

Summary
Carbon monoxide (CO) inhalation often leads to cardiac dysfunction, dysrhythmias, ischemia, infarction, and death. However, the underlying mechanism of CO toxicity is poorly understood. We hypothesize that inhaled CO interrupts myocardial oxidative phosphorylation by decreasing the activity of myocardial cytochrome oxidase (CcOX), the terminal oxidase of the electron transport chain. Male C57Bl6 mice were exposed to either 1000ppm (0.1%) CO or air for 3 hours. Cardiac ventricles were harvested and mitochondria were isolated. CcOX kinetics and heme aa3 content were measured. Vmax, Km, and turnover number were determined. Levels of CcOX subunit I message and protein were evaluated. Carboxyhemoglobin (COHb) levels were measured and tissue hypoxia was assessed with immunohistochemistry for pimonidazole hydrochloride. CO significantly decreased myocardial CcOX activity and Vmax without altering Km. Heme aa3 content and CcOX I protein levels significantly decreased following CO exposure while enzyme turnover number and CcOX I mRNA levels remained unchanged. CO exposure increased COHb levels without evidence of tissue hypoxia as compared to sham and hypoxic controls. Decreased CcOX activity following CO inhalation was likely due to decreased heme aa3 and CcOX subunit I content. Importantly, myocardial CcOX impairment could underlie CO induced cardiac dysfunction.
Keywords: cytochrome oxidase, carbon monoxide, heart, inhibition
INTRODUCTION
Carbon monoxide (CO) is the most common cause of lethal poisoning worldwide [1]. CO, a colorless, odorless and nonirritant gas, is responsible for over 10,000 deaths annually in the United States [2, 3]. CO exposure can cause cardiac dysfunction, dysrhythmias, myocardial ischemia, myocardial infarction, and cardiac arrest [4, 5]. Despite these potentially fatal consequences, the underlying mechanism of CO induced myocardial toxicity is poorly understood.
The most widely accepted mechanism of systemic CO toxicity is tissue hypoxia [1]. Because CO binds avidly and causes a conformational change in hemoglobin, it can reduce circulating oxygen content and impair tissue oxygen delivery [3]. However, this mechanism does not fully correlate with clinical response to CO exposure [6]. Thus, other mechanisms have been proposed. These include direct cellular damage by CO and binding to cellular proteins such as myoglobin and cytochrome oxidase [6, 7].
Cytochrome oxidase (CcOX) is the terminal enzyme in the mitochondrial electron transport chain [8]. CcOX subunit I, encoded by mitochondrial DNA, houses the heme aa3 binuclear center and is the active site of the enzyme [9]. CcOX catalyzes the reduction of molecular oxygen to water and couples this with proton translocation across the inner mitochondrial membrane [8]. Proton translocation is crucial in maintaining the motive force used by ATPase synthase to produce ATP [8].
Although prior work has shown that CO is capable of binding to CcOX, in vivo CcOX inhibition by CO has not been demonstrated [6]. We hypothesize that CO decreases myocardial CcOX activity, potentially interrupting oxidative phosphorylation and impairing cellular ATP production. Specifically, in the studies detailed here we evaluate the effects of inhaled CO on the ability of myocardial CcOX to oxidize its primary substrate, cytochrome c.
MATERIALS AND METHODS
CO Exposure
Experiments were approved by the Institutional Animal Care and Use Committee of The Children’s Hospital of Philadelphia, and all animals received care in compliance with the guidelines of the National Institute of Health. Six to eight week old male C57Bl/6 mice (Charles River Laboratories, Boston, MA) were individually placed in an airtight 7 liter Plexiglas chamber. Mice were exposed to 1000 parts per million (0.1%) of CO in air for 3 hours. The concentration of CO was chosen based on prior work [10]. Exposure to 3000 ppm CO was lethal, causing rapid acidosis, a profound reduction in blood pressure, and asystole. However, exposure to 1000 ppm CO was non-lethal and well tolerated resulting in moderate elevations of carboxyhemoglobin. Sham mice were exposed to compressed air for three hours. N = 5 per group per experiment. Following three hours of exposure, animals were immediately killed with 50 mg/kg of intraperitoneal pentobarbital.
CcOX Steady-state Kinetic Activity
Following euthanasia with intraperitoneal pentobarbital (50 mg/kg), the heart was excised; cardiac ventricles were washed out with injected saline and immediately placed in iced phosphate buffered saline. Ventricles were homogenized in H medium (70 mM sucrose, 220 mM mannitol, 2.5 mM Hepes, pH 7.4 and 2 mM EDTA). Mitochondria were isolated by differential centrifugation and protein concentration determined [11]. CcOX kinetics were assayed by the method of Smith in which the rate of oxidation of ferrocytochrome c is measured by following the decrease in absorbance at 550 nm [12, 13]. Assays were executed in a 1-mL reaction volume containing 50 mM PO4−2 (pH 7.0), 2% lauryl maltoside, and 1 μg of mitochondrial protein. Ferrocytochrome c was added at concentrations of 80, 40, 20, 10, 5, and 2 mM to initiate the reaction. First-order rate constants were calculated from mean values of three to four measurements at each ferrocytochrome c concentration. Specific activities were calculated using 21.1 mM−1 cm−1 as the extinction coefficient of ferrocytochrome c at 550 nm and Lineweaver-Burk plots were constructed. Vmax and Km were determined by nonlinear regression.
Measurement of Mitochondrial Heme aa3 Content
Total heme content of isolated mitochondria dispersed in 10% lauryl maltoside was determined using the extinction coefficient of 24 nM−1.cm−1 for the difference in spectra: reduced (605 nm) minus oxidized (630 nm) [11]. Turnover number (TN), which represents moles of substrate converted to product per unit time, is calculated by dividing CcOX specific activity by heme aa3 content [11].
Northern Blot Hybridization of CcOX subunit I
Total RNA was extracted from cardiac ventricles using the method of Chomczynski and Sacchi [14]. As previously described, 8 μg of denatured RNA were resolved by electrophoresis on 1.5% formaldehyde-containing agarose gels. Gels were transferred to nylon membrane and hybridized against purified, double-stranded 32P-labeled, CcOX subunit I cDNA probes. Autoradiography and densitometry were performed, and data were normalized to the density of the 18S ribosomal subunit.
Protein Immunoblotting
10 μg samples of mitochondrial protein were subjected to SDS-acrylamide gel electrophoresis and immunoblotting as previously described [12, 15]. Blots were labeled with a primary polyclonal antibody to mouse CcOX subunit I (Molecular Probes, Eugene, OR) and then were exposed to a rabbit antimouse IgG (Santa Cruz Biotechnology, Santa Cruz, CA). The signal was detected by enhanced chemiluminescence (ECL; Amersham Pharmacia Biotech, Piscataway, NJ) and scanning densitometry was used to measure density.
Carboxyhemoglobin Levels and Evaluation for Tissue Hypoxia
Following injection of intraperitoneal pentobarbital (50mg/kg) but before the mice became apneic, 200 μL of blood were obtained via intracardiac puncture and carboxyhemoglobin (COHb) levels were measured (Rapidlab 1265, Bayer Healthcare).
In a separate cohort of animals, pimonidazole hydrochloride (60 mg/kg) (Hypoxyprobe-1; Chemicon International Inc., Temucula, CA), a 2-nitroimidazole hypoxia marker, was injected into the peritoneal cavity of CO exposed and sham animals at the end of the 3 hour gas exposure. Mice exposed to 100% CO were used as hypoxic controls. Twenty minutes after injection, animals were killed with intraperitoneal pentobarbital (50 mg/kg), the heart was excised, and cardiac ventricles immediately placed in iced 10% formalin. Hearts were fixed in 10% formalin at 4°C overnight, paraffin embedded, and cut into 6 μM thick sections. Immunohistochemistry was performed with a primary monoclonal antibody to pimonidazole (Hypoxyprobe-1Mab1, Chemicon International Inc., Temucula, CA) followed by exposure to Texas red horse antimouse IgG (Vector Laboratories Inc. Burlingame, CA). A separate set of sections were exposed only to Texas red IgG for control. Fluorescence microscopy at 20X was performed using an inverted DM IRE2 HC fluo TCS 1-B-UV microscope coupled to a Leica TCS SP2 spectral confocal system/UV) (Leica Nussloch, Germany). Images were obtained at 596 nm excitation with emission at 620 nm.
Statistical analysis
Five animals were evaluated per group per experiment. Statistical significance was determined by using Student’s t-test with significance set at p < .05. Data are presented as the mean ± standard deviation.
RESULTS
CO Decreased Myocardial CcOX Activity
Exposure to 1000 ppm CO significantly reduced myocardial CcOX activity relative to sham exposure as demonstrated by a significantly increased slope in the Lineweaver-Burk plot (Fig. 1). CcOX maximum velocity (Vmax) decreased significantly to 33 +/− 5 μmoles cytochrome c·min−1·mg protein−1 following CO exposure compared to 44 +/− 8 μmoles cytochrome c·min−1·mg protein−1 following sham exposure (P < .03). Km, however, remained unchanged (41 +/− 11 μmoles cytochrome c following CO exposure versus 37 +/− 15 μmoles cytochrome c following sham exposure).
Figure 1. Lineweaver-Burk plot of myocardial CcOX kinetic activity.
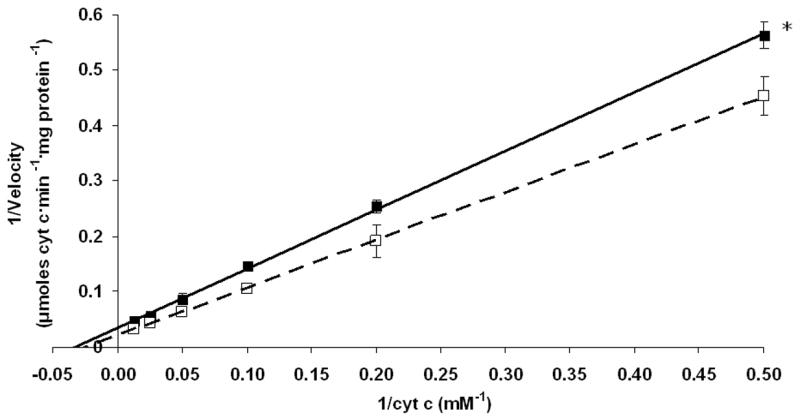
The plot depicts CcOX kinetic activity following exposure to 1000 ppm CO (closed squares with solid line) or compressed air (open squares with dashed line). Lines indicate best-fit linear regression for each data set: x- intercept represents -1/Km; y- intercept represents 1/Vmax. *p < 0.01.
CO Exposure Decreases Heme aa3 Content
Because CO can bind to the heme binuclear center within the active site of CcOX, we next measured heme aa3 content in isolated cardiac mitochondria. Relative to sham controls, CO exposure induced a significant, 50% decrease in heme aa3 levels (Fig. 2). However, because both myocardial CcOX activity and heme content were proportionately reduced following CO exposure, the calculated turnover number was statistically unchanged (1353 +/− 135 seconds−1 following CO exposure versus 1227 +/− 191 seconds−1 following sham exposure).
Figure 2. Myocardial heme aa3 content.
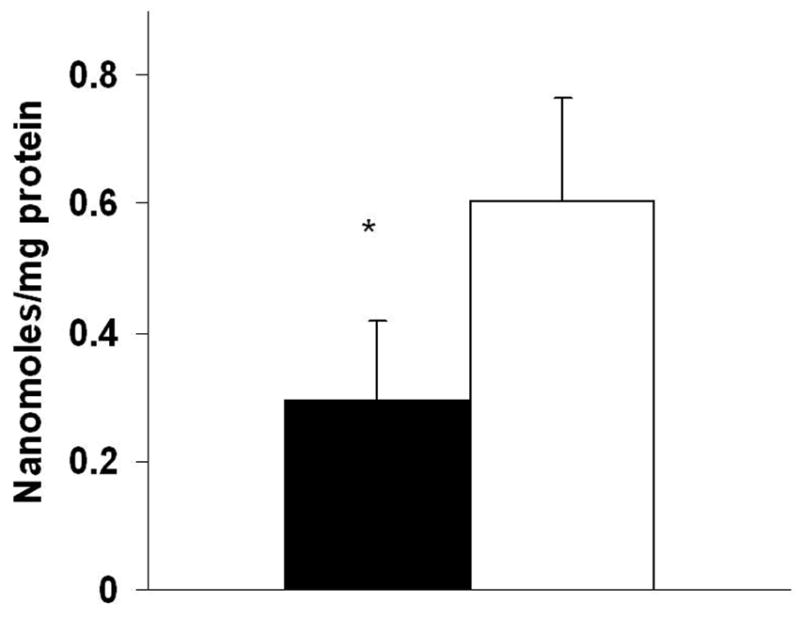
Closed bars = CO exposed mice; Open bars = sham mice. Data are mean +/− st dev. *p < 0.05.
CO Decreases Steady-state Levels of CcOX Subunit I Protein without Affecting Steady State mRNA Levels
CO induced inhibition of CcOX activity could result from a decrease in the amount enzyme present. Therefore, we examined the effect of CO on steady-state levels of myocardial CcOX I mRNA and protein using Northern blot analysis and immunoblotting, respectively. Following CO exposure, steady-state levels of CcOX I mRNA were unchanged compared to sham exposure (Fig. 3). In contrast, steady-state levels of CcOX I protein decreased by 40% following CO exposure relative to sham controls (Fig. 4).
Figure 3. Northern blot hybridization of CcOX I.
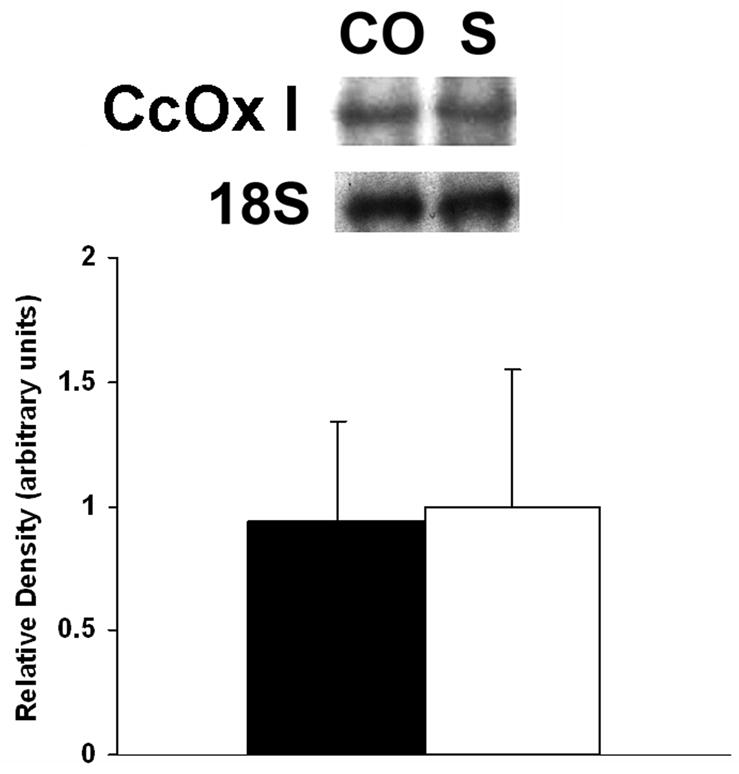
Above: A representative blot from one group of animals is depicted. CO = CO exposed group; S = sham exposure. 18S = ribosomal subunit. Below: graphic depiction of normalized signal densities. Closed bars = CO exposed mice; open bars =sham controls. Data are means +/− st dev. Values normalized to the density of the 18S ribosomal subunit. Sham densities arbitrarily set to 1. p = NS.
Figure 4. Western blot of CcOX I protein.
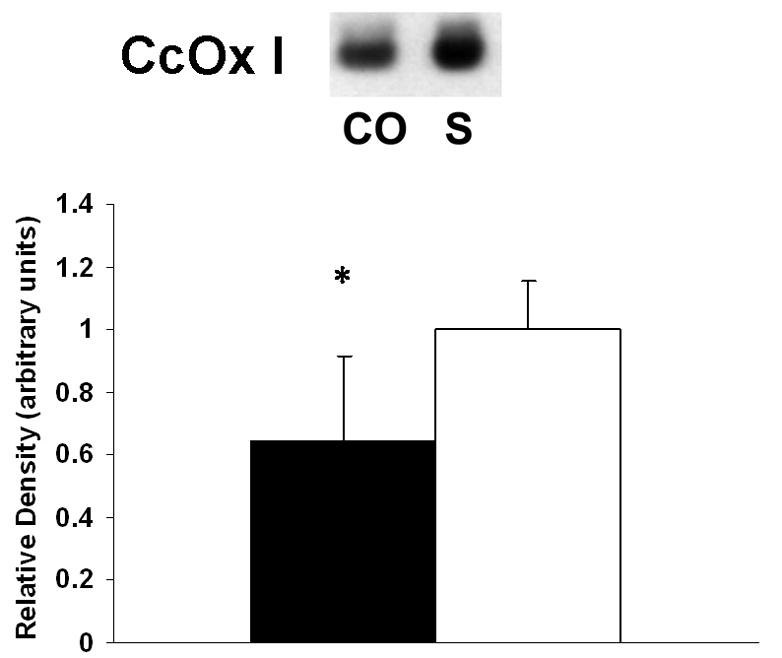
Above: Representative blot from one group of animals is depicted. CO = CO exposed group; S = sham exposure. Below: Graphic depiction of normalized signal densities. Closed bars = CO exposed mice; open bars = sham controls. Data are mean +/− st dev. Sham values arbitrarily set to 1. *p < 0.005.
CO Exposure (1000 ppm) Increases COHb Levels Without Causing Tissue Hypoxia
CO may cause cellular damage by binding to hemoglobin, limiting oxygen delivery and inducing tissue hypoxia. Therefore, we measured COHb levels following CO and sham exposure. Exposure to 1000 ppm of CO for 3 hours significantly increased COHb levels to 61 ±4% compared to 1 ±1% following exposure to compressed air (P < .001). To determine if this level of CO exposure resulted in cardiac tissue hypoxia, we injected a cohort of mice with pimonidazole hydrochloride, a commonly used hypoxic marker. Immunohistochemistry of cardiac sections from mice exposed to 1000ppm CO and sham controls yielded similar fluorescence patterns (Fig. 5). These fluorescent patterns, however, were in stark contrast to the bright fluorescence noted in sections from hypoxic controls (Fig. 5). These findings suggest the lack of tissue hypoxia in mice exposed to 1000 ppm CO and are consistent with prior work demonstrating that CO does not induce tissue hypoxia at COHb levels below 70% [1].
Figure 5. Assessment for myocardial tissue hypoxia using immunohistochemistry for pimonidazole hydrochloride.
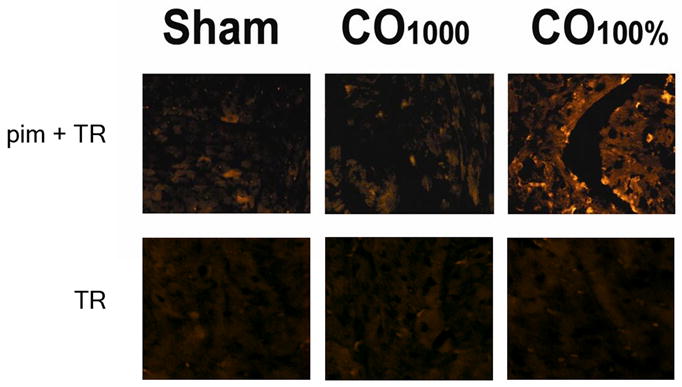
Representative sections from mice exposed to air (sham), mice exposed to 1000 ppm CO (CO0.1%), and from mice exposed to 100% CO (hypoxic controls, CO100%) are demonstrated. The upper panels represent sections exposed to primary antibody to pimonidazole and secondary antibody conjugated with Texas red (pim + TR). The lower panels are control sections from the representative groups exposed only to antibody conjugated with Texas red (TR). Sections were imaged using fluorescence microscopy at 20X with 596 nm excitation and 620 nm emission. Fluorescence patterns are similar between CO0.1% and normoxic sham controls. These were similar to TR control sections. However, regions of bright fluorescence are clearly seen in CO100% hypoxic controls.
DISCUSSION
In this work we demonstrated that inhaled CO significantly decreased the activity of myocardial CcOX, the terminal enzyme in the respiratory chain. This impairment was associated with significantly decreased CcOX I protein and heme aa3 content. Because there was no change in the turnover number of CcOX, it is likely that CO-dependent reduction of CcOX activity occurred on the basis of decreased enzyme and heme content. These changes occurred at moderate COHb levels in the absence of tissue hypoxia. These results are important because they suggest a previously unexplored cellular effect of CO inhalation: CO-induced loss of CcOX activity due to decreased heme aa3 and CcOX enzyme content. Although the mechanisms of reduced heme and enzyme content are unknown it is reasonable to postulate that the resultant decrease in CcOX activity could reduce aerobic ATP synthesis and contribute to or cause cellular and organ dysfunction. With respect to the heart, the effect would be altered cardiac contractility and relaxation. While no functional data are presented, these findings suggest that the effects of CO on cardiac function may be multifactorial and not exclusively due to the formation of COHb or resultant myocardial hypoxia [3].
Decreased steady-state levels of CcOX I protein in the setting of unchanged CcOX I mRNA levels suggests an abnormality in protein translation, decreased CcOX half life, or increased enzyme destruction. The latter is a likely explanation because CO avidly binds to heme groups and can result in reactive oxygen species production, oxidative stress, and subsequent protein destruction [16]. Additionally, it is possible that inhaled CO upregulates myocardial heme oxygenase (HO), the rate-limiting enzyme in heme catabolism [2, 16]. HO may reduce myocardial heme aa3 content, and, in concert with CcOX I destruction, could reduce overall CcOX activity. Since HO-1 induction and endogenous CO production are thought to be protective in the heart, alternative mechanisms of heme destruction during exogenous CO exposure may exist [17, 18]. Because tissue levels of CO during exogenous exposure far exceed those endogenously produced during heme turnover, the results reported here are more directly relevant to the toxic effect of inhaled CO as opposed to the physiologic or protective effect of endogenous CO [19]. However, consideration of potential effects of endogenous CO is warranted.
Endogenous CO is important for cellular homeostasis and signaling. CO is strikingly similar to another diatomic gas in this regard: nitric oxide (NO) [20]. Both CO and NO share similar molecular mass, water solubility, and basal rates of production [20]. In addition, both are believed to act as second messengers with similar effects on the cardiovascular system [20]. Furthermore, CO and NO can bind to heme and competitively inhibit CcOX [2, 21]. This physiologic regulation of CcOX is important. Notably, NO has been shown to target intracellular heme [22, 23]. In addition, endogenous NO impairs heme synthesis and enhances heme destruction by increasing HO activity [23]. Both of these effects lead to loss of cellular heme [23]. Therefore it is quite possible that CO-induced reduction of myocardial heme content occurs via similar mechanisms. Such an effect on heme content and biosynthesis might represent an important physiologic role for endogenous CO and NO while excess production or increased tissue levels following inhalation may lead to pathologically decreased cellular heme content. Reduced enzyme-bound heme results in impaired function of a variety of important enzymes such as catalase, cytochrome P450, and CcOX [23]. This could have dramatic effects on cell and organ function. Decreased CcOX activity, as we have shown, represents just one consequence of inhaled CO. The effects of endogenous and exogenous CO on heme biosynthesis and destruction, therefore, are important concepts in need of further investigation. It is likely that, as with NO, CO-induced effects on cellular heme are important physiologically. However, tipping the balance toward excess, as with exogenous exposure, may lead to reduced heme and resultant enzyme impairment and represents a previously unexplored mechanism of CO toxicity.
Acknowledgments
This work was supported by: NIH/NIGMS 1K08GM074117 (RJL), The Maria Fareri Children’s Hospital Foundation (RJL), NIH/NIGMS 2R01GM059930 (CSD).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gorman D, Drewry A, Huang YL, Sames C. The clinical toxicology of carbon monoxide. Toxicology. 2003;187:25–38. doi: 10.1016/s0300-483x(03)00005-2. [DOI] [PubMed] [Google Scholar]
- 2.Dulak J, Jozkowicz A. Carbon monoxide -- a “new” gaseous modulator of gene expression. Acta Biochim Pol. 2003;50:31–47. [PubMed] [Google Scholar]
- 3.Smithline HA, Ward KR, Chiulli DA, Blake HC, Rivers EP. Whole body oxygen consumption and critical oxygen delivery in response to prolonged and severe carbon monoxide poisoning. Resuscitation. 2003;56:97–104. doi: 10.1016/s0300-9572(02)00272-1. [DOI] [PubMed] [Google Scholar]
- 4.Gandini C, Castoldi AF, Candura SM, Locatelli C, Butera R, Priori S, Manzo L. Carbon monoxide cardiotoxicity. J Toxicol Clin Toxicol. 2001;39:35–44. doi: 10.1081/clt-100102878. [DOI] [PubMed] [Google Scholar]
- 5.Kao LW, Nanagas KA. Carbon monoxide poisoning. Med Clin North Am. 2005;89:1161–1194. doi: 10.1016/j.mcna.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 6.Brown SD, Piantadosi CA. In vivo binding of carbon monoxide to cytochrome c oxidase in rat brain. J Appl Physiol. 1990;68:604–610. doi: 10.1152/jappl.1990.68.2.604. [DOI] [PubMed] [Google Scholar]
- 7.Brown SD, Piantadosi CA. Reversal of carbon monoxide-cytochrome c oxidase binding by hyperbaric oxygen in vivo. Adv Exp Med Biol. 1989;248:747–754. doi: 10.1007/978-1-4684-5643-1_84. [DOI] [PubMed] [Google Scholar]
- 8.Saraste M. Oxidative phosphorylation at the fin de siecle. Science. 1999;283:1488–1493. doi: 10.1126/science.283.5407.1488. [DOI] [PubMed] [Google Scholar]
- 9.Parul D, Palmer G, Fabian M. Proton interactions with hemes a and a3 in bovine heart cytochrome c oxidase. Biochemistry. 2005;44:4562–4571. doi: 10.1021/bi048435c. [DOI] [PubMed] [Google Scholar]
- 10.Thom SR. Carbon monoxide-mediated brain lipid peroxidation in the rat. J Appl Physiol. 1990;68:997–1003. doi: 10.1152/jappl.1990.68.3.997. [DOI] [PubMed] [Google Scholar]
- 11.Vijayasarathy C, Biunno I, Lenka N, Yang M, Basu A, Hall IP, Avadhani NG. Variations in the subunit content and catalytic activity of the cytochrome c oxidase complex from different tissues and different cardiac compartments. Biochim Biophys Acta. 1998;1371:71–82. doi: 10.1016/s0005-2736(97)00278-2. [DOI] [PubMed] [Google Scholar]
- 12.Levy RJ, Vijayasarathy C, Raj NR, Avadhani NG, Deutschman CS. Competitive and noncompetitive inhibition of myocardial cytochrome C oxidase in sepsis. Shock. 2004;21:110–114. doi: 10.1097/01.shk.0000108400.56565.ab. [DOI] [PubMed] [Google Scholar]
- 13.Smith L. Spectrophotometric assay of cytochrome c oxidase. Methods Biochem Anal. 1955;2:427–434. doi: 10.1002/9780470110188.ch13. [DOI] [PubMed] [Google Scholar]
- 14.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 15.Weiss YG, Bouwman A, Gehan B, Schears G, Raj NR, Deutschman CS. Cecal ligation and double puncture impairs heat shock protein 70 (HSP-70) expression in the lungs of rats. Shock. 2000;13:19–23. doi: 10.1097/00024382-200013010-00004. [DOI] [PubMed] [Google Scholar]
- 16.Raub JA, Benignus VA. Carbon monoxide and the nervous system. Neurosci Biobehav Rev. 2002;26:925–940. doi: 10.1016/s0149-7634(03)00002-2. [DOI] [PubMed] [Google Scholar]
- 17.Bak I, Papp G, Turoczi T, Varga E, Szendrei L, Vecsernyes M, Joo F, Tosaki A. The role of heme oxygenase-related carbon monoxide and ventricular fibrillation in ischemic/reperfused hearts. Free Radic Biol Med. 2002;33:639–48. doi: 10.1016/s0891-5849(02)00913-9. [DOI] [PubMed] [Google Scholar]
- 18.Bak I, Szendrei L, Turoczi T, Papp G, Joo F, Das DK, de Leiris J, Der P, Juhasz B, Varga E, Bacskay I, Balla J, Kovacs P, Tosaki A. Heme oxygenase-1-related carbon monoxide production and ventricular fibrillation in isolated ischemic/reperfused mouse myocardium. FASEB J. 2003;17:2133–5. doi: 10.1096/fj.03-0032fje. [DOI] [PubMed] [Google Scholar]
- 19.Chan GCF, Lau YL, Yeung CY. End tidal carbon monoxide concentration in childhood haemolytic disorders. J Paediatr Child Health. 1998;34:447–450. doi: 10.1046/j.1440-1754.1998.00270.x. [DOI] [PubMed] [Google Scholar]
- 20.Hartsfiel CL. Cross talk between carbon monoxide and nitric oxide. Antioxid Redox Signal. 2002;4:301–307. doi: 10.1089/152308602753666352. [DOI] [PubMed] [Google Scholar]
- 21.Mason MG, Nicholls P, Wilson MT, Cooper CE. Nitric oxide inhibition of respiration involves both competitive (heme) and noncompetitive (copper) binding to cytochrome c oxidase. Proc Natl Acad Sci U S A. 2006;103:708–13. doi: 10.1073/pnas.0506562103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Henry Y, Lepoivre M, Drapier JC, Ducrocq C, Boucher JL, Guissani A. EPR characterization of molecular targets for NO in mammalian cells and organelles. FASEB J. 1993;7:1124–34. doi: 10.1096/fasebj.7.12.8397130. [DOI] [PubMed] [Google Scholar]
- 23.Kim YM, Bergonia HA, Muller C, Pitt BR, Watkins WD, Lancaster JR. Loss and degradation of enzyme-bound heme induced by cellular nitric oxide synthesis. J Biol Chem. 1995;270:5710–3. doi: 10.1074/jbc.270.11.5710. [DOI] [PubMed] [Google Scholar]