

Abstract
When the orphan nuclear receptors TR2 and TR4, the DNA-binding subunits of the DRED repressor complex, are forcibly expressed in erythroid cells of transgenic mice, embryos exhibit a transient mid-gestational anemia as a consequence of a reduction in the number of primitive erythroid cells. GATA-1 mRNA is specifically diminished in the erythroid cells of these TR2/TR4 transgenic embryos as it is in human CD34+ progenitor cells transfected with forcibly expressed TR2/TR4. In contrast, in loss-of-function studies analyzing either Tr2- or Tr4-germline-null mutant mice or human CD34+ progenitor cells transfected with force-expressed TR2 and TR4 short hairpin RNAs (shRNAs), GATA-1 mRNA is induced. An evolutionarily conserved direct repeat (DR) element, a canonical binding site for nuclear receptors, was identified in the GATA1 hematopoietic enhancer (G1HE), and TR2/TR4 binds to that site in vitro and in vivo. Mutation of that DR element led to elevated Gata1 promoter activity, and reduced promoter responsiveness to cotransfected TR2/TR4. Thus, TR2/TR4 directly represses Gata1/GATA1 transcription in murine and human erythroid progenitor cells through an evolutionarily conserved binding site within a well-characterized, tissue-specific Gata1 enhancer, thereby providing a mechanism by which Gata1 can be directly silenced during terminal erythroid maturation.
[Keywords: Primitive, definitive, erythropoiesis, DRED, G1HE]
In vertebrates, the hematopoietic compartment requires continual replenishment, and billions of red blood cells must be replaced daily in an adult human. This formidable biosynthetic task is achieved by robust differentiation induction from a relatively small number of pluripotent hematopoietic stem and progenitor cells into mature erythrocytes. Vertebrates exhibit two distinct waves of red blood cell production: primitive erythropoiesis, a transient phase stimulated in the extraembryonic yolk sac, and definitive erythropoiesis, which begins in the fetal liver but later moves to the bone marrow.
Over the past two decades, accumulated evidence has shown that transcription factor GATA-1 plays an essential role in erythroid differentiation (Ohneda and Yamamoto 2002). GATA-1 is the founding member of the GATA family of zinc finger transcription factors, and was originally identified by its ability to bind to a DNA regulatory motif (WGATAR) found in the promoters and/or enhancers of virtually all erythroid-specific genes. With the exception of Sertoli cells in the testis (Ito et al. 1993), where its function is still uncertain, GATA-1 expression is restricted to hematopoietic cells and their progenitors, where it fulfills multiple, vital differentiation functions. GATA-1 is expressed at low levels in primitive and definitive multipotential hematopoietic progenitor cells (Fujiwara et al. 1996), but becomes abundantly expressed in committed erythroid precursors, megakaryocytes (Martin et al. 1990), eosinophils, and mast cells (Zon et al. 1993).
Genetic studies have shown that GATA-1 is required for both primitive and definitive erythropoiesis. Mouse embryos lacking GATA-1 die of profound anemia between 10 and 11 d post-coitus (dpc) (Fujiwara et al. 1996). Gata1-null embryonic stem (ES) cells fail to generate primitive erythroid progenitors (Weiss et al. 1994), while definitive erythroid progenitors are normal in number but undergo developmental arrest and apoptosis at the proerythroblast stage (Weiss et al. 1994; Weiss and Orkin 1995). GATA-1 abundance is reduced in erythroblasts during the final steps of erythroid differentiation (Whyatt et al. 2000), but the mechanism by which GATA-1 synthesis is inhibited during the latter stages of erythropoiesis is unknown.
TR2 and TR4 are two closely related members of the nuclear receptor superfamily, with no known ligand (thus categorizing them as “orphans”). They are expressed in many tissues, including erythroid cells. TR2 and TR4 form homodimers or heterodimers with one another and bind to directly repeated (DR) AGGTCA sequences separated by 0- to 6-nucleotide spacing (DR0 to DR6) (Lee et al. 1998a), consensus binding sites for nonsteroidal nuclear receptors. Our previous studies implicated TR2 and TR4 as the core DNA-binding subunits of a large macromolecular repressor complex (DRED, for direct repeat erythroid-definitive) that suppresses fetal and embryonic β-type globin gene transcription in definitive erythroid cells (Tanimoto et al. 2000; Tanabe et al. 2002). A growing list of transcriptional cofactors has been shown to interact with TR2 and TR4: for example, histone deacetylases 1, 3, and 4, and the nuclear receptor coactivator/corepressor, RIP140 (Lee et al. 1998b; Franco et al. 2001). Depending on the context of their recognition sequences or on cellular conditions, TR2 and TR4 can both activate and repress cellular target genes (Lee et al. 2002). Recent gene ablation studies have also begun to reveal some in vivo functions of these receptors. Mice lacking Tr2 are viable and show no overt mutant phenotypes (Shyr et al. 2002), while Tr4 germline mutants display reproductive and neurological deficiencies (Collins et al. 2004; Mu et al. 2004; Chen et al. 2005).
Our previous experiments focused on the possibility that the DRED repressor, acting through its TR2/TR4 heterodimeric DNA-binding scaffold, directly regulates the human embryonic ε-globin and fetal γ-globin genes by binding to direct repeat (DR) elements in their promoters (Tanimoto et al. 2000; Tanabe et al. 2002, 2007). Here we show that transgenic mice in which either TR4 alone (TgTR4) or TR4 with TR2 (TgTR2/TR4) are forcibly expressed display a pronounced but transient mid-embryonic anemia. Progenitor assays conducted on hematopoietic precursors from these embryos revealed defects in primitive erythroid precursor formation, which could not be explained simply by the effects of TR2/TR4 on globin gene transcription.
To determine the molecular basis for the observed transient anemia in the TgTR2/TR4 mice, we first examined erythroid cells of TgTR2/TR4 embryos and quantified the abundance of several transcription factors that have been shown to be critical for erythroid development. Most of these transcription factors were unperturbed in amount, but GATA-1 transcript levels were specifically and significantly reduced in the embryonic blood and fetal livers of TgTR2/TR4 mice. Furthermore, GATA-1 mRNA levels were elevated (in comparison with wild type) in Tr2- or Tr4-null mutant mice. Similarly, GATA-1 mRNA abundance increased in in vitro differentiated human erythroblasts that were reduced in TR2 and TR4 mRNAs by RNA interference (RNAi)-mediated depletion. An evolutionarily conserved DR element was identified within the well-characterized GATA1 hematopoietic enhancer (G1HE) (Vyas et al. 1999b; Nishimura et al. 2000), and TR2 and TR4 bind to this site in vitro. Furthermore, chromatin immunoprecipitation (ChIP) assays revealed that TR2 and TR4 bind to the element in vivo in differentiated human erythroblasts, and we found that mutation of the G1HE DR site significantly reduced the effects of cotransfected TR2/TR4 on G1HE-dependent Gata1 transcription. Taken together, the data show that TR2 and TR4 are evolutionarily conserved transcriptional repressors of the murine and human GATA1 genes, and thus they directly affect erythroid differentiation. We conclude that the DRED repressor plays an important role in the regulation of Gata1 during erythroid differentiation, and serves as a direct upstream effector of this critical erythroid regulatory gene.
Results
Transient anemia in TR4 and in TR2/TR4 transgenic embryos
Transgenic mice were generated in which TR2 alone (TgTR2), TR4 alone (TgTR4), or both (TgTR2/TR4) were forcibly expressed in erythroid cells by insertion of both cDNAs into a Gata1-regulated expression cassette called G1HRD (GATA-1 hematopoietic regulatory domain) (Onodera et al. 1997; Tanabe et al. 2007). The TgTR2/TR4 line was generated by coinjection of the TR2 and TR4 transgenic constructs into mouse oocytes. The level of transgene (Tg)-derived TR2 or TR4 mRNA in 14.5-dpc fetal livers of each transgenic line was more than fivefold greater than that of endogenous TR2 or TR4 mRNA (Tanabe et al. 2007).
TgTR4 and TgTR2/TR4 embryos displayed pronounced anemia between 10.5 and 12.5 dpc (Fig. 1A–D). The fetal livers (where early definitive erythropoiesis originates) of TgTR4 and TgTR2/TR4 embryos were visibly paler in appearance between 10.5 and 13.5 dpc than their counterpart wild-type littermate controls, and TgTR2/TR4 embryos accumulated approximately one-third the number of primitive erythrocytes during this period (Table 1). Interestingly, however, there was essentially no residual anemia by the time definitive erythropoiesis was fully engaged at 15.5 dpc, and thus transgenic pups were born in a normal Mendelian ratio. All hematological parameters for 3-wk postnatal TgTR4 or TgTR2/TR4 mice were normal (data not shown). These data indicate that primitive (and possibly early definitive) erythropoiesis was significantly affected by forced erythroid-specific TR2 and TR4 expression.
Figure 1.

Transient embryonic anemia in TgTR4 and TgTR2/TR4 transgenic mice. The yolk sacs and embryos of 11.5-dpc TgTR4 (B,D) or their wild-type littermates (A,C) are shown. EryP colonies derived from the yolk sacs of wild-type (E) or TgTR2/TR4 (F) embryos are shown at the same magnification (see Materials and Methods). (G) Abundance of endogenous (open bars) or transgene-derived (shaded bars) TR2 and TR4 mRNAs in 11.5-dpc embryonic blood and 14.5-dpc fetal livers of the TgTR2/TR4 embryos or their wild-type littermates were determined by reverse transcription and real-time PCR, and were normalized to abundance of endogenous RNase inhibitor mRNA as the internal control (set at 100%). Data represent average with standard deviation of three to four embryos.
Table 1.
Transient embryonic anemia in TR2/TR4 transgenic embryos

Quantification of red blood cells (×105 per embryo) recovered from 10.5- to 15.5-dpc TgTR2/TR4 embryos in comparison with their wild-type littermates. Data represent the average, plus or minus standard deviations, of five to eight embryos representing each time point.
In order to address the mechanism underlying the transient anemia induced by TR2 and TR4 forced expression, we determined the abundance of endogenous and Tg-derived TR2 and TR4 mRNAs in both primitive (11.5-dpc circulating blood) and definitive (14.5-dpc fetal liver) erythroid cells by reverse transcription and real-time quantitative PCR (RT–PCR). The abundance of TR2 and TR4 mRNAs was normalized to endogenous RNase inhibitor mRNA as the internal control (Kolbus et al. 2003). In wild-type mice, the abundance of endogenous TR2 or TR4 mRNA in the 14.5-dpc fetal liver was sevenfold or fourfold higher than in 11.5-dpc primitive erythroid cells, respectively (Fig. 1G). The abundance of Tg-derived TR2 or TR4 mRNAs in 11.5-dpc primitive erythroid cells of the TgTR2/TR4 line was 40-fold or 30-fold higher than their respective abundance in wild-type littermates. In the 14.5-dpc fetal liver, Tg-derived TR2 mRNA increased about twofold from its abundance in 11.5-dpc primitive erythroid cells, whereas Tg-derived TR4 mRNA level was statistically the same in both erythroid populations. These data indicate that the transient embryonic anemia is not due to developmental stage-specific alteration in transgene expression.
Forced expression of TR2/TR4 affects primitive erythroid progenitor formation
We recently showed that mouse εy globin mRNA levels were reduced in both TgTR4 and TgTR2/TR4 mice (Tanabe et al. 2007). To investigate whether repression of mouse εy globin transcription was the sole cause of the transient anemic phenotype, we performed hematopoietic progenitor colony assays using 8.5-dpc yolk sacs from the TgTR2/TR4 embryos and nontransgenic littermates. The number of primitive erythroid (EryP) colonies that developed from the yolk sacs of TgTR2/TR4 embryos was ∼40% of wild-type controls, and the TgTR2/TR4 EryP colonies were noticeably smaller and less hemoglobinized (Fig. 1E,F; Table 2). These data indicate that TgTR2/TR4 embryos have defective primitive erythropoiesis, and that this defect is exhibited both in the number of progenitor colonies and in their differentiation. The data suggested that TR2 and TR4 might play a prominent role in erythroid differentiation.
Table 2.
Primitive erythroid progenitor number in TR2/TR4 transgenic embryos

Quantification of primitive erythroid (EryP), macrophage (Mac), or fibroblast colonies (per 1000 cells) derived from the yolk sacs of wild-type or TgTR2/TR4 embryos. Data represent the averages ± the standard deviation of six embryos of each genotype.
Gata1 expression is altered in TR2/TR4 gain-of-function and knockout mice
To identify possible regulatory molecules that could be direct or indirect transcriptional target(s) of TR2/TR4 in erythroid cells, we isolated mRNA from 11.5-dpc blood of TgTR2/TR4 embryos and their wild-type littermates and then analyzed the mRNA abundance of several key transcription factors that are known to play important roles in erythroid differentiation. EKLF, retinoic acid receptor, COUP-TFII, and Smad5 mRNAs, all previously implicated as potentially important effectors of erythropoiesis (Nuez et al. 1995; Perkins et al. 1995; Filipe et al. 1999; Collins 2002; Schmerer and Evans 2003), were essentially unchanged in abundance; however, GATA-1 mRNA abundance was profoundly inhibited in the TgTR2/TR4 transgenic embryos (Fig. 2A). In the embryonic blood of TgTR4 embryos, GATA-1 abundance was reduced to 20%–35% of wild type, while its level was not significantly altered in TgTR2 embryonic blood (Fig. 2B), consistent with the conclusion that TgTR2 mice do not exhibit a similar transient embryonic anemia that is observed in both TgTR4 and TgTR2/TR4 mice (data not shown). We also examined GATA-1 mRNA accumulation in a transgenic mouse line in which a previously established dominant-negative mutant of TR4 (TgdnTR4) was forcibly expressed (Tanabe et al. 2007). In conceptual harmony with the results analyzing the gain-of-function phenotypes, we found that GATA-1 mRNA was induced approximately twofold in the blood of TgdnTR4 embryos (Fig. 2B).
Figure 2.

Transcription factor expression in TR2/TR4 transgenic mice. (A) Expression of EKLF, RXR, RAR, COUP-TFII, Smad5, and GATA-1 mRNAs in 11.5-dpc circulating blood of TgTR2/TR4 mice (shaded bars) was determined by semiquantitative RT–PCR. The PCR signals were quantified and normalized to coamplified GAPDH signals. The averages of the relative expression levels, normalized to that of wild-type littermates (set at 1.0 and represented by open bars), are graphically depicted. All experiments were repeated twice with essentially the same results; representative data from one experiment are shown. (B) The abundance of the GATA-1 mRNA in 11.5-dpc blood cells of the TgTR2, TgTR4, TgTR2/TR4, and TgdnTR4 mice (shaded bars) was quantified as in A. Data represent the averages ± SD of blood samples taken from five to six different embryos. (C) The abundance of the GATA-1 mRNA in 14.5-dpc fetal liver cells of TgTR2, TgTR4, TgTR2/TR4, and TgdnTR4 mice (shaded bars) was determined as in A. Three animals of each genotype were used in the analysis. (*) P < 0.05; (**) P < 0.01 by Student t-test.
To investigate the possible regulatory effects of TR2/TR4 on Gata1 during definitive erythropoiesis, we determined the abundance of GATA-1 in 14.5-dpc fetal liver cells. In TgTR4 and in TgTR2/TR4 embryos, GATA-1 mRNA was reduced to 50%–65% of wild-type levels, while the abundance of GATA-1 transcript was not significantly altered in the fetal livers of TgTR2 embryos. In the dnTR4 transgenic embryos, GATA-1 abundance in the fetal liver was modestly induced (Fig. 2C). Thus, the effects of transgenic expression of the wild-type or mutant TR2 or TR4 on Gata1 transcription were diminished in fetal liver (definitive) erythroid cells in comparison with their effects on embryonic (primitive) erythropoiesis.
To further evaluate possible effects of TR2 and TR4 on Gata1 transcription, we examined GATA-1 mRNA accumulation in Tr2- or Tr4-germline-null mutant mice (Tanabe et al. 2007). In 11.5-dpc blood recovered from Tr2-null mutants, GATA-1 abundance was elevated approximately threefold in comparison with wild-type embryos; similarly, Tr4-null mutant mice accumulated some 2.5-fold more erythroid GATA-1 mRNA than their wild-type littermates (Fig. 3A). In hematopoietic cells purified from 14.5-dpc fetal livers of Tr2-null mutant embryos, the abundance of accumulated GATA-1 mRNA was 2.6-fold higher than in wild type, while the fetal livers of Tr4 mutants accumulated twofold more GATA-1 (Fig. 3B).
Figure 3.

Expression of GATA-1 in Tr2- and Tr4-null mutant mice. The abundance of GATA-1 transcript in 11.5-dpc circulating blood cells (A) or in 14.5-dpc fetal liver cells (B) of Tr2- or Tr4-null mutant mice (shaded bars) was determined by duplex semiquantitative RT–PCR, using GAPDH mRNA as the internal control. Generation of the conditionally null mutant mice from which the null mutants were generated (O. Tanabe, unpubl.) will be described elsewhere. The relative expression of GATA-1 mRNA normalized to wild-type littermate controls (set at 1.0, open bars) is graphically depicted with SD indicated. Three animals of each genotype were analyzed. (**) P < 0.01.
The TR2 and TR4 proteins share strong sequence similarity. They can form either homodimers or heterodimers with one another and both can bind to DR elements (Tanabe et al. 2007). It has been suggested that TR2 and TR4 may be at least partially redundant, and the fact that Tr2 and Tr4 mutant mice display only limited deficiencies whereas the compound null mutant embryos fail to survive beyond 7.5 dpc (i.e., prior to blood formation) (C. Chang, unpubl.) in the C57Bl/6 genetic background support this notion. Unfortunately, this fact also precludes the ability to experimentally determine GATA-1 levels in Tr2/Tr4 compound null mutant mice, where we would anticipate more pronounced (uncompensated) erythroid phenotypes.
Manipulation of TR2/TR4 abundance in definitive human erythroid progenitor cells alters GATA1 transcription
Cellular differentiation in the definitive erythroid lineage occurs over approximately a 2-wk time span in humans. The earliest clearly defined committed erythroid progenitor cell, the BFU-E (burst-forming unit erythroid), expresses the cell surface antigen CD34, as do all other early hematopoietic progenitors (Baines et al. 1988). The more mature erythroid progenitor, the CFU-E (colony forming unit erythroid), expresses the cell surface antigen CD36, but not CD34 (Tandon et al. 1989). CFU-E differentiate into erythroblasts, which begin to synthesize hemoglobin as well as the surface antigen glycophorin A, to eventually become mature red blood cells (Karhi et al. 1981).
To investigate whether or not the repressive effect of TR2/TR4 on GATA-1 is conserved between mice and humans, we examined the effects of TR2/TR4 loss- and gain-of-function on GATA-1 abundance in in vitro differentiated adult human hematopoietic progenitor cells. Purified CD34+ cells were transfected with TR2 and TR4 expression plasmids or RNAi vectors expressing short hairpin RNAs (shRNAs) together with a DsRed expression plasmid (to mark the transfected cells) and then subjected to in vitro differentiation (Giarratana et al. 2005). The cells were cultured in serum-free medium containing glucocorticoid, IL-3, stem cell factor (SCF), and erythropoietin for 8 d. By the end of day 8, the differentiated cells acquired characteristics typical of CFU-E, and began to express CD36. These cells were then transferred to murine OP9 stromal feeder cells in the presence of erythropoietin (alone) and grown for an additional 3 d (Nakano et al. 1994). By day 11, both glycophorin A and hemoglobin were being actively synthesized. In vitro erythroid differentiation was confirmed by flow cytometry (Fig. 4A); the differentiated cells displayed multiple surface markers that are typical of erythroid cells (data not shown).
Figure 4.

Altering the abundance of TR2 and TR4 in human erythroid progenitor cells affects GATA1 transcription. (A) Flow cytometric analysis of in vitro differentiated human CD34+ cells. (B) Expression of human GATA1 gene transcription in in vitro differentiated human erythroblasts transfected with TR2 and/or TR4 expression vectors or TR2/TR4 shRNA vectors was determined by duplex semiquantitative RT–PCR using GAPDH as the internal control. The relative expression levels of GATA-1 mRNA normalized to empty-vector controls (set at 1.0) are shown. Data represent the averages ± SD of three independent experiments. (mTR2/TR4) Transfection to promote forced expression of mouse TR2 and TR4; (TR2 RNAi, TR4 RNAi) shRNA expression vectors designed to specifically target and degrade human TR2 or TR4 mRNAs, respectively; (RNAi + mTR2/TR4) CD34+ cells were transfected with shRNA expression vectors targeting human TR2 and TR4 together with plasmids that forcibly express mouse TR2 and TR4; (vector only) empty expression vectors and RNAi vectors. (C) TR2 and TR4 depletion in shRNA-transfected and in vitro differentiated human erythroblasts. The abundance of TR2 and TR4 mRNA was determined by semiquantitative RT–PCR using GAPDH as the internal control, and normalized to vector-only controls (set at 1.0, open bars). Data represent the averages ± SD of three independent experiments.
After 11 d of differentiation in vitro, (DsRed-positive) transfected cells were collected by flow cytometric cell sorting. mRNA was extracted from the sorted cells and interrogated by radioactive duplex semiquantitative RT–PCR. In samples transfected with TR2 and TR4 expression vectors, GATA-1 expression was reduced to ∼25% of the vector-only transfection control (Fig. 4B, mTR2/TR4).
In loss-of-function experiments, RNAi-mediated knockdown achieved by a transfected TR2 shRNA expression vector led to significantly reduced TR2 mRNA accumulation (∼25% of wild-type levels) (Fig. 4C). In those same cells, GATA-1 mRNA abundance was induced ∼1.6-fold in comparison with the vector-only transfected control (Fig. 4B). Transfection of CD34+ cells with a TR4 shRNA forced expression construct reduced the amount of TR4 transcript to 20% of untransfected controls (Fig. 4C), and in the same samples, GATA-1 abundance increased approximately twofold. In cells cotransfected with both TR2 and TR4 shRNA expression vectors, GATA-1 abundance increased by approximately threefold over control levels (Fig. 4B). When the human TR2 and TR4 RNAi-treated samples were rescued by cotransfecting the cells with murine TR2 and TR4 cDNA expression constructs (which differed in nucleotide sequence and were therefore refractory to human shRNA-mediated mRNA degradation), GATA-1 production was restored to ∼75% of wild type abundance. These data, when considered together with the gain- and loss-of-function analyses in transgenic mice, show that TR2 and TR4 repress murine Gata1 and human GATA1 transcription in the erythroid cells of both species.
A conserved DR1 element in the Gata1/GATA1 erythroid enhancer
The mouse Gata1 gene consists of two alternative noncoding first exons/promoters (called IT and IE) and five coding exons (Fig. 5A, black boxes represent exons II–VI; Ito et al. 1993). The distal first exon (IT) is transcribed in Sertoli cells of the testis, whereas the proximal first exon (IE) specifies the majority of Gata1 transcription in hematopoietic cells. Of the cis-acting elements in the 5′ flanking region of the IE promoter, a double GATA motif (located at −680/−672) and a CACCC motif (at position −196/−192) are essential for hematopoietic lineage-specific promoter activity (Nicolis et al. 1991; Tsai et al. 1991; Ohneda et al. 2002). In addition to these proximal promoter elements, an evolutionarily conserved G1HE is located between −3.9 and −3.5 kb 5′ to the IE exon. Further dissection of that enhancer demonstrated that, while the entire enhancer is required for expression in megakaryocytes, only the 5′-most 62 base pairs (bp) are required for erythroid specificity (Vyas et al. 1999b; Nishimura et al. 2000). This 62-bp G1HE core sequence contains an essential GATA-binding site separated by 10 bp from an E-box motif (Fig. 5B). Previous studies have also shown that sequences within the first intron are also required to direct Gata1 gene expression during definitive hematopoiesis (Onodera et al. 1997). Thus, the promoter, containing at least 3.7-kb flanking exon IE as well as the first intron, are sufficient to direct hematopoietic Gata1 expression in both primitive and definitive erythrocytes (Onodera et al. 1997; Takahashi et al. 2000).
Figure 5.

The evolutionarily conserved DR element in the G1HE binds TR2/TR4 in vitro and in vivo. (A) Schematic structure of the murine Gata1 gene. Exons are depicted as black rectangles. The positions of three DR elements that bind to TR2/TR4 in vitro are indicated by arrows. GATA sites are indicated by open boxes, and the G1HE is shown as a shaded rectangle. (B) The G1HE DR element is conserved between mouse and human. (C) Ten micrograms of pCMV expression vector driving Flag-tagged TR2 or TR4 cDNAs were transfected separately or together into 293T cells for nuclear extract preparation and EMSA (top panel), or Western blotting with anti-Flag, anti-TR2, or anti-TR4 antibody (bottom panels). 32P-labeled oligonucleotides corresponding to the mouse or human G1HE DR sequence or the mutated sequences were used as probes. DRs are indicated by arrows, while only mutated nucleotides are shown in the mutant. Anti-TR2 or anti-TR4 antibodies or preimmune serum (control) was included in binding reactions where indicated. The arrow indicates the position of TR2 or TR4 proteins bound to the probe. (D) EMSA competitive binding assay. Nuclear extract from 293T cells cotransfected with pCMV-FlagTR2 and pCMV-FlagTR4 were incubated with 1 nM 32P-labeled ε distal DR probe, and 1, 5, or 25 nM unlabeled competitor oligonucleotides. (E) TR2 and TR4 ChIP of the G1HE in in vitro differentiated human CD34+ cells. Purified CD34+ hematopoietic progenitor cells were examined for TR2 or TR4 binding after in vitro differentiation for 0, 8, or 11 d (see Materials and Methods) using ChIP assays, employing either anti-TR2 or anti-TR4 rabbit antibodies (Tanabe et al. 2007) or preimmune rabbit serum (control) to test for binding of the proteins to regulatory sequences around the GATA1 gene promoter. Immunoprecipitated DNA was quantified by real-time quantitative PCR using specific primers within the human GATA1 locus located at −7.7, −3.7, +1.5, or +3.5 kb relative to the exon IE transcription initiation site, and normalized to input DNA. The −3.7 kb site corresponds to the DR element located within the G1HE that binds avidly to TR2/TR4 (shown in D), and represents the only preferentially enriched sequence in these experiments. Data represent the averages with SD of three experiments. Triangles with solid lines indicate data with anti-TR2 antibody, inverted triangles with dashed lines indicate data with anti-TR4 antibody, and circles with dotted lines indicate data with preimmune serum. Filled symbols represent ChIP data after 0 d of CD34+ cell differentiation, while open symbols represent the same experiment performed on cells differentiated in vitro for 11 d (differentiation for 8 d gave identical results to the 0 d control) (data not shown).
To ask whether TR2 and TR4 directly regulate Gata1 transcription, we searched for potential DR elements within 3.9 kb lying 5′ to the IE promoter, since TR2/TR4 repressed Gata1 in both primitive and definitive erythroid cells. Using an online bioinformatics tool (Sandelin and Wasserman 2005), 16 potential DR elements were identified. Oligonucleotides containing these 16 sites were synthesized and then tested directly in electrophoretic mobility shift assays (EMSA) using nuclear extracts from 293T cells transfected with TR2 and TR4 expression vectors.
Only three of the prospective Gata1 promoter DR elements bound to the TR2/TR4 receptor, and among these three, two of them (located at −1 or at −2.9 kb) (Fig. 5A, arrows) are in regions of the promoter that are not highly conserved between human and mouse. The single remaining avid binding site is a DR element with a one-nucleotide spacer (DR1) that is located 13 bp 3′ to the E-box motif in the G1HE, and is well conserved between human and mouse (Fig. 5B). While both the mouse and human DR1 elements bound to TR2/TR4 in EMSA, point mutations introduced into that sequence abrogated TR2/TR4 binding (Fig. 5C). All the DNA–protein complexes were inhibited by the addition of either an anti-TR2 or anti-TR4 antibody (Tanabe et al. 2007), indicating that TR2 and TR4 are both components of those complexes. Expression of TR2 alone in 293T cells led to a weaker signal for a DNA–protein complex than expression of TR4, either alone or together with TR2, even though protein expression with the TR2 vector alone is slightly higher than that with the TR4 vector alone or after TR2/TR4 cotransfection, as characterized by Western blotting with an anti-Flag antibody. These EMSA studies are consistent with the conclusion that the affinity of a DR element for the TR2 homodimer was lower than that for either the TR2/TR4 heterodimer or TR4 homodimer (Tanabe et al. 2007).
We next performed competition experiments to evaluate the affinity of the G1HE DR site for TR2/TR4 heterodimers in comparison with their affinity for the human ε- and γ-globin promoter DR sites. The equilibrium dissociation constants (Ki) of the ε-globin promoter distal DR and the single γ-globin promoter DR elements are 0.97 and 13 nM, respectively (Tanabe et al. 2007). A 32P-labeled oligonucleotide corresponding to the ε-globin distal DR element was used as a probe in these EMSA experiments, and various unlabeled oligonucleotides corresponding to the mouse or human Gata1/G1HE DR sites as well as DR sites in the human β-type globin gene promoters were examined as competitors. The results of these experiments show that the affinity of the murine G1HE DR site is lower than that of the ε-globin distal DR element but higher than that of the γ-globin promoter DR site, while the affinity of the human G1HE DR site was comparable with that of the γ-globin DR element (Fig. 5D). The data imply that the murine Gata1 enhancer DR site might be more sensitive to changes in TR2/TR4 signaling than the corresponding human DR site.
TR2 and TR4 bind to the G1HE in vivo
We next asked whether or not TR2 and/or TR4 directly associate with the GATA1 enhancer by ChIP assays. Human CD34+ cells were differentiated in vitro as described previously (Giarratana et al. 2005). Cells were collected after 0, 8, and 11 d of in vitro culture as described above. TR2- or TR4-specific antibodies (Tanabe et al. 2007) as well as preimmune rabbit serum were used to immunoprecipitate cross-linked chromatin. The relative amount of immunoprecipitated DNA compared with input DNA was determined by real-time quantitative PCR.
The −3.7 kb human G1HE, which contains the evolutionarily conserved DR element, is specifically enriched by the anti-TR2 and anti-TR4 antisera (when compared with the IgG control serum) in day 11 in vitro differentiated erythroblasts, but not in undifferentiated or in 8 d differentiated (data not shown) CD34+ cells (Fig. 5E). In contrast, irrelevant segments of the locus (at −7.7 kb or at two different sites 3′ to the G1HE enhancer) are not enriched by the TR2- or TR4-specific antisera. We conclude that TR2/TR4 binds to the −3.7 kb DR element within the G1HE, but only at late times (at the CFU-E stage or thereafter) during erythroid differentiation.
Mutation of the G1HE DR element induces Gata1 transcription
We finally examined the effect of mutation of the G1HE DR element on Gata1 transcription by mutagenesis and cotransfection assays. The reporter plasmid, pIE3.9intLuc, contains the entire mouse G1HRD (the IE promoter as well as the primitive and definitive enhancers) linked in cis to the firefly luciferase gene (Onodera et al. 1997; Nishimura et al. 2000). After cotransfection into a human erythroleukemia cell line (K562), forcibly coexpressed mouse TR2 and/or TR4 repressed reporter activity to 30% of the control level, while forced expression of nuclear receptors in the same family (RAR and RXR) had no effect (Fig. 6A). Approximately equal accumulation of the transfected TR2 and TR4 proteins in transfected K562 cells was confirmed by Western blotting (Fig. 6B). Cotransfection of this erythroid cell line with both TR2 and TR4 shRNA vectors increased reporter activity approximately fourfold, while reducing endogenous TR2 or TR4 abundance to ∼25% of their control levels (Fig. 6C). Adding back mouse TR2 and TR4 (by cotransfection with mTR2/mTR4 expression vectors) to the (human) RNAi-treated cells restored reporter activity to control levels. When the same clustered substitution mutation shown in Figure 5C was introduced into the pIE3.9intLuc plasmid, reporter activity increased by 2.5-fold. Forcibly expressed TR2 or TR4 affected the activity of the mutated reporter only modestly (to ∼60% of the control levels), and further, TR2/TR4 RNA inhibition did not significantly alter DR mutant reporter activity in comparison with the RNAi vector control (Fig. 6A). Collectively, the data show that TR2/TR4 binds to the G1HE DR site in vitro and in vivo, that the G1HE DR site functions as a GATA1 silencer element, and that repression of the GATA1 gene mediated through the G1HE DR site is specific for the nuclear orphan TR2 and TR4 receptors.
Figure 6.

G1HE DR mutation abrogates Gata1 transcriptional repression by TR2/TR4. (A) The wild-type or G1HE DR mutated IE3.9int Luc reporter (0.1 μg) was cotransfected with TR2, TR4, or RAR/RXR expression vectors (+, ++, and ++++ represent 0.4, 0.8, or 1.6 μg, respectively), or the TR2 and TR4 shRNA vectors into K562 cells. Firefly luciferase activities were normalized to cotransfected Renilla luciferase activities. Data represent the averages ± SD of three independent transfections. (B) Expression of Flag-tagged TR2 or TR4 in K562 cells after transfection with 9 or 18 μg of the pCMV/Flag/TR2 or pCMV/Flag/TR4 vector was examined by Western blotting with IRDye800-conjugated anti-Flag antibody. (C) Effect of RNAi on TR2 and TR4 expression in K562 cells. K562 cells (2 × 105) were transfected with shRNA vectors (shaded bars) or control vector (open bars) along with a DsRed expression plasmid; DsRed-positive (transfected) cells were collected by flow cytometry. The recovered transfected cells were used for RNA preparation. The abundance of TR2 and TR4 mRNA was determined by semiquantitative RT–PCR using GAPDH as the internal control. Data represent the averages ± SD of three independent transfection experiments.
Discussion
GATA-1 is among the most intensively studied transcription factors. Gata1 transcription is first activated in multipotential progenitor cells, and thereafter it is thought to autoactivate its own hematopoietic enhancer and promoter. During erythroid differentiation, Gata1 transcription significantly increases at the BFU-E stage, peaks at the polychromatic erythroblast stage, and is suppressed late during erythroid differentiation (Suzuki et al. 2003; Anguita et al. 2004). This suppression appears to be critical for GATA-1 function since persistent GATA-1 expression inhibits the final stages of erythropoiesis (Whyatt et al. 2000). Nevertheless, factors that control Gata1 repression at late erythroblast stages have not been identified.
We show here that transgenic erythroid-specific TR2/TR4 forced expression in mice repressed Gata1 transcription in both primitive and definitive erythroid cells. In contrast, both Tr2- or Tr4-null mutation and transgenic forced expression of a dominant-negative form of TR4 led to induced GATA-1 mRNA accumulation in both primitive and definitive erythroid cells. These results are both consistent with the hypothesis that TR2 and TR4 normally directly or indirectly repress Gata1 gene expression in vivo. Reduction in the abundance of GATA-1 mRNA by forced expression of TR2/TR4, or induction of GATA-1 mRNA by TR2/TR4 RNAi in differentiated definitive human erythroblasts, indicated that GATA1 repression by TR2/TR4 is an evolutionarily conserved mechanism in mice and humans. Although transgenic forced expression of GATA-1 causes erythroid maturation arrest (Whyatt et al. 2000), the induction of GATA-1 mRNA in Tr2- or Tr4-null mutant mice did not lead to an overt erythroid maturation defect. That may be due to the fact that the induction of Gata1 gene expression in the Tr2- or Tr4-null embryos led to at least an order of magnitude lower expression than the level achieved when the β-globin locus control region and promoter were used to direct expression in the GATA-1 transgenic mouse. While GATA-1 mRNA was induced by threefold or 2.5-fold in Tr2- or Tr4-null mutant embryos, GATA-1 protein level (determined by quantitative Western blotting using an anti-GATA-1 monoclonal antibody) in 14.5-dpc fetal liver of the same mutants was not significantly higher than that in wild-type embryos (data not shown). In contrast, the level of GATA-1 protein in the fetal livers of LCR-directed GATA-1 forced expression transgenic mice was at least fivefold higher than that in wild-type embryos (Whyatt et al. 2000). The reason for the more modest impact on GATA-1 protein levels than on mRNA levels after Tr2 or Tr4 ablation could be due to currently unknown compensatory mechanisms operating at a post-transcriptional level in the mutant mice.
The identification of a conserved DR1 element in the G1HE, and binding of TR2 and TR4 protein to the DR sequence in human erythroblasts (as demonstrated by ChIP assays using anti-TR2- and anti-TR4-specific antisera) strongly suggested that the repression of GATA1 transcription is mediated by direct TR2/TR4 binding to the G1HE DR element. Site-specific mutation of the DR sequence increased the transcriptional activity of G1HRD, and muted its response to forced expression or RNAi-mediated knockdown of TR2 and TR4 mRNA. These results demonstrated that TR2 and TR4 can exert repressor activity on the G1HRD through the −3.7 kb GATA1 enhancer DR element. The data collectively support the contention that the DR element within G1HRD is a major mediator of GATA1 repression, and therefore that TR2/TR4 (or more likely, the DRED repressor complex) is the first GATA1 transcriptional repressor to be identified.
Although TR2 and TR4 are expressed in undifferentiated human CD34+ hematopoietic cells (Tanabe et al. 2002), TR2/TR4 binding to the G1HE was not observed until the erythroblast stage at day 11 upon in vitro erythroid differentiation of the human hematopoietic progenitor cells. This differentiation stage-dependent binding may be due to direct changes in TR2/TR4 protein expression level, to (currently undefined) stage-dependent post-translational modification of TR2/TR4, or even to induction or repression of a differentiation-specific regulator of TR2/TR4 DNA binding.
Perhaps curiously, forced expression of TR2 alone did not affect GATA1 transcription, although expression of TR4 alone repressed GATA1. Since the level of transgene-derived mRNAs in the TgTR2 and TgTR4 lines is roughly equivalent, the difference in their phenotypes may be due to the lower affinity of the TR2 homodimer for DR elements in comparison with the TR4 homodimer or to the TR2/TR4 heterodimer (Tanabe et al. 2007).
The results from the analysis of TR2 or TR4 gain- and loss-of-function mice demonstrated that these orphan nuclear receptors repress Gata1 transcription in both primitive and definitive erythroid cells. Consistent with those observations, TR2 and TR4 also repressed GATA1 expression in definitive human erythroid progenitor cells. So how is it that TgTR4 and TgTR2/TR4 mice recover from the mid-embryonic anemia by 15.5 dpc and exhibit no postnatal phenotype? One hypothesis is that the mean expression of GATA-1 transcripts within individual clones of erythroid progenitors is indeed lower in TgTR4 and TgTR2/TR4 lines, but that recovery from the induced anemia could reflect in vivo selection of hematopoietic progenitors that happen to express the highest levels of GATA-1. Indeed it has been shown that mice bearing germline mutations in the Gata1 promoter exhibit lower overall GATA-1 mRNA accumulation and severe anemia. While most of these mice died before or shortly after birth, those that survive gradually express more GATA-1 and eventually develop normal blood indices (McDevitt et al. 1997). A second possibility lies in the observation that TR2 and TR4 are much more abundant in definitive than in primitive red blood cells, and consequently an increase in TR2 and TR4 abundance could exert a more profound effect in primitive than in definitive stage red blood cells. Possible autorepression of Gata1 promoter-directed TR2/TR4 transgenes by the forcibly expressed TR2/TR4 proteins cannot account for the transient nature of the embryonic anemia, because we did not observe any reduction in abundance of the Tg-derived TR2 and TR4 mRNAs in the fetal liver (definitive) versus yolk sac-derived (primitive) erythroid cells.
Forced transgenic expression of TR2 and TR4 in erythroid cells reduced the formation of primitive erythroid progenitors, and induced embryonic anemia. Previously, it was reported that Gata1-null hematopoietic stem cells failed to produce primitive erythroid precursors (Weiss et al. 1994). A second study demonstrated that mutant mice expressing GATA-1 mRNA at 20%–25% of wild-type levels exhibited anemia and defects in erythroid maturation during both the primitive and definitive stages (McDevitt et al. 1997). Taken together with those observations, the data presented here are consistent with the possibility that the anemic phenotype of TR2/TR4 transgenic embryos may be due to their transcriptional repression of Gata1. The ChIP assays indicate that TR2 and TR4 are bound to the −3.7 kb G1HE DR element in human erythroid progenitor cells following in vitro differentiation of CD34+ progenitors for 11 d. From the summary of these results, we hypothesize that TR2/TR4 plays a direct role in repressing GATA1 transcription in maturing erythroblasts, leading to GATA-1 down-regulation that is thought to be essential for terminal erythroid maturation (Whyatt et al. 2000).
Previous studies demonstrated that in addition to its roles in erythroid differentiation, GATA-1 is also indispensable for megakaryocyte development (Shivdasani et al. 1997). The absence of GATA-1 in megakaryocytes leads to increased proliferation and deficient maturation of megakaryocytic progenitors as well as to reduced numbers of mature circulating platelets. Furthermore, the platelets produced in GATA-1-deficient megakaryocytes are not fully functional and display abnormal morphology (Vyas et al. 1999a). GATA-1 is also required for the differentiation of eosinophils (Hirasawa et al. 2002; Yu et al. 2002) and mast cells (Harigae et al. 1998). The expression or function of TR2 and TR4 in hematopoietic lineages other than erythroid has not been examined, and therefore the question of whether or not TR2 and TR4 participate in regulating GATA-1 activities by repressing GATA-1 expression in myeloid cells is an issue that warrants further investigation.
Materials and methods
Mice
The transgenic mice forcibly expressing wild-type TR2 only (TgTR2)(line 2), TR4 only (TgTR4)(line 2), or both (TgTR2/TR4)(line 2), and the dominant-negative TR4 mutant (TgdnTR4) were described previously (Tanabe et al. 2007). For erythroid-specific expression of the transgenes, the mouse G1HRD cassette was used to direct expression of the murine TR2 and/or TR4 cDNAs (Onodera et al. 1997). Tr2- and Tr4-null mutant mice bearing deletions of the exons encoding the DNA-binding domains of the two receptors were generated by homologous recombination in ES cells and subsequent deletion of the Neo selection markers with Cre recombinase in mice (O. Tanabe, unpubl.). The details describing the generation of the Tr2 and Tr4 conditional null mutant mice will be described elsewhere.
Antibodies
Rabbit polyclonal anti-TR2 and anti-TR4 antibodies were described (Tanabe et al. 2007). PE-conjugated anti-mouse CD45, PE-conjugated anti-human CD36, and APC-conjugated anti-human glycophorin A were purchased from eBioscience Systems. The FITC-conjugated anti-human CD34 was from StemCell Technologies. IRDye800-conjugated rabbit anti-Flag antibody (Rockland Immunochemicals), rat anti-mouse Gata-1 (N6) antibody (Santa Cruz Biotechnology) with AlexaFluor680-conjugated goat anti-rat IgG secondary antibody (Invitrogen), or goat anti-mouse Lamin B (M-20) antibody (Santa Cruz Biotechnology) with IRDye800CW-conjugated donkey anti-goat IgG secondary antibody (Li-Cor) was used for Western blotting with the Odyssey infrared imaging system (Li-Cor).
Collection of embryonic and fetal blood cells
TgTR4 or TgTR2/TR4 transgenic embryos (10.5–15.5 dpc) and their wild-type littermates were separated from the uterus together with intact yolk sacs and placentas, and then rinsed with phosphate-buffered saline (PBS) several times to remove maternal blood. The macroscopic appearance of the embryos was determined immediately after rupturing the yolk sacs to minimize blood loss. To count blood cells, the 10.5- to 15.5-dpc embryos rinsed with PBS were placed in containers with 0.5 mL of PBS and then were detached from the yolk sac and placenta. Blood was collected from the embryos by piercing and compressing the heart with forceps. The number of erythrocytes in embryonic blood thus collected was determined with a hemocytometer.
Primitive erythroid progenitor assay
TR2/TR4 transgenic embryos (8.5 dpc) and their wild-type littermates were dissected under sterile conditions and were used for genotyping. Yolk sacs from embryos were incubated in Ca2+/Mg2+-free PBS with 20% fetal calf serum (FCS) and 0.1% collagenase (Sigma) for 1 h at 37°C. Cells were then disaggregated by passage through a syringe and 22-gauge needle (Wong et al. 1986). The yield from each yolk sac was 0.5 × 104 to 1 × 104 total cells. Subsequently, 1 × 103 cells from each yolk sac were plated in semisolid media to support the growth of all hematopoietic colonies (MethoCult GF M3434; StemCell Technology). After 3 d of culture, the number of primitive erythroid colonies (EryP), macrophage colonies (MAC), and fibroblast colonies was determined.
Collection of fetal liver hematopoietic cells
Fetal livers (14.5 dpc) were collected, rinsed, and placed in containers with 1 mL of PBS containing 4% fetal bovine serum (PBS/FBS). Hematopoietic and hepatic cells were released by dicing and repetitive pipeting, followed by passage through a cell strainer (Falcon). For antibody staining, 1 × 106 cells were suspended in 100 μL of chilled PBS/FBS and incubated with PE-conjugated anti-CD45 for 30 min on ice. After incubation, the cells were washed twice and suspended in 1 mL of ice-cold PBS/FBS. CD45-positive hematopoietic cells were collected by flow cytometry.
Semiquantitative RT–PCR
Total RNA was extracted from murine embryonic blood cells, murine fetal liver hematopoietic cells, or human erythroblasts that had been differentiated from CD34+ cells using Isogen (Nippon Gene). First-strand cDNA was synthesized with SuperScript II using 1 μg of total RNA in a 20-μL reaction volume. PCR was performed using 1 μL of cDNA in a 20-μL reaction volume (final) containing 20 mM Tris-HCl (pH 8.4), 50 mM KCl, 2.5 mM MgCl2, 200 μM each dNTP, 0.8 U of HotStart Taq DNA polymerase (Takara), 1 μCi of [α32P]-dCTP (ICN), and two gene-specific primer set (5 pmol) with temperature cycles of denaturation for 10 sec at 95°C, annealing for 10 sec at 58°C, and extension for 30 sec at 72°C. Optimal PCR cycle numbers within the exponential amplification range were experimentally determined for each primer set and each tissue. PCR reactions were electrophoresed on 8% polyacrylamide gels, dried, and quantified on a PhosphorImager (Molecular Dynamics). The primers used for PCR (m and h stand for murine and human, respectively) are listed in Table 3.
Table 3.
RT–PCR primers

Real-time PCR analysis for quantifying TR2 and TR4 cDNA
Real-time PCR analysis was performed with 0.1 μL of 11.5-dpc embryonic blood and 14.5-dpc fetal liver cDNAs in a 25-μL reaction using the ABI Prism 7000 Sequence Detection System and SYBR Green PCR Master Mix (Applied Biosystems). Sequences of the primers for endogenous and Tg-derived TR2 and TR4 cDNAs were described previously (Tanabe et al. 2007). Primer sequences for RNase inhibitor cDNA are listed in Table 3. All of the primer sets were designed to span introns. The abundance of each cDNA was determined based on its Ct (threshold cycle) value and an experimentally determined amplification efficiency for each primer set, and then normalized to the abundance of RNase inhibitor cDNA (as the internal control).
In vitro differentiation of human CD34+ cells
Human CD34+ cells purified from mobilized peripheral blood were obtained from the National Heart, Lung, and Blood Institute (NHLBI)-sponsored Program of Excellence in Gene Therapy National Core Center at the Fred Hutchinson Cancer Research Center. Cells were differentiated in vitro under conditions described previously (Giarratana et al. 2005), with only minor modifications. Cells were cultured in a serum-free medium (StemLine II; Sigma) supplemented with 120 μg/mL iron-saturated human transferrin, 900 ng/mL ferrous sulfate, 90 ng/mL ferric nitrate, and 10 μg/mL insulin (Sigma).
The expansion procedure was comprised of two steps. In the first step (days 0–8), 104 CD34+ cells per milliliter were cultured in the presence of 10−6 M glucocorticoid (Sigma), 100 ng/mL SCF, 5 ng/mL IL-3 (R&D Systems), and 3 IU/mL erythropoietin (SCF and erythropoietin were the generous gifts of Amgen, Inc.). On day 4, 1 vol of cell culture was diluted in 4 vol of fresh medium containing hydrocortisone, SCF, IL-3, and erythropoietin. For the second step (7 d after initiating the cultures) the cells were suspended at 2 × 105/mL and cultured on an adherent mouse OP9 stromal cell layer in fresh medium supplemented with erythropoietin. OP9 mouse stromal cells were cultured in MEMα medium (Gibco) containing 20% FBS and 100 U/mL penicillin/streptomycin. OP9 cells were split 1:3 every 3–4 d (Nakano et al. 1994).
For flow analysis of cell surface antigens, cells from different differentiation stages were collected and suspended in chilled PBS/FBS (1 × 106 cells per 100 μL). Fluorophore-conjugated anti-CD34, anti-CD36 and/or anti-glycophorin A were added to the cell suspension. After a 30-min incubation on ice, cells were washed twice, suspended in 1 mL of ice-cold PBS/FBS, and subjected to flow cytometric analysis.
Transfection of human CD34+ cells
Transfections were carried out following the CD34+ transfection protocol supplied by the Nucleofector manufacturer (Amaxa). For each transfection, 106 CD34+ cells and 4 μg of effector plasmid plus 1 μg of DsRed expression plasmid were used. After in vitro differentiation, transfected cells were collected by flow cytometric sorting for DsRed-positive cells.
RNAi
The pCS2DsRed2/U6 plasmid was used for expression of shRNAs under the control of the U6 promoter (Velkey and O’Shea 2003). The following 22-bp target sequences (specific for the human TR2 or TR4 mRNAs) were selected for RNAi-mediated knockdown: TR2E RNAi, 5′-AAAUCUUCCAACUGUGCCGC UU-3′; TR4D RNAi, 5′-AACCAAGCAAUUGUGCUGCUUC-3′.
EMSA
293T cells were transfected with pCMV Flag-mTR2, pCMV Flag-mTR4, or vector (12 μg of DNA per 10-cm plate) using Fugene6 (Roche). Nuclear extract preparation, DNA-binding reactions, and electrophoresis were performed as described (Tanimoto et al. 2000). The following blunt-ended oligonucleotides were used in the EMSA studies (only the sense strands are shown): mG1HE DR, 5′-tccctgctggcTGAACTgTGGCCAcagact tctgg-3′; mG1HE DR mutant, 5′-tccctgctggcTtAAaTgTaGCaA cagacttctgg-3′; hG1HE DR, 5′-tccttgctggcTGAACTgTCGCCG cagacttctga-3′; hG1HE DR mutant, 5′-tccttgctggcTtAAaTgTa GCaGcagacttctga-3′; ε-distal DR, 5′-CCCTGAGGACACAG GTCAGCCTTG-3′; γ DR, 5′-GCCTTGCCTTGACCAATAGC CTT-GACAA-3′; β, 5′-TAGGGTTGGCCAATCTACTCCC-3′.
For competitive binding experiments, the radioactive probe and unlabeled competitor oligonucleotides were mixed together before adding to the reaction.
ChIP
ChIP experiments were performed following the Upstate Biotechnology ChIP protocol with minor modifications: In vitro differentiated human erythroid progenitor cells (5 × 106) at different stages were harvested for each reaction. The cells were treated with 1% formaldehyde at room temperature for 12 min to cross-link protein to DNA. After sonication (12 × 5 sec, 70% power on a Branson Sonifier 450), a small fraction of the chromatin was uncross-linked by heating the mixture to 67°C for 4 h, and the average size of the DNA fragments (300–400 bp) was determined by gel electrophoresis to verify effective sonication. The rest of the cross-linked chromatin was immunoprecipitated using anti-TR2- or anti-TR4-specific antibodies overnight at 4°C. The supernatant from the no-antibody-added samples was saved to measure total input chromatin.
The cross-linked protein was next uncoupled from DNA by heating. DNA was purified using Qiaquick gel extraction columns (Qiagen) and resuspended in 50 μL of H2O. Finally, a Bio-Rad I-Cycler was used to perform quantitative real-time PCR. Two microliters of immunoprecipitated sample or 2 μL of diluted input DNA were amplified in a 20-μL reaction volume (final) containing 20 mM Tris-HCl (pH 8.4), 50 mM KCl, 2.5 mM MgCl2, 200 μM each dNTP, 0.8 U of HotStart Taq DNA polymerase (Takara), 100 nM FITC, 1 U of SYBR Green, and the sequence-specific primer pair (5 pmol) with temperature cycles of denaturation for 10 sec at 95°C, annealing for 10 sec at 58°C, and extension for 30 sec at 72°C.
The primer sequences used for this assay were hGATA-1 −7.7K, 5′-CCTAGCCACATTCTGGTTGTCC-3′ and 5′-ATG GGAGGATGTGGATTGATGTA-3′ (76-bp amplicon); hGATA-1 −3.7K, 5′-GACAGTTCAGCCAGCAAGGAG-3′ and 5′- GTCT CCCCCAAAGCCTGATCT-3′ (80-bp amplicon); hGATA-1 +1.5K, 5′-TCTGTCCGGAGAGTGATAACTGG-3′ and 5′-CA CCTCACACGTTGGTGAGTC-3′ (90-bp amplicon); hGATA-1 +3.5K, 5′-GACGGGGAGAATAAGAGGAAGTG-3′ and 5′-ATTTTCTCCTCCCCTCCAATTTC-3′ (114-bp amplicon).
Each primer pair was tested under the described PCR condition with serially diluted input DNA. The PCR efficiency (E) was derived by I-Cycler software. Finally, the relative abundance of DNA in each ChIP sample was calculated by the following equation:
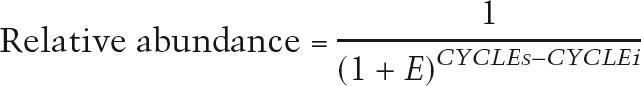 |
where CYCLEs is the threshold cycle for the ChIP sample and CYCLEi is the threshold cycle for the input DNA sample.
Luciferase reporter assay
The wild-type and mutant pIE3.9intLuc firefly luciferase reporter plasmids containing the entire G1HRD (Onodera et al. 1997) were used for transient transfection assay. The pRL-TK Renilla luciferase reporter plasmid (Promega) was used as an internal control. The eukaryotic expression plasmids used in this assay were pCMV Flag-mTR2, pCMV Flag-mTR4 (Tanabe et al. 2007), pCMV mRXR, and pCMV mRAR. The TR2 and TR4 pCS2DsRed2/U6 plasmids were also used. K562 cells were distributed in six-well plates (2 × 105/well), and transfected using Fugene6 (Roche) with a mixture of reporter plasmids (0.1 μg DNA) and eukaryotic expression plasmids (0.4–1.6 μg DNA). The total DNA amount was brought to 2 μg by the addition of pCDNA3.1 plasmid where needed. After 40 h of culture, the cells were harvested and subjected to dual luciferase assay (Promega) according to the manufacturer’s instructions. K562 cells were maintained in DMEM media containing 10% FBS and 100 U/mL penicillin/streptomycin.
To verify TR2/TR4 protein expression in transfected cells, K562 cells were divided among 10-cm plates (2 × 106 cells per plate) and transfected using Fugene6 as described above. The total DNA amount was 20 μg in each transfection reaction. Cells were harvested after 40 h of culture. Nuclear extracts were prepared and subjected to Western analysis using IRDye800-conjugated anti-Flag antibody.
Acknowledgments
We thank Xia Jiang for outstanding technical assistance, David Turner for the pCS2DsRed2/U6 RNAi plasmid and discussions, and David Engelke, Roland Kwok, Ray Trievel, Takashi Moriguchi, and Kim-Chew Lim for insightful discussions. We are extremely grateful to Tim Blauwkamp and Ken Cadigan for advice and help with the ChIP assays, and to Shelley Heimfeld (Fred Hutchinson Cancer Research Center) for the purified human CD34+ cells made available through the NHLBI Program of Excellence in Gene Therapy (HL66947). This work was supported by an award for the Anthony and Lillian Lu Fellowship to the Department of Biological Chemistry (to Y.S.) and research grants from the NIH (HL24415; to J.D.E.) and Amgen, Inc. (to O.T. and J.D.E.).
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.1593307
References
- Anguita E., Hughes J., Heyworth C., Blobel G.A., Wood W.G., Higgs D.R., Hughes J., Heyworth C., Blobel G.A., Wood W.G., Higgs D.R., Heyworth C., Blobel G.A., Wood W.G., Higgs D.R., Blobel G.A., Wood W.G., Higgs D.R., Wood W.G., Higgs D.R., Higgs D.R. Globin gene activation during haemopoiesis is driven by protein complexes nucleated by GATA-1 and GATA-2. EMBO J. 2004;23:2841–2852. doi: 10.1038/sj.emboj.7600274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines P., Mayani H., Bains M., Fisher J., Hoy T., Jacobs A., Mayani H., Bains M., Fisher J., Hoy T., Jacobs A., Bains M., Fisher J., Hoy T., Jacobs A., Fisher J., Hoy T., Jacobs A., Hoy T., Jacobs A., Jacobs A. Enrichment of CD34 (My10)-positive myeloid and erythroid progenitors from human marrow and their growth in cultures supplemented with recombinant human granulocyte-macrophage colony-stimulating factor. Exp. Hematol. 1988;16:785–789. [PubMed] [Google Scholar]
- Chen Y.T., Collins L.L., Uno H., Chang C., Collins L.L., Uno H., Chang C., Uno H., Chang C., Chang C. Deficits in motor coordination with aberrant cerebellar development in mice lacking testicular orphan nuclear receptor 4. Mol. Cell. Biol. 2005;25:2722–2732. doi: 10.1128/MCB.25.7.2722-2732.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins S.J. The role of retinoids and retinoic acid receptors in normal hematopoiesis. Leukemia. 2002;16:1896–1905. doi: 10.1038/sj.leu.2402718. [DOI] [PubMed] [Google Scholar]
- Collins L.L., Lee Y.F., Heinlein C.A., Liu N.C., Chen Y.T., Shyr C.R., Meshul C.K., Uno H., Platt K.A., Chang C., Lee Y.F., Heinlein C.A., Liu N.C., Chen Y.T., Shyr C.R., Meshul C.K., Uno H., Platt K.A., Chang C., Heinlein C.A., Liu N.C., Chen Y.T., Shyr C.R., Meshul C.K., Uno H., Platt K.A., Chang C., Liu N.C., Chen Y.T., Shyr C.R., Meshul C.K., Uno H., Platt K.A., Chang C., Chen Y.T., Shyr C.R., Meshul C.K., Uno H., Platt K.A., Chang C., Shyr C.R., Meshul C.K., Uno H., Platt K.A., Chang C., Meshul C.K., Uno H., Platt K.A., Chang C., Uno H., Platt K.A., Chang C., Platt K.A., Chang C., Chang C. Growth retardation and abnormal maternal behavior in mice lacking testicular orphan nuclear receptor 4. Proc. Natl. Acad. Sci. 2004;101:15058–15063. doi: 10.1073/pnas.0405700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipe A., Li Q., Deveaux S., Godin I., Romeo P.H., Stamatoyannopoulos G., Mignotte V., Li Q., Deveaux S., Godin I., Romeo P.H., Stamatoyannopoulos G., Mignotte V., Deveaux S., Godin I., Romeo P.H., Stamatoyannopoulos G., Mignotte V., Godin I., Romeo P.H., Stamatoyannopoulos G., Mignotte V., Romeo P.H., Stamatoyannopoulos G., Mignotte V., Stamatoyannopoulos G., Mignotte V., Mignotte V. Regulation of embryonic/fetal globin genes by nuclear hormone receptors: A novel perspective on hemoglobin switching. EMBO J. 1999;18:687–697. doi: 10.1093/emboj/18.3.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco P.J., Farooqui M., Seto E., Wei L.N., Farooqui M., Seto E., Wei L.N., Seto E., Wei L.N., Wei L.N. The orphan nuclear receptor TR2 interacts directly with both class I and class II histone deacetylases. Mol. Endocrinol. 2001;15:1318–1328. doi: 10.1210/mend.15.8.0682. [DOI] [PubMed] [Google Scholar]
- Fujiwara Y., Browne C.P., Cunniff K., Goff S.C., Orkin S.H., Browne C.P., Cunniff K., Goff S.C., Orkin S.H., Cunniff K., Goff S.C., Orkin S.H., Goff S.C., Orkin S.H., Orkin S.H. Arrested development of embryonic red cell precursors in mouse embryos lacking transcription factor GATA-1. Proc. Natl. Acad. Sci. 1996;93:12355–12358. doi: 10.1073/pnas.93.22.12355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giarratana M.C., Kobari L., Lapillonne H., Chalmers D., Kiger L., Cynober T., Marden M.C., Wajcman H., Douay L., Kobari L., Lapillonne H., Chalmers D., Kiger L., Cynober T., Marden M.C., Wajcman H., Douay L., Lapillonne H., Chalmers D., Kiger L., Cynober T., Marden M.C., Wajcman H., Douay L., Chalmers D., Kiger L., Cynober T., Marden M.C., Wajcman H., Douay L., Kiger L., Cynober T., Marden M.C., Wajcman H., Douay L., Cynober T., Marden M.C., Wajcman H., Douay L., Marden M.C., Wajcman H., Douay L., Wajcman H., Douay L., Douay L. Ex vivo generation of fully mature human red blood cells from hematopoietic stem cells. Nat. Biotechnol. 2005;23:69–74. doi: 10.1038/nbt1047. [DOI] [PubMed] [Google Scholar]
- Harigae H., Takahashi S., Suwabe N., Ohtsu H., Gu L., Yang Z., Tsai F.Y., Kitamura Y., Engel J.D., Yamamoto M., Takahashi S., Suwabe N., Ohtsu H., Gu L., Yang Z., Tsai F.Y., Kitamura Y., Engel J.D., Yamamoto M., Suwabe N., Ohtsu H., Gu L., Yang Z., Tsai F.Y., Kitamura Y., Engel J.D., Yamamoto M., Ohtsu H., Gu L., Yang Z., Tsai F.Y., Kitamura Y., Engel J.D., Yamamoto M., Gu L., Yang Z., Tsai F.Y., Kitamura Y., Engel J.D., Yamamoto M., Yang Z., Tsai F.Y., Kitamura Y., Engel J.D., Yamamoto M., Tsai F.Y., Kitamura Y., Engel J.D., Yamamoto M., Kitamura Y., Engel J.D., Yamamoto M., Engel J.D., Yamamoto M., Yamamoto M. Differential roles of GATA-1 and GATA-2 in growth and differentiation of mast cells. Genes Cells. 1998;3:39–50. doi: 10.1046/j.1365-2443.1998.00166.x. [DOI] [PubMed] [Google Scholar]
- Hirasawa R., Shimizu R., Takahashi S., Osawa M., Takayanagi S., Kato Y., Onodera M., Minegishi N., Yamamoto M., Fukao K., Shimizu R., Takahashi S., Osawa M., Takayanagi S., Kato Y., Onodera M., Minegishi N., Yamamoto M., Fukao K., Takahashi S., Osawa M., Takayanagi S., Kato Y., Onodera M., Minegishi N., Yamamoto M., Fukao K., Osawa M., Takayanagi S., Kato Y., Onodera M., Minegishi N., Yamamoto M., Fukao K., Takayanagi S., Kato Y., Onodera M., Minegishi N., Yamamoto M., Fukao K., Kato Y., Onodera M., Minegishi N., Yamamoto M., Fukao K., Onodera M., Minegishi N., Yamamoto M., Fukao K., Minegishi N., Yamamoto M., Fukao K., Yamamoto M., Fukao K., Fukao K., et al. Essential and instructive roles of GATA factors in eosinophil development. J. Exp. Med. 2002;195:1379–1386. doi: 10.1084/jem.20020170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito E., Toki T., Ishihara H., Ohtani H., Gu L., Yokoyama M., Engel J.D., Yamamoto M., Toki T., Ishihara H., Ohtani H., Gu L., Yokoyama M., Engel J.D., Yamamoto M., Ishihara H., Ohtani H., Gu L., Yokoyama M., Engel J.D., Yamamoto M., Ohtani H., Gu L., Yokoyama M., Engel J.D., Yamamoto M., Gu L., Yokoyama M., Engel J.D., Yamamoto M., Yokoyama M., Engel J.D., Yamamoto M., Engel J.D., Yamamoto M., Yamamoto M. Erythroid transcription factor GATA-1 is abundantly transcribed in mouse testis. Nature. 1993;362:466–469. doi: 10.1038/362466a0. [DOI] [PubMed] [Google Scholar]
- Karhi K.K., Andersson L.C., Vuopio P., Gahmberg C.G., Andersson L.C., Vuopio P., Gahmberg C.G., Vuopio P., Gahmberg C.G., Gahmberg C.G. Expression of blood group A antigens in human bone marrow cells. Blood. 1981;57:147–151. [PubMed] [Google Scholar]
- Kolbus A., Blazquez-Domingo M., Carotta S., Bakker W., Luedemann S., von Lindern M., Steinlein P., Beug H., Blazquez-Domingo M., Carotta S., Bakker W., Luedemann S., von Lindern M., Steinlein P., Beug H., Carotta S., Bakker W., Luedemann S., von Lindern M., Steinlein P., Beug H., Bakker W., Luedemann S., von Lindern M., Steinlein P., Beug H., Luedemann S., von Lindern M., Steinlein P., Beug H., von Lindern M., Steinlein P., Beug H., Steinlein P., Beug H., Beug H. Cooperative signaling between cytokine receptors and the glucocorticoid receptor in the expansion of erythroid progenitors: Molecular analysis by expression profiling. Blood. 2003;102:3136–3146. doi: 10.1182/blood-2003-03-0923. [DOI] [PubMed] [Google Scholar]
- Lee C.H., Chinpaisal C., Wei L.N., Chinpaisal C., Wei L.N., Wei L.N. A novel nuclear receptor heterodimerization pathway mediated by orphan receptors TR2 and TR4. J. Biol. Chem. 1998a;273:25209–25215. doi: 10.1074/jbc.273.39.25209. [DOI] [PubMed] [Google Scholar]
- Lee C.H., Chinpaisal C., Wei L.N., Chinpaisal C., Wei L.N., Wei L.N. Cloning and characterization of mouse RIP140, a corepressor for nuclear orphan receptor TR2. Mol. Cell. Biol. 1998b;18:6745–6755. doi: 10.1128/mcb.18.11.6745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y.F., Lee H.J., Chang C., Lee H.J., Chang C., Chang C. Recent advances in the TR2 and TR4 orphan receptors of the nuclear receptor superfamily. J. Steroid Biochem. Mol. Biol. 2002;81:291–308. doi: 10.1016/s0960-0760(02)00118-8. [DOI] [PubMed] [Google Scholar]
- Martin D.I.K., Zon L.I., Mutter G., Orkin S.H., Zon L.I., Mutter G., Orkin S.H., Mutter G., Orkin S.H., Orkin S.H. Expression of an erythroid transcription factor in megakaryocytic and mast cell lineages. Nature. 1990;344:444–447. doi: 10.1038/344444a0. [DOI] [PubMed] [Google Scholar]
- McDevitt M.A., Shivdasani R.A., Fujiwara Y., Yang H., Orkin S.H., Shivdasani R.A., Fujiwara Y., Yang H., Orkin S.H., Fujiwara Y., Yang H., Orkin S.H., Yang H., Orkin S.H., Orkin S.H. A ‘knockdown’ mutation created by cis-element gene targeting reveals the dependence of erythroid cell maturation on the level of transcription factor GATA-1. Proc. Natl. Acad. Sci. 1997;94:6781–6785. doi: 10.1073/pnas.94.13.6781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu X., Lee Y.F., Liu N.C., Chen Y.T., Kim E., Shyr C.R., Chang C., Lee Y.F., Liu N.C., Chen Y.T., Kim E., Shyr C.R., Chang C., Liu N.C., Chen Y.T., Kim E., Shyr C.R., Chang C., Chen Y.T., Kim E., Shyr C.R., Chang C., Kim E., Shyr C.R., Chang C., Shyr C.R., Chang C., Chang C. Targeted inactivation of testicular nuclear orphan receptor 4 delays and disrupts late meiotic prophase and subsequent meiotic divisions of spermatogenesis. Mol. Cell. Biol. 2004;24:5887–5899. doi: 10.1128/MCB.24.13.5887-5899.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano T., Kodama H., Honjo T., Kodama H., Honjo T., Honjo T. Generation of lymphohematopoietic cells from embryonic stem cells in culture. Science. 1994;265:1098–1101. doi: 10.1126/science.8066449. [DOI] [PubMed] [Google Scholar]
- Nicolis S., Bertini C., Ronchi A., Crotta S., Lanfranco L., Moroni E., Giglioni B., Ottolenghi S., Bertini C., Ronchi A., Crotta S., Lanfranco L., Moroni E., Giglioni B., Ottolenghi S., Ronchi A., Crotta S., Lanfranco L., Moroni E., Giglioni B., Ottolenghi S., Crotta S., Lanfranco L., Moroni E., Giglioni B., Ottolenghi S., Lanfranco L., Moroni E., Giglioni B., Ottolenghi S., Moroni E., Giglioni B., Ottolenghi S., Giglioni B., Ottolenghi S., Ottolenghi S. An erythroid specific enhancer upstream to the gene encoding the cell-type specific transcription factor GATA-1. Nucleic Acids Res. 1991;19:5285–5291. doi: 10.1093/nar/19.19.5285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura S., Takahashi S., Kuroha T., Suwabe N., Nagasawa T., Trainor C., Yamamoto M., Takahashi S., Kuroha T., Suwabe N., Nagasawa T., Trainor C., Yamamoto M., Kuroha T., Suwabe N., Nagasawa T., Trainor C., Yamamoto M., Suwabe N., Nagasawa T., Trainor C., Yamamoto M., Nagasawa T., Trainor C., Yamamoto M., Trainor C., Yamamoto M., Yamamoto M. A GATA box in the GATA-1 gene hematopoietic enhancer is a critical element in the network of GATA factors and sites that regulate this gene. Mol. Cell. Biol. 2000;20:713–723. doi: 10.1128/mcb.20.2.713-723.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuez B., Michalovich D., Bygrave A., Ploemacher R., Grosveld F., Michalovich D., Bygrave A., Ploemacher R., Grosveld F., Bygrave A., Ploemacher R., Grosveld F., Ploemacher R., Grosveld F., Grosveld F. Defective haematopoiesis in fetal liver resulting from inactivation of the EKLF gene. Nature. 1995;375:316–318. doi: 10.1038/375316a0. [DOI] [PubMed] [Google Scholar]
- Ohneda K., Yamamoto M., Yamamoto M. Roles of hematopoietic transcription factors GATA-1 and GATA-2 in the development of red blood cell lineage. Acta Haematol. 2002;108:237–245. doi: 10.1159/000065660. [DOI] [PubMed] [Google Scholar]
- Ohneda K., Shimizu R., Nishimura S., Muraosa Y., Takahashi S., Engel J.D., Yamamoto M., Shimizu R., Nishimura S., Muraosa Y., Takahashi S., Engel J.D., Yamamoto M., Nishimura S., Muraosa Y., Takahashi S., Engel J.D., Yamamoto M., Muraosa Y., Takahashi S., Engel J.D., Yamamoto M., Takahashi S., Engel J.D., Yamamoto M., Engel J.D., Yamamoto M., Yamamoto M. A minigene containing four discrete cis elements recapitulates GATA-1 gene expression in vivo. Genes Cells. 2002;7:1243–1254. doi: 10.1046/j.1365-2443.2002.00595.x. [DOI] [PubMed] [Google Scholar]
- Onodera K., Takahashi S., Nishimura S., Ohta J., Motohashi H., Yomogida K., Hayashi N., Engel J.D., Yamamoto M., Takahashi S., Nishimura S., Ohta J., Motohashi H., Yomogida K., Hayashi N., Engel J.D., Yamamoto M., Nishimura S., Ohta J., Motohashi H., Yomogida K., Hayashi N., Engel J.D., Yamamoto M., Ohta J., Motohashi H., Yomogida K., Hayashi N., Engel J.D., Yamamoto M., Motohashi H., Yomogida K., Hayashi N., Engel J.D., Yamamoto M., Yomogida K., Hayashi N., Engel J.D., Yamamoto M., Hayashi N., Engel J.D., Yamamoto M., Engel J.D., Yamamoto M., Yamamoto M. GATA-1 transcription is controlled by distinct regulatory mechanisms during primitive and definitive erythropoiesis. Proc. Natl. Acad. Sci. 1997;94:4487–4492. doi: 10.1073/pnas.94.9.4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins A.C., Sharpe A.H., Orkin S.H., Sharpe A.H., Orkin S.H., Orkin S.H. Lethal β-thalassaemia in mice lacking the erythroid CACCC-transcription factor EKLF. Nature. 1995;375:318–322. doi: 10.1038/375318a0. [DOI] [PubMed] [Google Scholar]
- Sandelin A., Wasserman W.W., Wasserman W.W. Prediction of nuclear hormone receptor response elements. Mol. Endocrinol. 2005;19:595–606. doi: 10.1210/me.2004-0101. [DOI] [PubMed] [Google Scholar]
- Schmerer M., Evans T., Evans T. Primitive erythropoiesis is regulated by Smad-dependent signaling in postgastrulation mesoderm. Blood. 2003;102:3196–3205. doi: 10.1182/blood-2003-04-1094. [DOI] [PubMed] [Google Scholar]
- Shivdasani R.A., Fujiwara Y., McDevitt M.A., Orkin S.H., Fujiwara Y., McDevitt M.A., Orkin S.H., McDevitt M.A., Orkin S.H., Orkin S.H. A lineage-selective knockout establishes the critical role of transcription factor GATA-1 in megakaryocyte growth and platelet development. EMBO J. 1997;16:3965–3973. doi: 10.1093/emboj/16.13.3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyr C.R., Collins L.L., Mu X.M., Platt K.A., Chang C., Collins L.L., Mu X.M., Platt K.A., Chang C., Mu X.M., Platt K.A., Chang C., Platt K.A., Chang C., Chang C. Spermatogenesis and testis development are normal in mice lacking testicular orphan nuclear receptor 2. Mol. Cell. Biol. 2002;22:4661–4666. doi: 10.1128/MCB.22.13.4661-4666.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki N., Suwabe N., Ohneda O., Obara N., Imagawa S., Pan X., Motohashi H., Yamamoto M., Suwabe N., Ohneda O., Obara N., Imagawa S., Pan X., Motohashi H., Yamamoto M., Ohneda O., Obara N., Imagawa S., Pan X., Motohashi H., Yamamoto M., Obara N., Imagawa S., Pan X., Motohashi H., Yamamoto M., Imagawa S., Pan X., Motohashi H., Yamamoto M., Pan X., Motohashi H., Yamamoto M., Motohashi H., Yamamoto M., Yamamoto M. Identification and characterization of 2 types of erythroid progenitors that express GATA-1 at distinct levels. Blood. 2003;102:3575–3583. doi: 10.1182/blood-2003-04-1154. [DOI] [PubMed] [Google Scholar]
- Takahashi S., Shimizu R., Suwabe N., Kuroha T., Yoh K., Ohta J., Nishimura S., Lim K.C., Engel J.D., Yamamoto M., Shimizu R., Suwabe N., Kuroha T., Yoh K., Ohta J., Nishimura S., Lim K.C., Engel J.D., Yamamoto M., Suwabe N., Kuroha T., Yoh K., Ohta J., Nishimura S., Lim K.C., Engel J.D., Yamamoto M., Kuroha T., Yoh K., Ohta J., Nishimura S., Lim K.C., Engel J.D., Yamamoto M., Yoh K., Ohta J., Nishimura S., Lim K.C., Engel J.D., Yamamoto M., Ohta J., Nishimura S., Lim K.C., Engel J.D., Yamamoto M., Nishimura S., Lim K.C., Engel J.D., Yamamoto M., Lim K.C., Engel J.D., Yamamoto M., Engel J.D., Yamamoto M., Yamamoto M. GATA factor transgenes under GATA-1 locus control rescue germline GATA- 1 mutant deficiencies. Blood. 2000;96:910–916. [PubMed] [Google Scholar]
- Tanabe O., Katsuoka F., Campbell A.D., Song W., Yamamoto M., Tanimoto K., Engel J.D., Katsuoka F., Campbell A.D., Song W., Yamamoto M., Tanimoto K., Engel J.D., Campbell A.D., Song W., Yamamoto M., Tanimoto K., Engel J.D., Song W., Yamamoto M., Tanimoto K., Engel J.D., Yamamoto M., Tanimoto K., Engel J.D., Tanimoto K., Engel J.D., Engel J.D. An embryonic/fetal β-type globin gene repressor contains a nuclear receptor TR2/TR4 heterodimer. EMBO J. 2002;21:3434–3442. doi: 10.1093/emboj/cdf340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe O., McPhee D., Kobayashi S., Shen Y., Brandt W., Jiang X., Campbell A.D., Chen Y.T., Chang C., Yamamoto M., McPhee D., Kobayashi S., Shen Y., Brandt W., Jiang X., Campbell A.D., Chen Y.T., Chang C., Yamamoto M., Kobayashi S., Shen Y., Brandt W., Jiang X., Campbell A.D., Chen Y.T., Chang C., Yamamoto M., Shen Y., Brandt W., Jiang X., Campbell A.D., Chen Y.T., Chang C., Yamamoto M., Brandt W., Jiang X., Campbell A.D., Chen Y.T., Chang C., Yamamoto M., Jiang X., Campbell A.D., Chen Y.T., Chang C., Yamamoto M., Campbell A.D., Chen Y.T., Chang C., Yamamoto M., Chen Y.T., Chang C., Yamamoto M., Chang C., Yamamoto M., Yamamoto M., et al. Embryonic and fetal β-globin gene repression by the orphan nuclear receptors, TR2 and TR4. EMBO J. 2007;26:2295–2306. doi: 10.1038/sj.emboj.7601676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tandon N.N., Lipsky R.H., Burgess W.H., Jamieson G.A., Lipsky R.H., Burgess W.H., Jamieson G.A., Burgess W.H., Jamieson G.A., Jamieson G.A. Isolation and characterization of platelet glycoprotein IV (CD36) J. Biol. Chem. 1989;264:7570–7575. [PubMed] [Google Scholar]
- Tanimoto K., Liu Q., Grosveld F., Bungert J., Engel J.D., Liu Q., Grosveld F., Bungert J., Engel J.D., Grosveld F., Bungert J., Engel J.D., Bungert J., Engel J.D., Engel J.D. Context-dependent EKLF responsiveness defines the developmental specificity of the human ε-globin gene in erythroid cells of YAC transgenic mice. Genes & Dev. 2000;14:2778–2794. doi: 10.1101/gad.822500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai S.-F., Strauss E., Orkin S.H., Strauss E., Orkin S.H., Orkin S.H. Functional analysis and in vivo footprinting implicate the erytroid transcription factor GATA-1 as a positive regulator of its own promoter. Genes & Dev. 1991;5:919–931. doi: 10.1101/gad.5.6.919. [DOI] [PubMed] [Google Scholar]
- Velkey J.M., O’Shea K.S., O’Shea K.S. Oct4 RNA interference induces trophectoderm differentiation in mouse embryonic stem cells. Genesis. 2003;37:18–24. doi: 10.1002/gene.10218. [DOI] [PubMed] [Google Scholar]
- Vyas P., Ault K., Jackson C.W., Orkin S.H., Shivdasani R.A., Ault K., Jackson C.W., Orkin S.H., Shivdasani R.A., Jackson C.W., Orkin S.H., Shivdasani R.A., Orkin S.H., Shivdasani R.A., Shivdasani R.A. Consequences of GATA-1 deficiency in megakaryocytes and platelets. Blood. 1999a;93:2867–2875. [PubMed] [Google Scholar]
- Vyas P., McDevitt M.A., Cantor A.B., Katz S.G., Fujiwara Y., Orkin S.H., McDevitt M.A., Cantor A.B., Katz S.G., Fujiwara Y., Orkin S.H., Cantor A.B., Katz S.G., Fujiwara Y., Orkin S.H., Katz S.G., Fujiwara Y., Orkin S.H., Fujiwara Y., Orkin S.H., Orkin S.H. Different sequence requirements for expression in erythroid and megakaryocytic cells within a regulatory element upstream of the GATA-1 gene. Development. 1999b;126:2799–2811. doi: 10.1242/dev.126.12.2799. [DOI] [PubMed] [Google Scholar]
- Weiss M.J., Orkin S.H., Orkin S.H. GATA transcription factors: Key regulators of hematopoiesis. Exp. Hematol. 1995;23:99–107. [PubMed] [Google Scholar]
- Weiss M.J., Keller G., Orkin S.H., Keller G., Orkin S.H., Orkin S.H. Novel insights into erythroid development revealed through in vitro differentiation of GATA-1 embryonic stem cells. Genes & Dev. 1994;8:1184–1197. doi: 10.1101/gad.8.10.1184. [DOI] [PubMed] [Google Scholar]
- Whyatt D., Lindeboom F., Karis A., Ferreira R., Milot E., Hendriks R., de Bruijn M., Langeveld A., Gribnau J., Grosveld F., Lindeboom F., Karis A., Ferreira R., Milot E., Hendriks R., de Bruijn M., Langeveld A., Gribnau J., Grosveld F., Karis A., Ferreira R., Milot E., Hendriks R., de Bruijn M., Langeveld A., Gribnau J., Grosveld F., Ferreira R., Milot E., Hendriks R., de Bruijn M., Langeveld A., Gribnau J., Grosveld F., Milot E., Hendriks R., de Bruijn M., Langeveld A., Gribnau J., Grosveld F., Hendriks R., de Bruijn M., Langeveld A., Gribnau J., Grosveld F., de Bruijn M., Langeveld A., Gribnau J., Grosveld F., Langeveld A., Gribnau J., Grosveld F., Gribnau J., Grosveld F., Grosveld F., et al. An intrinsic but cell-nonautonomous defect in GATA-1-overexpressing mouse erythroid cells. Nature. 2000;406:519–524. doi: 10.1038/35020086. [DOI] [PubMed] [Google Scholar]
- Wong P.M., Chung S.W., Reicheld S.M., Chui D.H., Chung S.W., Reicheld S.M., Chui D.H., Reicheld S.M., Chui D.H., Chui D.H. Hemoglobin switching during murine embryonic development: Evidence for two populations of embryonic erythropoietic progenitor cells. Blood. 1986;67:716–721. [PubMed] [Google Scholar]
- Yu C., Cantor A.B., Yang H., Browne C., Wells R.A., Fujiwara Y., Orkin S.H., Cantor A.B., Yang H., Browne C., Wells R.A., Fujiwara Y., Orkin S.H., Yang H., Browne C., Wells R.A., Fujiwara Y., Orkin S.H., Browne C., Wells R.A., Fujiwara Y., Orkin S.H., Wells R.A., Fujiwara Y., Orkin S.H., Fujiwara Y., Orkin S.H., Orkin S.H. Targeted deletion of a high-affinity GATA-binding site in the GATA-1 promoter leads to selective loss of the eosinophil lineage in vivo. J. Exp. Med. 2002;195:1387–1395. doi: 10.1084/jem.20020656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zon L.I., Yamaguchi Y., Yee K., Albee E.A., Kimura A., Bennett J.C., Orkin S.H., Ackerman S.J., Yamaguchi Y., Yee K., Albee E.A., Kimura A., Bennett J.C., Orkin S.H., Ackerman S.J., Yee K., Albee E.A., Kimura A., Bennett J.C., Orkin S.H., Ackerman S.J., Albee E.A., Kimura A., Bennett J.C., Orkin S.H., Ackerman S.J., Kimura A., Bennett J.C., Orkin S.H., Ackerman S.J., Bennett J.C., Orkin S.H., Ackerman S.J., Orkin S.H., Ackerman S.J., Ackerman S.J. Expression of mRNA for the GATA-binding proteins in human eosinophils and basophils: Potential role in gene transcription. Blood. 1993;81:3234–3241. [PubMed] [Google Scholar]