

Abstract
High-resolution crystal structures of α-l-arabinofuranosidase from Geobacillus stearothermophilus T-6, a family 51 glycosidase, are described. The enzyme is a hexamer, and each monomer is organized into two domains: a (β/α)8-barrel and a 12-stranded β sandwich with jelly-roll topology. The structures of the Michaelis complexes with natural and synthetic substrates, and of the transient covalent arabinofuranosyl– enzyme intermediate represent two stable states in the double displacement mechanism, and allow thorough examination of the catalytic mechanism. The arabinofuranose sugar is tightly bound and distorted by an extensive network of hydrogen bonds. The two catalytic residues are 4.7 Å apart, and together with other conserved residues contribute to the stabilization of the oxocarbenium ion-like transition state via charge delocalization and specific protein–substrate interactions. The enzyme is an anti-protonator, and a 1.7 Å electrophilic migration of the anomeric carbon takes place during the hydrolysis.
Keywords: arabinofuranosidase/clan GH-A glycosidase/enzyme mechanism/glycoside hydrolase family 51/X-ray crystallography
Introduction
Most of the biomass synthesized by photosynthetic CO2 fixation is stored in the plant cell wall as polymeric carbohydrates, mainly in the form of cellulose and hemicellulose. The degradation of these polymers is a key step in the carbon cycle, and is mediated by microorganisms that produce specific enzymes, i.e. cellulases and hemicellulases (Figure 1A) (Shallom and Shoham, 2003). α-l-Arabinofuranosidases (EC 3.2.1.55) are hemicellulases that hydrolyze the arabinofuranosyl substitutions in hemicellulose. Some of these enzymes exhibit broad substrate specificity, acting on arabinofuranoside moieties at O-2 or O-3 as a single substituent, as well as from O-2 and O-3 doubly substituted xylans, xylooligomers and α-1,5-linked arabinans (Saha, 2000). The glycosidic bond between two sugars is one of the most stable bonds in nature, and its enzymatic hydrolysis, carried out by glycoside hydrolases, provides an acceleration rate (kcat/kuncat) which can be as high as 1017-fold (Wolfenden et al., 1998). The key elements in this remarkable catalysis are the finely tuned positions of the catalytic residues, and the distortion of the sugar ring so as to allow the stabilization of an oxocarbenium-ion-like transition state, as well as the movement of the hydrogen on the anomeric carbon to allow a direct nucleophilic attack (Zechel and Withers, 1999). Many glycosidases are modular proteins, and in addition to their catalytic domains include other functional modules, mainly carbohydrate-binding modules (CBMs) (Bourne and Henrissat, 2001). Currently, >8500 glycosidase sequences are known, and the sequence-based classification of their catalytic domains into glycoside hydrolase (GH) families and clans is available on the continuously updated Carbohydrate-Active Enzymes (CAZy) server (http://afmb.cnrs-mrs.fr/CAZY). The different bacterial, fungal and plant α-l-arabinofuranosidases are members of GH families 3, 43, 51, 54 and 62.

Fig. 1. (A) Upper, the basic structural components of xylan, and the hemicellulases responsible for its degradation; lower, the natural and synthetic substrates of α-l-arabinofuranosidases used in this work: Ara-α(1,3)-Xyl and 4-nitrophenyl-Ara. (B) The double-displacement reaction mechanism for retaining glycosidases (the Koshland mechanism).
The enzymatic hydrolysis of glycosidic bonds results in either an overall inversion or retention of the anomeric configuration, and about two-thirds of the characterized GH families are retaining enzymes. Most retaining glycosidases cleave the glycosidic bond using two carboxylic acids, acting as a nucleophile and an acid/base. The hydrolysis proceeds through a two-step double-displacement mechanism, in which a covalent glycosyl–enzyme intermediate is formed amid the two reaction steps (Davies et al., 1998a; Sinnott, 1990) (Figure 1B). Crystal structures of native or mutant enzymes in complex with substrates, products, non-hydrolyzable substrate-analogues and transition-state analogues provide valuable detailed information regarding the specificity, binding mechanism and transition-state stabilization in retaining glycosidases, as recently reviewed by Vasella et al. (2002).
The α-l-arabinofuranosidase from Geobacillus stearothermophilus T-6 (AbfA) belongs to the retaining GH-51 family, and its catalytic residues were recently identified, Glu175 is the acid/base, and Glu294 is the nucleophile (Shallom et al., 2002a,b). In this study, we describe high-resolution (1.2–2.0 Å) crystal structures of native and catalytic mutant of AbfA in complex with different substrates (Figure 1A). These structures include the Michaelis complexes with natural and synthetic substrates and the transient covalent arabinofuranosyl-enzyme intermediate with a non-fluorinated substrate. The structures allow thorough examination of the catalytic mechanism, including the two stable states of the glycosylation step, the interactions mediating substrate distortion and the features governing substrate specificity.
Overall fold and oligomeric structure
The crystal structure of the native AbfA was determined at 1.75 Å resolution (Table I). The structure contains all of the 502 amino acid residues, except the first four residues at the N-terminus, for which the electron density was not sufficiently clear. Figure 2 shows a representative electron density map demonstrating the quality and reliability of the model. The final refined models exhibit excellent stereochemistry and the bound ligand atoms refine well with average thermal factors comparable to the values for all protein atoms (Table I).
Table I. Statistics of data collection, structure solution and refinement.
| Data set | MAD Se peak | MAD inflection | MAD remote | MIR Hg | MIR Pt | Native wild type | 4-nitrophenyl-Ara E175A | Covalent Ara E175A | Ara-Xyl E175A |
|---|---|---|---|---|---|---|---|---|---|
| Wavelength (Å) | 0.97948 | 0.97971 | 0.9645 | 1.54178 | 1.54178 | 1.54178 | 0.84590 | 1.54178 | 1.54178 |
| Space group | R3 | R3 | R3 | R3 | R3 | R3 | R3 | R3 | R3 |
| a,b (Å) | 177 838 | 177 897 | 177 921 | 178 319 | 177 865 | 178 322 | 179 311 | 179 430 | 178 871 |
| c (Å) | 100 583 | 100 639 | 100 649 | 100 242 | 100 642 | 100 410 | 100 397 | 100 231 | 100 404 |
| Unique reflections | 135 163 | 131 318 | 132 006 | 65 700 | 72 203 | 119 711 | 370 260 | 75 814 | 107 887 |
| Resolution range (Å) | 20.0–1.68 | 20.0–1.68 | 20.0–1.68 | 20.0–2.10 | 20.0–2.05 | 20.0–1.75 | 50.0–1.20 | 20.0–2.0 | 20.0–1.80 |
| Completeness (final shell) (%) | 100.0 (99.9) | 97.1 (98.4) | 97.6 (98.7) | 94.9 (96.0) | 97.3 (96.9) | 99.7 (99.7) | 99.1 (97.9) | 93.3 (96.2) | 96.9 (92.4) |
| <I>/<σ> (final shell) | 20.4 (5.9) | 18.5 (3.4) | 18.0 (3.5) | 5.9 (3.1) | 15.0 (2.9) | 26.5 (2.8) | 16.3 (1.7) | 13.4 (1.7) | 16.9 (1.9) |
| Rsym (%) (final shell) | 5.5 (27.0) | 4.5 (35.7) | 4.4 (31.1) | 13.5 (29.5) | 6.5 (31.8) | 4.7 (39.5) | 4.9 (38.3) | 6.2 (37.5) | 5.5 (41.0) |
| FOM solve/resolve | 0.57/0.81a | 0.57/0.81a | 0.57/0.81a | 0.41/0.66b | 0.41/0.66b | ||||
| R-factor (Rfree) | 17.1 (20.4) | 16.0 (17.9) | 16.9 (21.5) | 17.4 (21.3) | |||||
| Resolution range | 20.0–1.75 | 50.0–1.20 | 20.0–2.00 | 20.0–1.80 | |||||
| R.m.s. deviations from ideality | |||||||||
| Bonds (Å) | 0.014 | 0.014 | 0.018 | 0.013 | |||||
| Angles (°) | 1490 | 1563 | 1667 | 1474 | |||||
| Average B-factors (Å)2 | |||||||||
| Main chain | 23.5 | 15.9 | 40.9 | 28.9 | |||||
| Side chain | 26.3 | 18.7 | 43.4 | 31.3 | |||||
| Solvent | 37.8 | 33.3 | 48.2 | 39.2 | |||||
| Ligand | 29.3 | 22.3 | 43.7 | 43.2 |
aMAD data to 2.5 Å resolution, before and after density modification.
bBoth derivatives to 3.0 Å resolution, before and after density modification.

Fig. 2. Stereo view of the 2DFo – mFc electron density maps of a representative portion of the native AbfA at 1.75 Å, contoured at 2.0 σ. The area shown is that of the active-site residues Glu29 and Glu294, and the non-proline cis-peptide bond between residues Ala350 and Gln351. Selected hydrogen bonds between the protein and the water molecule are shown in dotted lines. Color coding: red, oxygen; blue, nitrogen; yellow, carbon.
AbfA is a homohexamer with a D3 point symmetry (Hövel et al., 2003). The asymmetric unit of the R3 crystals contains two AbfA monomers with dimeric structure. Each AbfA monomeric subunit is organized into two domains: a (β/α)8 barrel (TIM barrel), and a 12-stranded β sandwich with a jelly-roll topology (Figure 3). The (β/α)8-barrel domain (residues 20–383) has an elliptical cross-section with the major axis of the distorted barrel running from β-strands 1 to 5, as observed for other (β/α)8 glycosidases (Jenkins et al., 1995). There are several deviations from the basic (β/α)8-barrel motif. An additional subdomain, composed of three α-helices and one anti-parallel β-sheet, is inserted between β-strand 2 and α-helix 2. Another α-helix is present between α-helix 3 and β-strand 4 and a short α-helix is inserted between β-strand 7 and α-helix 7. Furthermore, an anti-parallel β-sheet is found between β-strand 8 and α-helix 8.

Fig. 3. Overall structure of AbfA. (A) Two views of the AbfA monomer related by a 90° rotation. (B) The hexameric enzyme is shown along the crystallographic 3-fold axis. All non-schematic figures were prepared with Molscript (Kraulis, 1991), Bobscript (Esnouf, 1997) and Raster3D (Merritt and Bacon, 1997).
GH families with overall similar fold can be further grouped into clans. Based on sequence analysis, the GH-51 arabinofuranosidases were classified as part of the GH-A clan, together with another 16 GH families (Henrissat et al., 1995; Zverlov et al., 1998). Within clan GH-A, the structures of enzymes from families 1, 2, 5, 10, 17, 26, 42 and 53 have been described, and their catalytic domains all share the same overall (β/α)8-topology and similar active site architecture. Thus, the (β/α)8 fold of AbfA, as well as the location of the catalytic residues (see below), are consistent with the classification of GH-51 within the GH-A clan.
The second domain of AbfA is comprised of 12 β-strands with an uncommon jelly-roll topology. The eight β-strands of the jelly roll are arranged in two sheets that are packed against each other, stabilized by hydrophobic interactions between the upper and lower β-sheets. The eighth β-strand of the jelly roll is composed of the N-terminal residues 6–9, whereas the C-terminal residues 384–501 build up the remaining part of the domain. This domain shows structural similarity to domain C of several α-amylases and also to cellulose binding domains (Tormo et al., 1996; Fujimoto et al., 1998; Kamitori et al., 1999). It is possible that this domain formerly functioned as a carbohydrate-binding domain that lost its function during evolution. In this context it is worth noting that in our hands, AbfA did not exhibit binding ability to xylan (D.Shallom and Y.Shoham, unpublished data). The similarity to α-amylases relates also to the interactions between the jelly-roll domain and the (β/α)8-barrel. Several hydrogen-bonds are found between residues around the sixth and seventh α-helices of the (β/α)8-barrel, and residues from the adjacent sheet of the jelly roll domain. Similar interface architecture is found between domain C and the (β/α)8-barrel of the α-amylase from Thermoactinomyces vulgaris R-47, where it was proposed that the jelly-roll domain stabilizes the TIM-barrel structure (Kamitori et al., 1999).
The oligomeric structure of AbfA can be described as a trimer of dimers. The hexameric architecture along the 3-fold axis is illustrated in Figure 3B. This molecular axis coincides with the crystallographic 3-fold axis resulting in a special position of the hexamer in the crystallographic unit cell. In the interface of each dimer, residues at the N-terminus of α-helix 5 and residues at the C-terminal end of α-helix 6 participate in the main interactions between the two monomers. The structural motifs that deviate from the basic (β/α)8-barrel and residues from α-helices 2, 3 and 4 of the barrel are all involved in the main interactions forming the hexamer. To date, only very few glycosidases have been suggested to be hexameric (Iriyama et al., 2000), and the structure of AbfA is the first crystal structure described for a hexameric glycosidase.
The Michaelis complexes and substrate binding
The kinetic characteristics of the acid/base E175A AbfA mutant have made it possible to obtain enzyme complexes with several substrates. For substrates with relatively poor leaving groups, such as 4-nitrophenyl-α-l-arabinofuranoside(4-nitrophenyl-Ara) and the natural substrate derived from xylan, arabinofuranosyl-α-(1,3)-xylose [Ara-α(1,3)-Xyl], the E175A mutant exhibits at least 103-fold lower activity than the wild-type enzyme (Shallom et al., 2002b). After soaking the crystal of the AbfA E175A mutant with each of the substrates 4-nitrophenyl-Ara and Ara-α(1,3)-Xyl, structures of the corresponding Michaelis complexes with the intact substrates were obtained at 1.2 and 1.8 Å resolution, respectively. The superimposition of the native enzyme structure with those of the Michaelis complexes reveals no significant change in the overall protein structure upon substrate binding (r.m.s.d. = 0.13 Å for all main chain atoms).
Figure 4A shows the hydrogen bonds network at the active site of the wild-type enzyme. Interestingly, a glycerol molecule (originating from the cryo solution) is located in the active site, where the hydroxyls of the glycerol take similar positions to the hydroxyls of the substrates, as described below. Figure 4B and C shows the active site of the E175A mutant Michaelis complexes with the intact substrates 4-nitrophenyl-Ara and Ara-α(1,3)-Xyl, respectively. In both structures, the arabinofuranose moiety at the –1 subsite is bound by a large number of hydrogen bonds: each of its hydroxyls can form two possible hydrogen bonds with the enzyme. In the Ara-α(1,3)-Xyl complex, the xylose at the +1 subsite can form only one hydrogen bond and therefore is far less tightly bound than the arabinofuranose at the –1 subsite. This may explain the capability of AbfA to bind and hydrolyze substrates with structurally very different leaving groups (Shallom et al., 2002b). An additional electron density was observed in the +2 subsite, resulting from a second xylose connected to the O4 of the first xylose in some of the molecules, originating from the heterogeneity of the substrate (see Material and methods). The density map, however, was not clear enough, and the second xylose was not built into the final model.

Fig. 4. Interactions between ligands and key residues in the active site. Hydrogen bonds are shown as dotted black lines, their length in Ångströms is indicated. Color coding: yellow, carbon atoms of protein residues; green, carbon atoms of ligands; red, oxygen; blue, nitrogen. (A) Native AbfA with a glycerol molecule in the active site. The β-strands of the (β/α)8-barrel are shown schematically in light grey. (B) The Michaelis complex of the E175A mutant with 4-nitrophenyl-Ara. (C) The Michaelis complex of the E175A mutant with Ara-α(1,3)-Xyl. The insets in (B) and (C) show the two energetically close conformations of the arabinofuranose ring at the –1 subsite. (D) The covalent arabinofuranosyl–enzyme complex, obtained by using the E175A mutant with 2,5-dinitrophenyl-Ara. The inset shows the strong hydrogen bond between the sugar 2-hydroxyl and the nucleophile (Glu294).
The crystal structures of the native and Michaelis complexes reveal nine key residues responsible for catalysis and substrate binding interactions: Glu29, Arg69, Asn74, Asn174, Glu175, His244, Tyr246, Glu294 and Gln351 (Figure 4). The location and function of many of these residues is conserved in clan GH-A (Sakon et al., 1996). The catalytic acid/base Glu175 is involved in a hydrogen bond to the conserved His244 at the end of β-strand 6 (not shown). In other GH-A clan enzymes, this position is occupied by homologous residues that can be a His or Tyr (family 5), Asn (families 1, 2, and 17), Asp (family 26), Gln (family 10), and a Ser (family 53) (Ryttersgaard et al., 2002). In AbfA, Asn174 preceding the proton donor at the end of β-strand 4 is involved in a hydrogen bond to the C2 sugar hydroxyl group of the arabinofuranosyl moiety (3.4 Å) (Figure 4C). This residue is invariant in the GH-A clan members and was shown to have a critical structural and functional role in maintaining the conformation and protonation state of the active site residues. For example, in the GH-5 family, a substitution of the homologous Asn by Asp led to nearly complete loss of enzyme activity (Navas and Beguin, 1992). Additionally, this residue seems to have an important role in assisting the formation of the covalent glycosyl–enzyme intermediate (Ryttersgaard et al., 2002). Arg69 of AbfA is located at the bottom of the active site (end of β-strand 2) and is conserved in families 1, 2, 5, 17 and 53 (not shown). This residue seems to keep the catalytic nucleophile Glu294 deprotonated, as Oε2 of Glu294 is within hydrogen bond distance to Nη1 of Arg69 (3.1 Å). In addition, Oε1 of Glu294 can form a hydrogen bond also with Tyr246, and this conserved residue can form two more hydrogen bonds, to the endocyclic O4 and to the O5 of the arabinofuranosyl at the –1 subsite. The mechanistic implications of these hydrogen bonds will be discussed later.
A single non-proline cis-peptide bond is present in AbfA at the end of β-strand 8, between Ala350 and Gln351 (Figure 2). This unusual conformation constrains the orientation of the Gln351 side chain, thus enabling it to form a hydrogen bond with the 5-OH group of the arabinofuranosyl moiety (Figure 4B–D). This unique cis-peptide bond occurs in at least six different GH families with a (β/α)8-topology (1, 2, 5, 17, 18 and 42) (Sakon et al., 1996; Juers et al., 1999). Unlike AbfA, in most GH-A clan members an aromatic residue, often a tryptophan, forms the cis-peptide bond with the next residue. Similarly to AbfA, in these enzymes the cis-peptide bond constrains the position of the tryptophan and enables it to form hydrogen bonds to the substrate. The occurrence of this rare type of bond in the same location and for similar purposes in glycosidases with structurally very different substrates, gives further support for the proposed common evolutionary relationship between the glycosidases families sharing the (β/α)8-topology (Juers et al., 1999).
The furanose 3-OH is within a hydrogen bond distance (2.9 Å) from the backbone nitrogen of Asn74 (Figure 4B). This residue is located above the C-terminal end of β-strand 2, in a similar position to a His residue, which is conserved in families 1, 5, 10 and 26. The His residue, absent in the structure of AbfA, is located at the end of β-strand 3 in the other GH-A clan members (Ryttersgaard et al., 2002), forming a hydrogen bond to the C3 hydroxyl of the glycosyl moiety. An intriguing feature of the active site in AbfA is Glu29 at the end of β-strand 1, which forms a direct contact with the substrate at subsite –1. Strong hydrogen bonds are observed between the Oε1 of Glu29 and the furanose C3 hydroxyls of the 4-nitrophenyl-Ara and Ara-α(1,3)-Xyl (Figure 4B and C). Replacing this residue in the GH-51 α-l-arabinofuranosidase from Thermobacillus xylanilyticus led to a reduction in the catalytic activity of three to four orders of magnitude, and it was therefore suggested to be a third catalytic residue (Debeche et al., 2002). From the current structure, it is clear that Glu29 is too far from the scissile bond to be directly involved in catalysis. However, it plays a key role in substrate binding: in addition to the strong hydrogen bond to the substrate, Glu29 can form hydrogen bonds to Asn74 and Gln351, which also participate in substrate binding. The elimination of Glu29 probably alters the hydrogen bonding network at the non-reducing end of the substrate, thus resulting in a severe drop in activity.
Substrate specificity
In this work we present the structures of a GH-A glycosidase in complex with a furanosidic substrate. The comparison of these structures to those of other GH-A glycosidases in complex with pyranosidic substrates can provide insight into the binding mechanism governing the substrate specificity of this related group of enzymes. The conformations of the arabinofuranose rings at the two Michaelis complexes of AbfA seem to be almost identical: a 4E conformation (an envelope with C4 above the plane) in the 4-nitrophenyl-Ara complex (Figure 4B), and the energetically close 4T0 conformation (an asymmetrical twist with C4 above and the endocyclic O4 beneath the plane) in the Ara-α(1,3)-Xyl complex (Figure 4C). These conformations place the glycosidic bond in a more quasi-axial orientation than in the E3 conformation (an envelope with C3 below the plane) of free l-arabinofuranoses, allowing an in-line attack of the nucleophile Glu294 at the anomeric carbon, and placing the glycosidic oxygen closer to the acid/base Glu175. In pyranose-specific retaining glycosidases, the distortion of the substrate from its relaxed 4C1 conformation is one of the most important characteristics of substrate binding, and therefore of catalysis itself (Tews et al., 1996; Davies et al., 1998b; Notenboom et al., 1998a; Sidhu et al., 1999; Vocadlo et al., 2001; Ducros et al., 2002). In furanoses, however, the energy barriers between the various envelope and twist conformations are small relative to those involved in the conformational inversion of pyranoses (Collins and Ferrier, 1995).
The catalytic domain of AbfA shows resemblance to that of the endoglucanase Cel5A from Bacillus agaradhaerens. Cel5A is a retaining GH-5 family enzyme (grouped also as a clan GH-A glycosidase), which hydrolyzes β-1,4-glycosidic bonds in cello-oligosaccharides (Varrot and Davies, 2003). The structural similarity of AbfA and Cel5A made it possible to superimpose the structures, and compare the binding of glucopyranosidic substrates versus arabinofuranosidic substrates at the active site of these two GH families. The superposition of AbfA including its furanosidic substrate 4-nitrophenyl-Ara, together with the structure of the endoglucanase Cel5A with its pyranosidic substrate 2,4-dinitrophenyl-2-deoxy-2-fluoro-β-d-cellobioside is shown in Figure 5. The d-glucose ring fits well in the active site of AbfA, except for the C6 and 6-OH, which appear to be too close to Trp298 (1.3 Å from the 6-OH to the tryptophan). This residue is conserved in most of the GH-51 arabinofuranosidases, but not in the only two GH-51 endoglucanases that hydrolyze glucopyranosydic substrates. Therefore, it appears that Trp298 is responsible for the discrimination against glucopyranosidic substrates in the GH-51 family. In a similar manner, the C5 and 5-OH of the l-arabinofuranose do not allow the binding in the endoglucanases active site, because of their close proximity to the conserved aromatic residue at the end of β-strand 8, Trp262 in Cel5A (1.8 Å from the 5-OH to the tryptophan). In AbfA, Gln351 is present in the equivalent position but, as described before, the preceding cis-peptide bond allows this residue to form a hydrogen bond to the 5-OH of the l-arabinofuranose. Thus, it appears that only two residues govern the distinction between d-glucopyranosidic and l-arabinofuranosidic substrates in these enzymes. It is tempting to speculate that the reciprocal replacements of these residues in the two enzymes will change their substrate specificity; experiments testing this hypothesis are currently under way.
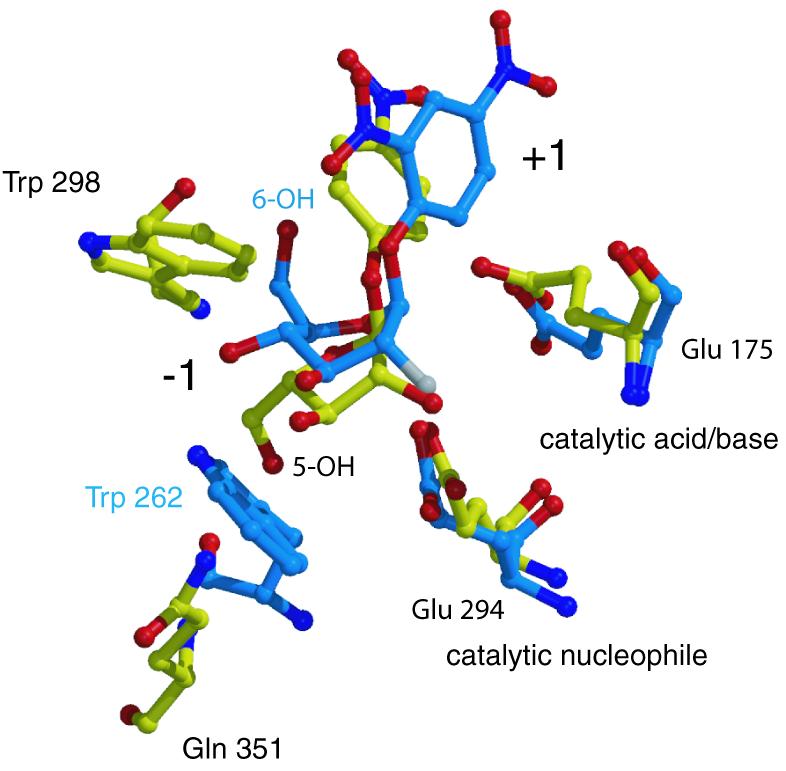
Fig. 5. Substrate specificity of AbfA and Cel5A. Superposition of the Michaelis complexes of AbfA with 4-nitrophenyl-Ara and Cel5A endoglucanase with 2,4-dinitrophenyl-2-deoxy-2-fluoro-β-d-cellobioside (PDB code: 1H2J). Only the catalytic residues and the residues responsible for the discrimination between the d-glucopyranosidic and l-arabinofuranosidic substrates are shown. Color coding: red, oxygen; blue, nitrogen; white, fluorine; yellow, carbon atoms of AbfA; light blue, carbon atoms of Cel5A.
Interestingly, although belonging to the arabinofuranose-specific GH-51, AbfA can accommodate xylopyranosidic substrates. d-Xylopyranose and l-arabino furanose share spatial similarity, and the xylopyranoses do not have the C6 and 6-OH that cause the steric hindrance with Trp298 (as in glucose). This similarity rationalizes the existence of bifunctional α-l-arabinofuranosidases/β-d-xylosidases in GH-3 and GH-43 (Utt et al., 1991; Sakka et al., 1993; Lee et al., 2003). AbfA hydrolyzes the pyranosidic synthetic substrates aryl β-d-xylopyranosides with 2–30-fold lower activity (kcat) and 60–280-fold lower specificity (kcat/Km) compared with the corresponding aryl α-l-arabinofuranosides (Table II) (Shallom et al., 2002a,b). As mentioned earlier, the furanosidic substrates bind with relatively low distortion. Apparently, AbfA can accommodate xylopyranose in the active site, but without the necessary distortion required for the efficient catalysis of six-membered rings. This explains in part the lower specificity of AbfA towards the xylopyranosidic substrates.
Table II. AbfA kinetic parameters for the hydrolysis of aryl-α-l-arabinofuranosides and aryl-β-d-xylopyranosidesa.
| Phenol substituent (pKa) | Glyconb | kcat (s–1) | Ratio kcat [AF/Xyl] | Km (mM) | Ratio Km [AF/Xyl] | kcat/Km (s–1 mM–1) | Ratio kcat/Km [AF/Xyl] |
|---|---|---|---|---|---|---|---|
| 2,5-Dinitro (5.15) | AF | 190 | 2.6 | 0.35 | 1/23 | 540 | 60 |
| Xyl | 74 | 8.0 | 9.3 | ||||
| 3,4-Dinitro (5.36) | AF | 340 | 34 | 0.53 | 1/8 | 640 | 280 |
| Xyl | 10 | 4.4 | 2.3 | ||||
| 4-Nitro (7.18) | AF | 87 | 4.3 | 0.65 | 1/23 | 130 | 100 |
| 2-Nitro (7.22) | Xyl | 20 | 15.3 | 1.3 |
aValues taken from Shallom et al. (2002a,b).
bAF, arabinofuranosyl; Xyl, xylopyranosyl.
Trapping the transient covalent intermediate
In retaining glycosidases, structures demonstrating the covalent bond between the catalytic nucleophile and the glycosyl moiety provide essential evidence for the catalytic mechanism in which the hydrolysis proceeds via a covalent glycosyl–enzyme intermediate. Although this mechanism was accepted for most retaining glycosidases since first proposed by Koshland in 1953 (Koshland, 1953), it was only in 2001 that this mechanism was verified for one of the most studied retaining glycosidases, the GH-22 hen egg-white lysozyme (Vocadlo et al., 2001).
Most structures of covalent intermediates of glycosidases were obtained by using modified 2-fluoro-2-deoxy sugar substrates with good leaving groups, together with wild-type enzymes or acid/base mutants (Wicki et al., 2002). The use of these mechanism-based inhibitors results in the accumulation of the glycosyl–enzyme intermediate, which can be further identified by mass-spectrometry or X-ray crystallography. A major drawback of structures obtained with this technique is that they are missing the strong hydrogen bond between the 2-hydroxyl of the sugar substrate and the nucleophile. Such interaction was found to contribute at least 10 kcal/mol to the transition state stabilization in several retaining glycosidases (Namchuk and Withers, 1995; White et al., 1996; Zechel and Withers, 1999).
To date there are only two crystal structures of non-fluoro glycosyl–enzyme intermediates reported, although there are several examples of identifying the intermediates by mass spectrometry (Vocadlo et al., 2001; Li et al., 2002). The structures of the covalent intermediate complexes of the GH-10 glycosidase Cex and the GH-13 cyclodextrin glycosyltransferase were obtained by using their acid/base mutants together with substrates bearing good leaving groups (Notenboom et al., 1998a; Uitdehaag et al., 1999). Indeed, in the structure of Cex, an unusually short hydrogen bond (2.37 Å) was measured between the sugar 2-hydroxyl and the nucleophile, which was naturally not present in the fluoro-substituted complexes of the same enzyme (White et al., 1996; Notenboom et al., 1998b).
In this work we followed a similar strategy: in an attempt to isolate the covalent glycosyl–enzyme intermediate of AbfA, we used the highly reactive substrate 2′,5′-dinitrophenyl α-l-arabinofuranoside (2,5-dinitrophenyl-Ara) with the acid/base mutant E175A. With this substrate, deglycosylation is the rate-limiting step of the acid/base mutant, leading to an accumulation of the glycosyl–enzyme intermediate (Shallom et al., 2002b). Indeed, after briefly soaking the crystals of the acid/base mutant E175A with 2,5-dinitrophenyl-Ara at 4°C, followed by flash freezing with liquid nitrogen, the electron density at 2.0 Å resolution unambiguously showed the covalent bond (1.5 Å long) between the nucleophile Glu294 and the anomeric carbon of the arabinofuranose (Figure 4D). A strong hydrogen bond (2.6 Å) is observed between the nucleophile Glu294 (Oε2) and the furanose 2-hydroxyl, suggesting that this interaction is essential also in the GH-51 arabinofuranosidases. As in the Michaelis complexes, no significant change in the protein structure was observed upon the intermediate formation (r.m.s.d. = 0.18 Å for all main chain atoms). The formation of the covalent intermediate was also confirmed by electrospray ionization mass spectrometry (ESI-MS). The experimental mass difference between the free E175A mutant protein (Mr 57 021) and the covalent intermediate (Mr 57 154) was 133, exactly the molecular weight of the arabinofuranosyl moiety (Supplementary figure 1).
Mechanistic implications
The catalytic residues of AbfA, the acid/base Glu175 and the nucleophile Glu294, are located at the C-terminal ends of β-strands 4 and 7 in the (β/α)8-barrel fold, respectively, as expected for a GH-A clan enzyme (Figure 4A) (Henrissat and Davies, 1997; Zverlov et al., 1998). The average trans residual distance between the four carboxylic oxygens of the catalytic residues is 4.7 Å, typical to retaining glycosidases (Zechel and Withers, 1999). The exact position of the catalytic residues relative to the bound substrate can be visualized by superimposing the structures of the native enzyme with that of the Ara-α(1,3)-Xyl Michaelis complex (Figure 6). The carboxyl oxygen of Glu175 is located 3.0 Å from the glycosidic oxygen of Ara-α(1,3)-Xyl, allowing the required acid catalysis and the direct protonation of the departing aglycon by Glu175. The nucleophile Glu294 is at an appropriate distance (3.2 Å) for nucleophilic attack on the anomeric carbon and for formation of the covalent glycosyl–enzyme intermediate. In the second step of catalysis, a water molecule should be activated by the acid/base residue in order to accomplish the hydrolysis of the glycosyl–enzyme intermediate. Indeed, in the superposition of Glu175 with the covalent intermediate structure, Glu175 is located 3.9 Å from the anomeric carbon of the arabinofuranosyl, a suitable distance to accommodate and deprotonate the nucleophilic water molecule that attacks the anomeric carbon (superposition not shown).
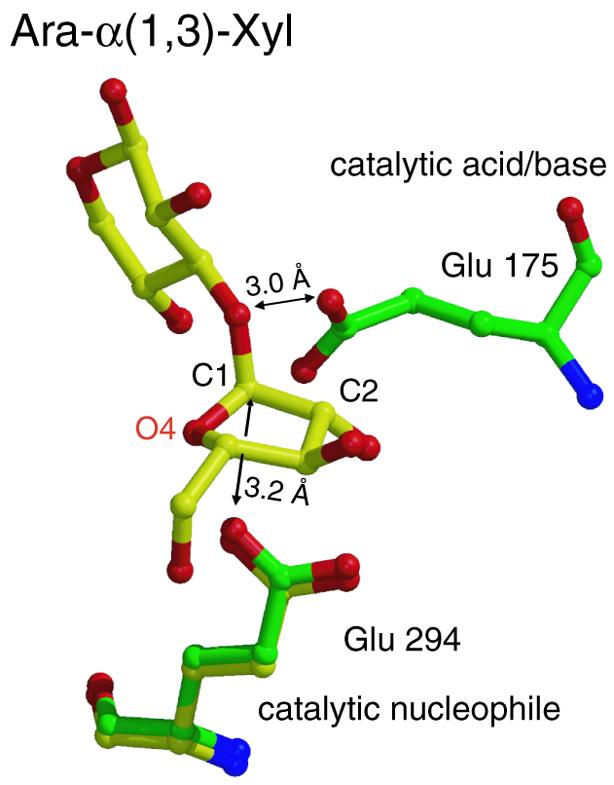
Fig. 6. Superposition of the structures of the native enzyme (green) with that of the Michaelis complex (yellow). Glu175 is located 3.0 Å from the glycosidic oxygen of Ara-α(1,3)-Xyl, which is a hydrogen-bonding distance, allowing the direct protonation of the departing aglycon. Glu294 is in an appropriate distance (3.2 Å) for a nucleophilic attack on the anomeric carbon.
In the structures of both the Michaelis complexes and of the covalent intermediate, the conserved Tyr246 can form three possible hydrogen bonds: to the O5 of the sugar at –1 and to the Glu294 Oε1 oxygen (both 2.6–2.7 Å long in the different structures), and also to the endocyclic O4 (3.1–3.3 Å). At the oxocarbenium ion-like transition state, the hydrogen bond donated by Tyr246 probably favors Glu294 and the O5, thus allowing electrostatic or dipolar interaction between the Tyr246 Oη and the positively charged endocyclic O4. This dipolar interaction also improves the proton donation of Tyr246, which further stabilizes the partial negative charge on the Glu294 Oε1. Similar hydrogen bonds network architecture is found also in the GH-11 xylanase from Bacillus circulans, where the replacement of the corresponding Tyr by a Phe residue led to complete loss of activity (Wakarchuk et al., 1994; Sidhu et al., 1999).
Retaining glycosidases can be categorized according to their protonation trajectories (Vasella et al., 2002): in anti-protonators, the proton donor is positioned anti to the sugar endocyclic O4–C1 bond, while in syn-protonators, the proton donor is placed syn to that bond (Heightman and Vasella, 1999). The protonation trajectory of glycosidases is crucial when designing selective inhibitors. Optimal inhibition is achieved when the inhibitor mimics the shape, charge, and in glycosidases, also the protonation trajectory of the transition state. For example, xylobiose-derived aza-sugars inhibit GH-10 xylanases (anti-protonators) much better than GH-11 xylanases (syn-protonators), in part since these inhibitors do not contain a syn lone-pair in the position corresponding to the substrate’s glycosidic oxygen (Notenboom et al., 2000). In AbfA, the acid/base Glu175 is located closer to the C2 side of the furanose rather than to the endocyclic O4 side (Figure 6), and thus AbfA is an anti-protonator. To date, all the GH families from clan GH-A for which the protonation trajectory was determined (GH-1, 2, 5, 10 and 26), were found to be anti-protonators (Heightman and Vasella, 1999; Ducros et al., 2002; Vasella et al., 2002). It is likely therefore, that all clan GH-A families, furanose- and pyranose-specific, use the anti-protonation trajectory.
In the covalent arabinofuranosyl–enzyme intermediate structure, the bound sugar is present in a twisted 2T1 conformation. When superimposing the structures of the bound sugars in the Michaelis and covalent intermediate complexes, it seems that the furanosidic hydroxyls 2-OH, 3-OH and 5-OH are approximately in the same position, and the major change involved in the covalent intermediate formation includes the 1.7 Å movement of C1 towards the nucleophile (Figure 7). Assuming that during the transition-state formation the furanosidic hydroxyls are tightly bound in the same places, then the position of the C1 in the transition state would have to be somewhere between its positions in the two observed structures. This will allow the sugar ring to reach an almost flat conformation, in which the C4, O4, C1 and C2 are co-planar, as required from an oxocarbenium ion-like transition state. The movement of the C1, termed ‘electrophilic migration’, was described previously for the GH-22 hen egg-white lysozyme and the GH-5 endoglucanase Cel5A, both with pyranosidic substrates (Vocadlo et al., 2001; Varrot and Davies, 2003). Apparently, electrophilic migration takes place also in furanose-hydrolyzing glycosidases.

Fig. 7. Electrophilic migration of C1. (A) Overlay of the two critical states of the glycosylation step: Michaelis complex (yellow) and covalent intermediate (green). C1 migrates ∼1.7 Å to form the covalent bond with the nucleophilic oxygen. (B) Simplified schematic representation of the electrophilic migration of C1.
Electrophilic migration mechanism of the anomeric carbon of furanoses was extensively studied in N-ribosyl-transferase such as purine nucleoside phosphorylases, which catalyze the reversible cleavage of bonds between the ribose and the purine as part of the purine salvage system (Schramm and Shi, 2001; Schramm, 2002). As in glycosidases, the catalytic pathway of these enzymes proceeds through an oxocarbenium ion-like transition state. But in contrast to glycosidases, in which the two catalytic residues directly participate in the transition state stabilization, in the N-ribosyl-transferase enzymes there are no enzymatic anions sufficiently close to the scissile bond to stabilize the transition state. With these enzymes, the catalytic acceleration is achieved by placing the nucleophile (phosphate, water or pyrophosphate) near the ribosyl group, and by excluding water molecules from the active site to prevent hydrolysis in the phosphotransferases. Combination of crystal structures of purine nucleoside phosphorylases in complex with transition state analogue inhibitor, together with kinetic isotope effect analyses, showed that the formation of the transition state involves the migration of the anomeric carbon from the purine ring toward the phosphate, while all the other parts of the reactants are more restricted (Fedorov et al., 2001).
Conclusions
Taken together, the complex structures described provide an example for the visualization of the stable states along the reaction coordinate of AbfA, and of GH-51 glycosidases in general (Figure 8). The furanosidic substrate binds into the active site of the free enzyme, where the two catalytic residues are positioned in optimized conformations so as to allow the nucleophilic attack at the anomeric carbon, and the protonation of the leaving aglycon (Figure 8A and B). In the oxocarbenium ion-like transition state, the bond between the glycosidic oxygen and the anomeric carbon lengthens and pushes the C1 toward the nucleophile, collapsing into the covalent glycosyl–enzyme intermediate (Figure 8C). The latter is then hydrolyzed by the incoming water molecule, being deprotonated by the catalytic acid/base residue.

Fig. 8. Snapshots along the reaction pathway obtained from the X-ray structures (upper), and a schematic representation of the corresponding stages of the glycosylation step (lower). (A) The catalytic residues of the native AbfA. (B) Michaelis complex of the intact substrate Ara-α(1,3)-Xyl located in subsites –1 and +1. The absence of electron density at this contour level for the 4-OH of the xylose at the +1 subsite results from the heterogeneity of the substrate. The second xylose connected to only part of the substrate molecules probably distorts the O4 of the first xylose. (C) Structure of the trapped covalent arabinofuranosyl–enzyme intermediate. The 2DFo – mFc electron density maps are contoured at 1.5 σ.
The structures of the Michaelis and the covalent intermediate complexes reveal that AbfA have similar mechanistic features to those found in other glycosidases. The arabinofuranosyl sugars in the active site are tightly bound and distorted by an extensive network of hydrogen bonds. The two catalytic residues are 4.7 Å apart, and together with other conserved residues they contribute to the stabilization of the oxocarbenium ion-like transition state via charge delocalization and specific protein–substrate interactions. AbfA is an anti-protonator, and the 1.7 Å electrophilic migration of the anomeric carbon during the glycosylation suggest an almost flat conformation of the arabinofuranose ring in the transition state.
Materials and methods
Materials
The native α-l-arabinofuranosidase from G.stearothermophilus T-6 (AbfA), its Se-Met derivative and its E175A mutant were cloned and purified as previously reported (Shallom et al., 2002b; Hövel et al., 2003). 4-Nitrophenyl-Ara and all other chemicals and reagents were obtained from Aldrich or Sigma. 2,5-Dinitrophenyl-Ara was synthesized according to published procedure (Kelly et al., 1988). The natural substrate for the enzyme, Ara-α(1,3)-Xyl was prepared as follows: an aqueous solution (2% w/v) of Oat-spelts xylan (Sigma) was digested extensively with recombinant xylanase and β-xylosidase both from G.stearothermophilus T-6, 0.1 mg/ml of each, for 12 h at 50°C. The reaction mixture was concentrated 10-fold, followed by centrifugation (14 000 g for 15 min). The soluble products in the supernatant were separated using a BioGel P-2 (BioRad, Richmond, CA) gel filtration column (100 × 2 cm) with water as the mobile phase at room temperature. The fractions were analyzed by TLC, and fractions containing sugars hydrolyzable by AbfA were collected. Partial 1H-NMR analysis has identified the sugars as mixtures of short xylo-oligomers containing arabinofuranosyl substitutions at different positions (data not shown).
Preparing substrate complexes and trapping the covalent reaction intermediate for X-ray analysis
The E175A mutant was crystallized under similar conditions as described for the native and Se-Met AbfA (Hövel et al., 2003). Crystals of the E175A mutant were grown from solutions containing 23% (w/v) PEG 3350, 0.2 M NH4F, 5% (v/v) 2-propanol and 0.1 M Tris–HCl pH 8.0. For preparation of the 4-nitrophenyl-Ara complex, the E175A mutant crystals were soaked for 15 min in the cryo solution [12.5% (v/v) glycerol, 23% (w/v) PEG 3350, 0.2 M NH4F, 5% (v/v) 2-propanol and 0.1 M Tris–HCl pH 8.0] enriched by 20 mM of 4-nitrophenyl-Ara. The Ara-α(1,3)-Xyl complex was obtained by soaking the E175A mutant crystals for at least 12 h in the cryo solution enriched by 20 mM of the natural substrate. The covalent reaction intermediate was trapped at 4°C by briefly soaking the E175A mutant crystals in the cryo solution containing 20 mM of 2,5-dinitrophenyl-Ara, and then direct immersion in liquid nitrogen.
X-ray data collection
A complete MAD (Hendrickson and Ogata, 1997) data set was collected at 100 K to 1.68 Å resolution using a MAR-CCD (165 mm) detector on beamline BW7A at the EMBL outstation, Hamburg. High resolution data of the 4-nitrophenyl-Ara complex was measured with a MAR 345 imaging plate on beamline BW7B at the EMBL outstation, Hamburg. All other X-ray diffraction data were collected at 100 K on a MAR 345 image plate detector using CuKα radiation from a Rigaku RU200 X-ray generator. All data were processed and scaled with DENZO and SCALEPACK (Otwinowski and Minor, 1997). Selected data collection parameters are shown in Table I.
Phasing, model building and refinement
The structure of AbfA was solved both from the anomalous signal of Se by MAD phasing (Hendrickson and Ogata, 1997) and by MIR phasing using platinum and mercury derivatives. The platinum and mercury data used for MIR phasing were collected from native enzyme crystals that were soaked in 10 mM K2PtCl4 for 10 h and 10 mM HgCl2 for 5 days. The automated protocol of SOLVE (Terwilliger and Berendzen, 1999) was used for determination of the heavy metal atom substructures, calculation of initial phases and electron density maps. Both phasing approaches independently resulted in interpretable electron density maps. After density modification with RESOLVE (Terwilliger, 2001) a figure of merit of 0.81 was obtained for the MAD data (0.66 for the MIR data) and the excellent quality of the map permitted ARP/wARP (Perrakis et al., 1999) to auto-build ∼95% of the model. Subsequently, all model building and refinement was carried out with ‘O’ (Jones et al., 1991) and REFMAC5 (Pannu et al., 1998). Native and E175A mutant structures were refined with ‘O’ and REFMAC5 using the MAD structure (R-factor of 17.2%) as a starting model. In the Glu175Ala mutant structures, clear electron density maps were observed for the bound substrates, 4-nitrophenyl-Ara and Ara-α(1,3)-Xyl, and for the covalent glycosyl–enzyme intermediate after the first refinement step. The restraints for sugar bond lengths and bond angles were varied during refinement, but the torsion angles were always left unrestrained to study the various sugar conformations. Anisotropic displacement factors were included for the 4-nitrophenyl-Ara data. Selected refinement statistics are shown in Table I.
The coordinates and structure factors of the final models of native AbfA, 4-nitrophenyl-Ara, Ara-α(1,3)-Xyl and covalent intermediate have been deposited in the Protein Data Bank with PDB codes 1PZ3, 1QW9, 1QW8 and 1PZ2, respectively.
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We thank E.Pohl and co-workers (EMBL Outstation Hamburg) for excellent support during measurements; A.Admon and T.Ziv for the ESI-MS analysis; and N.Adir, J.Abendroth and B.Henrissat for critical reading of the manuscript. This study was supported by The Israel Science Foundation (676/00 to G.S. and Y.S.) and by the French–Israeli Association for Scientific and Technological Research (Y.S.). Additional support was provided by the Otto Meyerhof Center for Biotechnology at the Technion, established by the Minerva Foundation (Munich, Germany). V.B. acknowledges financial support from the Center of Absorption in Science, the Ministry of Immigration Absorption and the Ministry of Science and Arts, Israel (Kamea Program).
References
- Bourne Y. and Henrissat,B. (2001) Glycoside hydrolases and glycosyltransferases: families and functional modules. Curr. Opin. Struct. Biol., 11, 593–600. [DOI] [PubMed] [Google Scholar]
- Collins P. and Ferrier,R. (1995) Monosaccharides, their Chemistry and their Roles in Natural Products. John Wiley and Sons Ltd, Chichester, UK. [Google Scholar]
- Davies G., Sinnott,M.L. and Withers,S.G. (1998a) Glycosyl Transfer. In Sinnott,M.L. (ed.), Comprehensive Biological Catalysis. Academic Press Limited, London, UK, Vol. 1, pp. 119–209. [Google Scholar]
- Davies G.J., Mackenzie,L., Varrot,A., Dauter,M., Brzozowski,A.M., Schulein,M. and Withers,S.G. (1998b) Snapshots along an enzymatic reaction coordinate: analysis of a retaining β-glycoside hydrolase. Biochemistry, 37, 11707–11713. [DOI] [PubMed] [Google Scholar]
- Debeche T., Bliard,C., Debeire,P. and O’Donohue,M.J. (2002) Probing the catalytically essential residues of the α-l-arabinofuranosidase from Thermobacillus xylanilyticus. Protein Eng., 15, 21–28. [DOI] [PubMed] [Google Scholar]
- Ducros V.M., Zechel,D.L., Murshudov,G.N., Gilbert,H.J., Szabo,L., Stoll,D., Withers,S.G. and Davies,G.J. (2002) Substrate distortion by a β-mannanase: snapshots of the Michaelis and covalent-intermediate complexes suggest a B(2,5) conformation for the transition state. Angew. Chem. Int. Ed., 41, 2824–2827. [DOI] [PubMed] [Google Scholar]
- Esnouf R.M. (1997) An extensively modified version of MolScript that includes greatly enhanced coloring capabilities. J. Mol. Graph. Model., 15, 132–134. [DOI] [PubMed] [Google Scholar]
- Fedorov A. et al. (2001) Transition state structure of purine nucleoside phosphorylase and principles of atomic motion in enzymatic catalysis. Biochemistry, 40, 853–860. [DOI] [PubMed] [Google Scholar]
- Fujimoto Z., Takase,K., Doui,N., Momma,M., Matsumoto,T. and Mizuno,H. (1998) Crystal structure of a catalytic-site mutant α-amylase from Bacillus subtilis complexed with maltopentaose. J. Mol. Biol., 277, 393–407. [DOI] [PubMed] [Google Scholar]
- Heightman T.D. and Vasella,A.T. (1999) Recent insights into inhibition, structure and mechanism of configuration-retaining glycosidases. Angew. Chem. Int. Ed., 38, 750–770. [DOI] [PubMed] [Google Scholar]
- Hendrickson W.A. and Ogata,C.M. (1997) Phase determination from multiwavelength anomalous diffraction measurements. Methods Enzymol., 276, 494–523. [DOI] [PubMed] [Google Scholar]
- Henrissat B. and Davies,G. (1997) Structural and sequence-based classification of glycoside hydrolases. Curr. Opin. Struct. Biol., 7, 637–644. [DOI] [PubMed] [Google Scholar]
- Henrissat B., Callebaut,I., Fabrega,S., Lehn,P., Mornon,J.P. and Davies,G. (1995) Conserved catalytic machinery and the prediction of a common fold for several families of glycosyl hydrolases. Proc. Natl Acad. Sci. USA, 92, 7090–7094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hövel K., Shallom,D., Niefind,K., Baasov,T., Shoham,G., Shoham,Y. and Schomburg,D. (2003) Crystallization and preliminary X-ray analysis of a family 51 glycoside hydrolase, the α-l-arabinofuranosidase from Geobacillus stearothermophilus T-6. Acta Crystallogr. D, 59, 913–915. [DOI] [PubMed] [Google Scholar]
- Iriyama N., Takeuchi,N., Shiraishi,T., Izumi,K., Sawada,M.T., Takahashi,N., Furuhata,K., Ogura,H. and Uda,Y. (2000) Enzymatic properties of sialidase from the ovary of the starfish, Asterina pectinifera. Comp. Biochem. Physiol. B Biochem. Mol. Biol., 126, 561–569. [DOI] [PubMed] [Google Scholar]
- Jenkins J., Lo Leggio,L., Harris,G. and Pickersgill,R. (1995) β-Glucosidase, β-galactosidase, family A cellulases, family F xylanases and two barley glycanases form a superfamily of enzymes with 8-fold β/α architecture and with two conserved glutamates near the carboxy-terminal ends of β-strands four and seven. FEBS Lett., 362, 281–285. [DOI] [PubMed] [Google Scholar]
- Jones T.A., Zou,J.Y., Cowan,S.W. and Kjeldgaard. (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A, 47, 110–119. [DOI] [PubMed] [Google Scholar]
- Juers D.H., Huber,R.E. and Matthews,B.W. (1999) Structural comparisons of TIM barrel proteins suggest functional and evolutionary relationships between β-galactosidase and other glycohydrolases. Protein Sci., 8, 122–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamitori S., Kondo,S., Okuyama,K., Yokota,T., Shimura,Y., Tonozuka,T. and Sakano,Y. (1999) Crystal structure of Thermoactinomyces vulgaris R-47 α-amylase II (TVAII) hydrolyzing cyclodextrins and pullulan at 2.6 Å resolution. J. Mol. Biol., 287, 907–921. [DOI] [PubMed] [Google Scholar]
- Kelly M.A., Sinnott,M.L. and Widdows,D. (1988) Preparation of some aryl α-l-arabinofuranosides as substrates for arabinofuranosidase. Carbohydr. Res., 181, 262–266. [Google Scholar]
- Koshland D.E. (1953) Stereochemistry and the mechanism of enzymatic reactions. Biol. Rev. Camb. Phil. Soc., 28, 416–436. [Google Scholar]
- Kraulis P.J. (1991) MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr., 24, 946–650. [Google Scholar]
- Lee R.C., Hrmova,M., Burton,R.A., Lahnstein,J. and Fincher,G.B. (2003) Bifunctional family 3 glycoside hydrolases from barley with α-l-arabinofuranosidase and β-d-xylosidase activity. Character ization, primary structures and COOH-terminal processing. J. Biol. Chem., 278, 5377–5387. [DOI] [PubMed] [Google Scholar]
- Li Y.K., Chir,J., Tanaka,S. and Chen,F.Y. (2002) Identification of the general acid/base catalyst of a family 3 β-glucosidase from Flavobacterium meningosepticum. Biochemistry, 41, 2751–2759. [DOI] [PubMed] [Google Scholar]
- Merritt E.A. and Bacon,D.J. (1997) Raster3D: Photorealistic Molecular Graphics. Methods Enzymol., 277, 505–524. [DOI] [PubMed] [Google Scholar]
- Namchuk M.N. and Withers,S.G. (1995) Mechanism of Agrobacterium β-glucosidase: kinetic analysis of the role of noncovalent enzyme/substrate interactions. Biochemistry, 34, 16194–16202. [DOI] [PubMed] [Google Scholar]
- Navas J. and Beguin,P. (1992) Site-directed mutagenesis of conserved residues of Clostridium thermocellum endoglucanase CelC. Biochem. Biophys. Res. Commun., 189, 807–812. [DOI] [PubMed] [Google Scholar]
- Notenboom V., Birsan,C., Nitz,M., Rose,D.R., Warren,R.A. and Withers,S.G. (1998a) Insights into transition state stabilization of the β-1,4-glycosidase Cex by covalent intermediate accumulation in active site mutants. Nat. Struct. Biol., 5, 812–818. [DOI] [PubMed] [Google Scholar]
- Notenboom V., Birsan,C., Warren,R.A., Withers,S.G. and Rose,D.R. (1998b) Exploring the cellulose/xylan specificity of the β-1,4-glycanase cex from Cellulomonas fimi through crystallography and mutation. Biochemistry, 37, 4751–4758. [DOI] [PubMed] [Google Scholar]
- Notenboom V., Williams,S.J., Hoos,R., Withers,S.G. and Rose,D.R. (2000) Detailed structural analysis of glycosidase/inhibitor interactions: complexes of Cex from Cellulomonas fimi with xylobiose-derived aza-sugars. Biochemistry, 39, 11553–11563. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor,W. (1997) Processing of X-Ray Diffraction Data Collection in Oscillation Mode. Methods Enzymol., 276, 407–426. [DOI] [PubMed] [Google Scholar]
- Pannu N.S., Murshudov,G.N., Dodson,E.J. and Read,R.J. (1998) Incorporation of prior phase information strengthens maximum-likelihood structure refinement. Acta Crystallogr. D, 54, 1285–1294. [DOI] [PubMed] [Google Scholar]
- Perrakis A., Morris,R. and Lamzin,V.S. (1999) Automated protein model building combined with iterative structure refinement. Nat. Struct. Biol., 6, 458–463. [DOI] [PubMed] [Google Scholar]
- Ryttersgaard C., Lo Leggio,L., Coutinho,P.M., Henrissat,B. and Larsen,S. (2002) Aspergillus aculeatus β-1,4-galactanase: substrate recognition and relations to other glycoside hydrolases in clan GH-A. Biochemistry, 41, 15135–15143. [DOI] [PubMed] [Google Scholar]
- Saha B.C. (2000) α-l-Arabinofuranosidases: biochemistry, molecular biology and application in biotechnology. Biotechnol. Adv., 18, 403–423. [DOI] [PubMed] [Google Scholar]
- Sakka K., Yoshikawa,K., Kojima,Y., Karita,S., Ohmiya,K. and Shimada,K. (1993) Nucleotide sequence of the Clostridium stercorarium xylA gene encoding a bifunctional protein with β-d-xylosidase and α-l-arabinofuranosidase activities and properties of the translated product. Biosci. Biotechnol. Biochem., 57, 268–272. [DOI] [PubMed] [Google Scholar]
- Sakon J., Adney,W.S., Himmel,M.E., Thomas,S.R. and Karplus,P.A. (1996) Crystal structure of thermostable family 5 endocellulase E1 from Acidothermus cellulolyticus in complex with cellotetraose. Biochemistry, 35, 10648–10660. [DOI] [PubMed] [Google Scholar]
- Schramm V.L. (2002) Development of transition state analogues of purine nucleoside phosphorylase as anti-T-cell agents. Biochim. Biophys Acta, 1587, 107–117. [DOI] [PubMed] [Google Scholar]
- Schramm V.L. and Shi,W. (2001) Atomic motion in enzymatic reaction coordinates. Curr. Opin. Struct. Biol., 11, 657–665. [DOI] [PubMed] [Google Scholar]
- Shallom D. and Shoham,Y. (2003) Microbial hemicellulases. Curr. Opin. Microbiol., 6, 219–228. [DOI] [PubMed] [Google Scholar]
- Shallom D., Belakhov,V., Solomon,D., Gilead-Gropper,S., Baasov,T., Shoham,G. and Shoham,Y. (2002a) The identification of the acid- base catalyst of α-arabinofuranosidase from Geobacillus stearothermophilus T-6, a family 51 glycoside hydrolase. FEBS Lett., 514, 163–167. [DOI] [PubMed] [Google Scholar]
- Shallom D., Belakhov,V., Solomon,D., Shoham,G., Baasov,T. and Shoham,Y. (2002b) Detailed kinetic analysis and identification of the nucleophile in α-l-arabinofuranosidase from Geobacillus stearothermophilus T-6, a family 51 glycoside hydrolase. J. Biol. Chem., 277, 43667–43673. [DOI] [PubMed] [Google Scholar]
- Sidhu G., Withers,S.G., Nguyen,N.T., McIntosh,L.P., Ziser,L. and Brayer,G.D. (1999) Sugar ring distortion in the glycosyl-enzyme intermediate of a family G/11 xylanase. Biochemistry, 38, 5346–5354. [DOI] [PubMed] [Google Scholar]
- Sinnott M.L. (1990) Catalytic mechanisms of enzymic glycosyl transfer. Chem. Rev., 90, 1171–1202. [Google Scholar]
- Terwilliger T.C. (2001) Maximum-likelihood density modification using pattern recognition of structural motifs. Acta Crystallogr. D, 57, 1755–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger T.C. and Berendzen,J. (1999) Evaluation of macromolecular electron-density map quality using the correlation of local r.m.s. density. Acta Crystallogr. D, 55, 1872–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tews I., Perrakis,A., Oppenheim,A., Dauter,Z., Wilson,K.S. and Vorgias,C.E. (1996) Bacterial chitobiase structure provides insight into catalytic mechanism and the basis of Tay–Sachs disease. Nat. Struct. Biol., 3, 638–648. [DOI] [PubMed] [Google Scholar]
- Tormo J., Lamed,R., Chirino,A.J., Morag,E., Bayer,E.A., Shoham,Y. and Steitz,T.A. (1996) Crystal structure of a bacterial family-III cellulose-binding domain: a general mechanism for attachment to cellulose. EMBO J., 15, 5739–5751. [PMC free article] [PubMed] [Google Scholar]
- Uitdehaag J.C., Mosi,R., Kalk,K.H., van der Veen,B.A., Dijkhuizen,L., Withers,S.G. and Dijkstra,B.W. (1999) X-ray structures along the reaction pathway of cyclodextrin glycosyltransferase elucidate catalysis in the α-amylase family. Nat. Struct. Biol., 6, 432–436. [DOI] [PubMed] [Google Scholar]
- Utt E.A., Eddy,C.K., Keshav,K.F. and Ingram,L.O. (1991) Sequencing and expression of the Butyrivibrio fibrisolvens xylB gene encoding a novel bifunctional protein with β-d-xylosidase and α-l-arabinofuranosidase activities. Appl. Environ. Microbiol., 57, 1227–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varrot A. and Davies,G.J. (2003) Direct experimental observation of the hydrogen-bonding network of a glycosidase along its reaction coordinate revealed by atomic resolution analyses of endoglucanase Cel5A. Acta Crystallogr. D, 59, 447–452. [DOI] [PubMed] [Google Scholar]
- Vasella A., Davies,G.J. and Bohm,M. (2002) Glycosidase mechanisms. Curr. Opin. Chem. Biol., 6, 619–629. [DOI] [PubMed] [Google Scholar]
- Vocadlo D.J., Davies,G.J., Laine,R. and Withers,S.G. (2001) Catalysis by hen egg-white lysozyme proceeds via a covalent intermediate. Nature, 412, 835–838. [DOI] [PubMed] [Google Scholar]
- Wakarchuk W.W., Campbell,R.L., Sung,W.L., Davoodi,J. and Yaguchi,M. (1994) Mutational and crystallographic analyses of the active site residues of the Bacillus circulans xylanase. Protein Sci., 3, 467–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White A., Tull,D., Johns,K., Withers,S.G. and Rose,D.R. (1996) Crystallographic observation of a covalent catalytic intermediate in a β-glycosidase. Nat. Struct. Biol., 3, 149–154. [DOI] [PubMed] [Google Scholar]
- Wicki J., Rose,D.R. and Withers,S.G. (2002) Trapping covalent intermediates on β-glycosidases. Methods Enzymol., 354, 84–105. [DOI] [PubMed] [Google Scholar]
- Wolfenden R., Lu,X. and Young,G. (1998) Spontaneous hydrolysis of glycosides. J. Am. Chem. Soc., 120, 6814–6815. [Google Scholar]
- Zechel D.L. and Withers,S.G. (1999) Glycosidase mechanisms: anatomy of a finely tuned catalyst. Acc. Chem. Res., 33, 11–18. [DOI] [PubMed] [Google Scholar]
- Zverlov V.V., Liebl,W., Bachleitner,M. and Schwarz,W.H. (1998) Nucleotide sequence of arfB of Clostridium stercorarium and prediction of catalytic residues of α-L-arabinofuranosidases based on local similarity with several families of glycosyl hydrolases. FEMS Microbiol. Lett., 164, 337–343. [DOI] [PubMed] [Google Scholar]