

Abstract
Focal adhesion kinase (FAK) becomes activated upon integrin-mediated cell adhesion and controls cellular responses to the engagement of integrins, including cell migration and survival. We show here that a coordinated signaling by integrins and growth factor receptors induces expression of suppressor of cytokine signaling-3 (SOCS-3) and subsequent interaction between endogenous FAK and SOCS-3 proteins in 3T3 fibroblasts. Cotransfection studies demonstrated that SOCS-3, and also SOCS-1, interact with FAK in a FAK-Y397-dependent manner, and that both the Src homology 2 (SH2) and the kinase inhibitory region (KIR) domains of the SOCS proteins contribute to FAK binding. SOCS-1 and SOCS-3 were found to inhibit FAK-associated kinase activity in vitro and tyrosine phosphorylation of FAK in cells. The SOCS proteins also promoted polyubiquitination and degradation of FAK in a SOCS box-dependent manner and inhibited FAK-dependent signaling events, such as cell motility on fibronectin. These studies suggest a negative role of SOCS proteins in FAK signaling, and for a previously unidentified regulatory mechanism for FAK function.
Keywords: adhesion/integrin/kinase/migration/signaling
Introduction
Focal adhesion kinase (FAK) is a ubiquitously expressed non-receptor protein tyrosine kinase that has emerged as a crucial molecule in integrating signals from integrins and receptor tyrosine kinases in processes such as cell survival, proliferation and motility (Schlaepfer et al., 1999; Schaller, 2001). Genetic ablation of FAK results in early embryonic lethality in mice, and cells derived from knock-out embryos demonstrate severe migration and survival defects (Ilic et al., 1995; Ilic et al., 1998; Sieg et al., 1998; Owen et al., 1999). Conversely, enhanced FAK signaling increases cell motility and promotes cell survival in an anchorage-independent manner (Cary et al., 1996; Frisch et al., 1996). Along these lines, a number of groups have reported that FAK protein levels and/or its activity are upregulated in invasive cancer cells (Jones et al., 2000).
Ligand binding of integrins results in catalytic activation of FAK and in its autophosphorylation at Y397, which serves as a binding site for several Src homology 2 (SH2) domain-containing proteins, including Src kinases. The FAK–Src dual kinase complex leads to further phosphorylation of FAK, and also to phosphorylation and activation of a number of cytoskeleton-linked proteins, which transduce integrin-generated signals to downstream pathways, such as the mitogen-activated protein (MAP) kinase cascades, and to control of cell survival, motility and proliferation (Hanks and Polte, 1997; Schlaepfer et al., 1999; Schaller, 2001). At present, the molecular mechanisms that control FAK activation remain largely unknown.
Suppressors of cytokine signaling (SOCS) are a family of proteins that act in a feedback loop to inhibit cytokine responses and activation of the JAK/STAT pathway in hematopoietic cells (Naka et al., 1999; Yasukawa et al., 2000; Krebs and Hilton, 2001). Evidence indicates that SOCS proteins modulate other signaling pathways, as well. Cacalano et al. (2001) reported recently that SOCS-3 participates in regulation of signaling by various growth factors in non-hematopoietic cells. The SOCS-proteins are structurally characterized by distinct domains that include a central SH2 domain and a carboxyl-terminal homology region termed SOCS box. The SH2 domain of SOCS-proteins interacts with phosphorylated tyrosine residues in tyrosine kinases and, in many cases, negatively regulates their activity (Yasukawa et al., 2000; Krebs and Hilton, 2001). The SOCS boxes of SOCS-1 and SOCS-3 have been found to mediate interaction with the elongin B/C complex (Kamura et al., 1998; Zhang et al., 1999). The elongin B/C complex was originally identified as a component of the von Hippel-Lindau tumor suppressor E3 ligase complex, which also contains RING finger protein Rbx1 and Cullin-2 (Ivan and Kaelin, 2001). Thus, the SOCS box may act as a bridge between SOCS-SH2 interacting proteins and E3 ubiquitin ligases, and regulate protein turnover by targeting proteins for polyubiquitination and proteasome-mediated degradation. For SOCS-1, there is direct evidence that it can ubiquitinate and regulate the half life of Vav (De Sepulveda et al., 2000) and the JAK tyrosine kinases (Frantsve et al., 2001; Kamizono et al., 2001; Ungureanu et al., 2002).
We report here that a coordinated signaling by integrins and growth factor receptors results in upregulation of SOCS protein expression and subsequent interaction of SOCS proteins with FAK in 3T3 fibroblasts. Our studies further demonstrate that SOCS-1 and SOCS-3 bind to tyrosine-phosphorylated FAK in a FAK-Y397-dependent manner, and inhibit FAK-associated kinase activity in vitro and tyrosine phosphorylation of FAK in cells. SOCS-1 and SOCS-3 were also found to facilitate ubiquitination and degradation of FAK in a SOCS box-dependent manner and to inhibit FAK-induced MAPK activation and cell motility. These studies identify SOCS proteins as potential novel regulators of FAK signaling.
Results
Coordinate signaling by integrins and growth factor receptors results in SOCS expression and SOCS–FAK interaction
It was reported recently that treatment of adherent cells, including NIH 3T3 fibroblasts, with growth factors such as PDGF and EGF results in a rapid induction of SOCS-3 (Cacalano et al., 2001). We also noticed a report in which Pyk2, a close homolog of FAK, had been tested in a yeast-two hybrid experiment and found to interact with SOCS-1 and SOCS-3, but not with several other members of the SOCS family (Masuhara et al., 1997). These observations prompted us to examine the intriguing possibility that SOCS proteins might have a previously unidentified role in regulating FAK signaling. To this end, serum-starved monolayer cultures of NIH 3T3 cells were detached, stimulated or not with PDGF, and either kept in suspension or replated for 1 h on dishes that had been coated with the integrin ligand fibronectin (FN). As shown in Figure 1A (upper panels), simultaneous activation of integrin and growth factor pathways resulted in a rapid induction of SOCS-3 protein levels. While unavailability of suitable antibodies prohibited us from examining the protein levels of SOCS-1, a robust, 5-fold induction in the SOCS-1 mRNA levels was observed in 3T3 cells that had been plated on FN and treated with PDGF, compared with cells that had been kept in suspension (Figure 1B). In order to examine the potential interaction between endogenous FAK and SOCS-3 proteins, anti-FAK immunoprecipitation was carried out followed by immunoblotting with anti-SOCS-3 antibodies. A coordinate signaling by integrins and PDGF resulted in an interaction between the endogenous FAK and SOCS-3 proteins in NIH 3T3 cells (Figure 1A).

Fig. 1. (A) A coordinate signaling by integrins and PDGF receptor results in SOCS expression and SOCS–FAK interaction. Serum-starved NIH 3T3 cells were detached, stimulated or not with 50 ng/ml PDGF and either kept in suspension or replated on FN-coated dishes for 1 h. Cell lysates were subjected to anti-SOCS-3 immunoblot analysis, and to anti-tubulin immunoblotting to confirm equal loading (top two panels). Cell lysates prepared as above were also subjected to immunoprecipitation analysis with anti-FAK antibody followed by immunoblot analysis with anti-SOCS-3 antibody. The same membrane was stripped and reprobed with anti-FAK antibody to confirm equal loading (bottom two panels). TCL, total cell lysate. (B) Integrin and PDGF receptor signaling enhances SOCS-1 mRNA levels. Cells were treated as above, and total RNA was isolated. The levels of SOCS-1 mRNA, and GAPDH levels as a control, were analyzed by a semi-quantitative RT–PCR analysis as described in Materials and methods.
In order to study the mechanism of SOCS-3 and FAK interaction in more detail, Myc-tagged SOCS-3 was transiently expressed in COS-7 cells, and its interaction with FAK in suspended and FN-plated cells was examined. We also examined the potential interaction between transfected, Myc-tagged SOCS-1 and FAK. As shown in Figure 2A, transfected SOCS-1 and SOCS-3 readily interacted with FAK in cells that had been plated on FN, but not in cells that had been kept in suspension. Another SOCS protein, CIS1, failed to interact with FAK in both suspended and adherent cells (Figure 2B). Thus, our results demonstrate that integrin-mediated signaling contributes to the induction of SOCS family members upon growth factor stimulation, and also regulates the interaction between FAK and exogenously expressed SOCS-1 and SOCS-3 proteins.

Fig. 2. Adhesion-dependent interaction of SOCS-1 and SOCS-3 with FAK. (A) COS-7 cells were transiently transfected with the indicated plasmids (0.2 µg of HA-FAK, 0.5 µg of Myc-SOCS-1/3). Twenty-four hours after transfection, cells were serum-starved for additional 24 h, detached, kept in suspension for 30 min (‘susp.’), and replated or not on FN for the indicated times. Alternatively, cells were lysed as a monolayer culture 48 h after transfection without prior additional manipulation (‘cult.’). Cell lysates were immunoprecipitated with anti-HA antibody, and the precipitates were analyzed by immunoblotting with antibodies against Myc, phosphotyrosine (pTyr) or HA. Total cell lysates were subjected to immunoblotting with anti-Myc antibodies to confirm SOCS-protein expression levels. (B) COS-7 cells were transiently transfected with the indicated plasmids (0.2 µg of HA-FAK, 0.5 µg of Myc-SOCS-1/3 or CIS1). Twenty-four hours after transfection, CIS1-FAK-cotransfected cells were serum-starved for additional 24 h, detached, kept in suspension for 30 min, and replated or not on FN for 1 h. FAK-SOCS-1/3-cotransfected cells were treated as described above for monolayer culture. Cell lysates were immunoprecipitated with anti-HA antibody, and the precipitates were analyzed by immunoblotting with antibodies against Myc, pTyr or HA. An aliquot of total cell lysate from CIS1-transfected cells was included as a control in the anti-Myc immunoblot of anti-HA immunoprecipitates to determine the motility of the CIS1 protein relative to the IgG band. Total cell lysates of all samples were subjected to immunoblot with anti-Myc antibody to confirm SOCS-protein expression levels.
Y397 in FAK and the KIR- and SH2-domains of SOCS proteins participate in the FAK–SOCS interaction
Previous studies have indicated that the SH2 domain and an adjacent N-terminal region named the KIR domain mediate the binding of SOCS-proteins to activated and tyrosine-phosphorylated JAK kinases (Nicholson et al., 1999; Sasaki et al., 1999; Yasukawa et al., 1999). Ligation of integrins with matrix proteins results in a rapid activation and tyrosine phosphorylation of FAK, which, as shown in Figure 2A, correlated with the binding of FAK to SOCS-1 and SOCS-3. This finding suggested that similar to JAKs, FAK may interact with SOCS proteins in an activation- and tyrosine phosphorylation-dependent manner. To investigate this further, a form of FAK in which the main autophosphorylation site, Y397, had been mutated to phenylalanine (FAK-Y397F) was transfected into COS cells and its interaction with cotransfected SOCS-1 and SOCS-3 was studied. As shown in Figure 3A, FAK-Y397F demonstrated little tyrosine phosphorylation in adherent COS cells, and in contrast to wild-type FAK, it failed to interact with cotransfected SOCS-1 and SOCS-3 proteins.


Fig. 3. Y397 in FAK regulates the FAK–SOCS interaction. (A) COS-7 cells were transiently transfected with the indicated plasmids (0.2 µg of HA-FAK or HA-FAK-Y397F, 0.5 µg of Myc-SOCS-1/3). Forty-eight hours after transfection, cell lysates were immunoprecipitated with anti-HA antibody, and the precipitates were analyzed by immunoblotting with antibodies against Myc, pTyr or HA. Total cell lysates were subjected to immunoblotting with anti-Myc antibody to confirm SOCS protein expression levels. (B) Denatured lysates of HA-FAK or HA-FAK-Y397F-transfected COS-7 cells were incubated with purified GST or with GST–SOCS-3-SH2 fusion proteins conjugated to glutathione–Sepharose beads. Associated proteins were examined by anti-HA immunoblotting (top panel). Ponceau S staining of the same blot is shown on the bottom panel. (C) GST fusion proteins coding for the N-terminal domain (amino acids 1–406) of wild-type or Y397-mutant of FAK were produced in the bacterial TK strain expressing an active tyrosine kinase. The produced proteins were separated by SDS–PAGE and transferred onto PVDF membrane and analyzed by immunoblotting with the indicated antibodies (upper left panel), or by far-western blotting with the indicated fusion proteins as described in Materials and methods (lower panel). Coomassie Blue staining of the fusion proteins used as probes is shown on the upper right panel.
That SOCS proteins would interact with a tyrosine-phosphorylated form of FAK was further supported by two additional lines of investigation. First, we found that a GST fusion protein of the SH2 domain of SOCS-3 readily precipitated the wild-type, but not the Y397F-mutant form of FAK in denatured cell lysates, while a SOCS-3 fusion protein with an inactive SH2-domain (R71E) failed to do so (Figure 3B). Second, we carried out a far-western blot analysis to further demonstrate that the FAK–SOCS interaction is dependent on the FAK-Y397 and the SH2 domain of the SOCS protein, and to explore the possibility that the two proteins might interact directly. As shown in Figure 3C, this analysis indicated that the wild-type form of SOCS-3-SH2, but not the R71E-mutant form was capable of binding to bacterially produced and tyrosine-phosphorylated FAK N-terminal fusion protein (FAK-NT, amino acids 1–406). The SH2 domain of SOCS-3 failed to associate with the Y397F mutant form of FAK-NT, suggesting that at least in vitro, the SH2-domain of SOCS-3 directly interacts with FAK in Y397-dependent manner.
We next expressed forms of SOCS-1 or SOCS-3 in which either the KIR domain (F59E and F25A mutants, respectively) or the SH2-domain (R105K and R71E mutants, respectively) had been inactivated. Similar to what has been reported previously, we found that intact KIR- and SH2-domains are necessary for JAK2 to interact with SOCS-1 and SOCS-3 (Figure 4A). When coexpressed with FAK, the mutant SOCS-proteins demonstrated a greatly reduced, but detectable interaction with FAK. Importantly, however, double KIR/SH2-mutant forms of SOCS-1 and SOCS-3 failed to interact with FAK (Figure 4B), suggesting that the KIR- and SH2-domains of SOCS-proteins mediate binding to FAK. Whether these results are reflective of a quantitative and/or qualitative difference in the mechanism by which SOCS proteins interact with JAK and FAK kinases remains to be determined.

Fig. 4. The KIR- and SH2-domains of SOCS proteins participate in SOCS–FAK interaction. (A and B) COS-7 cells were transiently transfected with indicated plasmids (0.2 µg of HA-FAK or HA-JAK2, 0.5 µg of Myc-SOCS-1/3). Forty-eight hours after transfection, cell lysates were immunoprecipitated with anti-HA antibody, and the precipitates were analyzed by immunoblotting with antibodies against Myc or HA. Total cell lysates were subjected to immunoblot with anti-Myc antibody to confirm SOCS protein expression levels. The relative FAK-SOCS interaction was determined by a densitometric analysis and the NIH Image 1.62 program. The KIR-mutant form of SOCS-1 (SOCS-1-F59EdN) and the SOCS-1 double-mutant (SOCS-1-F59E, R105K) contain a deletion of the first 51 amino acids that has been shown to stabilize the expressed protein without changing its binding affinity or specificity (Yasukawa et al., 1999).
SOCS proteins inhibit FAK-associated kinase activity in vitro, and tyrosine phosphorylation of FAK in cells
SOCS-1 and SOCS-3 are known to inhibit the catalytic activity of JAK2 by binding through the KIR and SH2-domains (Nicholson et al., 1999; Sasaki et al., 1999; Yasukawa et al., 1999);we therefore examined the effect of SOCS proteins on the catalytic activity and phosphorylation of FAK in vitro and in cells. FAK was coexpressed or not with SOCS-1 and SOCS-3 and FAK immunoprecipitates were subjected to an in vitro kinase assay. As shown in Figure 5A, the capability of immunoprecipitated FAK to catalyze its own phosphorylation or phosphorylation of the exogenous substrate poly(Glu:Tyr) was greatly reduced upon coexpression of SOCS-1 or SOCS-3. The reduced in vitro catalytic activity correlated with reduced tyrosine phosphorylation of FAK in cells. When increasing amounts of SOCS-1 and SOCS-3 proteins were expressed in COS cells, a dose-dependent decrease in the tyrosine phosphorylation of FAK was observed (Figure 5B). Importantly, the KIR- and SH2-mutant forms of SOCS-1 and SOCS-3 failed to have a significant effect on FAK tyrosine phosphorylation (Figure 5C). Thus, these studies suggest that SOCS-1 and SOCS-3 regulate FAK activity and phosphorylation both in vitro and in cells, and that this regulation correlates with the capability of the SOCS proteins to interact with FAK.


Fig. 5. SOCS-proteins inhibit FAK-associated kinase activity in vitro and tyrosine phosphorylation of FAK in cells. (A) COS-7 cells were transfected with HA-FAK (0.5 µg) with or without Myc-SOCS-1 or SOCS-3 (1.0 µg). Twenty-four hours after transfection, cells were serum-starved for an additional 24 h, lysed and the lysates were subjected to anti-HA immunoprecipitation. An in vitro kinase assay was carried out as described in Materials and methods. Immunoblotting of the anti-HA immunoprecipitates with anti-HA antibody, and immunoblotting of total cell lysates with anti-Myc antibody are shown in the bottom panels. (B and C) COS-7 cells were transiently transfected with 0.2 µg of HA-FAK with or without increasing concentrations of wild-type Myc-SOCS-1 or Myc-SOCS-3 (0, 0.2, 0.6 and 1.0 µg) (B), or 1.0 µg of the indicated SOCS mutants (C). Twenty-four hours after transfection, cells were serum-starved for an additional 24 h, lysed and the lysates were subjected to anti-HA immunoprecipitation. Equal amounts of cell lysates were immunoprecipitated with anti-HA antibody, followed by immunoblotting with anti-pTyr and anti-HA antibodies. The relative FAK phosphorylation was determined in (B) as above. Total cell lysates were subjected to immunoblot with anti-Myc antibody to confirm SOCS protein expression levels.
SOCS proteins induce proteasome-dependent degradation and SOCS box-dependent ubiquitination of FAK
As noted above, SOCS proteins have been reported to induce proteasome-dependent degradation of their target proteins. During the course of these studies, we observed that when wild-type SOCS-1 or SOCS-3 proteins were overexpressed with FAK, the amounts of FAK protein in cell lysates were reduced (see e.g. Figure 5B). Metabolic labeling and pulse–chase experiments were therefore conducted to investigate the effect of SOCS-1 and SOCS-3 on the turnover of FAK. As shown in Figure 6, the turnover of the 35S-labeled wild-type FAK protein was significantly enhanced upon coexpression of SOCS-1 and SOCS-3, while treatment of the cells with β-lactacystin, which is an irreversible proteasome inhibitor, stabilized the protein. The turnover of the FAK-Y397F mutant protein, which is incapable of interacting with SOCS-1 and SOCS-3, was not affected by the coexpression of the SOCS-proteins. These data suggest that SOCS proteins promote a proteasome-dependent degradation of FAK, and this effect correlates with the capability of the SOCS proteins to interact with FAK.

Fig. 6. SOCS-1 and SOCS-3 enhance the turnover of wild-type FAK but not of FAK-Y397F protein. (A) COS-7 cells were transfected with HA-FAK or HA-FAK-Y397F (0.5 µg) with or without Myc-SOCS-1 or Myc-SOCS-3 (1.0 µg). The transfected cells were pulse-labeled as described in Materials and methods followed by a chase for the indicated time periods. The 35S-labeled FAK proteins were immunoprecipitated with anti-HA antibody, and subjected to SDS–PAGE followed by autoradiography. Where indicated, the proteasome inhibitor β-lactacystin at 25 µM was added to the cells 1 h before labeling and maintained in the medium throughout the pulse–chase experiment. A representative experiment is shown in (A). (B) Densitometric analysis of the blots of three individual experiments was carried out by using the NIH-Image 1.62 program. The relative protein amounts at time point zero were denoted as 100.
To investigate whether SOCS-induced FAK degradation was due to SOCS-mediated ubiquitination of FAK, we cotransfected FAK with wild-type and SOCS box-deleted forms of SOCS-1 and SOCS-3. To enhance tyrosine phosphorylation and subsequent interaction of FAK with the SOCS-proteins, the cells were pretreated with the phosphatase inhibitor pervanadate for 60 min; similar treatment was performed when ubiquitination of JAK2 by SOCS-1 was studied (Ungureanu et al., 2002). In some experiments, treatment of cells with β-lactacystin was carried out to inhibit proteolytic degradation of ubiquitinated proteins. After immunoprecipitation, FAK immunocomplexes were dissociated followed by FAK reimmunoprecipitation to remove putative coimmunoprecipitated proteins from the complex. As shown in Figure 7A, little polyubiquitination of FAK was observed in the absence of coexpression of SOCS proteins. Importantly, coexpression of wild-type SOCS-1 and SOCS-3, but not their SOCS box-deleted forms, resulted in enhanced polyubiquitination of FAK, which was further augmented by pretreatment of the cells with β-lactacystin. SOCS box-dependent stimulation of FAK ubiquitination was also observed in in vitro ubiquitination assays, in which wild-type SOCS proteins but not their SOCS box-deletion mutants catalyzed polyubiquitination of either immunoprecipitated (Figure 7B) or in vitro transcribed and translated (Figure 7C) wild-type FAK protein, but not of the mutant FAK-Y397F protein.


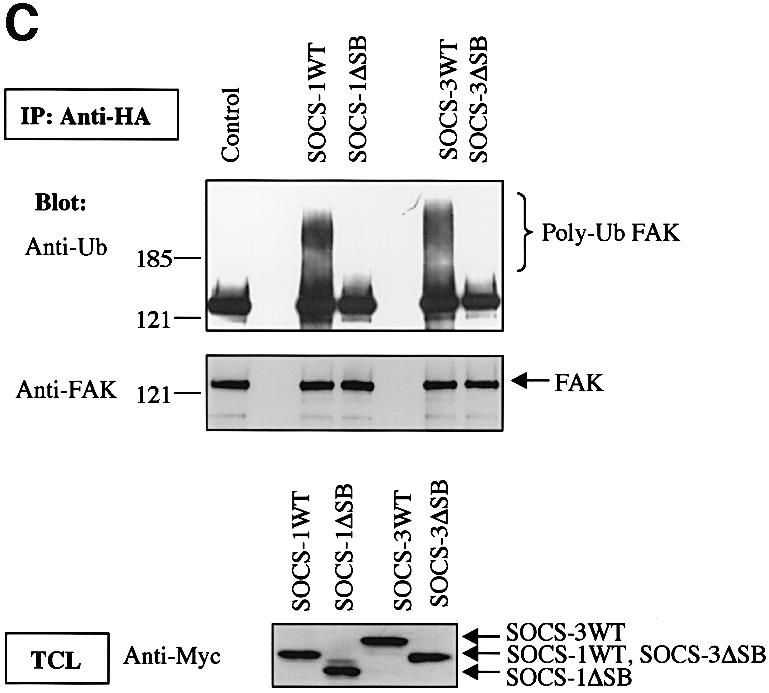

Fig. 7. SOCS-proteins promote polyubiquitination of FAK. (A) COS-7 cells were transiently transfected with HA-FAK (0.5 µg) alone or with wild-type forms of Myc-SOCS-1 or SOCS-3 or their SOCS box-deletion mutants (SOCSΔSB) (1.0 µg). Twenty-four hours after transfection the cells were serum-starved overnight and pretreated with or without β-lactacystin (25 µM) for 4 h prior to pervanadate treatment (50 µM) for 1 h. FAK was immunoprecipitated with an anti-HA antibody, followed by a dissociation and reprecipitation with the same antibody. FAK immunoprecipitates were analyzed by immunoblotting with antibodies against ubiquitin and HA. Total cell lysates were subjected to immunoblotting with anti-Myc antibody to confirm SOCS-protein expression levels. (B) In vitro ubiquitination reactions on immunoprecipitated wild-type FAK or FAK-Y397F mutant were performed as described in the Materials and methods in the presence or absence of cell lysates containing the indicated SOCS proteins. The reaction products were separated on SDS–PAGE, transferred onto PVDF membrane, and the blots were probed with anti-ubiquitin antibody (upper panel). Equal loading of the various SOCS proteins was verified by anti-Myc immunoblotting of total cell lysates (lower panel). (C) HA-tagged wild-type FAK was generated by a coupled in vitro transcription/translation system. An in vitro ubiquitination assay was performed on the Sepharose-purified FAK as in (B). (D) NIH 3T3 cells were transfected with 0.5 µg of plasmid coding for ubiquitin with or without 1.0 µg of SOCS-3ΔSB. Twenty-four hours after transfection, the cells were serum-starved overnight, detached, stimulated or not with PDGF, and either kept in suspension or replaced on FN for 1 or 4 h in the presence of 50 µM pervanadate and 25 µM proteasome inhibitor MG132. Cell lysates were subjected to immunoprecipitation with anti-FAK antibody followed by immunoblotting with antibodies against ubiquitin, FAK, pTyr, SOCS-3 and Myc (upper panels). An isotype-matched IgG was used as a control to demonstrate that anti-FAK antibodies do not unspecifically precipitate SOCS-3 (middle panel). Total cell lysates were subjected to immunoblot with anti-SOCS-3, anti-Myc and anti-tubulin antibodies (bottom three panels).
We next examined whether the endogenous FAK protein is a target for SOCS-mediated polyubiquitination. Ubiquitin-transfected 3T3 cells were serum-starved and pretreated with pervanadate and MG132 to enhance FAK tyrosine phosphorylation and to inhibit proteolytic degradation of ubiquitinated proteins, respectively. The cells were then stimulated or not with PDGF, and either kept in suspension or replated on FN. As shown in Figure 7D, simultaneous stimulation of cells by integrin ligation and PDGF resulted in polyubiquitination of the endogenous FAK protein, and this coincided with induction of SOCS-3 protein expression and interaction between endogenous FAK and SOCS-3 proteins. Importantly, exogenously expressed SOCS-3ΔSB abolished polyubiquitination of FAK in FN-plated, PDGF-stimulated cells in a dominant-negative manner. Taken together, these results demonstrate that SOCS-1 and SOCS-3 induce polyubiquitination of FAK in cells in a SOCS box-dependent manner, and that this modification likely targets FAK for proteasome-mediated degradation.
SOCS proteins inhibit FAK-induced Erk activity and cell migration
As a corollary of the above findings, we next examined whether SOCS proteins regulate FAK-dependent cellular functions. It has been demonstrated previously that exogenous expression of FAK results in activation of the Erk MAP kinase cascade (Schlaepfer and Hunter, 1997), and we utilized this assay as a biochemical read out to examine the effect of SOCS proteins on FAK signaling. Similar to what was reported previously, we found that exogenous expression of FAK induced activation of Erk as measured by anti-phospho-MAPK immunoblotting (Figure 8A). Coexpression of SOCS-1 or SOCS-3 in turn significantly inhibited the capability of FAK to activate Erk. In a control experiment, SOCS-1 and SOCS-3 had no effect on the basal phosphorylation of Erk1 in COS cells.



Fig. 8. SOCS proteins inhibit FAK-induced Erk activity and cell migration. (A) COS-7 cells were cotransfected with GST–Erk1 (1.0 µg) and HA-FAK (0.5 µg) with or without Myc-SOCS-1 or SOCS-3 (1.0 µg). Cell lysates were prepared 48 h after transfection, precipitated with anti-GST antibody, and the precipitates were analyzed by immunoblotting with anti-phospho-Erk antibody. The membrane was stripped and reprobed with anti-GST antibody to confirm equal loading. Total cell lysates were immunoblotted with antibodies against GST, HA and Myc (lower panels). Student’s t-test demonstrated P < 0.02 between Erk activity in cells transfected with HA-FAK and HA-FAK + Myc-SOCS-1/3 in three independent experiments. (B) NIH 3T3 cells were transfected with an empty pcDNA3 vector, or with HA-FAK (0.5 µg) together with or without Myc-SOCS-1 or SOCS-3 (1.0 µg). A plasmid encoding β-galactosidase (0.2 µg) was included in all transfections. Haptotactic cell migration assay on FN was carried out as described in Materials and methods. (C) NIH 3T3 cells were transfected with pcDNA3 vector, HA-FRNK (1.0 µg), or increasing amounts of Myc-SOCS-1 or SOCS-3 (0.2, 0.6 or 1.0 µg). A vector coding for β-galactosidase (0.2 µg) was included in all transfections. Cells were added to the migration chambers and the number of cells that migrating towards PDGF (10 ng/ml) was determined 3 h later as described in Materials and methods.
As an additional functional read-out, we examined the capability of SOCS-proteins to modulate FAK-dependent cell motility. FAK was transiently expressed in NIH 3T3 cells and as demonstrated before (Cary et al., 1996), its overexpression induced cell motility on FN. As shown in Figure 8B, coexpression of SOCS-1 and SOCS-3 significantly inhibited FAK-stimulated cell motility. As an additional line of investigation, we studied the capability of SOCS-proteins to modulate PDGF-induced cell motility on FN. It has been shown previously that FAK integrates PDGF receptor and integrin signals to promote cell migration (Sieg et al., 2000), and we found the same here by demonstrating that the dominant-negative molecule of FAK known as FRNK (Gilmore and Romer, 1996; Richardson and Parsons, 1996) inhibits PDGF-induced cell motility on FN (Figure 8C). Importantly, exogenous expression of SOCS-1 and SOCS-3 similarly resulted in a dose-dependent inhibition of PDGF-stimulated cell migration. Taken together, these studies demonstrate that exogenous expression of SOCS proteins results in an efficient inhibition of FAK-mediated biochemical and cell biological signaling events.
Discussion
In contrast to the rapid progress that has been made in elucidating the FAK downstream signaling pathways, relatively little is known about the mechanisms of regulation of FAK activity. Our studies in this report demonstrate that the SOCS proteins may have a previously unidentified role in providing a negative regulatory mechanism for FAK function.
The expression of SOCS proteins is induced in response to cytokine stimulation, and overexpression of these proteins results in inhibition of cytokine signaling (Naka et al., 1999; Yasukawa et al., 2000; Krebs and Hilton, 2001). Our studies demonstrate an essential role for integrin-mediated cell adhesion in contributing to the rapid upregulation of SOCS-3 protein levels by PDGF in 3T3 fibroblasts. Thus, these results identify a novel mechanism for cross-talk between integrins and receptor tyrosine kinases; it remains to be determined whether integrin ligation regulates SOCS protein expression in response to growth factor and cytokine stimulation in other cell types. The pathways downstream of integrins and PDGF receptor that lead to the induction of SOCS-3 are presently unknown.
Our studies demonstrate that integrin-mediated cell adhesion regulates the interaction between FAK and the SOCS proteins. We found that the interaction is dependent on the major autophosphorylation site, Y397, in FAK, suggesting that SOCS-1 and SOCS-3 specifically interact with and target activated FAK. We further found that the SH2-domain of SOCS-3 was able to interact with FAK in denatured cell lysates and in a far-western experiment. These results are suggestive of a direct interaction between FAK and SOCS proteins. Our studies further demonstrated that the KIR domain within the SOCS proteins also binds to FAK (or FAK protein complex), but the exact mechanism of this interaction remains to be determined.
It has been shown that autophosphorylation of FAK on Y397, subsequent binding of Src kinase to this site, and further phosphorylation of FAK in the activation loop by Src are required for full FAK activation (Calalb et al., 1995). Conceivably, binding of SOCS proteins to Y397 could prevent full activation of FAK by Src, which could result in the observed decrease in FAK kinase activity and phosphorylation upon SOCS protein expression. Alternatively (or at the same time), SOCS protein-mediated displacement of Src from FAK in and of itself could account for the decreased activity observed in the kinase assay, decreased phosphorylation of FAK in cells, and decreased activation of FAK-dependent biochemical and biological signaling events. Previous studies have demonstrated that FAK acts as a docking protein to recruit Src to phosphorylate intracellular substrate proteins (Ruest et al., 2001), and in many cell systems, it is the FAK–Src dual-kinase complex, rather than FAK activity per se, that is functionally significant (Schlaepfer et al., 1999).
Our studies do not preclude the possibility that SOCS-proteins could interact with other sites in FAK, which become phosphorylated in a Y397-dependent manner in vivo. Along these lines, tyrosine phosphorylation of FAK at Y397 is required for Src-mediated phosphorylation of Y576/577 in the activation loop of FAK (Calalb et al., 1995). Interestingly, SOCS-1 and SOCS-3 have been shown to bind to a phosphorylated Y1007 in the activation loop of JAK2 and inhibit its kinase activity (Nicholson et al., 1999; Sasaki et al., 1999; Yasukawa et al., 1999); a similar mechanism of binding and inhibition could apply to FAK. Additionally, Y397-dependent phosphorylation of FAK is known to result in binding of FAK to various SH2-domain containing proteins (Schlaepfer et al., 1999; Schaller, 2001). Some of these proteins, such as Grb2, have been shown to interact with SOCS-proteins (De Sepulveda et al., 1999) and could therefore function as a bridge between FAK and SOCS proteins.
Our results indicate that SOCS proteins not only regulate tyrosine phosphorylation of FAK, but they also promote its proteasome-mediated degradation. To our knowledge, these studies are the first to demonstrate that FAK is a target for polyubiquitination-induced degradation. Our functional studies indicated that exogenous expression of SOCS proteins results in an efficient downregulation of FAK-mediated cellular functions, such as cell motility; whether this downregulation requires both the kinase inhibitory and the polyubiquitination activity of SOCS proteins remains to be determined. At present, little is known about the molecular mechanisms that regulate FAK function in vivo. A novel protein inhibitor FIP200 has been recently shown to bind to the kinase domain of FAK and to inhibit its kinase activity upon coexpression (Abbi et al., 2002). Numerous phosphatases have been suggested as regulators of FAK, and FAK has also been shown to be a target for proteolytic cleavage by caspases and calpain (Schlaepfer et al., 1999; Schaller, 2001). Whether cleavage of FAK by these proteases simply terminates signaling or generates fragments that transmit aberrant signals remains to be determined.
In addition to integrin-mediated cell adhesion, a number of other extracellular stimuli are known to induce activation and tyrosine phosphorylation of FAK (Schlaepfer et al., 1999; Schaller, 2001). Interestingly, several of these stimuli, such as treatment of cells with prolactin, also induce SOCS protein expression (Naka et al., 1999; Yasukawa et al., 2000; Krebs and Hilton, 2001). FAK may therefore be a target for regulation by SOCS proteins in other signaling pathways, as well. Our preliminary studies indicate that signaling by the FAK-family member Pyk2 is also suppressed by SOCS proteins (data not shown). Pyk2 becomes activated by a number of cytokines that are known to regulate SOCS proteins, such as tumor necrosis factor-α (TNF-α) and interferon-γ (IFN-γ) (Schlaepfer et al., 1999; Avraham et al., 2000). Of interest, Pyk2 has been suggested to be an important mediator of IFN-γ signaling (Takaoka et al., 1999), and to mediate TNF-α-induced apoptosis (Avdi et al., 2001). In this context, the analysis of mice lacking SOCS-1 has revealed that SOCS-1 is critical in the negative regulation of IFN-γ and in suppressing TNF-α-induced cell death in fibroblasts (Metcalf, 1999; Morita et al., 2000). Additional future research is needed to clarify the physiological significance of FAK family proteins and their interactions with SOCS proteins in various signaling pathways.
Materials and methods
Cell culture and treatment
COS-7 and NIH 3T3 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM/l glutamine, 100 U/ml penicillin and streptomycin. For cell adhesion experiments, COS-7 cells were serum-starved for 24 h. The cells were detached, maintained in suspension in DMEM/0.5% bovine serum albumin (BSA) at 37°C for 30 min, and then either kept in suspension for 30 min, or replated on FN-coated dishes (10 µg/ml) for the indicated time periods. When studying SOCS-3 expression (Figure 1), NIH 3T3 cells were serum-starved for 24 h in DMEM containing 1% FBS. The cells were detached, stimulated or not with 50 ng/ml hPDGF-BB (Roche Molecular Biochemicals), and either kept in suspension or replated on FN-coated dishes for 1 h. For ubiquitination experiments, cells were pretreated with 50 µM pervanadate for 60 min before analysis. Where indicated, the proteasome inhibitors MG132 or β-lactacystin (Sigma) at 25 µM were added to the cells 4 h before pervanadate treatment and maintained throughout the experiment.
Plasmids and transient transfections
Hemagglutinin (HA) epitope-tagged wild-type FAK and its autophosphorylation site mutant FAK-Y397F in the pcDNA3 vector were obtained from Dr David Schlaepfer. Myc-tagged wild-type SOCS-1, SOCS-3, CIS-1 and their single SH2- and KIR-domain mutants were kind gifts from Dr Akihiko Yoshimura. The SOCS-1 SH2-KIR-double mutant was generated by ligation of a 424 bp XcmI–XbaI fragment of SOCS-1-R105K, which contained the R105K mutation in the SH2 domain, to XcmI–XbaI-digested SOCS-1-F59EdN. The SOCS-3 SH2-KIR-double mutant was generated by creating a F25A mutation in the KIR domain of the SOCS-3-R71E construct by using QuikChange Site-Directed Mutagenesis Kit (Stratagene). HA-JAK2 was obtained from Dr Olli Silvennoinen, GST–Erk1 from Dr Gen-Sheng Feng, HA-FRNK from Dr Jun-Lin Guan, and HA-ubiquitin from Dr Dirk Bohmann. The plasmid pCMV-β-Gal coding for β-galactosidase was used in all migration assays to monitor the transfection efficiency. Transient transfection experiments were carried out in serum-free medium with the indicated amounts of plasmids by using Lipofectamine (Invitrogen).
Immunoprecipitations and immunoblot analysis
Cells were washed twice with ice-cold phosphate-buffered saline (PBS) and lysed in a modified radioimmunoprecipitation assay (RIPA) lysis buffer [20 mM HEPES pH 7.3, 1% NP-40, 0.1% SDS, 150 mM NaCl, 50 mM NaF, 10 mM sodium pyrophosphate, 0.5% sodium deoxycholate, 1 mM Na3VO4, 1 mM PMSF and Protease Inhibitor Cocktail (Roche)]. Clarified cell extracts were immunoprecipitated with the indicated antibodies followed by SDS–PAGE analysis and immunoblotting coupled with enhanced chemiluminescence detection (ECL). For re-immunoprecipitation experiments, FAK immunecomplexes were eluted by boiling in 2% SDS, 150 mM NaCl for 3 min, and diluted to a final concentration of 0.1% SDS using lysis buffer and re-immunoprecipitated with the anti-HA antibody. The following antibodies were used in this study: anti-SOCS-3 (Immuno-Biological Laboratories, Japan), anti-pTyr (Transduction Laboratories), anti-FAK-pY397 (Biosource International), anti-FAK (N17, C-20), anti-Myc, anti-GST, anti-HA (all from Santa Cruz Biotechnology), anti-ubiquitin (Zymed), anti-phospho-MAPK (Cell Signaling Technology), and anti-Tubulin (Calbiochem).
SH2 domain binding assay
In vitro association experiments were performed with the GST fusion proteins of SOCS-3 covering the SH2-domain (amino acids V34–L182) with or without an R71E point mutation. The constructs were generated by PCR and subcloned to pGEX-4T-1 (Amersham). The fusion proteins were expressed in Escherichia coli and purified as described previously (Vuori et al., 1996). Four hundred micrograms of precleared cell lysate was incubated for 2 h at 4°C with 5 µg of GST alone or of the GST fusion proteins which had been immobilized on glutathione–Sepharose beads. The beads were collected and washed extensively with RIPA lysis buffer and the bound proteins were released by boiling in sample buffer and then subjected to SDS–PAGE followed by immunoblot analysis.
Far-western blotting
Oligonucleotides (5′-CTCGGATCCATGGCAGCTTATCTTG-3′ and 5′-GTTCTCGAGTGTCTTCCTCATCG-3′) were used to amplify sequences coding for amino acids 1–406 of murine wild-type FAK and FAK-Y397F to generate GST–FAK-NT (WT) and GST–FAK-NT (Y397F) constructs in pGEX-4T-1, respectively. The constructs were transformed into the TKX1 strain (Stratagene), which harbors a plasmid-encoded, inducible tyrosine kinase gene. The induction of protein expression was carried out according to the manufacturer’s protocols. Glutathione Sepharose-purified FAK-NT (WT) and FAK-NT (Y397F) were run on SDS–PAGE and transferred onto PVDF membrane. The membranes were blocked with binding buffer (10 mM Tris–HCl pH 7.4, 1 mM EDTA, 150 mM NaCl, 1 mM DTT and 0.5% BSA) for 2 h. The membranes were probed for 4 h at room temperature in the binding buffer with 1 µg/ml EZ-Link™ Sulfo-NHS-LC-Biotin (Pierce)-labeled GST–SH2 fusion proteins of either Src (Vuori et al., 1996), SOCS-3 or SOCS-3-R71E. The blots were washed four times with binding buffer and developed with peroxidase-conjugated ExtrAvidin (Sigma) and ECL.
RT–PCR
NIH 3T3 cells were washed twice with ice-cold PBS, and lysed in Trizol (Invitrogen). Total RNA was isolated as per manufacturer’s instructions and subjected to reverse transcription using Superscript II Reverse Transcriptase (Invitrogen). Primers used for SOCS-1 amplification were 5′-ACACTCACTTCCGCACCTTC-3′ and 5′-GAAGCCATCTTCACG CTGAG-3′. Primers used for GAPDH amplification were 5′-TTC ATTGACCTCAACTACATGG-3′ and 5′-GTGGCAGTGATGGCAT GGAC-3′. PCRs were carried out with Expand High Fidelity enzyme (Roche) (30 s denaturing at 94°C, 30 s annealing at 55°C and 30 s extension at 72°C). The linearity of the reactions was verified by removing sample aliquots after 25, 30 and 35 cycles.
In vitro kinase assay
HA-tagged FAK was immunoprecipitated from transiently transfected COS-7 cells and the immunocomplexes were washed extensively in kinase assay buffer (20 mM HEPES pH 7.4, 10 mM MnCl2, 100 µM vanadate, 0.05% Triton X-100), and then incubated in kinase assay buffer containing 0.25 µCi/µl of [γ-32P]ATP (6000 Ci/mmol; DuPont-NEN) for 15 min at 30°C. In some experiments, 20 µg of poly(Glu:Tyr) (4:1; Sigma) was added to FAK immunoprecipitates as an exogenous substrate, and kinase reactions were initiated by ATP addition (final concentrations: 3 µM ATP, 3 µCi/sample of [γ-32P]ATP). The reactions were stopped by addition of an equal volume of 2× SDS–PAGE sample buffer, and analyzed by SDS–PAGE followed by autoradiography.
Pulse–chase experiments
Twenty-five micromolar β-lactacystin was added or not to transiently transfected COS-7 cells 1 h prior to cell manipulations where indicated and maintained throughout the experiment. The cells were then starved in methionine- and cysteine-free DMEM (Gibco-BRL) for 15 min, pulsed with medium containing 100 µCi/ml [35S]methionine and [35S]cysteine (Promix; Amersham) for 30 min, and chased for 1, 2 or 4 h. Equivalent amounts of protein extracts, as determined by the Bradford assay, were immunoprecipitated with anti-HA antibody to recover FAK. The immunoprecipitates were separated on SDS–PAGE, and the gels were treated with Amplify (Amersham), dried and visualized by autoradiography.
In vitro ubiquitination assay
HA-tagged wild-type FAK and FAK-Y397F from either transfected COS-7 cells, or HA-tagged wild-type FAK from a coupled in vitro transcription/translation reaction (TnT, Promega) were immunoprecipitated with anti-HA antibody and purified by using protein A-Sepharose beads. After washing extensively with lysis buffer, the beads were resuspended in 50 µl reaction buffer containing 20 mM HEPES pH 7.3, 10 mM MgCl2, 1 mM DTT, 2 mM ATP, 25 mM MG132, 5 µg/ml ubiquitin, 20 µg/ml ubiquitin aldehyde and 100 µg of lysates from COS-7 cells transfected with either wild-type SOCS-1, SOCS-1ΔSB, wild-type SOCS-3, SOCS-3ΔSB or a control vector. Five microliters of crude rabbit reticulocyte lysates were added to each reaction. To deplete endogenous ATP, 1 mg/ml of hexokinase and 20 mM 2-deoxyglucose were also added. The reaction mixture was incubated for 1.5 h at 30°C, the Sepharose beads were washed extensively with lysis buffer containing 25 mM MG132, protease and phosphatase inhibitors, resuspended in SDS loading buffer, separated on SDS–PAGE and transferred onto PVDF membrane. The blots were probed with anti-ubiquitin antibody.
Migration assays
Cell migration assays were performed using 24-well Transwell chambers (Costar). In haptotactic migration assays, the underside of the membrane was coated with 10 µg/ml FN in PBS for 2 h at 37°C and blocked with 0.5% BSA in PBS. In chemotactic migration assays, both sides of the membrane were coated with FN, and 10 ng/ml hPDGF-BB was added to the bottom chamber. Serum-starved cells were detached with Hanks’ balanced salt solution containing 5 mM EDTA, 25 mM HEPES, pH 7.2, and 0.01% trypsin, washed twice with migration buffer (fibroblast basal medium with 0.5% BSA), and resuspended in the same buffer at 106 cells/ml. Cells (1 × 105) were added to the upper migration chamber and allowed to migrate for 3 h at 37°C, the non-migratory cells on the upper membrane surface were removed with a cotton tip, and the migratory cells attached to the lower membrane surface were fixed and analyzed for β-galactosidase activity using X-gal as a substrate. Cell migration values were determined by counting stained cells (cells/field using a 40× objective). Each determination represents the average of three individual wells, and error bars represent the standard deviation. Background values of cell migration to 0.5% BSA were <1% of values obtained with FN. Data presented are representative of three independent experiments.
Acknowledgments
Acknowledgements
We are grateful to Drs Dirk Bohmann, Gen-Sheng Feng, Jun-Lin Guan, David Schlaepfer, Olli Silvennoinen and Akihiko Yoshimura for providing reagents used in this study. We thank Kathy Becherer and Eric Lau for technical assistance, and the members of the Vuori lab for useful discussions. This work was supported by grants from the National Institutes of Health (to K.V.). J.-F.C. is a recipient of a Post-Doctoral Fellowship from the Canadian Institute of Health Research.
References
- Abbi S., Ueda,H., Zheng,C., Cooper,L.A., Zhao,J., Christopher,R. and Guan,J.L. (2002) Regulation of focal adhesion kinase by a novel protein inhibitor FIP200. Mol. Biol. Cell, 13, 3178–3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avdi N.J., Nick,J.A., Whitlock,B.B., Billstrom,M.A., Henson,P.M., Johnson,G.L. and Worthen,G.S. (2001) Tumor necrosis factor-α activation of the c-Jun N-terminal kinase pathway in human neutrophils. Integrin involvement in a pathway leading from cytoplasmic tyrosine kinases apoptosis. J. Biol. Chem., 276, 2189–2199. [DOI] [PubMed] [Google Scholar]
- Avraham H., Park,S.Y., Schinkmann,K. and Avraham,S. (2000) RAFTK/Pyk2-mediated cellular signalling. Cell. Signal., 12, 123–133. [DOI] [PubMed] [Google Scholar]
- Cacalano N.A., Sanden,D. and Johnston,J.A. (2001) Tyrosine-phosphorylated SOCS-3 inhibits STAT activation but binds to p120 RasGAP and activates Ras. Nat. Cell Biol., 3, 460–465. [DOI] [PubMed] [Google Scholar]
- Calalb M.B., Polte,T.R. and Hanks,S.K. (1995) Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: a role for Src family kinases. Mol. Cell. Biol., 15, 954–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cary L.A., Chang,J.F. and Guan,J.L. (1996) Stimulation of cell migration by overexpression of focal adhesion kinase and its association with Src and Fyn. J. Cell Sci., 109, 1787–1794. [DOI] [PubMed] [Google Scholar]
- De Sepulveda P., Okkenhaug,K., Rose,J.L., Hawley,R.G., Dubreuil,P. and Rottapel,R. (1999) Socs1 binds to multiple signalling proteins and suppresses steel factor-dependent proliferation. EMBO J., 18, 904–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Sepulveda P., Ilangumaran,S. and Rottapel,R. (2000) Suppressor of cytokine signaling-1 inhibits VAV function through protein degradation. J. Biol. Chem., 275, 14005–14008. [DOI] [PubMed] [Google Scholar]
- Frantsve J., Schwaller,J., Sternberg,D.W., Kutok,J. and Gilliland,D.G. (2001) Socs-1 inhibits TEL-JAK2-mediated transformation of hematopoietic cells through inhibition of JAK2 kinase activity and induction of proteasome-mediated degradation. Mol. Cell. Biol., 21, 3547–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch S.M., Vuori,K., Ruoslahti,E. and Chan-Hui,P.Y. (1996) Control of adhesion-dependent cell survival by focal adhesion kinase. J. Cell Biol., 134, 793–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore A.P. and Romer,L.H. (1996) Inhibition of focal adhesion kinase (FAK) signaling in focal adhesions decreases cell motility and proliferation. Mol. Biol. Cell, 7, 1209–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanks S.K. and Polte,T.R. (1997) Signaling through focal adhesion kinase. BioEssays, 19, 137–145. [DOI] [PubMed] [Google Scholar]
- Ilic D. et al. (1995) Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature, 377, 539–544. [DOI] [PubMed] [Google Scholar]
- Ilic D., Almeida,E.A., Schlaepfer,D.D., Dazin,P., Aizawa,S. and Damsky,C.H. (1998) Extracellular matrix survival signals transduced by focal adhesion kinase suppress p53-mediated apoptosis. J. Cell Biol., 143, 547–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivan M. and Kaelin,W.G.,Jr (2001) The von Hippel-Lindau tumor suppressor protein. Curr. Opin. Genet. Dev., 11, 27–34. [DOI] [PubMed] [Google Scholar]
- Jones R.J., Brunton,V.G. and Frame,M.C. (2000) Adhesion-linked kinases in cancer; emphasis on src, focal adhesion kinase and PI 3-kinase. Eur. J. Cancer, 36, 1595–1606. [DOI] [PubMed] [Google Scholar]
- Kamizono S. et al. (2001) The SOCS box of SOCS-1 accelerates ubiquitin-dependent proteolysis of TEL-JAK2. J. Biol. Chem., 276, 12530–12538. [DOI] [PubMed] [Google Scholar]
- Kamura T., Sato,S., Haque,D., Liu,L., Kaelin,W.G.,Jr, Conaway,R.C. and Conaway,J.W. (1998) The Elongin BC complex interacts with the conserved SOCS-box motif present in members of the SOCS, ras, WD-40 repeat and ankyrin repeat families. Genes Dev., 12, 3872–3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs D.L. and Hilton,D.J. (2001) SOCS proteins: negative regulators of cytokine signaling. Stem Cells, 19, 378–387. [DOI] [PubMed] [Google Scholar]
- Masuhara M. et al. (1997) Cloning and characterization of novel CIS family genes. Biochem. Biophys. Res. Commun., 239, 439–446. [DOI] [PubMed] [Google Scholar]
- Metcalf D. (1999) The SOCS-1 story. Exp. Hematol., 27, 1715–1723. [DOI] [PubMed] [Google Scholar]
- Morita Y., Naka,T., Kawazoe,Y., Fujimoto,M., Narazaki,M., Nakagawa,R., Fukuyama,H., Nagata,S. and Kishimoto,T. (2000) Signals transducers and activators of transcription (STAT)-induced STAT inhibitor-1 (SSI-1)/suppressor of cytokine signaling-1 (SOCS-1) suppresses tumor necrosis factor α-induced cell death in fibroblasts. Proc. Natl Acad. Sci. USA, 97, 5405–5410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naka T., Fujimoto,M. and Kishimoto,T. (1999) Negative regulation of cytokine signaling: STAT-induced STAT inhibitor. Trends Biochem. Sci., 24, 394–398. [DOI] [PubMed] [Google Scholar]
- Nicholson S.E. et al. (1999) Mutational analyses of the SOCS proteins suggest a dual domain requirement but distinct mechanisms for inhibition of LIF and IL-6 signal transduction. EMBO J., 18, 375–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen J.D., Ruest,P.J., Fry,D.W. and Hanks,S.K. (1999) Induced focal adhesion kinase (FAK) expression in FAK-null cells enhances cell spreading and migration requiring both auto- and activation loop phosphorylation sites and inhibits adhesion-dependent tyrosine phosphorylation of Pyk2. Mol. Cell. Biol., 19, 4806–4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson A. and Parsons,T. (1996) A mechanism for regulation of the adhesion-associated proteintyrosine kinase pp125FAK. Nature, 380, 538–540. [DOI] [PubMed] [Google Scholar]
- Ruest P.J., Shin,N.Y., Polte,T.R., Zhang,X. and Hanks,S.K. (2001) Mechanisms of CAS substrate domain tyrosine phosphorylation by FAK and Src. Mol. Cell. Biol., 21, 7641–7652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki A., Yasukawa,H., Suzuki,A., Kamizono,S., Syoda,T., Kinjyo,I., Sasaki,M., Johnston,J.A. and Yoshimura,A. (1999) Cytokine-inducible SH2 protein-3 (CIS3/SOCS3) inhibits Janus tyrosine kinase by binding through the N-terminal kinase inhibitory region as well as SH2 domain. Genes Cells, 4, 339–351. [DOI] [PubMed] [Google Scholar]
- Schaller M.D. (2001) Biochemical signals and biological responses elicited by the focal adhesion kinase. Biochim. Biophys. Acta, 1540, 1–21. [DOI] [PubMed] [Google Scholar]
- Schlaepfer D.D. and Hunter,T. (1997) Focal adhesion kinase overexpression enhances ras-dependent integrin signaling to ERK2/mitogen-activated protein kinase through interactions with and activation of c-Src. J. Biol. Chem., 272, 13189–13195. [DOI] [PubMed] [Google Scholar]
- Schlaepfer D.D., Hauck,C.R. and Sieg,D.J. (1999) Signaling through focal adhesion kinase. Prog. Biophys. Mol. Biol., 71, 435–478. [DOI] [PubMed] [Google Scholar]
- Sieg D.J., Ilic,D., Jones,K.C., Damsky,C.H., Hunter,T. and Schlaepfer,D.D. (1998) Pyk2 and Src-family protein-tyrosine kinases compensate for the loss of FAK in fibronectin-stimulated signaling events but Pyk2 does not fully function to enhance FAK- cell migration. EMBO J., 17, 5933–5947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieg D.J., Hauck,C.R., Ilic,D., Klingbeil,C.K., Schaefer,E., Damsky,C.H. and Schlaepfer,D.D. (2000) FAK integrates growth-factor and integrin signals to promote cell migration. Nat. Cell Biol., 2, 249–256. [DOI] [PubMed] [Google Scholar]
- Takaoka A. et al. (1999) Protein tyrosine kinase Pyk2 mediates the Jak-dependent activation of MAPK and Stat1 in IFN-γ, but not IFN-α, signaling. EMBO J., 18, 2480–2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungureanu D., Saharinen,P., Junttila,I., Hilton,D.J. and Silvennoinen,O. (2002) Regulation of Jak2 through the ubiquitin-proteasome pathway involves phosphorylation of Jak2 on Y1007 and interaction with SOCS-1. Mol. Cell. Biol., 22, 3316–3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuori K., Hirai,H., Aizawa,S. and Ruoslahti,E. (1996) Induction of p130cas signaling complex formation upon integrin-mediated cell adhesion: a role for Src family kinases. Mol. Cell. Biol., 16, 2606–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasukawa H. et al. (1999) The JAK-binding protein JAB inhibits Janus tyrosine kinase activity through binding in the activation loop. EMBO J., 18, 1309–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasukawa H., Sasaki,A. and Yoshimura,A. (2000) Negative regulation of cytokine signaling pathways. Annu. Rev. Immunol., 18, 143–164. [DOI] [PubMed] [Google Scholar]
- Zhang J.G., et al. (1999) The conserved SOCS box motif in suppressors of cytokine signaling binds to elongins B and C and may couple bound proteins to proteasomal degradation. Proc. Natl Acad. Sci. USA, 96, 2071–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]