

Abstract
Saccharomyces cerevisiae are able to convert proline to glutamate so that it may be used as a source of nitrogen. Here, we show that the activator of the proline utilization genes, Put3p, is transcriptionally inert in the absence of proline but transcriptionally active in its presence. The activation of Put3p requires no additional yeast proteins and can occur in the presence of certain proline analogues: an unmodified pyrrolidine ring is able to activate Put3p as efficiently as proline itself. In addition, we show that a direct interaction occurs between Put3p and proline. These data, which represent direct control of transcriptional activator function by a metabolite, are discussed in terms of the regulation of proline-specific genes in yeast and as a general mechanism of the control of transcription.
Keywords: gene regulation/genetic switch/in vitro transcription/proline/Put3p
Introduction
Yeast cells have the capacity to use proline from their surroundings as a source of nitrogen. The action of the products of the proline utilization genes PUT1 and PUT2 (encoding proline oxidase and Δ1-pyrroline-5-carboxylate dehydrogenase, respectively) catalyse the conversion of proline to glutamate (Brandriss, 1987). PUT1 and PUT2 are only transcribed at high levels if yeast cells are grown on proline and they have a functional copy of the PUT3 gene (Marczak and Brandriss, 1991). Put3p is a transcriptional activator, composed of 979 amino acids, that has an overall architecture typical of the Zn(II)2Cys6 binuclear cluster family of proteins. It possesses an N-terminal DNA-binding and dimerization domain and a highly acidic C-terminal end that is required for transcriptional activation (des Etages et al., 1996). In vivo footprinting clearly demonstrates that Put3p is bound to DNA upstream of its target genes, whether or not the cells are grown on proline (Axelrod et al., 1991). The transcriptional activity of Put3p, but not its DNA-binding activity, is increased 50-fold when yeast cells are grown on proline as the sole nitrogen source (Brandriss, 1987; Axelrod et al., 1991). The activation domain of Put3p is not responsive to proline. Hybrid activators in which the C-terminal activation domain of Put3p has been fused to an heterologous DNA-binding domain are constitutively active, suggesting that regulation of activation function occurs elsewhere within the protein (des Etages et al., 1996). Genetic analysis has, however, failed to locate the proline-responsive region of the protein. Put3p has been identified as a phosphoprotein, and the extent of phosphorylation has been correlated with transcriptional activity (Huang and Brandriss, 2000; Saxena et al., 2003). However, the phosphorylation of Put3p does not appear to be dependent on the presence of proline, but rather on the quality of the nitrogen source available to the cells. This suggests that there are at least two signals impinging on Put3p activity (proline induction and the quality of nitrogen source) that act synergistically to allow maximum expression of the proline utilization pathway genes (Saxena et al., 2003).
Much is known about both the structure of the DNA-binding domain of Put3p (Swaminathan et al., 1997; Walters et al., 1997) and its mechanism of action (Reece and Ptashne, 1993). Despite a wealth of genetic information (Brandriss, 1987; Marczak and Brandriss, 1989, 1991; Siddiqui and Brandriss, 1989), relatively little is known about the function of the remainder of the polypeptide. It has been reported that the addition of proline to immunoprecipitated Put3p results in increased resistance to certain proteases (des Etages et al., 2001). Proline may therefore induce a conformational change within the protein that alters its accessibility to proteases. These data support the notion that the activation domain of Put3p may be uncoupled from an inhibitory region elsewhere in the protein in the presence of proline. However, the nature of the interaction between the protein and the amino acid, and the molecular consequences of this interaction, are unclear. We have established a system to produce and purify large amounts of Put3p. Here, we show that the purified protein will activate transcription in vitro, using either yeast or human nuclear extracts, provided that proline is present in the assays. In addition, we define the regions of the proline molecule required for the activation of Put3p and show that an unmodified pyrrolidine ring will function indistinguishably from proline itself. Finally, we show that proline and Put3p interact directly with each other. Hence, we show that the direct physical binding of a metabolite to an activator regulates gene expression. These results are discussed in terms of the mechanism of transcriptional control exerted by small molecules on activators.
Results
Purification of Put3p
A recombinant baculovirus expressing full-length PUT3 from the polyhedrin promoter was constructed and used to infect High Five insect cells. Western blotting was used to monitor the production of recombinant protein (Figure 1A). While Put3p could not be detected in uninfected insect cells (Figure 1A, lanes 1 and 2) or in cells infected with a wild-type virus (data not shown), at 2 days post-infection the Put3p recombinant virus gave rise to a band of ∼116 kDa, corresponding to the expected size of the full-length tagged protein. Put3p accumulated within the cells 48–72 h post-infection (Figure 1A, lanes 7–10). At later times, the amount of full-length Put3p within the cells diminished, although the protein could be detected in the media in an intact form (Figure 1A, lane 14). Based on these data, cells were harvested 3 days post-infection, before the onset of cell lysis. Put3p was purified from infected cells by nickel-affinity chromatography (Figure 1B). Buffer containing 30 mM imidazole (Figure 1B, lanes 3–5) eluted a significant number of contaminants from the purification column, while Put3p was eluted from the column with buffer containing 250 mM imidazole (Figure 1B, lane 6). Put3p produced in this way is estimated to be >90% pure, with ∼0.2 mg of protein being obtained from 1 × 108 insect cells in adherent cell culture.
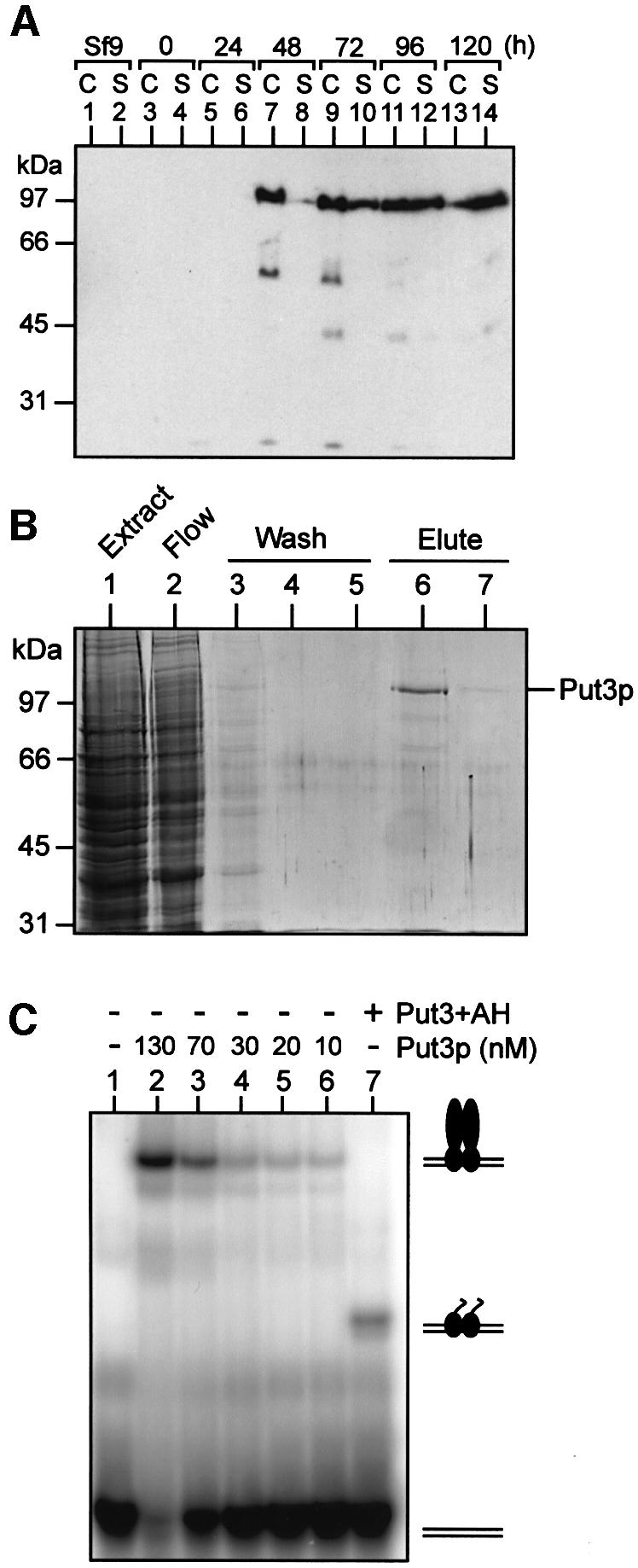
Fig. 1. Over-production and purification of functional Put3p. (A) Time course of Put3p expression in baculovirus-infected insect cells. Cultures of High Five insect cells (50% confluent) were infected at time 0 with recombinant baculovirus containing the PUT3 gene under the control of the polyhedrin promoter. Samples of the cells (C) or the culture supernatant (S) were taken at the times indicated and analysed by western blotting. (B) Purification of Put3p. Insect cells infected with recombinant PUT3 baculovirus were harvested 3 days post-infection. Cell extracts were prepared and soluble protein applied to a Ni2+-NTA agarose column. The column flow-through is shown in lane 2. The column was washed with buffer containing 30 mM imidazole (lanes 3–5) and then Put3p was eluted with buffer containing 250 mM imidazole (lanes 6 and 7). Samples of each fraction were run on an SDS–polyacrylamide gel that was subsequently stained with Coomassie Blue. The sizes of molecular weight standards (in kDa) are indicated. (C) DNA-binding properties of purified Put3p. Electrophoretic mobility shift assays were performed as described in Materials and methods. Reactions contained 32P-labelled DNA comprising a single Put3p-binding site and, as indicated, either purified full-length Put3p, at the concentrations shown, or Put3+AH (500 nM). The positions of the free DNA and the protein–DNA complexes are indicated.
DNA-binding activity of purified Put3p
The DNA-binding activity of Put3p is proline independent (Axelrod et al., 1991), and it has previously been shown that the DNA-binding domain resides within amino acids 31–100 of the protein (des Etages et al., 1996). Fragments corresponding to this portion of the protein are capable of binding to a consensus Put3 DNA-binding site with an affinity (Kd) of ∼30 nM (Reece and Ptashne, 1993; des Etages et al., 1996). The DNA-binding activity of purified full-length Put3p is shown in Figure 1C. The protein possessed a similar affinity for a Put3 DNA-binding site as the isolated DNA-binding domain. A protein composed of the DNA-binding domain of Put3p (amino acids 1–100) fused to the artificial activating sequence AH (designated Put3+AH) was also capable of binding to this site (Figure 1C, lane 7), although the DNA-binding activity of this chimeric activator was reduced in comparison with either the full-length protein (Figure 1C) or versions of the protein to which AH had not been fused (des Etages et al., 1996). The addition of AH may have had a deleterious effect on binding by either reducing the ability of the Put3p DNA-binding domain to form dimers or by some direct DNA inference mechanism. However, the ability to produce large amounts of the fusion protein in Escherichia coli allowed the DNA-binding sites for in vitro transcription templates (see below) to be saturated such that transcriptional activity could be monitored.
Transcriptional activity of purified Put3p
The transcriptional activity of Put3p was investigated using a yeast nuclear extract based in vitro transcription system. Plasmid DNA bearing four Put3p-binding sites upstream of the E4 gene (diagrammed above Figure 2) was incubated with nuclear extract, and the transcripts from the E4 gene were analysed by primer extension. If this experiment was performed using a nuclear extract prepared from a yeast strain containing a functional copy of PUT3, then significant levels of proline-specific activation of transcription could be observed in the absence of any added recombinant Put3p (data not shown). This suggests that the nuclear extracts produced from wild-type strains contain Put3p. Using nuclear extracts prepared from a yeast strain deleted for PUT3, and in the absence of other added protein, the transcript pattern obtained was not altered by the presence of l-glycine, l-valine or l-proline (Figure 2, lanes 2–4). The ability of the DNA template containing Put3p-binding sites to promote transcription was assessed using the chimeric activator Put3+AH. This protein was able to activate transcription (Figure 2, lane 5), and this activation was unaffected by the presence of l-proline, l-valine or l-glycine (Figure 2, lanes 6–8).

Fig. 2. Transcriptional activity of purified Put3p. Yeast in vitro transcription reactions contained, where indicated, either Put3+AH (200 nM) or Put3p (15 nM) and were supplemented with compounds at the concentrations indicated (in mM). Transcription products from the template shown above the figure were analysed by primer extension. The positions of the E4 extension products (indicated by the asterisk) and the primer are indicated, and the sizes of the primer and extension products (in numbers of nucleotides) are shown.
Purified full-length Put3p was found to be unable to promote transcriptional activation (Figure 2, lane 9) unless the reactions were supplemented with l-proline (Figure 2, lanes 12–14). Neither l-glycine nor l-valine were able to substitute for proline to elicit such a response (Figure 2, lanes 10 and 11). The size of the predominant primer extension product produced by Put3+AH and full-length Put3p were found to be different (Figure 2, compare lanes 8 and 14). While Put3+AH predominately resulted in the production of a 73 nucleotide primer extension product, Put3p produced a larger 90 nucleotide product. This indicated that Put3p promoted the initiation of transcription at a position 17 nucleotides before that mediated by Put3+AH. Mapping these start sites onto the template DNA indicates that, for full-length Put3p, transcription initiates 30 nucleotides downstream of the TATA box, while Put3+AH initiates transcription 47 nucleotides downstream of the TATA box.
Activation of Put3p requires no additional yeast proteins
The activation of transcription we observed using the yeast nuclear extract system (Figure 2) suggests that proline may exert a direct effect on the ability of Put3p to activate transcription. It is conceivable, however, that an additional protein in the yeast nuclear extract may mediate the interaction between the amino acid and the activator. We therefore performed in vitro transcription reactions using a HeLa nuclear extract (Figure 3). Using this system, Put3+AH resulted in proline-independent transcriptional activation (Figure 3, lane 1), while full-length Put3p could only elicit transcriptional activation in the presence of l-proline (Figure 3, lanes 2–6). A similar concentration of proline was required to generate a transcription response as observed using the yeast in vitro transcription system. The transcripts produced by Put3+AH and the full-length protein are, again, qualitatively different (Figure 3, compare lanes 1 and 4). The full-length protein produces a longer transcript using human extracts, again indicating that the transcript initiates closer to the TATA box in comparison with that initiated by Put3+AH. The ability of l-proline to convert Put3p from a transcriptionally inert state into an active one in both yeast and human transcription extracts (Figures 2 and 3, respectively) strongly suggests that a direct interaction occurs between the amino acid and the activator and that no other proteins are involved in the process.
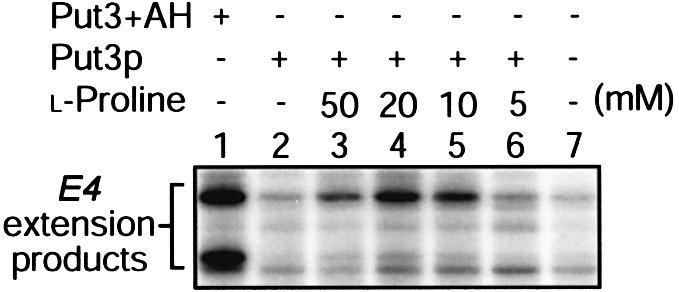
Fig. 3. In vitro transcription of Put3p using HeLa nuclear extracts. Reactions contained the DNA template diagrammed above Figure 2 together with Put3+AH (100 nM) or full-length Put3p (15 nM) where indicated. Reactions were supplemented with proline at the concentrations shown. Transcription products were analysed by primer extension.
Activation of Put3p by proline analogues
To further define the interaction between Put3p and the amino acid, we tested a series of proline analogues (Figure 4) for their ability to substitute for proline as inducers of Put3p. For each of the analogues used, its addition to an in vitro transcription reaction in the absence of Put3p failed to elicit a transcriptional response (data not shown). d-Proline was able to support Put3p-dependent activation of transcription, although at a level ∼50% of that observed for the l-enantiomer (Figure 5, compare lanes 2 and 3). The l- and d-enantiomers of prolinol were also able to support Put3p-dependent transcriptional activation, and once again the l-enantiomer was more active than the d-enantiomer (Figure 5, lanes 5 and 6). Similarly, both pyrrolidine and pyrrolidinone were capable of converting Put3p into a transcriptional activator, with pyrrolidine acting as efficiently as l-proline itself (Figure 5, lane 8) while pyrrolidinone was found to act more weakly (Figure 5, lane 7). Other analogues of proline, such as those bearing a hydroxyl group at either the 3 or 4 positions of the pyrrolidine ring (Figure 5, lanes 4 and 9), or those in which the pyrrolidine ring contains one or more double bonds (Figure 5, lanes 10–12), were unable to convert Put3p into a transcriptional activator. The inability of hydroxyproline to convert Put3p into a transcriptionally active form in vitro reflects its effects observed in vivo (des Etages et al., 2001). The analogues of proline that were unable to support Put3p-mediated transcriptional activation were also screened for their ability to inhibit the ability of proline to function as an inducer. The non-functional derivatives used in Figure 5 were unable to affect the ability of proline to activate Put3p (data not shown).

Fig. 4. The molecular structures of proline and the proline analogues used in this study.

Fig. 5. Alternative inducers of Put3p. In vitro transcription reactions, mediated by a yeast nuclear extract, each contained Put3p (15 nM) and were supplemented with the indicated compounds (each at a final concentration of 10 mM). Transcription products were analysed by primer extension, and the positions of the primer and of the E4 extension products are indicated.
Direct interaction between Put3p and proline
The data presented above are consistent with a mechanism of transcriptional activation in which proline directly interacts with Put3p. Given the relatively high concentration of proline required to activate Put3p in our assays, it is likely that the interaction between the protein and the amino acid is weak. We have been unable to detect any significant interaction between the two by direct methods, e.g. equilibrium gel filtration (data not shown). We therefore attempted to observe interactions between the protein and immobilized forms of proline. This approach proved unsuccessful using commercially available proline resins, e.g. proline agarose in which the proline was attached to the gel matrix through the nitrogen of the pyrrolidine ring (data not shown). However, based on the data we obtained regarding the ability of proline analogues to activate Put3p (Figure 5), it was apparent that a number of changes could be made to the carboxyl group of proline without affecting its ability to activate Put3p. We therefore constructed an affinity column matrix bearing proline attached via its carboxyl group, and tested whether Put3p could bind (Figure 6). Put3p, but not the contaminants in the protein preparation, was able to bind to this matrix (Figure 6A, lane 2) and could be eluted from the column with a buffer containing l-proline (Figure 6A, lane 3). However, Put3p was unable to bind to similarly produced matrices containing a different amino acid, e.g. l-glycine (data not shown). Additionally, Put3p was unable to adhere to the proline-affinity column if the protein had been previously incubated with l-proline (data not shown). Other yeast transcriptional activator proteins are unable to interact with the proline-affinity column. For example, Ppr1p, the activator of the pyrimidine biosynthetic genes in yeast, is a similar size to Put3p and shares a similar overall architecture in that it possesses an N-terminal Zn(II)2Cys6 zinc cluster DNA-binding domain and a C-terminal acidic activation domain (Kammerer et al., 1984). Ppr1p was unable to associate with the proline-affinity column and was found exclusively in the wash (Figure 6A, lane 5). Put3p was eluted from the column at a proline concentration between 25 and 50 mM (Figure 6B). This is a level comparable with that of the metabolite that was required to elicit a response in our in vitro transcription assays (Figures 2 and 3). Together, the data above provide compelling evidence for a direct and specific interaction between Put3p and proline that regulates activated transcription.
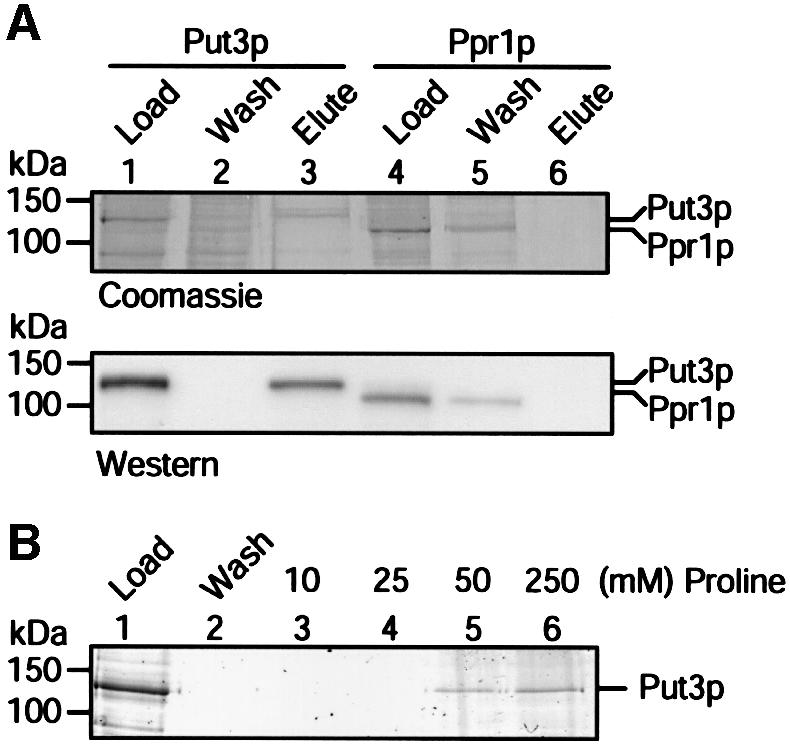
Fig. 6. Direct interaction between Put3p and proline. (A) Samples of purified Put3p or Ppr1p were applied to a l-proline–EAH Sepharose column. The flow-through from each column (wash) and the material eluted in the presence of 250 mM l-proline were separated by SDS–PAGE and either stained with Coomassie Blue (top) or visualized as a western blot to detect the polyhistidine tag of the protein (bottom). The sizes of molecular weight standards (in kDa) are indicated. (B) Elution of Put3p from a l-proline–EAH Sepharose column using different concentrations of l-proline. Samples of purified protein were applied, in parallel, to proline-affinity columns. Unbound proteins were washed from the column and then bound protein eluted using l-proline at the concentration indicated. Samples were separated by SDS–PAGE and subjected to silver staining.
Discussion
The work presented here shows that purified Put3p is able to activate transcription, using a variety of in vitro systems, in a proline-dependent fashion. The process of activation can be mediated by certain proline analogues, but not by others. Finally, we show that a direct and specific interaction occurs between the transcriptional activator and the metabolite whose utilization pathway it controls.
During our yeast in vitro transcription experiments, we noted that in the absence of added recombinant protein, the DNA template containing the Put3p-binding sites generated a 56 nucleotide primer extension band (Figure 2, lane 1). Other similarly constructed templates containing either Gal4p- or Ppr1p-DNA-binding sites do not result in the formation of this band in the absence of activator (Carey et al., 1990; Platt and Reece, 1998; Flynn and Reece, 1999). The origins of this band and the reasons for its formation are not clear. The transcript begins 64 nucleotides downstream of the TATA box, in a region of the plasmid not previously identified as an initiation site using either yeast or HeLa nuclear extracts. Given that we only observe this band using DNA templates bearing Put3p-binding sites and that its appearance is not dependent upon Put3p itself, it is likely that another protein from the nuclear extract is binding to the multimerized Put3p-binding sites (either to the sites themselves or to the intervening DNA sequence) and promoting the initiation of transcription at this point. However, the activation of transcription mediated by both Put3p and Put3+AH are easily distinguishable from this constitutively produced band.
The predominant transcripts produced in vitro by full-length Put3p were found to be 17 nucleotides longer than those produced by other activators. Put3p activation resulted in the formation of transcripts that initiated 30 bp downstream of the TATA box, while those initiated by Put3+AH initiated 47 bp downstream of the TATA box. This latter pattern of in vitro transcript formation is commonly observed with activators containing the DNA-binding domain of Gal4p, e.g. Gal4p+AH, Gal4p+VP16 (Carey et al., 1990) and Gal4p bearing its own activation domain (Platt and Reece, 1998). Other full-length yeast activators, e.g. Ppr1p, have also been found to give patterns of transcript initiation similar to that observed for Put3+AH when measured from DNA templates bearing their own binding sites (Flynn and Reece, 1999). The genes naturally controlled by Put3p within the yeast cell, PUT1 and PUT2, contain two and one binding sites for Put3p, respectively. In the promoter of the PUT1 gene, these two sites are separated by 12 bp and are located 146 bp upstream of the TATA box (Wang and Brandriss, 1987). Transcriptional initiation has been mapped to positions 72–80 bp downstream of the TATA box itself. The single Put3p-binding site in the PUT2 promoter is located 27 bp upstream of several potential TATA boxes (Siddiqui and Brandriss, 1988), and numerous initiation sites of RNA synthesis occur 50–80 bp downstream from these (Krzywicki and Brandriss, 1984). It is therefore not clear whether the unusual transcript pattern produced by full-length Put3p in our in vitro assays represents a novel function of the protein, or is simply an artifact of the assay process. It is possible that Put3p directs the polymerase machinery to alternative start sites, but in vivo analysis of the start sites of the PUT1 and PUT2 genes whose expression is driven by other activators will be required to address this issue.
A variety of proline analogues were found to able to support transcriptional activation mediated through Put3p. There appears to be some preference of the l-form of the amino acid over the d-form in terms of its ability to activate Put3p, but proline analogues in which additions have been made to either the 3 or 4 positions of the pyrrolidine ring were unable to modulate the function of Put3p. Additionally, desaturation of the pyrrolidine ring negates Put3p activation. These data, combined with the lack of activation observed by either l-glycine or l-valine, suggest that the integrity of the pyrrolidine ring is essential for the activation of Put3p. The carboxyl group of proline is unlikely to be involved in interaction with the protein. It may be altered or deleted without severely impairing the ability to induce transcription and can be used as a linking point for the construction of a proline-affinity column that has been successfully used to trap the protein. An affinity column produced by connecting the amino acid to the matrix via its nitrogen atom was unable to bind Put3p, suggesting that the nitrogen may be important in the process of metabolite-protein recognition.
During the course of this work, we have shown a direct, physical interaction between Put3p and proline (Figure 6). In yeast, a number of genetic switches have been identified that are regulated in response to changes in the availability of metabolites. For example, Leu3p is transcriptionally active only in the presence of α-isopropylmalate, an intermediate of leucine biosynthesis (Sze et al., 1992); Ppr1p is transcriptionally active only in the presence of either dihydroorotic acid or orotic acid, both of which are intermediates of pyrimidine biosynthesis (Flynn and Reece, 1999); the activation of the arginine anabolic and catabolic genes have been correlated to the promotion of DNA binding of a complex of proteins via the interaction between arginine and ARGRII (Amar et al., 2000); and the Gal4p complex of proteins is activated in the presence of galactose and ATP (Zenke et al., 1996; Platt and Reece, 1998; Peng and Hopper, 2002). In each of these cases, the interaction between the metabolite and its presumed protein sensor has been described as being weak, and direct interactions between the two have not been reported. For example, a vast array of circumstantial evidence indicates that Gal3p, the ligand sensor of the GAL genetic switch, interacts with both galactose and ATP (Reece, 2000). However, the weak nature of the interaction has precluded direct binding experiments using biochemical techniques. Based on the concentrations of proline that are required to observe activated transcription (Figure 2), we also believe that the interaction between Put3p and the amino acid is also weak. It is possible, of course, that a weak interaction is required so that the genetic switch is only activated in vivo at relatively high levels of the metabolite.
Together, the data presented here are consistent with a model for Put3p activation in which a DNA-bound version of the activator acts directly as a proline sensor. Under appropriate conditions (the absence of an alternative source of nitrogen) the interaction between proline and Put3p is sufficient to bring about a transcriptional response. It has previously been suggested that two separate signals act on Put3p to control its activity—the presence of proline and the phosphorylation state of the protein in response to the quality of the available nitrogen source (Huang and Brandriss, 2000; Saxena et al., 2003). Species of Put3p that migrate faster through gels, possibly representing under-phosphorylated forms of the protein, are observed when yeast cells are grown on rich nitrogen sources. It has been suggested that these may be refractory to activation by proline, whereas more highly phosphorylated forms of the protein, which are found in poor nitrogen sources, may be activatable by proline (Huang and Brandriss, 2000). Put3p produced in baculovirus-infected insect cells appears to be largely unphosphorylated. It migrates as a tight single band on conventional SDS–PAGE (Figure 1B) and also under conditions that have been previously been used (Huang and Brandriss, 2000) to observe phosphorylated forms of the protein (data not shown). Additionally, attempts to use phosphatase enzymes to convert the recombinant protein to a faster migrating species have proved unsuccessful (Sellick and Reece, unpublished observations). We therefore believe that proline is able to act on unphosphorylated Put3p to convert it into a functional transcriptional activator. However, the precise mechanism by which this is achieved is unclear. An attractive model is that, in a mechanism similar to that described for Leu3p (Wang et al., 1999), the presence of proline unmasks the activation domain of Put3p from an inhibitory region elsewhere within the protein. This model is also supported by protease protection experiments in which the presence of proline promotes altered accessibility to certain proteases (des Etages et al., 2001). The ability to bind the full-length protein to a proline-affinity column will allow the subsequent detection and assignment of the proline-binding portion of the protein. This, together with information regarding the nature of the interaction between the protein and proline itself, is essential to understand the molecular mechanism of gene control by metabolites. The work presented in this paper represents a case of the modulation of gene expression that is directly linked to the binding of a metabolite to an activator protein.
Materials and methods
Media and strains
Escherichia coli strain DH5α was used for all DNA manipulations, and strain BL21(DE3) was used for protein expression from the T7 promoter (Studier et al., 1990). Saccharomyces cerevisiae strain MC2 (MATa, trp1, ura3-52, leu2-3, prc1-407, prb1-112, pep4-3) (Platt et al., 2000) was deleted for the PUT3 gene using a SnaBI restriction enzyme fragment of the plasmid pDB101 (a gift of Marjorie Brandriss, New Jersey Medical School) (Marczak and Brandriss, 1991). Yeast nuclear extract was prepared from MC2::ΔPUT3 cells grown in YPD medium (Ohashi et al., 1994). Spodoptera frugiperda Sf9 insect cells (Invitrogen) were grown in Grace’s insect medium (Invitrogen) supplemented with 10% (v/v) fetal calf serum at 27°C. High Five insect cells (Invitrogen) were grown in Ultimate Insect serum-free medium (Invitrogen) at 27°C.
Construction of recombinant baculovirus
The coding region of the PUT3 gene was amplified (oligonucleotide sequences available on request) from yeast genomic DNA (Promega) and cloned into the PstI–NcoI sites of pBlueBacHisB (Invitrogen). The resulting plasmid (pRJR235) contains the entire coding sequence for Put3p, fused to an N-terminal MRGSH6-tag, under the control of the polyhedrin promoter. The coding region was sequenced to confirm that no mutations had arisen (data not shown). The plasmid was cotransfected with wild-type, linearized Autographa californica nuclear polyhedrosis virus DNA into Sf9 insect cells using a Bac-N-Blue transfection kit (Invitrogen) according to the manufacturer’s instructions. Recombinant viruses were identified as those yielding blue plaques on plates containing 5-bromo-4-chloro-3-indolyl β-d-galactopyranoside. PCR analysis of the viral DNA confirmed the presence of the PUT3 gene (data not shown) and the pure, recombinant virus was used to create a high-titre viral stock using Sf9 cells (3 × 108 p.f.u./ml).
Construction of Put3+AH
DNA fragments encoding amino acids 1–100 of Put3p (des Etages et al., 1996), and the 15 amino acids of the artificial AH transcriptional activator (Giniger and Ptashne, 1987) were fused together using a two-step PCR strategy (oligonucleotide sequences available on request) and cloned into pET16b (Novagen) such that the resulting plasmid (pRJR177) expressed the fusion gene under the control of the T7 promoter.
Protein purification
Full-length Put3p was purified from High Five insect cells grown in monolayer culture in 225 cm2 tissue culture flasks (Costar) infected with Put3p recombinant baculovirus at an m.o.i. of 5 and harvested 2–3 days post-infection. Cell pellets were resuspended in 1 ml per 107 infected cells of Buffer A [20 mM HEPES-KOH pH 7.8, 300 mM NaCl, 10% (v/v) glycerol] containing the Complete protease inhibitor cocktail (Boehringer Mannheim), sonicated and centrifuged at 30 000 g for 20 min at 4°C. Put3p was purified by nickel-affinity chromatography using Pro-Bond resin (Invitrogen) equilibrated in Buffer A. Following washing with Buffer A, and Buffer A containing 500 mM NaCl, 10 mM β-mercaptoethanol, 30 mM imidazole, protein was eluted from the column with Buffer A containing 250 mM imidazole. The protein was then dialysed into several changes of Buffer B [20 mM HEPES-KOH pH 7.8, 150 mM NaCl, 1 mM EDTA, 20 µM ZnSO4, 1.4 mM β-mercaptoethanol, 10% (v/v) glycerol] at 4°C overnight. Aliquots were subsequently frozen in liquid nitrogen and stored at –80°C. Ppr1p was produced as described previously (Flynn and Reece, 1999).
Put3(1–100)+AH was purified from BL21(DE3) cells transformed with pRJR177. Cells were grown at 37°C in the presence of 100 µg/ml ampicillin and 20 µM ZnSO4 to an OD600 of 0.6. After induction with 1 mM IPTG for 3 h, cells were harvested by centrifugation and resuspended in Buffer C [20 mM HEPES-KOH pH 7.8, 150 mM NaCl, 10% (v/v) glycerol, 20 µM ZnSO4, 1.4 mM β-mercaptoethanol]. Following sonication, Put3+AH was purified on a FAST S column (Pharmacia Biotech) equilibrated with Buffer C and washed extensively with Buffer C. The protein was eluted with a linear gradient of 150 mM to 1 M NaCl in Buffer C. Aliquots were subsequently frozen in liquid nitrogen and stored at –80°C.
Western blotting
Proteins were separated on 10% SDS–polyacrylamide gels and then transferred to a nitrocellulose membrane using a wet blotter (Bio-Rad). Membranes were washed in phosphate-buffered saline (PBS) and blocked with PBS containing 5% (w/v) non-fat dried milk and 0.2% (v/v) Tween 20. The anti-His primary antibody (Santa Cruz) was detected with goat anti-mouse immunoglobulin-peroxidase conjugate (Sigma) and visualized using SuperSignal West Dura extended duration substrate (Pierce) and a Fluor-S MultiImager (Bio-Rad).
Mobility shift assay
The PUT3 probe used was a double-stranded oligonucleotide (5′-GATCCCCGGGAAGCGCTTCCCGGGAAGCT-3′), representing a high-affinity Put3p-binding site (shown in bold). Reaction mixes (10 µl) containing 20 mM HEPES-KOH pH 8.0, 150 mM NaCl, 10% (v/v) glycerol, 5 mM MgCl2, 10 µM ZnSO4, 10 pM 32P-labelled probe DNA, and protein at the indicated concentrations were incubated for 30 min at room temperature and then subjected to electrophoresis through a pre-run 5% polyacrylamide gel containing 0.5× TBE, 1% (v/v) glycerol for 120 min at 150 V. Gels were analysed by autoradiography.
In vitro transcription
In vitro transcription using yeast nuclear extract were performed as described previously (Flynn and Reece, 1999) using the plasmid pPUT34E4 (a gift of Josh Brickman, University of Edinburgh) as a template. This plasmid contains four Put3 DNA binding sites (Reece and Ptashne, 1993) cloned 23 bp upstream of the adenoviral E4 TATA box and ∼250 bp of coding sequence. Reactions were supplemented with proline or its analogues (Sigma) at the concentrations indicated, and with the appropriate Put3p derivative. RNA produced was detected by primer extension. In vitro transcription reactions using HeLa nuclear extracts were performed as follows. Reaction mixes (50 µl) containing 6 mM MgCl2, 0.5 mM nucleoside triphosphates, 3 nM pPUT34E4 and 25 µl HeLa nuclear extract (10 mg/ml) (a gift of Stefan Roberts, The University of Manchester), supplemented with l-proline at the concentrations indicated, were incubated with Put3p or Put3+AH for 60 min at 30°C. Primer extension analysis of the RNA was then performed.
Proline-affinity chromatography
Proline was coupled to EAH Sepharose 4B (Amersham Biosciences) through its carboxyl group. Briefly, 5 ml of EAH Sepharose was mixed with l-proline (0.65 M final concentration) and N-ethyl-N′-(3-dimethylaminopropyl)carbodiimide hydrochloride (0.65 M final concentration). The mixture was adjusted to pH 4.5 and rocked gently overnight at 4°C. Coupling efficiency was determined using trace amounts of 14C proline. The matrix was then washed for four cycles with 0.1 M acetate buffer (pH 4.0) containing 0.5 M NaCl and 0.1 M Tris–HCl (pH 8.0) containing 0.5 M NaCl. The matrix was then equilibrated in Buffer D (20 mM HEPES-KOH pH 7.8, 150 mM NaCl, 1 mM EDTA, 20 µM ZnSO4) prior to the addition of protein. Proteins were washed through the column in Buffer D, and eluted with Buffer D containing l-proline at various concentrations, as indicated.
Acknowledgments
Acknowledgements
We thank Stefan Roberts for the gift of HeLa nuclear extract and for useful discussions; Josh Brickman and Marjorie Brandriss for the gift of materials; and David Timson, Noel Curtis and Cristina Merlotti for many helpful comments and suggestions. We also thank Paul Flynn for his help during the initial stages of this project. This work was supported by grants from The Wellcome Trust and the Biotechnology and Biological Sciences Research Council to R.J.R.
References
- Amar N., Messenguy,F., El Bakkoury,M. and Dubois,E. (2000) ArgRII, a component of the ArgR–Mcm1 complex involved in the control of arginine metabolism in Saccharomyces cerevisiae, is the sensor of arginine. Mol. Cell. Biol., 20, 2087–2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelrod J.D., Majors,J. and Brandriss,M.C. (1991) Proline-independent binding of PUT3 transcriptional activator protein detected by footprinting in vivo. Mol. Cell. Biol., 11, 564–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandriss M.C. (1987) Evidence for positive regulation of proline utilization in Saccharomyces cerevisiae. Genetics, 117, 429–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey M., Leatherwood,J. and Ptashne,M. (1990) A potent GAL4 derivative activates transcription at a distance in vitro. Science, 247, 710–712. [DOI] [PubMed] [Google Scholar]
- des Etages S.G., Falvey,D.A., Reece,R.J. and Brandriss,M.C. (1996) Functional analysis of the PUT3 transcriptional activator of the proline utilization pathway in Saccharomyces cerevisiae. Genetics, 142, 1069–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- des Etages S.G., Saxena,D., Huang,H.L., Falvey,D.A., Barber,D. and Brandriss,M.C. (2001) Conformational changes play a role in regulating the activity of the proline utilization pathway-specific regulator in Saccharomyces cerevisiae. Mol. Microbiol., 40, 890–899. [DOI] [PubMed] [Google Scholar]
- Flynn P.J. and Reece,R.J. (1999) Activation of transcription by metabolic intermediates of the pyrimidine biosynthetic pathway. Mol. Cell. Biol., 19, 882–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giniger E. and Ptashne,M. (1987) Transcription in yeast activated by a putative amphipathic α helix linked to a DNA binding unit. Nature, 330, 670–672. [DOI] [PubMed] [Google Scholar]
- Huang H.L. and Brandriss,M.C. (2000) The regulator of the yeast proline utilization pathway is differentially phosphorylated in response to the quality of the nitrogen source. Mol. Cell. Biol., 20, 892–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kammerer B., Guyonvarch,A. and Hubert,J.C. (1984) Yeast regulatory gene PPR1. I. Nucleotide sequence, restriction map and codon usage. J. Mol. Biol., 180, 239–250. [DOI] [PubMed] [Google Scholar]
- Krzywicki K.A. and Brandriss,M.C. (1984) Primary structure of the nuclear PUT2 gene involved in the mitochondrial pathway for proline utilization in Saccharomyces cerevisiae. Mol. Cell. Biol., 4, 2837–2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marczak J.E. and Brandriss,M.C. (1989) Isolation of constitutive mutations affecting the proline utilization pathway in Saccharomyces cerevisiae and molecular analysis of the PUT3 transcriptional activator. Mol. Cell. Biol., 9, 4696–4705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marczak J.E. and Brandriss,M.C. (1991) Analysis of constitutive and noninducible mutations of the PUT3 transcriptional activator. Mol. Cell. Biol., 11, 2609–2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi Y., Brickman,J.M., Furman,E., Middleton,B. and Carey,M. (1994) Modulating the potency of an activator in a yeast in vitro transcription system. Mol. Cell. Biol., 14, 2731–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng G. and Hopper,J.E. (2002) Gene activation by interaction of an inhibitor with a cytoplasmic signaling protein. Proc. Natl Acad. Sci. USA, 99, 8548–8553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt A. and Reece,R.J. (1998) The yeast galactose genetic switch is mediated by the formation of a Gal4p/Gal80p/Gal3p complex. EMBO J., 17, 4086–4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt A., Ross,H.C., Hankin,S. and Reece,R.J. (2000) The insertion of two amino acids into a transcriptional inducer converts it into a galactokinase. Proc. Natl Acad. Sci. USA, 97, 3154–3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reece R.J. (2000) Molecular basis of nutrient controlled gene expression in Saccharomyces cerevisiae. Cell. Mol. Life Sci., 57, 1161–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reece R.J. and Ptashne,M. (1993) Determinants of binding-site specificity among yeast C6 zinc cluster proteins. Science, 261, 909–911. [DOI] [PubMed] [Google Scholar]
- Saxena D., Kannan,K.B. and Brandriss,M.C. (2003) Rapamycin treatment results in GATA factor-independent hyperphosphorylation of the proline utilization pathway activator in Saccharomyces cerevisiae. Eukaryot. Cell, 2, 552–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui A.H. and Brandriss,M.C. (1988) A regulatory region responsible for proline-specific induction of the yeast PUT2 gene is adjacent to its TATA box. Mol. Cell. Biol., 8, 4634–4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui A.H. and Brandriss,M.C. (1989) The Saccharomyces cerevisiae PUT3 activator protein associates with proline-specific upstream activation sequences. Mol. Cell. Biol., 9, 4706–4712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier F.W., Rosenberg,A.H., Dunn,J.J. and Dubendorff,J.W. (1990) Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol., 185, 60–89. [DOI] [PubMed] [Google Scholar]
- Swaminathan K., Flynn,P.J., Reece,R.J. and Marmorstein,R. (1997) Crystal structure of the PUT3–DNA complex reveals a novel mechanism for DNA recognition by a protein containing a Zn2Cys6 binuclear cluster. Nat. Struct. Biol., 4, 751–759. [DOI] [PubMed] [Google Scholar]
- Sze J.-Y., Woontner,M., Jaehning,J.A. and Kohlhaw,G.B. (1992) In vitro transcriptional activation by a metabolic derivative: activation of Leu3 depends on α-isopropylmalate. Science, 258, 1143–1145. [DOI] [PubMed] [Google Scholar]
- Walters K.J., Dayie,K.T., Reece,R.J., Ptashne,M. and Wagner,G. (1997) Structure and mobility of the PUT3 dimer: a DNA pincer. Nat. Struct. Biol., 4, 744–750. [DOI] [PubMed] [Google Scholar]
- Wang D., Zheng,F., Holmberg,S. and Kohlhaw,G.B. (1999) Yeast transcriptional regulator Leu3p. Self-masking, specificity of masking and evidence for regulation by the intracellular level of Leu3p. J. Biol. Chem., 274, 19017–19024. [DOI] [PubMed] [Google Scholar]
- Wang S.S. and Brandriss,M.C. (1987) Proline utilization in Saccharomyces cerevisiae: sequence, regulation and mitochondrial localization of the PUT1 gene product. Mol. Cell. Biol., 7, 4431–4440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenke F.T., Engles,R., Vollenbroich,V., Meyer,J., Hollenberg,C.P. and Breunig,K.D. (1996) Activation of Gal4p by galactose-dependent interaction of galactokinase and Gal80p. Science, 272, 1662–1665. [DOI] [PubMed] [Google Scholar]