

Abstract
The LHX3 LIM-homeodomain transcription factor is required for correct development of the mammalian pituitary gland and spinal motoneurons. Mutations in the LHX3 gene underlie complex diseases featuring combined anterior pituitary hormone deficiency and, in specific cases, loss of neck rotation considered to result from nervous system abnormalities. The molecular basis for LHX3 protein actions in both normal and aberrant pituitary and nervous system development is poorly understood. In this study, the gene regulatory abilities of mutant LHX3 proteins associated with distinct types of diseases (LHX3a A210V, LHX3a E173Ter, and LHX3a W224Ter) were investigated. The capacity of these proteins to activate pituitary hormone and transcription factor gene promoters, nervous system target genes, and to localize to the nucleus of pituitary cells was measured. Consistent with the symptoms of patients with these mutations, the abnormal proteins displayed diminished capacities to activate the promoters of genes expressed in the pituitary gland. On nervous system promoters, several mutant proteins retained some activity. The ability of the mutant proteins to concentrate in the nucleus of pituitary cells was correlated with the retention of defined nuclear localization signals in the protein sequence, except for the E173Ter protein which unexpectedly localizes to the nucleus, likely due to the insertion of cryptic nuclear localization signals by a frame shift caused by the mutation. This study extends the molecular characterization of the severe neuroendocrine diseases associated with LHX3 gene mutations.
Keywords: homeodomain, LIM, hormone, growth, development
1. Introduction
The LHX3 transcription factor is a member of the LIM-homeodomain (LIM-HD) family of gene regulatory proteins (Hunter and Rhodes, 2005; Mullen et al., 2007). LIM-HD proteins have two amino terminal LIM domains that are used for interactions with partner proteins and a central HD that binds to recognition elements in target genes (Fig. 1A). The human LHX3 gene produces three protein isoforms: LHX3a and LHX3b, which have alternate amino termini, and a shorter protein, M2-LHX3 (Fig. 1A) (Sloop et al., 1999; Sloop et al., 2001a). LHX3 proteins have a strong activation domain in their carboxyl terminus that is required for activating pituitary-expressed genes and LHX3a has significantly higher activity on these target genes compared to LHX3b and M2-LHX3 (Sloop et al., 1999; Sloop et al., 2001a; West et al., 2004).
Fig. 1.

Proteins encoded by mutated LHX3 genes display impaired gene activation properties. A. Schematic showing wild type (WT) and mutant LHX3 proteins: a, b = LHX3a- and LHX3b-specific amino terminal domains; L1, L2 = LIM domains; HD = homeodomain; C-AD = carboxyl activation domain. Black box = “missense” protein sequence resulting from frame shift. B. Expression vectors for wild type and mutant LHX3 proteins were transiently cotransfected into pituitary GHFT1 cells with a luciferase reporter gene under the control of the FSHβ promoter. Promoter activity was assayed by measuring luciferase activity 48 hr after transfection. Negative controls (Control) received equivalent amounts of empty expression vector plasmid. Activities are mean [light units/10 seconds/μg total protein] of triplicate assays ± S.E.M. A representative experiment of at least three experiments is depicted.
Studies of knockout mice and humans with recessive LHX3 gene mutations have established that LHX3 is required during development of the pituitary gland and the nervous system (Sheng et al., 1996; Sheng et al., 1997; Sharma et al., 1998; Netchine et al., 2000; Sloop et al., 2001b; Bhangoo et al., 2006; Pfaeffle et al., 2007). Human patients with LHX3 gene mutations have combined pituitary hormone deficiency (CPHD) diseases featuring losses of growth hormone (GH), prolactin (PRL), thyroid-stimulating-hormone (TSH), follicle-stimulating hormone (FSH), and luteinizing hormone (LH). In addition, patients with mutations that affect both the amino and carboxyl termini of the protein, or that disable the protein by affecting the DNA-binding domain, have a limited rotation of the neck that is hypothesized to result from deficits in motor neuron development (Netchine et al., 2000; Sobrier et al., 2004; Bhangoo et al., 2006). A recent description of patients with novel LHX3 gene mutations included a nonsense mutation that preserved the amino terminus and HD of the protein (LHX3a W224Ter; Fig. 1A) (Pfaeffle et al., 2007). Patients with this specific genetic alteration have CPHD but fail to display the neck dysfunction previously associated with all LHX3 mutations. These findings are consistent with the hypothesis that the roles of LHX3 in pituitary development and function require the carboxyl terminus but the amino terminus and HD are sufficient for nervous system development (Sloop et al., 2001a; Thaler et al., 2002).
In this study, we investigated the ability of unique mutant LHX3 proteins to activate representative pituitary hormone gene promoters, pituitary transcription factor promoters, nervous system target genes, and to localize to the nucleus in the context of the pituitary cell microenvironment.
2. Materials and methods
2.1. Plasmid construction and site-directed mutagenesis
The construction of LHX3 cDNA expression plasmids in pcDNA3-based vectors (Invitrogen, Carlsbad, CA) has been described (Sloop et al., 1999; Sloop et al., 2001b; Pfaeffle et al., 2007). Expression constructs for ISL1, NeuroM, and E47 were generous gifts from Dr. Gordon Gill (University of California San Diego), Dr. Teri Belecky-Adams (IUPUI), and Dr. Stephen Konieczny (Purdue University), respectively. The FSHβ, TSHβ, PIT1 EP0.5 enhancer/promoter, and LHX3 consensus binding site (3x LBC) luciferase reporter plasmids were as described (Drolet et al., 1991; Rhodes et al., 1993; Bridwell et al., 2001; West et al., 2004). The M250 region of the Hb9 gene (Thaler et al., 2002) was amplified from C57black6 mouse genomic DNA by the PCR using the following primers: 5′-cgcggatcctttctcgcaacacttccaggctcag-3′, 5′-gaagatctcccctactctccctacagtctctgg-3′. This amplicon was ligated into the BamHI site of the 36 bp PRL minimal promoter/luciferase reporter gene (Bridwell et al., 2001). A similar Hb9 enhancer plasmid was a generous gift from Dr. Samuel Pfaff (Salk Institute). Wild type and mutant expression vectors for enhanced cyan fluorescent protein (CFP)-LHX3 fusion proteins were generated by cloning cDNAs into pECFP-C1 (Clontech, Palo Alto, CA). The integrity of all plasmids was confirmed by DNA sequencing on both strands (Biochemistry Biotechnology Facility, Indiana University School of Medicine).
2.2. Cell culture and transfection assays
Mouse GHFT1 pituitary cells and mouse embryonal carcinoma P19 cells were kind gifts of Pamela Mellon (University of California San Diego) and Andrew Lindsley (Indiana University School of Medicine), respectively. Cells were cultured in 6 × 35 mm dishes as described (Skerjanc, 1999; Sloop et al., 2001b). Expression vectors and luciferase reporter genes were transiently transfected into the cells using Lipofectamine 2000 (Invitrogen). 3–5 × 105 cells in a 35 mm well were transfected with 0.5 μg of reporter gene plasmid and 0.25–1 μg of expression vector. Parallel control samples received equivalent amounts of empty expression vector DNA. Assays were performed in triplicate. Forty-eight hours following transfection, cells were lysed in assay buffer and luciferase activity was measured using a Beckman Coulter LD400 plate reader/luminometer (Beckman Coulter, Fullerton, CA) as described (Sloop et al., 2001a). Following determination of total protein levels by the Bradford method (BioRad, Hercules, CA), luciferase activities were normalized to protein concentration. Experiments were repeated at least three times.
2.3. Confocal and fluorescence microscopy
GHFT1 cells were plated on chamber slide flaskettes (Nalge Nunc, Naperville, IL) and were transiently transfected with expression vectors as described above. Forty-eight hours post-transfection, cells were fixed in 2% formaldehyde in phosphate-buffered saline (PBS), pH 7.7. To detect wild type and mutant CFP-LHX3, the nuclei of cells were counterstained with Hoechst 33258 dye and then visualized by fluorescence microscopy using a Nikon Eclipse TE 200-U inverted microscope or by confocal microscopy using a Zeiss LSM510 microscope with Coherent Enterprises UV and argon lasers.
3. Results and Discussion
3.1. Transcriptional properties of disease-associated LHX3 proteins
A recent study has significantly extended the known types of LHX3 gene mutations (Pfaeffle et al., 2007). In 7 patients with CPHD from 4 consanguineous families, 4 novel, recessive mutations were identified: a deletion of the entire gene (del/del), nonsense mutations causing truncated proteins (E173Ter, W224Ter), and a transition mutation causing a substitution in the HD (A210V) (Fig. 1A). Whereas all other known LHX3 mutations are associated with limited neck rotation, patients with the W224Ter mutation, that preserves the LIM domains and the HD, do not exhibit this symptom (Pfaeffle et al., 2007). To better understand the molecular properties of the aberrant LHX3 proteins associated with these diseases, we performed gene regulation experiments using hormone and transcription factor promoters that are expressed in a pituitary-specific fashion.
CPHD patients with mutations in LHX3 present with deficiencies in circulating gonadotropin hormones (LH and FSH). The aberrant proteins resulting from the mutations have been shown to impact LHX3 regulation of the alpha glycoprotein (αGSU) gene that encodes the common protein subunits of the LH and FSH hormones (Sloop et al., 2001b; Pfaeffle et al., 2007). Recently, LHX3 has been shown to directly increase transcription from the FSHβ gene that encodes the specific beta subunit of the FSH hormone but to not affect transcription from the LHβ promoter (West et al., 2004). In order to better understand the molecular origins of the FSH deficiency observed in the patients, cotransfections with a FSHβ-luciferase reporter gene and LHX3 expression vectors were conducted in pituitary GHFT1 cells. In addition to wild type protein, vectors encoding the E173Ter, A210V, and W224Ter proteins were examined. A short protein lacking the HD and the carboxyl terminus (1–150 amino acids only) also was included as a comparative negative control. LHX3a activated the FSHβ promoter, while the mutant LHX3 proteins had poor or insignificant activities (Fig. 1B). Similar observations were made using LHX3b proteins, except that activation of FSHβ by wild type LHX3b is not as striking as that by LHX3a (data not shown). These data are consistent with loss of LHX3 gene function resulting in loss of gonadotropin hormone production in these patients.
The TSHβ gene encodes the beta subunit of the TSH pituitary metabolic hormone. The αGSU protein provides the common subunit to complete the functional hormone. Patients with LHX3 gene mutations have deficiencies of TSH (Pfaeffle et al., 2007). We therefore examined regulation of the TSHβ promoter by the “mutant” LHX3 proteins. Activation of the TSHβ promoter is complex, requiring combinations of pituitary transcription factors such as LHX3 and PIT1 (Bach et al., 1995). Wild type and mutant LHX3 proteins were expressed in pituitary GHFT1 cells with a −1.2 kb TSHβ reporter gene. All of the mutant LHX3a proteins had significant reductions in activation levels using this promoter (Fig. 2A). Similar data were recorded in experiments testing an equivalent series of LHX3b isoform proteins (Fig. 2B). Comparable data have been reported for regulation of the PRL gene promoter, which has a similar mechanism of regulation by PIT1 and LHX3 (Pfaeffle et al., 2007). The inability of the aberrant LHX3 proteins encoded by the mutant genes to activate this reporter gene correlates with the thyroid hormone deficiencies in the patients (Pfäffle et al., 2007).
Fig. 2.
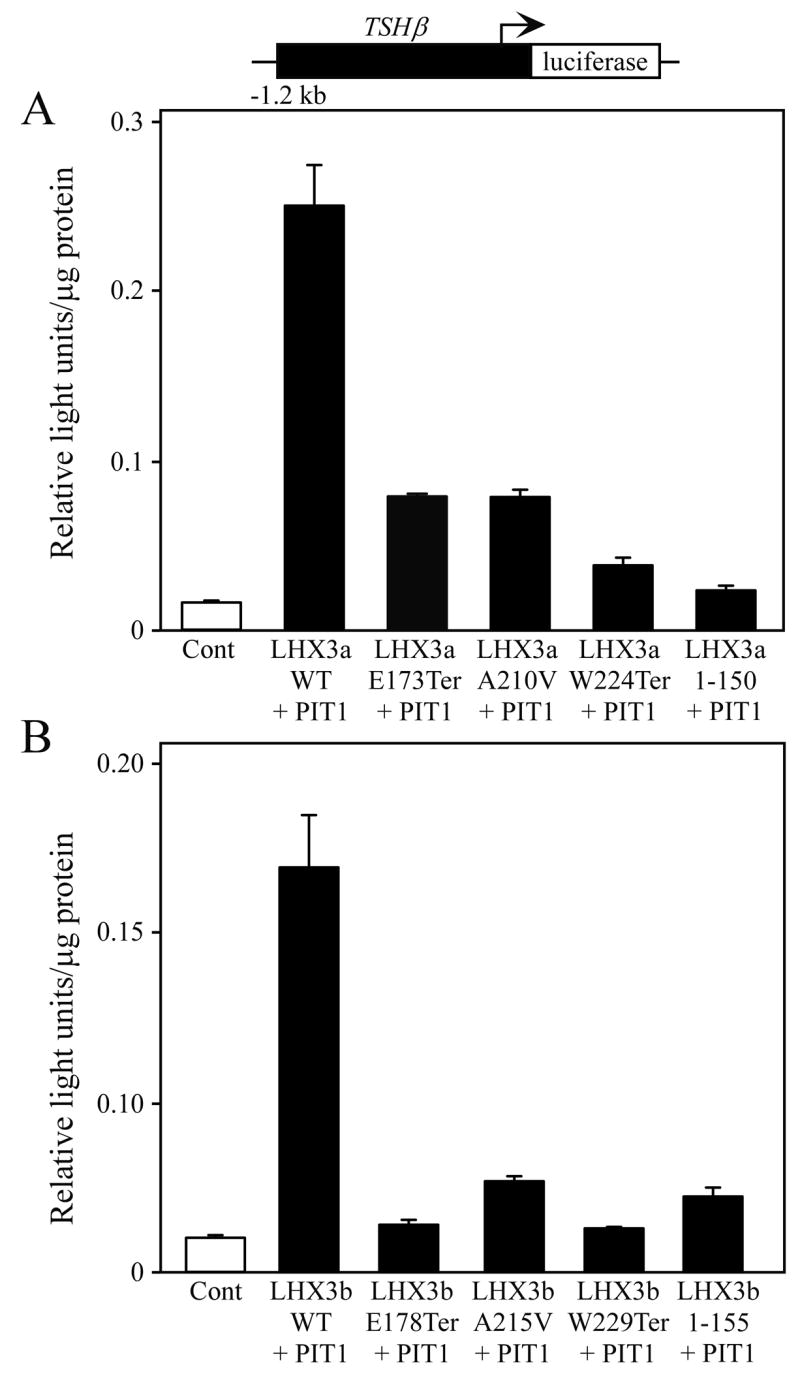
Activation of a luciferase reporter gene containing the TSHβ gene promoter (black boxes) by normal and mutant LHX3 protein isoforms. The indicated PIT1, LHX3a (panel A), LHX3b (panel B) or control expression vectors were transiently co-transfected into mouse pituitary GHFT1 cells. Promoter activity was assayed by measurement of luciferase activity after 48 hours. Activities are mean [light units/10 seconds/μg total protein] of triplicate assays ± S.E.M. WT = wild type.
We next tested the ability of the aberrant proteins to activate the PIT1 pituitary transcription factor gene. PIT1 lies downstream of LHX3 in models of the pathways controlling pituitary development (Mullen et al., 2007). In CPHD patients with loss of PIT1 function there is a specific loss of GH, PRL, and TSH hormones, consistent with PIT1 having a later, more restricted, role than LHX3 in pituitary organogenesis (reviewed by Dattani, 2005; Zhu et al., 2005). As for TSHβ, regulation of the PIT1 gene involves the combinatorial actions of multiple transcription factors interacting with both enhancer and promoter elements, including autoregulatory functions (Rhodes et al., 1993; Bach et al., 1995). Whereas wild type LHX3a activated a reporter gene containing the proximal promoter and the distal enhancer of the PIT1 locus, each of the aberrant LHX3a proteins were notably compromised in gene activation, as was the control 1–150 amino acid protein that lacks the HD and carboxyl terminus (Fig. 3A). Similar activity patterns were observed with the LHX3b proteins (Fig. 3B).
Fig. 3.
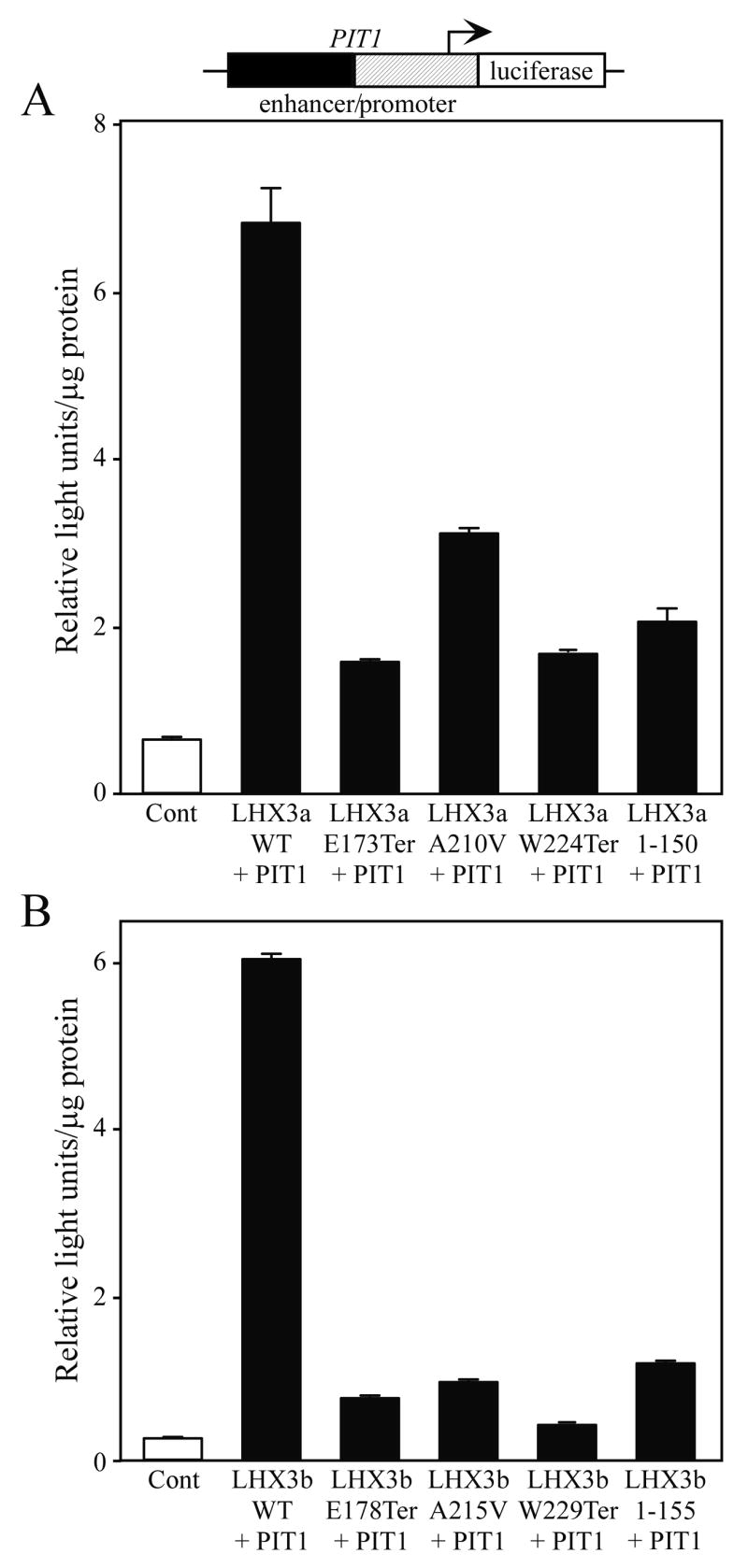
Activation of a luciferase reporter gene containing the PIT1 transcription factor gene enhancer/promoter (black/hatched boxes) by normal and mutant LHX3 protein isoforms. The indicated PIT1, LHX3a (panel A), LHX3b (panel B) or control expression vectors were transiently co-transfected into pituitary GHFT1 cells. Promoter activity was assayed by measurement of luciferase activity after 48 hours. Activities are mean [light units/10 seconds/μg total protein] of triplicate assays ± S.E.M. WT = wild type.
LHX3 proteins recognize a similar AT-rich DNA site, the LHX3 binding consensus (or LBC; Bridwell et al., 2001; Yaden et al., 2005). We also tested the abilities of wild type and mutant LHX3 proteins to activate a simple, synthetic reporter gene containing three copies of a LBC element. None of the mutant proteins activated this reporter gene, except the M2-LHX3 A77V point mutant that displayed low levels of activity (Fig. 4).
Fig. 4.

Activation of a synthetic luciferase reporter gene containing a minimal PRL gene promoter (black box) and 3 copies of the LHX3 consensus binding element (LBC) by normal and mutant LHX3 protein isoforms. The indicated LHX3 or control expression vectors were transiently co-transfected into pituitary GHFT1 cells. A. LHX3a isoforms. B. LHX3b isoforms. C. M2-LHX3 isoforms. Promoter activity was assayed by measurement of luciferase activity after 48 hours. Activities are mean [light units/10 seconds/μg total protein] of triplicate assays ± S.E.M. WT = wild type.
Overall, these data show that the E174Ter, A210V, W224Ter LHX3 proteins have impaired activation of pituitary hormone and transcription factor genes consistent with the CPHD diseases displayed by patients that are homozygous for the affected alleles encoding these aberrant proteins. The arrest of pituitary development at an early stage in mice with a null mutation of the Lhx3 gene and the pituitary gland morphological defects in human patients with LHX3 gene mutations indicate that LHX3 also is required for early events in pituitary development (Sheng et al., 1996; Netchine et al., 2000; Bhangoo et al., 2006; Pfaeffle et al., 2007). It is likely that the described mutations also significantly impact the activities of LHX3 on yet-to-be-defined early target genes resulting in incomplete or aberrant development of the gland.
3.2. Combinatorial activation of nervous system genes
The specification of motor neurons in the developing nervous system requires the combinatorial actions of many transcription factors, including multiple members of the LIM-HD family forming a “LIM-HD code” (Thaler et al., 2002; Thaler et al., 2004; Hunter and Rhodes, 2005). LHX3 has been shown to participate in the differentiation of motor neurons and V2 interneurons (Thaler et al., 2002). For V2 interneuron specification, LHX3 forms a tetrameric complex comprised of two LHX3 proteins and two copies of the LIM-interacting protein NLI. In order to specify motoneurons, for example on an enhancer of the Hb9 gene, LHX3 participates in a multiprotein gene regulatory complex involving the ISL1, NLI, and the NeuroM and E47 basic helix-loop-helix factors (Thaler et al., 2002). To test whether the mutant human LHX3 proteins could function in this complex, we reconstituted the Hb9 gene enhancer assay described by Thaler at al. (Thaler et al., 2002) in mouse P19 embryonal carcinoma cells. Only when ISL1, NeuroM, E47, and LHX3a were transfected (NLI is present at high endogenous levels), was strong transcription induced from a reporter gene containing the Hb9 gene enhancer and a minimal promoter (Fig. 5A). Transfections testing transcription factors individually gave readouts less than assays involving two or more factors (data not shown). Of the three LHX3 protein isoforms, LHX3a has the most potent actions on pituitary hormone gene promoters and LHX3b is alone a comparatively poor activator of most pituitary hormone promoters (Sloop et al., 1999; Sloop et al., 2001a). Using this assay, we were for the first time able to test the hypothesis that perhaps LHX3b had notable activity on nervous system gene targets compared to LHX3a. However, activation by LHX3b-containing complexes was not as robust as those involving LHX3a (Fig. 5A). The LIM-deficient M2-LHX3 isoform had even less activity: this result was not surprising because part of the protein-protein interactions involved in the proposed model for Hb9 gene activation include actions by the LIM domains of LHX3 as well as the HD (Thaler et al., 2002).
Fig. 5.

Mutant LHX3 proteins contribute poorly to combinatorial activation of a nervous system transcription unit. A. A luciferase reporter gene containing a minimal PRL promoter under the control of the enhancer of the Hb9 transcription factor gene (Thaler et al., 2002) was transiently co-transfected into mouse P19 embryonal teratocarcinoma cells with the indicated combinations of wild type human LHX3 isoforms, NeuroM (NeuM), E47, and ISL1 transcription factor expression vectors. Promoter activity was assayed by measurement of luciferase activity after 48 hours. Activities are mean [light units/10 seconds/μg total protein] of triplicate assays ± S.E.M. The dotted line indicates activity with all factors except LHX3. B. Similar experiment testing mutant LHX3 proteins.
We next compared the actions of the mutant LHX3 proteins with wild type LHX3a. The truncated E173Ter protein was not active but the A210V and W224Ter proteins displayed activity in this assay (Fig. 5B). The residual activity of the W224Ter protein is consistent with the hypothesis that the amino terminus and HD are required for the nervous system actions of LHX3 (Thaler et al., 2002) and the missing carboxyl terminus contains an activation domain required for pituitary gene transcription functions (Sloop et al., 2001a). However, the A210V protein also has similar activity in this assay. The amino acid substitution in the HD of this protein reduces DNA binding but does not abolish it (Pfaeffle et al., 2007). The intact LIM domains and residual DNA binding in this protein may be sufficient to have some activity in this assay but overall the impact on DNA binding function of the protein results in deleterious effects on pituitary gland and nervous system development.
3.3. Nuclear localization
In order to function as transcription factors, proteins such as LHX3 must enter the nuclear compartment. Nuclear localization of LHX3 proteins is complex, requiring the combined actions of four nuclear localization signals (NLSs) located within and just after the HD (Parker et al., 2000). To test whether the mutant LHX3 proteins were able to enter the nucleus, we created expression vectors for enhanced cyan fluorescent protein (CFP)-LHX3 fusion proteins. These plasmids were transfected into GHFT1 pituitary cells and the localization of the proteins was visualized by confocal microscopy. In parallel control experiments, CFP was found in both the cytoplasmic and nuclear compartments (Fig. 6A) but CFP-LHX3a was highly enriched in the nucleus of the cells (Fig. 6B). As expected, the LHX3a W224Ter protein that retains all four NLSs was observed in the nucleus (Fig. 6E). The LHX3a A210V protein has a single mutation in the third NLS (known as the “basic 3” or B3 NLS). However, this substitution affects a non-basic amino acid residue and may not affect the function of this NLS. Further, even if the B3 NLS is impaired, nuclear localization may be possible considering the known combinatorial properties of LHX3 NLSs (Parker et al., 2000). Indeed, this protein retained nuclear localization (Fig. 6D). Intriguingly, the LHX3a E173Ter protein that has a short “missense” sequence of amino acids (instead of the second LIM domain, the HD and the carboxyl terminus) due to a translation frame shift (Fig. 1A), was unexpectedly found to be concentrated in the nucleus. This result is likely not due to the small size of the protein because the smaller LHX3a 1–150 protein that lacks all four NLSs was not restricted to the nucleus in parallel controls (Fig. 1F). Instead, examination of the missense amino acid sequence introduced by the frame shift suggests an alternate explanation. This sequence is VLSRPRRWCAAPRTSCTTCTALPASCASGSWPRATSSTSWRTAGSCARRTTKPPSSERP RPRPSGRARPSPPSSWRR, a sequence rich in basic amino acids that may form cryptic NLSs. Indeed, testing of this sequence by the ESLpred and LOCtree programs that predict intracellular targeting of proteins (Bhasin and Raghava, 2004; Nair and Rost, 2005), reveals that this sequence is likely to confer nuclear localization (data not shown). These data indicate that the aberrant LHX3 proteins retain the ability to localize to the nucleus and that the lack of transcriptional activity is likely due to loss of DNA binding and trans-activation functions (Pfäffle et al., 2007; and data not shown). It is not possible to examine the expression of LHX3 proteins in the pituitaries of affected patients. For the E173Ter and W224Ter mRNAs, the position of the premature termination codons suggests that these RNAs might be targeted for nonsense-mediated decay, causing an absence of LHX3 proteins. However, there are exceptions to the nonsense-mediated decay rules (Holbrook et al., 2004). Further, the delayed and different syndrome of the homozygous W224Ter patients suggests that some LHX3 function is retained in these individuals.
Fig. 6.
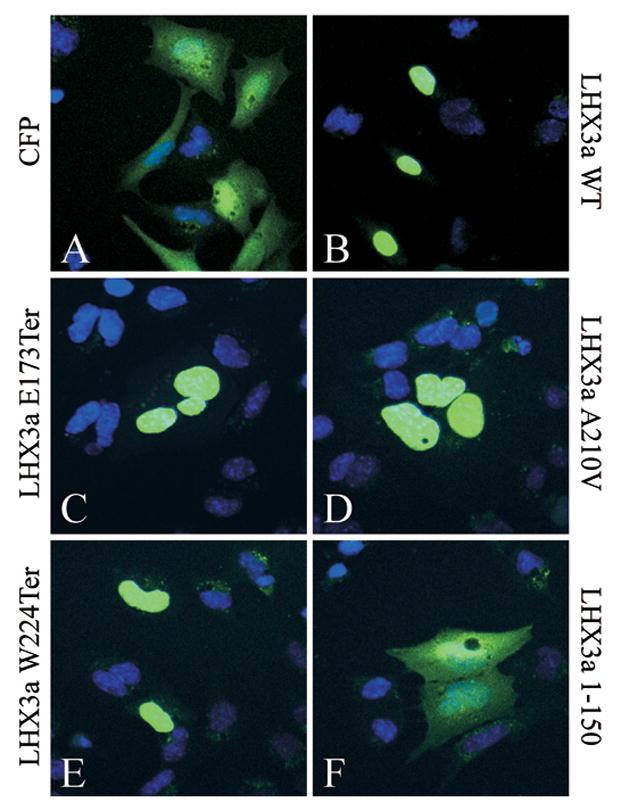
Intracellular localization of wild type (WT) and mutant LHX3a proteins fused to the cyan fluorescent protein (CFP). The indicated expression vectors were transiently co-transfected into pituitary GHFT1 cells. Forty-eight hours post-transfection, cells were fixed and the nuclei of cells were counterstained with Hoechst 33258 dye (blue color). Fluorescence was visualized by confocal microscopy. Representative fields are shown.
4. Conclusions
Consistent with the symptoms of patients carrying the corresponding mutations in the LHX3 gene, the E174Ter, A210V, W224Ter LHX3 proteins have impaired activation of pituitary hormone genes, pituitary transcription factor genes, and a synthetic reporter gene containing a high affinity LHX3 binding element.
On the Hb9 nervous system transcription factor gene enhancer, LHX3a has greater activity than LHX3b or M2-LHX3.
LHX3a E174Ter cannot participate in combinatorial transcription factor activation of the Hb9 gene enhancer, but LHX3a A210V and LHX3a W224Ter retain some function.
The mutant LHX3 proteins retain the critical nuclear localization signals and are therefore found in the nucleus of pituitary cells. A missense protein sequence introduced by the E173Ter mutations contains basic residues that apparently mimic nuclear localization signals.
Acknowledgments
We are very grateful to Drs. T. Belecky-Adams, G. Gill, S. Konieczny, A. Lindsley, P. Mellon, and S. Pfaff for providing reagents. Supported by grants to SJR from the National Institutes of Health (HD42024) and the National Science Foundation (IBN 0131702). JJS was an Elizabeth Steele Creveling Memorial Scholar during this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bach I, Rhodes SJ, Pearse RV, 2nd, Heinzel T, Gloss B, Scully KM, Sawchenko PE, Rosenfeld MG. P-Lim, a LIM homeodomain factor, is expressed during pituitary organ and cell commitment and synergizes with Pit-1. Proc Natl Acad Sci U S A. 1995;92:2720–4. doi: 10.1073/pnas.92.7.2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhangoo AP, Hunter CS, Savage JJ, Anhalt H, Pavlakis S, Walvoord EC, Ten S, Rhodes SJ. Clinical case seminar: a novel LHX3 mutation presenting as combined pituitary hormonal deficiency. J Clin Endocrinol Metab. 2006;91:747–53. doi: 10.1210/jc.2005-2360. [DOI] [PubMed] [Google Scholar]
- Bhasin M, Raghava GP. ESLpred: SVM-based method for subcellular localization of eukaryotic proteins using dipeptide composition and PSI-BLAST. Nucleic Acids Res. 2004;32:W414–9. doi: 10.1093/nar/gkh350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridwell JA, Price JR, Parker GE, McCutchan Schiller A, Sloop KW, Rhodes SJ. Role of the LIM domains in DNA recognition by the Lhx3 neuroendocrine transcription factor. Gene. 2001;277:239–50. doi: 10.1016/s0378-1119(01)00704-1. [DOI] [PubMed] [Google Scholar]
- Dattani MT. Growth hormone deficiency and combined pituitary hormone deficiency: does the genotype matter? Clin Endocrinol (Oxf) 2005;63:121–30. doi: 10.1111/j.1365-2265.2005.02289.x. [DOI] [PubMed] [Google Scholar]
- Drolet DW, Scully KM, Simmons DM, Wegner M, Chu KT, Swanson LW, Rosenfeld MG. TEF, a transcription factor expressed specifically in the anterior pituitary during embryogenesis, defines a new class of leucine zipper proteins. Genes Dev. 1991;5:1739–53. doi: 10.1101/gad.5.10.1739. [DOI] [PubMed] [Google Scholar]
- Holbrook JA, Neu-Yilik G, Hentze MW, Kulozik AE. Nonsense-mediated decay approaches the clinic. Nat Genet. 2004;36:801–8. doi: 10.1038/ng1403. [DOI] [PubMed] [Google Scholar]
- Hunter CS, Rhodes SJ. LIM-homeodomain genes in mammalian development and human disease. Mol Biol Rep. 2005;32:67–77. doi: 10.1007/s11033-004-7657-z. [DOI] [PubMed] [Google Scholar]
- Mullen RD, Colvin SC, Hunter CS, Savage JJ, Walvoord EC, Bhangoo AP, Ten S, Weigel J, Pfaeffle RW, Rhodes SJ. Roles of the LHX3 and LHX4 LIM-homeodomain factors in pituitary development. Mol Cell Endocrinol. 2007;265–266:190–5. doi: 10.1016/j.mce.2006.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair R, Rost B. Mimicking cellular sorting improves prediction of subcellular localization. J Mol Biol. 2005;348:85–100. doi: 10.1016/j.jmb.2005.02.025. [DOI] [PubMed] [Google Scholar]
- Netchine I, Sobrier ML, Krude H, Schnabel D, Maghnie M, Marcos E, Duriez B, Cacheux V, Moers A, Goossens M, Gruters A, Amselem S. Mutations in LHX3 result in a new syndrome revealed by combined pituitary hormone deficiency. Nat Genet. 2000;25:182–6. doi: 10.1038/76041. [DOI] [PubMed] [Google Scholar]
- Parker GE, Sandoval RM, Feister HA, Bidwell JP, Rhodes SJ. The homeodomain coordinates nuclear entry of the Lhx3 neuroendocrine transcription factor and association with the nuclear matrix. J Biol Chem. 2000;275:23891–8. doi: 10.1074/jbc.M000377200. [DOI] [PubMed] [Google Scholar]
- Pfaeffle RW, Savage JJ, Hunter CS, Palme C, Ahlmann M, Kumar P, Bellone S, Schoenau E, Korsch E, Brämswig J, Stobbe H, Blum WF, Rhodes SJ. Four novel mutations of the LHX3 gene cause combined pituitary hormone deficiencies with or without limited neck rotation. J Clin Endocrinol Metab. 2007 Feb 27; doi: 10.1210/jc.2006-2177. In Press. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Rhodes SJ, Chen R, DiMattia GE, Scully KM, Kalla KA, Lin SC, Yu VC, Rosenfeld MG. A tissue-specific enhancer confers Pit-1-dependent morphogen inducibility and autoregulation on the pit-1 gene. Genes Dev. 1993;7:913–32. doi: 10.1101/gad.7.6.913. [DOI] [PubMed] [Google Scholar]
- Sharma K, Sheng HZ, Lettieri K, Li H, Karavanov A, Potter S, Westphal H, Pfaff SL. LIM homeodomain factors Lhx3 and Lhx4 assign subtype identities for motor neurons. Cell. 1998;95:817–28. doi: 10.1016/s0092-8674(00)81704-3. [DOI] [PubMed] [Google Scholar]
- Sheng HZ, Moriyama K, Yamashita T, Li H, Potter SS, Mahon KA, Westphal H. Multistep control of pituitary organogenesis. Science. 1997;278:1809–12. doi: 10.1126/science.278.5344.1809. [DOI] [PubMed] [Google Scholar]
- Sheng HZ, Zhadanov AB, Mosinger B, Jr, Fujii T, Bertuzzi S, Grinberg A, Lee EJ, Huang SP, Mahon KA, Westphal H. Specification of pituitary cell lineages by the LIM homeobox gene Lhx3. Science. 1996;272:1004–7. doi: 10.1126/science.272.5264.1004. [DOI] [PubMed] [Google Scholar]
- Skerjanc IS. Cardiac and skeletal muscle development in P19 embryonal carcinoma cells. Trends Cardiovasc Med. 1999;9:139–43. doi: 10.1016/s1050-1738(99)00017-1. [DOI] [PubMed] [Google Scholar]
- Sloop KW, Dwyer CJ, Rhodes SJ. An isoform-specific inhibitory domain regulates the LHX3 LIM homeodomain factor holoprotein and the production of a functional alternate translation form. J Biol Chem. 2001a;276:36311–9. doi: 10.1074/jbc.M103888200. [DOI] [PubMed] [Google Scholar]
- Sloop KW, Meier BC, Bridwell JL, Parker GE, Schiller AM, Rhodes SJ. Differential activation of pituitary hormone genes by human Lhx3 isoforms with distinct DNA binding properties. Mol Endocrinol. 1999;13:2212–25. doi: 10.1210/mend.13.12.0395. [DOI] [PubMed] [Google Scholar]
- Sloop KW, Parker GE, Hanna KR, Wright HA, Rhodes SJ. LHX3 transcription factor mutations associated with combined pituitary hormone deficiency impair the activation of pituitary target genes. Gene. 2001b;265:61–9. doi: 10.1016/s0378-1119(01)00369-9. [DOI] [PubMed] [Google Scholar]
- Sobrier ML, Attie-Bitach T, Netchine I, Encha-Razavi F, Vekemans M, Amselem S. Pathophysiology of syndromic combined pituitary hormone deficiency due to a LHX3 defect in light of LHX3 and LHX4 expression during early human development. Gene Expr Patterns. 2004;5:279–84. doi: 10.1016/j.modgep.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Thaler JP, Koo SJ, Kania A, Lettieri K, Andrews S, Cox C, Jessell TM, Pfaff SL. A postmitotic role for Isl-class LIM homeodomain proteins in the assignment of visceral spinal motor neuron identity. Neuron. 2004;41:337–50. doi: 10.1016/s0896-6273(04)00011-x. [DOI] [PubMed] [Google Scholar]
- Thaler JP, Lee SK, Jurata LW, Gill GN, Pfaff SL. LIM factor Lhx3 contributes to the specification of motor neuron and interneuron identity through cell-type-specific protein-protein interactions. Cell. 2002;110:237–49. doi: 10.1016/s0092-8674(02)00823-1. [DOI] [PubMed] [Google Scholar]
- West BE, Parker GE, Savage JJ, Kiratipranon P, Toomey KS, Beach LR, Colvin SC, Sloop KW, Rhodes SJ. Regulation of the follicle-stimulating hormone beta gene by the LHX3 LIM-homeodomain transcription factor. Endocrinology. 2004;145:4866–79. doi: 10.1210/en.2004-0598. [DOI] [PubMed] [Google Scholar]
- Yaden BC, Savage JJ, Hunter CS, Rhodes SJ. DNA recognition properties of the LHX3b LIM homeodomain transcription factor. Mol Biol Rep. 2005;32:1–6. doi: 10.1007/s11033-004-4069-z. [DOI] [PubMed] [Google Scholar]
- Zhu X, Lin CR, Prefontaine GG, Tollkuhn J, Rosenfeld MG. Genetic control of pituitary development and hypopituitarism. Curr Opin Genet Dev. 2005;15:332–40. doi: 10.1016/j.gde.2005.04.011. [DOI] [PubMed] [Google Scholar]