growth factor expression during epidermal morphogenesis. Proc. Natl. Acad. Sci. USA 101:14114-14119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wilson, D. J., K. A. Fortner, D. H. Lynch, R. R. Mattingly, I. G. Macara, J. A. Posada, and R. C. Budd. 1996. JNK, but not MAPK, activation is associated with Fas-mediated apoptosis in human T cells. Eur. J. Immunol. 26:989-994. [DOI] [PubMed] [Google Scholar]
- 40.Wooten, R. M., Y. Ma, R. A. Yoder, J. P. Brown, J. H. Weis, J. F. Zachary, C. J. Kirschning, and J. J. Weis. 2002. Toll-like receptor 2 is required for innate, but not acquired, host defense to Borrelia burgdorferi. J. Immunol. 168:348-355. [DOI] [PubMed] [Google Scholar]
- 41.Yang, D. D., D. Conze, A. J. Whitmarsh, T. Barrett, R. J. Davis, M. Rincon, and R. A. Flavell. 1998. Differentiation of CD4+ T cells to Th1 cells requires MAP kinase JNK2. Immunity 9:575-585. [DOI] [PubMed] [Google Scholar]