

Abstract
Malignant gliomas have been shown to release glutamate, which kills surrounding brain cells, creating room for tumor expansion. This glutamate release occurs primarily via system , a Na+-independent cystine-glutamate exchanger. We show here, in addition, that the released glutamate acts as an essential autocrine/paracrine signal that promotes cell invasion. Specifically, chemotactic invasion and scrape motility assays each show dose-dependent inhibition of cell migration when glutamate release was inhibited using either S-(4)-CPG or sulfasalazine, both potent blockers of system . This inhibition could be overcome by the addition of exogenous glutamate (100 μmol/L) in the continued presence of the inhibitors. Migration/invasion was also inhibited when Ca2+-permeable α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors (AMPA-R) were blocked using GYKI or Joro spider toxin, whereas CNQX was ineffective. Ca2+ imaging experiments show that the released glutamate activates Ca2+-permeable AMPA-R and induces intracellular Ca2+ oscillations that are essential for cell migration. Importantly, glioma cells release glutamate in sufficient quantities to activate AMPA-Rs on themselves or neighboring cells, thus acting in an autocrine and/or paracrine fashion. System and the appropriate AMPA-R subunits are expressed in all glioma cell lines, patient-derived glioma cells, and acute patient biopsies investigated. Furthermore, animal studies in which human gliomas were xenographed into scid mice show that chronic inhibition of system –mediated glutamate release leads to smaller and less invasive tumors compared with saline-treated controls. These data suggest that glioma invasion is effectively disrupted by inhibiting an autocrine glutamate signaling loop with a clinically approved candidate drug, sulfasalazine, already in hand.
Introduction
The majority of primary brain tumors derive from glial cells and are collectively called gliomas. They are among the most challenging cancers to treat and carry a poor prognosis due to their exceptional ability to infiltrate normal brain, often along blood vessels or nerve fibers (1, 2). This feature makes complete surgical resection virtually impossible (3). Although tumor invasion has been intensely investigated, many aspects of this biology remain poorly understood. Previous findings from our laboratory (4) and others (5, 6) implicate glutamate as an important player in the growth and invasion of gliomas.
By binding to α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA)/kainate, N-methyl d-aspartate (NMDA), or metabotropic glutamate receptors, glutamate mediates excitatory neurotransmission. Because sustained activation of these receptors can cause excitotoxic neuronal cell death (7, 8), [glutamate]o is tightly regulated by uptake into nonmalignant glia through Na+-dependent transport systems of the excitatory amino acid transporter (EAAT) family (9, 10). Surprisingly, glioma cells lack functional EAAT transporters (11) and release glutamate rather than take it up (4). The released glutamate has been suggested to promote tumor growth as it causes peritumoral excitotoxic neuronal cell loss (12). This may explain the peritumoral seizures that are frequently observed in patients with malignant gliomas (13).
Interestingly, glutamate release from glioma cells occurs as an obligatory by-product of cellular cystine uptake via system (4), an abundantly expressed but poorly investigated amino acid transporter. System is a heterodimeric protein complex consisting of a catalytic light chain (xCT), which confers specificity, and a regulatory heavy chain (4F2hc), which localizes the transporter in the membrane (14). This antiporter imports cystine in exchange for the release of glutamate (15, 16). Cystine, in turn, serves as a precursor for the synthesis of the reducing agent glutathione (GSH). Pharmacologic inhibition of system inhibits GSH production and greatly attenuates tumor growth in vivo (17).
Glutamate seems to also play an important role in neuronal migration during brain development. Specifically, activation of NMDA receptors in migratory granule cells induced intracellular Ca2+ oscillations that were highly synchronous with cell movements (18). In this study, we examine whether invading glioma cells use similar signaling mechanisms observed in migratory neurons during development. We show that glioma cells are, indeed, stimulated to migrate in response to glutamate. In contrast to cerebellar neurons, however, glutamate acts on Ca2+-permeable AMPA receptors (AMPA-R). Most importantly, glutamate released from the same or neighboring glioma cells drives the process making it an autocrine or paracrine signal, thus increasing the ability of the glioma cell to invade.
Materials and Methods
Cell culture
Experiments were conducted using the glioma cell lines STTG-1, U251-MG, U87-MG [glioblastoma multiforme (GBM), WHO grade 4, American Tissue Culture Collection], and D54-MG (glioblastoma multiforme, WHO grade 4; Dr. D.D. Bigner, Duke University, Durham, NC); and two patient-derived acute GBM cultures, passages 4 to 20, labeled GBM 50 and GBM 62. Glioma cells were maintained in DMEM/F12 (Media Prep, University of Alabama at Birmingham Media Preparation Facility) with 7% fetal bovine serum (Aleken Biologicals) and were supplemented with 2 mmol/L glutamine. Primary, postnatal day 0, Sprague-Dawley rat cortical astrocytes were used as a nonglioma control at 10 to 14 days in culture. Human fetal astrocytes (22 weeks gestation, passage 2, Cambrex Corporation) were grown with medium provided with the cells. These cells are passed two to nine times only, according to the instructions. Unless otherwise stated, all reagents were purchased from Sigma. Cells were treated in all experiments with the two available reagents to inhibit system , S(4)-CPG (refs. 4, 19; Tocris) and sulfasalazine (ref. 20; Sigma).
Immunofluorescence microscopy
Glioma cells and primary cultures of rat cortical astrocytes were cultured on glass coverslips. Cryotome sections (8 μm; Zeiss HM505E; Zeiss) of fresh frozen patient glioma tissues and brain sections of mice were mounted on microscope slides. The patient specimen experiments were approved by the Institutional Review Board of the University of Alabama at Birmingham (IRB F971030027). Cells were fixed in 4% paraformaldehyde followed by incubation for 1 h in PBS and 10% normal goat serum plus 0.1% Triton X-100. Cells were incubated overnight with rabbit or mouse anti-4F2hc (1:200) from Santa Cruz Biotechnology (CD98); rabbit anti-xCT (1:100; CosmoBio); guinea pig anti–GLT-1 (1:1,000; Chemicon/Millipore); glutamate receptor subunits GluR1, GluR2, GluR3, GluR4; and NMDA subunit NR1 (1:500; Upstate Biologicals/Millipore). After removing the primary antibodies, the cells were incubated with secondary IgG antibodies, and guinea pig, rabbit, or mouse Alexa 546 and Alexa 488 (Molecular Probes/Invitrogen). Cells were incubated with 4′,6-diamidino-2-phenylindole (DAPI) and mounted onto slides with Permamount solution. Cell staining was examined on a Zeiss Axiovert 200M microscope with a ×5, ×20, or ×40 oil objective, and images were collected with Axiovision software (Zeiss). Exposures to each wavelength of secondary antibody fluorescence were compared with the secondary-only control in every case.
Western blot analysis
The expression of 4F2hc, xCT, GLT-1, GluR1, GluR2, GluR3, GluR4, and NMDA subunit NR1 in glioma cells and patient samples were assessed by standard Western blot analysis (17). Affinity-purified anti-4F2hc antibody (1:1,000), anti–glutamate receptor antibodies (1:500), and anti-actin antibody (1:2,000) were diluted in TBS-T supplemented with 0.5% nonfat dry milk and 1% bovine serum albumin. Appropriate horseradish peroxidase–conjugated goat anti-rabbit or mouse or guinea pig IgG (Amersham) diluted 1:1,000 was applied as secondary antibodies. Blots were visualized with enhanced chemiluminescence (ECL) and exposed on hypersensitive ECL film. Blots were stripped and then reprobed with antibodies to GluR1, GluR2, GluR4, and NMDA subunit NR1.
Reverse transcription-PCR
Total RNAs were isolated from cell lines and brain biopsies using RNAqueous (Ambion) according to the instructions of the manufacturer (Qiagen). For the detection of xCT, 4F2hc, and actin RNA transcripts, OneStep reverse transcription-PCR (RT-PCR) kit was used according to manufacturer’s instructions. The following pairs of primers were used: xCT, GCTGGCTGGTTTTACCTC (5′ primer) and TGAAAGGACGATGCATATC (3′ primer); 4F2hc, GCTGCTGCTCTTCTGGCTC (5′ primer) and GCCAGTGGCATTCAAATAC (3′ primer); actin, CATGCCATCCTGCGTC (5′ primer) and CTCCTTCTGCATCCTGTC (3′ primer). Expected amplification product sizes were as follows: 525 bp for xCT, 726 bp for 4F2hc, and 431 bp for actin. The reverse transcription reactions were done at 50°C for 30 min with 250 ng RNA, followed by 30 cycles of PCR amplification (20 s at 94°C, 20 s at 50°C, and 1 min at 72°C).
Glutamate release assays: bioluminescence
The bioluminescence method was used for detection of glutamate in solution as described by the Fosse group (21). Glioma cell supernatant was incubated and transferred to a 96-well white cliniplate (Labsystem). The glutamate-correlated luminescence was measured by a luminescence plate reader equipped with automatic solution pumps (LUMIstar; BMG LabTechnologies). This method is sensitive to glutamate concentrations as low as 20 nmol/L. We determined that all drugs, except for sulfasalazine, used in these studies did not interfere with the bioluminescence assay at the concentration used. Glutamate standards used for calibration were measured at both the beginning and end of each plate. [Glutamate]i was calculated from the amount of [glutamate]i normalized to the total amount of protein and expressed in nmol/mg protein. [Glutamate]o was either expressed as an absolute concentration (μmol/L) or multiplied by the volume and was then normalized to cellular protein levels and expressed in nmol/mg protein.
[3H]Glutamate release
Uptake procedures were based on previous protocols (4). l-[3H]glutamate was used as a tracer to study high-affinity, Na+-dependent uptake and release. Cells were washed twice with uptake solution, l-[3H]glutamate (0.4 μCi/mL) was mixed with 200 μmol/L cold glutamate and incubated for 10 min, then washed with ice-cold uptake solutions to stop the activity and remove excess hot and cold glutamate. Cells rested for 10 min in the presence of 200 μmol/L cystine before glutamate released into the supernatant was collected and counted. 3H activity was detected in a liquid scintillation counter (Beckman Instruments). Total glutamate release was normalized to protein content according to the Bradford method using a Bio-Rad protein assay kit.
Invasion/migration assays
Transwell migration assays were used as an in vitro model for standard invasive migration (22). Drugs were added to both sides of the filter 30 min after plating cells. After 6-h migration, cells were fixed and stained with an ethanol/crystal violet solution. Cells were wiped away from the top of transwell filters before counting cells on the bottom (i.e., those cells with nuclei that had migrated across the filter). Cells were counted immediately after staining and stored at 4°C in PBS. A Zeiss microscope with the ×20 objective was used with transmitted light to capture images of the bottom of transwell filters to count cells. An investigator blinded to the identity of the transwell filter counted cells from six random fields in each of three wells per treatment. All counts per treatment were averaged and SE values were calculated. These experiments were repeated thrice, data were pooled, and statistics were done within the graphing software, Origin.
Ratiometric [Ca2+]i measurements
D54-MG or U251-MG cells were plated on 35-mm glass bottom dishes (MatTek, Inc.) at 140 × 103 per dish and cultured for 2 days. Cells were loaded in serum-free culture medium for 30 min with the ratiometric Ca2+ dye Fura-2-acetoxymethylester (5 μmol/L; TEFLABS) reconstituted in 20% w/v pluronic acid in DMSO. Cells were rinsed with serum-free feeding medium and allowed to rest in 7% serum-containing medium for 30 min at 37°C. The glass-bottomed dishes were placed in an environmental chamber mounted on a Zeiss Axiovert microscope. Cells were allowed to equilibrate in the chamber for 15 min before calcium images were collected. Glutamate, cystine, or any inhibitors were added and allowed to equilibrate for 15 min. A new dish of sister cells was used for every application. Recordings were obtained with a fluorescent imaging microscope (Zeiss), where cells were alternately excited at 340 and 380 nm using a monochromatic light source. Emitted light was collected at >520 nm. Images were digitized online, and 340:380 nm ratios were obtained every 10 to 15 s.
Animal studies
D54-MG glioma cells, 2.5 × 105 in 10 μL methylcellulose, were stereotactically implanted through a small burr hole using a 30-gauge Hamilton syringe into the cranium of a female nude mouse as previously described (23). After 7 days, animals were randomized into three groups of five animals each. One group received 1 mL i.p. saline injections twice daily for 3 weeks. The two test groups received a 4 or 8 mg/mL dose of sulfasalazine in 1 mL saline twice daily for 3 weeks. Tumor growth and animal health were monitored. Mouse brains were collected, fixed in 4% paraformaldehyde overnight, rinsed, and placed in 30% sucrose until saturated. Brains were stored at −80°C until cryosectioned. This experiment was repeated at least thrice.
Results
The central hypothesis of this article is that invading glioma cells use the neurotransmitter glutamate as a signal to stimulate and coordinate tumor cell invasion. Glutamate release occurs through a cystine-glutamate transporter (system ) and activates glutamate receptors either on the same or adjacent cells, hence acting in either autocrine or paracrine fashion.
Glioma cells and human biopsies express the system cystine-glutamate transporter
In a first series of experiments, we investigated expression of glutamate transporters in glioma cell lines, as well as glioma cells acutely derived from patients and compared the expression with nonmalignant samples. RT-PCR, Western blot analysis, and immunocytochemistry were used specifically to search for transcripts and proteins of the two transporter families potentially involved in glutamate transport. These are the excitatory amino acid transporter EAAT2, also known as GLT-1 (24), and the system cystine-glutamate transporter (14), composed of a catalytic (xCT) and regulatory (4F2hc) subunit. Figure 1A illustrates the presence of PCR transcripts for both subunits of system in several human glioma cell lines and rat astrocytes. Only a subset of the tested glioma samples is shown in the figures, yet all investigated glioma cell lines and acute biopsy samples showed prominent expression of xCT and 4F2hc transcripts. The expression of the system transporter was confirmed at the protein level by Western blot analysis (Fig. 1B). The regulatory subunit 4F2hc was expressed prominently in all samples, even those with low xCT expression. Importantly, both the regulatory (4F2hc) and catalytic (xCT) subunits were prominently expressed in all five GBM patient samples investigated (Fig. 1C). As in previous studies (11), we observed an absence of expression of GLT-1 glutamate transporters in all investigated glioma cells, yet GLT-1 is highly expressed in rat astrocytes (25), nonmalignant brain (9), and to a lesser extent in human fetal astrocytes (Fig. 1B).
Figure 1.
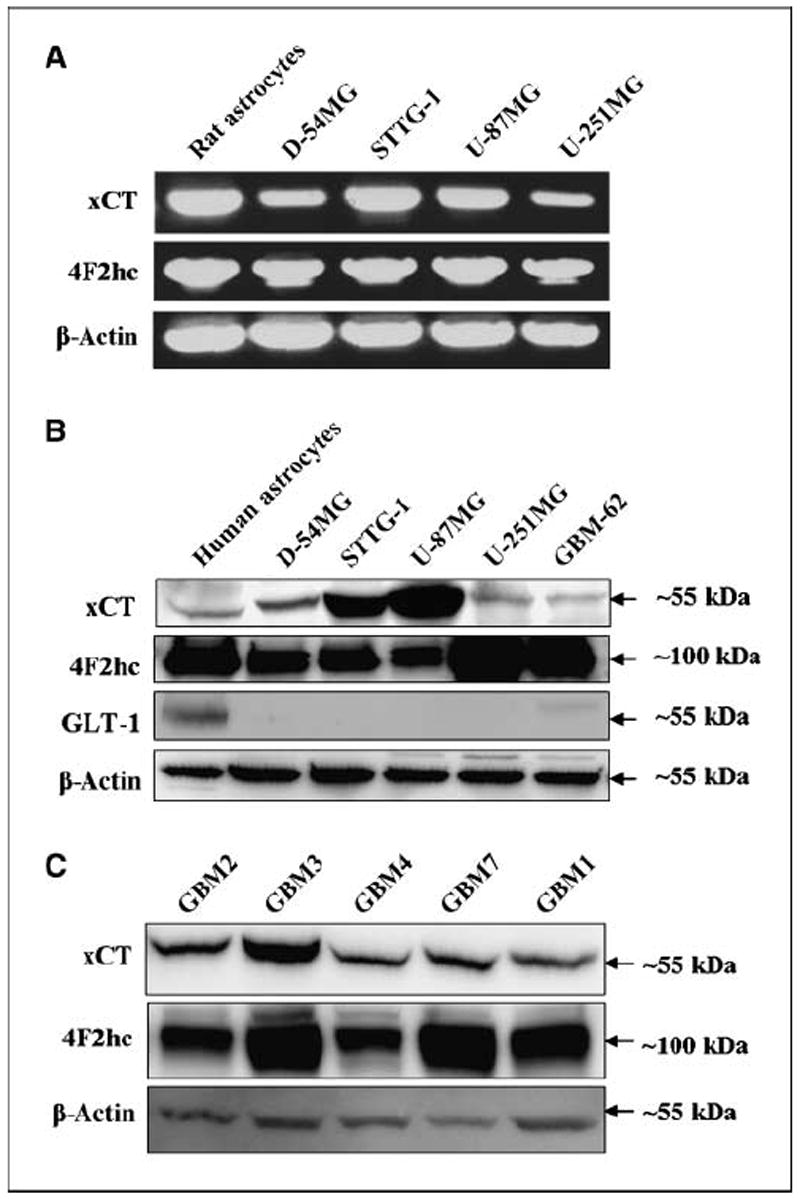
Subunits of system are expressed in glioma cell lines and patient tumor samples. A, RT-PCR indicated that transcripts of catalytic subunit xCT and the regulatory subunit 4F2hc of system are expressed in primary cultures of rat astrocytes and all glioma cell lines tested; four lines are shown. B, protein expression of xCT and 4F2hc are shown in human astrocytes and five human glioma cell lines. This is compared with the glutamate transporter GLT-1, expressed robustly in mature astrocytes but less so in human fetal astrocytes (immature). C, protein expression of system subunits in five GBM patient tissues are shown from individuals aged 51 to 72 y. β-Actin is shown as a control for protein loading efficiency. The approximate molecular weights of the proteins are indicated to the right as each blot image was cropped for clarity.
Next, we used specific antibodies to xCTand 4F2hc to examine the surface expression and subcellular localization of the cystine-glutamate transporter in gliomas. Figure 2A and B show representative examples of two glioma cell lines, Fig. 2C is a patient-derived cell sample, and Fig. 2D is a patient glioma section. The images of triple-immunostained fields illustrate the regulatory subunit alone (4F2hc) in the first panels and the catalytic subunit (xCT) in the second panels, both shown in black and white for clarity. The third panels show merged color images that include DAPI-labeled blue nuclei. These examples show colocalization of xCT and 4F2hc for the two glioma cell lines and colocalization in the majority of cells in the acute patient-derived GBM cell culture, GBM-50. Finally, prominent membrane-associated labeling for xCT and 4F2hc is shown for an acute biopsy section GBM (Fig. 2D). Note that there is some heterogeneity in the expression of xCT and 4F2hc in glioma cultures as seen in GBM 50 cells (Fig. 2), which may account for varying protein expression in the Western blot analysis. Taken together, these data indicate that glioma cells and glioma biopsy sections prominently express the two subunits that compose the system transporter.
Figure 2.
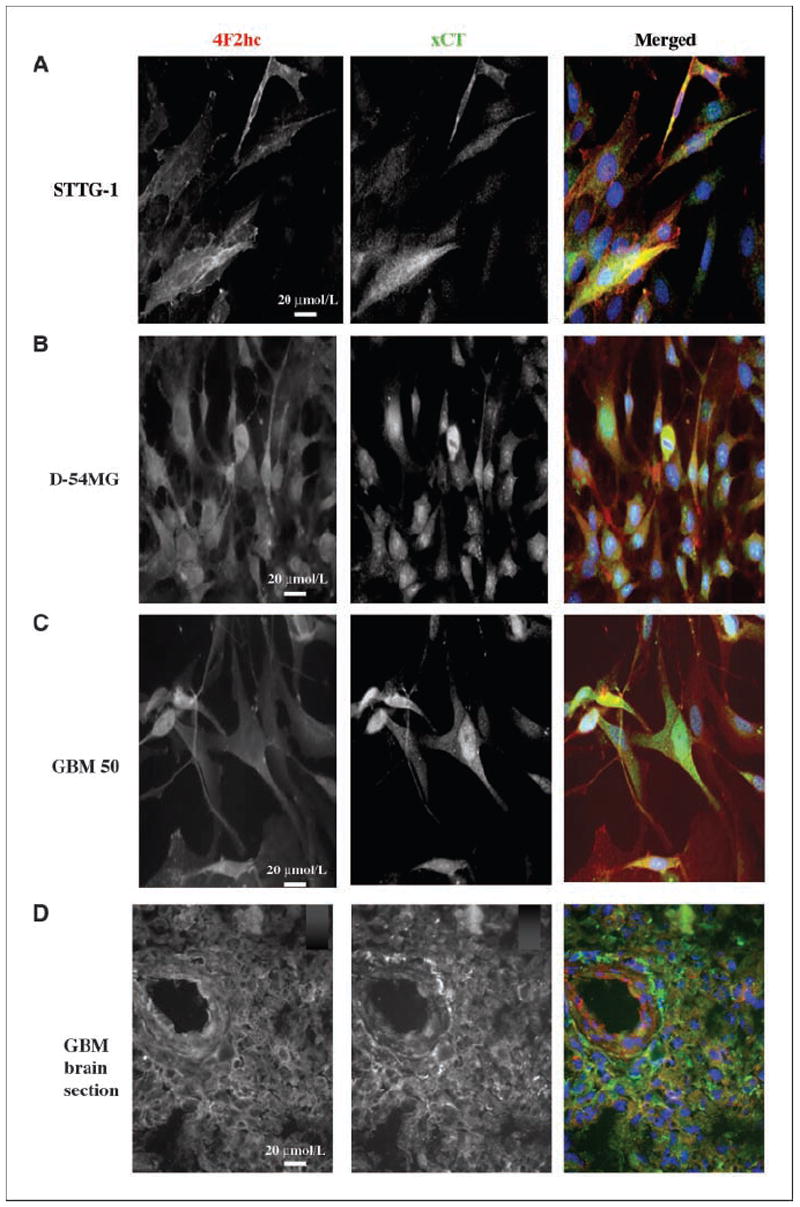
Specificity of system subunits show antibody binding in the membranes of glioma cells. A, sister coverslips of glioma cell lines were stained with mouse anti-4F2hc and rabbit anti-xCT. Both fluorescence channels are shown separately in black and white until merged in the third color panels. A to D, merged images: red, xCT; green, 4F2hc; blue, DAPI, a nuclear stain. The GBM-stained patient section is shown in D, where xCT is green and 4F2hc is red. Merged images were taken at the exposure time appropriate for each wavelength. All images were taken at ×40 magnification.
Glioma cells release glutamate via system
To assess the functional expression of system transporters and to ascertain its possible involvement in glutamate release from glioma cells, we used a combination of radiotracer release assays and enzymatic detection of extracellular glutamate. We took advantage of the fact that system function is Na+ independent and inhibited by sulfasalazine (20) and S-(4)-CPG (4, 19). Figure 3A shows a direct comparison of glutamate release among two glioma cell lines, two patient-derived cell samples, and primary cortical rat astrocytes as determined by glutamate bioluminescence. All glioma cells released glutamate at ~100 to 300 nmol/mg protein over a 4-h time period. As expected for a Na+-independent transport system, this glutamate release was not affected by removal of extracellular Na+ (data not shown).
Figure 3.
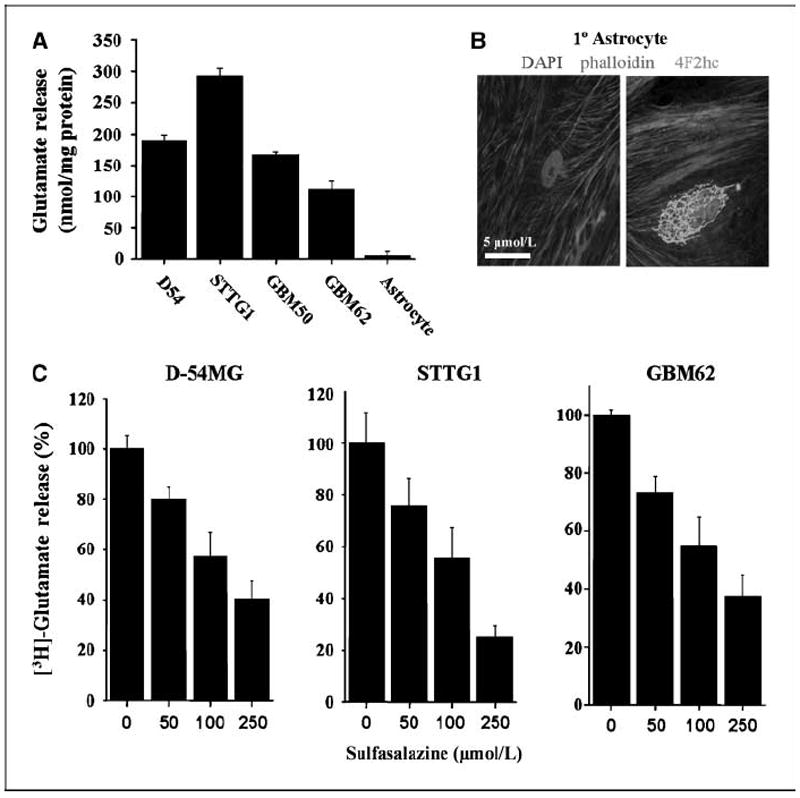
Glutamate release by glioma cells is inhibited by sulfasalazine in a dose-dependent manner. A, glutamate released by four glioma cell lines were compared with primary cultured astrocytes using an enzyme-based bioluminescent signal normalized to protein. B, images of primary cultures of rat astrocytes show a control (left) and positive staining for 4F2hc on the right, labeling only the Golgi, in green. DAPI-labeled nuclei and phalloidin (a cytoskeleton protein conjugated with Alexa 546) are included. System is nonfunctional in astrocyte cultures. C, glutamate release is shown in the presence of increasing concentrations of sulfasalazine using [3H]glutamate-loaded glioma cell lines to measure release. Columns, percent control; bars, SE. All experiments were done with at least n = 3.
In comparison with the glioma cells shown in Fig. 2, where both xCT and 4F2hc are shown on the surface of the cells, nonmalignant astrocytes consistently expressed 4F2hc in a perinuclear region (Fig. 3B) that was shown to be Golgi localization by counterstaining with GP130, a Golgi-specific antibody (data not shown). This deficiency in membrane localization of system in astrocytes explains a lack of glutamate release by this system (Fig. 3A).
We also measured glutamate release in response to sulfasalazine, an inhibitor of system in glioma cell lines by [3H]glutamate release. Sulfasalazine inhibited glutamate release in a dose-dependent manner as illustrated in Fig. 3C. Similar data were obtained with S-(4)-CPG (data not shown). Furthermore, inhibition of system by sulfasalazine and S-(4)-CPG (both at 250 μmol/L) decreased [35S]cystine uptake by 60% in gliomas, but was without effect on astrocytic cystine uptake (Supplementary Fig. S1). This is not surprising, as normal astrocytes take up the monomeric cysteine as a precursor for glutathione synthesis through Na+-dependent transporters, most likely GLT-1 (26). These data clearly show that glioma cells robustly release glutamate through the sulfasalazine-sensitive system transporter.
Glutamate release through system causes intracellular Ca2+ oscillations
Because glutamate release through system must be coupled to an equimolar uptake of cystine, we should be able to drive glutamate release with increasing extracellular cystine concentrations. This is illustrated in Fig. 4A, giving us the experimental tool to selectively activate glutamate release from gliomas using a physiologic stimulus, namely cystine. We hypothesized that glutamate release, in turn, may activate glutamate receptors (27), causing changes in intracellular Ca2+. To examine this, glioma cells were loaded with the ratiometric Ca2+ dye FURA2-AM (Fig. 4B) and imaged over 1 to 2 h. Under these conditions, application of 100 μmol/L cystine (Fig. 4C) caused Ca2+ oscillations with a periodicity of ~3 min in 19.5 ± 3.17% of cells and representative responses are shown in Fig. 4C. However, when cystine-glutamate exchange was inhibited with either 250 μmol/L sulfasalazine or S-(4)-CPG in the continued presence of cystine, these oscillations disappeared (2.04 ± 1.27% and 0.0%, respectively, in the total number of cells examined). These experiments suggest that glutamate release was indeed due to the activity of cystine-glutamate exchange. Interestingly, Ca2+ oscillations of the magnitude and frequency observed here are almost identical to those previously seen in migratory cerebellar granule cells where they correlated absolutely with the ability of the cells to migrate (18). Therefore, we next sought to examine whether glutamate release via system may be functionally involved in glioma cell migration.
Figure 4.
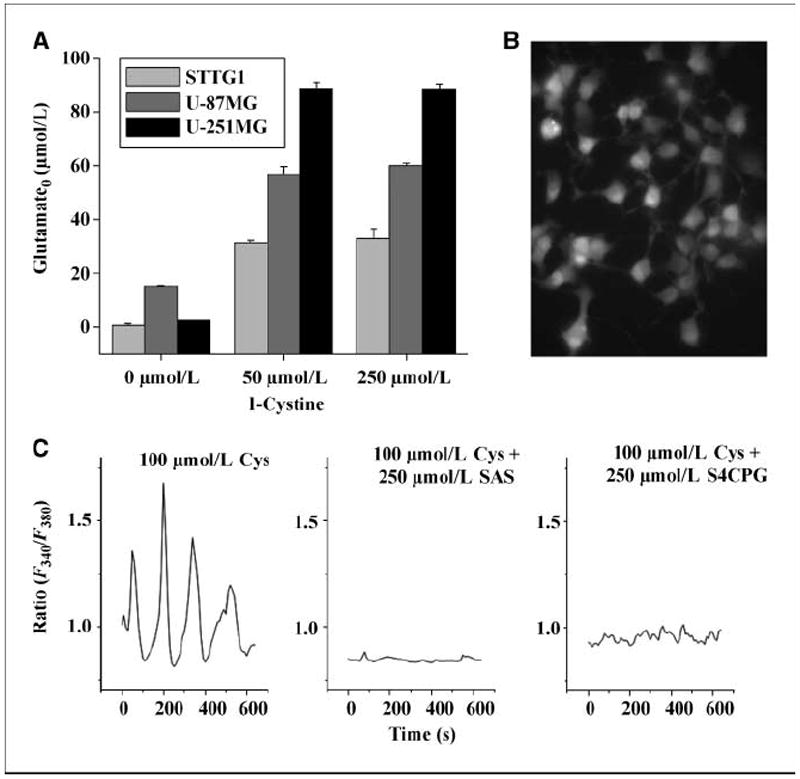
Glutamate released through system caused glioma Ca2+ oscillations. A, increasing amounts of the cystine were added to three glioma cell lines, thus inducing the release of glutamate in a dose-dependent manner as determined by the highly sensitive enzyme-based bioluminescent assay. B, FURA2-AM–loaded D54-MG cells are shown in a representative field before measuring calcium oscillations at ×40 magnification. C, three representative cells from three coverslips were imaged every 15 s in the presence of cystine or with cystine but in the presence of one of the two system inhibitors, sulfasalazine (SAS) or S-(4)-CPG.
Glutamate release promotes glioma invasion/migration
To mimic the spatial constraints that would be encountered by a glioma cell invading the brain, we used transwell migration/invasion assays. These have the advantage of examining drug effects in a highly reproducible manner. Cells isolated from a patient biospsy (GBM 62) were allowed to migrate 6 h before being analyzed. Cumulative data from at least three separate experiments are shown in the first part of Fig. 5A, indicating the invasion effects of control, glutamate alone, sulfasalazine, and sulfasalazine plus glutamate. Inhibition of system with 250 μmol/L sulfasalazine consistently reduced Transwell migration by 40% to 50% in every glioma cell line investigated (Supplementary Fig. S3). Importantly, this inhibition by sulfasalazine was overcome by adding 1 μmol/L exogenous glutamate in the presence of the inhibitor. Glutamate alone caused an insignificant increase in invasion, indicating that the normal glutamate accumulation (4) of glioma cells was sufficient for maximal receptor activation. Similar results were obtained when system was inhibited using 250 μmol/L S-(4)-CPG (not shown).
Figure 5.
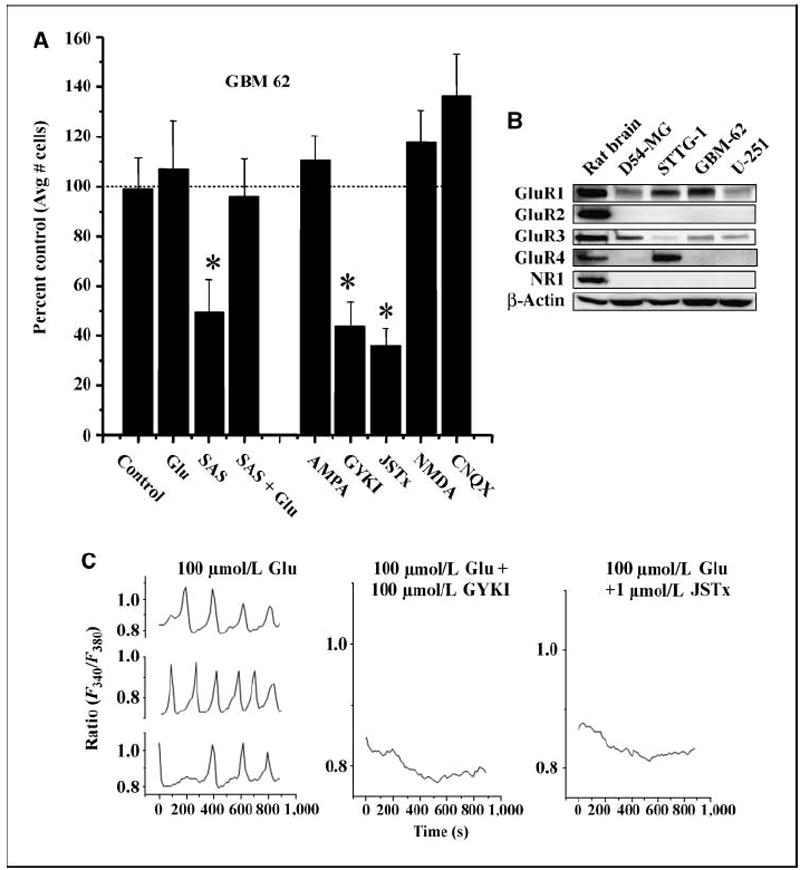
Glutamate induces AMPA-R–dependent calcium oscillations in glioma cells. A, the invasion results of patient-derived glioma cells (GBM 62) are shown in the presence of sulfasalazine (250 μmol/L SAS) and SAS + glutamate (100 μmol/L). Using sister wells, in the second half of A, glioma cells were allowed to invade the filter pores in the presence of the AMPA-R agonist AMPA (100 μmol/L); AMPA-R antagonists GYKI (100 μmol/L) and Joro spider toxin (JSTx; 1 μmol/L); NMDA-R agonist NMDA (100 μmol/L); and AMPA/KA-R antagonist, CNQX (100 μmol/L). Each experiment had three similarly treated inserts where six fields per insert were imaged and data averaged. This was repeated thrice. Bars, SE * P ≤ 0.05. B, glioma cells express Ca2+-permeable AMPA-Rs as shown by Western blot analysis for four glioma cell lines with whole rat brain lysates as a positive control. Glutamate subunits of AMPA-Rs were probed with antibodies to GluR1-4. β-Actin is shown as a control for protein loading efficiency. C, representative traces from three glioma cells loaded with FURA2-AM showed oscillatory changes in [Ca2+]i in response to 100 μmol/L glutamate. Single representative traces are shown following simultaneous application of 100 μmol/L glutamate and the AMPA-R blockers GYKI (100 μmol/L) and Joro spider toxin showing that [Ca2+]i oscillations stimulated by glutamate are inhibited. Each experiment was repeated three independent times.
Ca2+-permeable AMPA-Rs mediate glutamate-induced Ca2+ oscillations and migration in human glioma cells
We next sought to identify the target receptor through which glutamate acts to enhance migration. We therefore evaluated the subunit expression of ionotropic glutamate receptors in glioma cells by Western blot (Fig. 5B). All glioma lines and patient-derived gliomas (GBM 62) lacked the NR1 subunit, which is a required subunit for NMDA receptors. However, all of the glioma cells examined expressed the AMPA-R subunit GluR1, in combination with either GluR3 or GluR4, but consistently lacked the GluR2 subunit. Interestingly, most neuronal AMPA-Rs contain the GluR2 subunit, which prevents calcium permeation through the channel pore (28). Early in development, GluR2 is absent in some neuronal glutamatergic receptors (29), allowing Ca2+ to permeate the channel. The lack of GluR2 subunit in glioma cells suggests that they express similar Ca2+-permeable AMPA-Rs. As such, Western blot analysis (Fig. 5B) suggests that the main ionotropic glutamate receptor expressed by glioma cells is a specific variety of AMPA-R that is permeable to Ca2+. This finding was confirmed by immunocytochemistry (not shown) demonstrating the same expression patterns of glutamate receptors.
To further show that the above effects of glutamate on glioma invasion were mediated by Ca2+-permeable AMPA-Rs, we studied glioma invasion in the presence and absence of specific inhibitors of ionotropic glutamate receptors. Figure 5A, the second set of columns, summarizes pooled data for the patient-derived glioma cells (GBM 62) showing a ~60% reduction of cell migration in the presence of either 100 μmol/L GYKI, a broad-spectrum AMPA-R inhibitor, or 1 μmol/L Joro spider toxin, a more specific blocker of the Ca2+-permeable AMPA-R (30). As expected, this noncompetitive inhibition was not rescued with the exogenous addition of glutamate (not shown). Consistent with the Western blot data, both 100 μmol/L NMDA and kainate, agonists of NMDA receptor (NMDA-R) and kainate receptor, respectively, were ineffective in enhancing invasion. These data are consistent with the lack of protein expression of the NR1 subunit in the glioma cell line as determined by the above Western blot analysis. Similarly, CNQX, an antagonist to the kainate/AMPA-R, which is not permeable to Ca2+, had no inhibitory effect on invasion. Taken together, these data suggest that activation of Ca2+-permeable AMPA-Rs by glutamate was responsible for the observed enhancement of glioma invasion. Indeed, this evidence supports the postulated model whereby glioma cells release glutamate via system , which then acts on AMPA-R on the same cell (autocrine) and/or on neighboring cells (paracrine) to enhance migration.
We examined whether the observed Ca2+ oscillations in response to exogenous and accumulated glutamate were due to Ca2+-permeable AMPA-R. Similar to Fig. 4C, where 100 μmol/L cystine was used to induce calcium oscillations, exogenous glutamate showed identical Ca2+ oscillations in 21 ± 2.07% of glioma cells (Fig. 5C). These glutamate-induced Ca2+ oscillations were completely inhibited by either 100 μmol/L GYKI or 1 μmol/L Joro spider toxin (Fig. 5C), both blockers of Ca2+-permeable AMPA-Rs, suggesting that Ca2+ increases were caused by glutamate activation of the Ca2+-permeable AMPA-Rs (1.57 ± 1.57% and 1.27 ± 1.27%, respectively). In addition, oscillations did not occur without extracellular Ca2+ nor in the presence of calcium chelators (i.e., BAPTA; data not shown), confirming the source of calcium is external rather than internal. None of these cells appeared to be proliferating or undergoing cell death. Thus far, the above data are indicative of a system in which extracellular glutamate accumulation through system act via Ca2+-permeable AMPA-Rs to both cause Ca2+ oscillations and enhance migration.
Tumor invasion, in vivo, is inhibited by sulfasalazine
To determine if these in vitro assays have any bearing on tumor invasion in vivo, we used a frequently used mouse glioma model in which D54-MG cells were xenografted into the cerebrum of scid mice as described in our previous studies(17). We compared a total of 15 animals, in three separate experiments, in each treatment. Each of the two groups was injected twice daily i.p. with 4 mg/mL sulfasalazine or saline for the control animals. Representative ×0.75 magnification coronal brain sections are shown in Fig. 6A. Multiple tumors of varying sizes were seen in saline-treated mice at the end of 30 to 45 days posttumor injection (Fig. 6A, first column), whereas tumors in the sulfasalazine-treated mice had smaller tumors, no satellite tumors, and a clear demarcation of the tumor boundary as seen in Fig. 6A (second column). Control animals consistently showed large (average 5.0 mm, ranging from 2 to 8 mm) and highly invasive tumors with satellite tumors at distant sites. Microscopically, invading cells could be identified at the tumor boundaries of the normal brain by DAPI labeling where each labeled spot represent the nucleus of a single cell (×5 images in Fig. 6B). Multiple pathways of invading cells are readily visible in Fig. 6B (left). By comparison, sulfasalazine-treated animals showed smaller tumors overall (average 2.57-mm diameter ranging from <1 to 6 mm) and few, if any, satellite tumors. The tumor boundaries were well demarcated without evidence for cellular invasion (Fig. 6B, right). To highlight the invasiveness further, we stained consecutive sections with antibodies to GFAP and GLT-1. GFAP prominently labeled astrocytes in normal brain, which also expressed GLT-1. GLT-1 is absent in the tumors (Fig. 6C) but creates a sharp border at the tumor in mice brains (GLT-1–positive astrocytes, Fig. 6C, red). To further show that the tumor cells in sulfasalazine-treated animals still exhibits high levels of system expression, we show labeling for both transporter subunits stained in a representative brain section from a sulfasalazine-treated mice in Fig. 6D. Taken together, these animal experiments support the in vitro data above, suggesting that, indeed, tumor cell invasion is compromised in vivo when glutamate release via system is inhibited.
Figure 6.
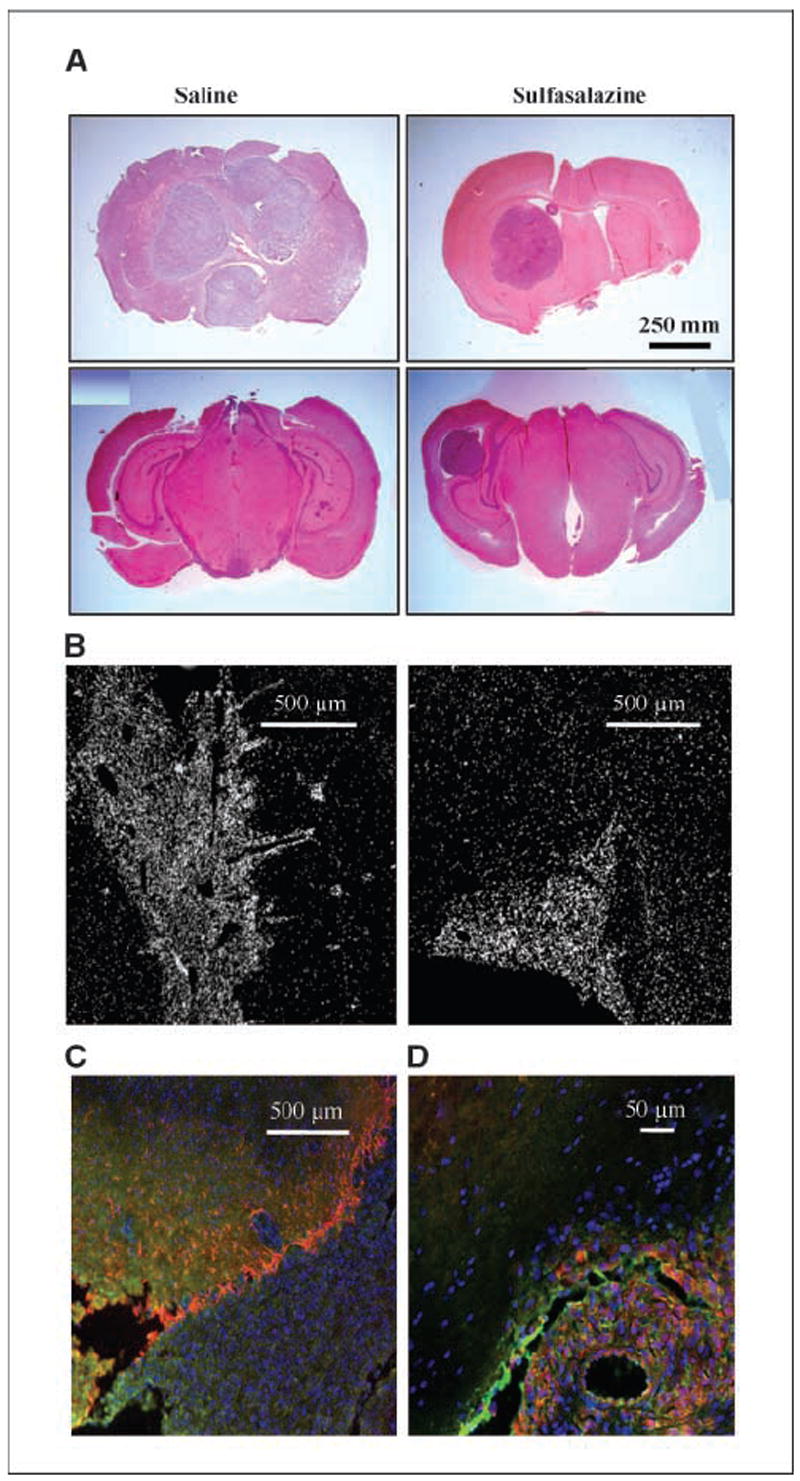
Glioma invasion of tumor growth in the mouse glioma model is inhibited by sulfasalazine. A, glioma xenographed mice brains were sectioned for H&E staining to identify tumor regions. Left, tumor formation in two saline-treated mice. Right, two SAS-treated brains. Top row, tumor formation at 25 d posttumor injection; bottom row, tumor formation at 21 d posttumor injection. B, fluorescent images of DAPI-labeled nuclei show densely populated tumor formation in brain sections of saline and SAS-treated mice. C, merged GLT-1–, GFAP-, and DAPI-stained brain slice shows astrocytes lining the tumor perimeter. D, merged system staining in tumor is shown in sulfasalazine-treated mouse brain section.
Discussion
In this study, we present evidence that glioma cells use glutamate as an autocrine or paracrine signal to promote cell migration/invasion (Supplementary Fig. S4). This model is supported by the finding that glioma cells release glutamate that both induces [Ca2+]i oscillations and enhances glioma migration via activation of Ca2+-permeable AMPA-Rs. Furthermore, in vivo treatment of scid mice with sulfasalazine significantly reduced tumor invasion.
In past studies, we (4) found an unusual release of glutamate from gliomas that has been confirmed by others (2, 5). We initially proposed that the main purpose for such release is to inflict excitotoxic injury to vacate room for tumor expansion. This assumption was supported by the finding that C6 cell lines deficient in glutamate release fail to grow solid tumors in vivo (2). Additionally, pharmacologic inhibition of system slows tumor growth and extends the life of tumor-bearing animals (17). The findings in this study now suggest a second role for glutamate: Glutamate acts as an important promoter for cell migration; that is, as a “motogen.” Such a role for glutamate in cell migration has been previously described for immature neurons in the cerebellum but had not been envisioned as a mechanism for cancer metastasis. Specifically, granule cells migrate from the external granular layer to their final destination in the internal granular layer, and, as they move, they display spontaneous Ca2+ oscillations that are highly synchronous with cell movement (18). Although the role of Ca2+ in regulating cell migration is not fully understood, it is without question that [Ca2+]i changes are a prerequisite for movement of cells or their processes to occur. One likely way in which [Ca2+]i increases facilitate cell movement pertains to their role in regulating actin-myosin contractions (31) and focal adhesion sites (32). As cells move, they must form new attachments at their leading edge while detaching on the trailing end. In U-87 glioma cells, calcium oscillations indeed trigger the disassembly of focal adhesions (33). Clearly, the molecular pathways that transduce Ca2+ oscillations to cell movement warrant further study because this phenomenon appears universally and intrinsically associated with cell movement.
In a previous study, we showed extracellular glutamate concentration of up to 400 μmol/L (4). In the proposed autocrine/paracrine signaling mode, glutamate may become an important motogen and chemoattractant for glioma cells as they invade. Glioma cells often migrate along blood vessels, where cells display chain migration; that is, cells follow a single leading cell. In vivo, a well-known characteristic of gliomas is a breakdown of the blood-brain barrier. Hence, there is a disruption of the tight junctions formed by brain endothelial cells. A recent study now shows that glutamate, acting through both NMDA-Rs and AMPA-Rs on endothelial cells, causes a down-regulation of occludins and, in turn, a loss of barrier function of the underlying endothelial cells (34). Quite possibly, glioma cells that migrate along vessels and release glutamate could induce such a loss of endothelial tight junctions. This would allow them to gain access to nutrients and amino acids enriched in plasma. This may indeed be one reason these tumor cells seek out vessels as they migrate.
Our imaging experiments and amino acid transport studies initially evaluated cultured glioma cells and cells isolated from acute patient biopsies. These consistently showed an up-regulation of the system transporter along with expression of subunits that compose the Ca2+-permeable AMPA-Rs and suggest that the observed mechanisms are not restricted to cultured cells, but are also operant in vivo. To support this conclusion, we used a mouse glioma model in which human gliomas were xenografted into scid mice. Control mice showed tumors that were diffusely invasive at the tumor margins and consistently produced satellite tumors. However, when treated with the system inhibitor sulfasalazine by i.p. injection, tumors failed to establish satellites and microscopically did not show diffuse invasion observed in controls. These studies provide proof-of-principle evidence for the utility of sulfasalazine, a Food and Drug Administration–approved drug, to inhibit glioma invasion in vivo and offer a well-defined and possibly short path to clinical application.
Supplementary Material
Acknowledgments
Grant support: NIH grants R01NSO52634 and P50-CA97247.
We thank the Cooperative Human Tissue Network, the University of Alabama at Birmingham Brain Specialized Programs of Research Excellence under the direction of Dr. Yancey Gillespie, and the Alzheimer’s Disease Research Center under the direction of Dr. Stephen Carroll for the generous supply of human brain tissues.
References
- 1.Laerum OD, Bjerkvig R, Steinsvag S, de Ridder L. Invasiveness of primary brain tumors. Cancer Metastasis Rev. 1984;3:223–36. doi: 10.1007/BF00048386. [review] [91 refs] [DOI] [PubMed] [Google Scholar]
- 2.Takano T, Lin JH, Arcuino G, Gao Q, Yang J, Nedergaard M. Glutamate release promotes growth of malignant gliomas. Nat Med. 2001;7:1010–5. doi: 10.1038/nm0901-1010. [DOI] [PubMed] [Google Scholar]
- 3.Kaba SE, Kyritsis AP. Recognition and management of gliomas. Drugs. 1997;53:235–44. doi: 10.2165/00003495-199753020-00004. [DOI] [PubMed] [Google Scholar]
- 4.Ye ZC, Sontheimer H. Glioma cells release excitotoxic concentrations of glutamate. Cancer Res. 1999;59:4383–91. [PubMed] [Google Scholar]
- 5.Ishiuchi S, Tsuzuki K, Yoshida Y, et al. Blockage of Ca(2+)-permeable AMPA receptors suppresses migration and induces apoptosis in human glioblastoma cells. Nat Med. 2002;8:971–8. doi: 10.1038/nm746. [DOI] [PubMed] [Google Scholar]
- 6.Behrens PF, Langemann H, Strohschein R, Draeger J, Hennig J. Extracellular glutamate and other metabolites in and around RG2 rat glioma: an intracerebral micro-dialysis study. J Neurooncol. 2000;47:11–22. doi: 10.1023/a:1006426917654. [DOI] [PubMed] [Google Scholar]
- 7.Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;1:623–34. doi: 10.1016/0896-6273(88)90162-6. review. [DOI] [PubMed] [Google Scholar]
- 8.Olney JW. New mechanisms of excitatory transmitter neurotoxicity. J Neural Transm Suppl. 1994;43:47–51. [review] [12 refs] [PubMed] [Google Scholar]
- 9.Bar-Peled O, Ben-Hur H, Biegon A, et al. Distribution of glutamate transporter subtypes during human brain development. J Neurochem. 1997;69:2571–80. doi: 10.1046/j.1471-4159.1997.69062571.x. [DOI] [PubMed] [Google Scholar]
- 10.Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- 11.Ye ZC, Rothstein JD, Sontheimer H. Compromised glutamate transport in human glioma cells: reduction-mislocalization of sodium-dependent glutamate transporters and enhanced activity of cystine-glutamate exchange. J Neurosci. 1999;19:10767–77. doi: 10.1523/JNEUROSCI.19-24-10767.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sontheimer H. Malignant gliomas: perverting glutamate and ion homeostasis for selective advantage. Trends Neurosci. 2003;26:543–9. doi: 10.1016/j.tins.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 13.Oberndorfer S, Schmal T, Lahrmann H, Urbanits S, Lindner K, Grisold W. [The frequency of seizures in patients with primary brain tumors or cerebral metastases. An evaluation from the Ludwig Boltzmann Institute of Neuro-Oncology and the Department of Neurology, Kaiser Franz Josef Hospital, Vienna] Wien Klin Wochenschr. 2002;114:911–6. [PubMed] [Google Scholar]
- 14.Sato H, Tamba M, Ishii T, Bannai S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem. 1999;274:11455–8. doi: 10.1074/jbc.274.17.11455. [DOI] [PubMed] [Google Scholar]
- 15.McBean GJ. Cerebral cystine uptake: a tale of two transporters. Trends Pharmacol Sci. 2002;23:299–302. doi: 10.1016/s0165-6147(02)02060-6. [DOI] [PubMed] [Google Scholar]
- 16.McBean GJ, Flynn J. Molecular mechanisms of cystine transport. Biochem Soc Trans. 2001;29:6–22. doi: 10.1042/0300-5127:0290717. [DOI] [PubMed] [Google Scholar]
- 17.Chung WJ, Lyons SA, Nelson GM, et al. Inhibition of cystine uptake disrupts the growth of primary brain tumors. J Neurosci. 2005;25:7101–10. doi: 10.1523/JNEUROSCI.5258-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Komuro H, Rakic P. Intracellular Ca2+ fluctuations modulate the rate of neuronal migration. Neuron. 1996;17:275–85. doi: 10.1016/s0896-6273(00)80159-2. [DOI] [PubMed] [Google Scholar]
- 19.Patel SA, Warren BA, Rhoderick JF, Bridges RJ. Differentiation of substrate and non-substrate inhibitors of transport system : an obligate exchanger of l-glutamate and l-cystine. Neuropharmacology. 2004;46:273–84. doi: 10.1016/j.neuropharm.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 20.Gout PW, Buckley AR, Simms CR, Bruchovsky N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)– cystine transporter: a new action for an old drug. Leukemia. 2001;15:1633–40. doi: 10.1038/sj.leu.2402238. [DOI] [PubMed] [Google Scholar]
- 21.Fosse VM, Kolstad J, Fonnum F. A bioluminescence method for the measurement of l-glutamate: applications to the study of changes in the release of l-glutamate from lateral geniculate nucleus and superior colliculus after visual cortex ablation in rats. J Neurochem. 1986;47:340–9. doi: 10.1111/j.1471-4159.1986.tb04507.x. [DOI] [PubMed] [Google Scholar]
- 22.Ransom CB, O’Neal JT, Sontheimer H. Volume-activated chloride currents contribute to the resting conductance and invasive migration of human glioma cells. J Neurosci. 2001;21:7674–83. doi: 10.1523/JNEUROSCI.21-19-07674.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soroceanu L, Gillespie Y, Khazaeli MB, Sontheimer H. Use of chlorotoxin for targeting of primary brain tumors. Cancer Res. 1998;58:4871–9. [PubMed] [Google Scholar]
- 24.Gegelashvili G, Dehnes Y, Danbolt C, Schousboe A. The high-affinity glutamate transporters GLT1, GLAST, EAAT4 are regulated via different signalling mechanisms. Neurochemistry International. 2000;37:163–70. doi: 10.1016/s0197-0186(00)00019-x. [DOI] [PubMed] [Google Scholar]
- 25.Gegelashvili G, Civenni G, Racagni G, Danbolt NC, Schousboe I, Schousboe A. Glutamate receptor agonists up-regulate glutamate transporter GLAST in astrocytes. Neuroreport. 1996;8:261–5. doi: 10.1097/00001756-199612200-00052. [DOI] [PubMed] [Google Scholar]
- 26.Anderson CM, Swanson RA. Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia. 2000;32:1–14. [PubMed] [Google Scholar]
- 27.Rakic P, Komuro H. The role of receptor/channel activity in neuronal cell migration. J Neurobiol. 1995;26:299–315. doi: 10.1002/neu.480260303. [DOI] [PubMed] [Google Scholar]
- 28.Hollmann M, Hartley M, Heinemann S. Ca2+ permeability of KA-AMPA-gated glutamate receptor channels depends on subunit composition. Science. 1991;252:851–3. doi: 10.1126/science.1709304. [DOI] [PubMed] [Google Scholar]
- 29.Pellegrini-Giampietro DE, Bennett MV, Zukin RS. Are Ca(2+)-permeable kainate/AMPA receptors more abundant in immature brain? Neurosci Let. 1992;144:65–9. doi: 10.1016/0304-3940(92)90717-l. [DOI] [PubMed] [Google Scholar]
- 30.Deng W, Rosenberg PA, Volpe JJ, Jensen FE. Calcium-permeable AMPA/kainate receptors mediate toxicity and preconditioning by oxygen-glucose deprivation in oligodendrocyte precursors. Proc Natl Acad Sci U S A. 2003;100:6801–6. doi: 10.1073/pnas.1136624100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rakic P, Cameron RS, Komuro H. Recognition, adhesion, transmembrane signaling and cell motility in guided neuronal migration. Curr Opin Neurobiol. 1994;4:63–9. doi: 10.1016/0959-4388(94)90033-7. Review. [DOI] [PubMed] [Google Scholar]
- 32.Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Bio. 2000;1:11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- 33.Uhm JH, Gladson CL, Rao JS. The role of integrins in the malignant phenotype of gliomas. Front Biosci. 1999;4:59–61. doi: 10.2741/uhm. [DOI] [PubMed] [Google Scholar]
- 34.András IE, Deli MA, Veszelka S, et al. The NMDA and AMPA/KA receptors are involved in glutamate-induced alterations of occludin expression and phosphorylation in brain endothelial cells. J Cereb Blood Flow Metab. 2007;8:1431–43. doi: 10.1038/sj.jcbfm.9600445. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.