

Abstract
Mycobacterium tuberculosis places an enormous burden on the welfare of humanity. Its ability to grow and its pathogenicity are linked to sulfur metabolism, which is considered a fertile area for the development of antibiotics, particularly because many of the sulfur acquisition steps in the bacterium are not found in the host. Sulfite reduction is one such mycobacterium-specific step and is the central focus of this paper. Sulfite reduction in Mycobacterium smegmatis was investigated using a combination of deletion mutagenesis, metabolite screening, complementation, and enzymology. The initial rate parameters for the purified sulfite reductase from M. tuberculosis were determined under strict anaerobic conditions [kcat = 1.0 (±0.1) electron consumed per second, and Km(SO3−2) = 27 (±1) μM], and the enzyme exhibits no detectible turnover of nitrite, which need not be the case in the sulfite/nitrite reductase family. Deletion of sulfite reductase (sirA, originally misannotated nirA) reveals that it is essential for growth on sulfate or sulfite as the sole sulfur source and, further, that the nitrite-reducing activities of the cell are incapable of reducing sulfite at a rate sufficient to allow growth. Like their nitrite reductase counterparts, sulfite reductases require a siroheme cofactor for catalysis. Rv2393 (renamed che1) resides in the sulfur reduction operon and is shown for the first time to encode a ferrochelatase, a catalyst that inserts Fe2+ into siroheme. Deletion of che1 causes cells to grow slowly on metabolites that require sulfite reductase activity. This slow-growth phenotype was ameliorated by optimizing growth conditions for nitrite assimilation, suggesting that nitrogen and sulfur assimilation overlap at the point of ferrochelatase synthesis and delivery.
The human genome does not encode the sulfur reduction and cysteine-biosynthetic enzymes found in many pathogenic bacteria. The species specificity of these enzymes and the essential metabolic nature of sulfur recommend them as potential targets for antimicrobial development. A considerable literature links sulfur metabolism to the pathogenicity and antibiotic susceptibility of Mycobacterium tuberculosis. Specific sulfolipids correlate well with the virulence of M. tuberculosis (16, 17, 19, 20, 26) and are reported to inhibit phagosome-lysosome fusion (18), which is critical for the survival of the bacterium in macrophages (9, 15). More-recent literature calls into question whether the segregation of sulfolipid across virulent and avirulent strains is a manifestation, rather than a root cause, of virulence (33, 41), and efforts to trace this lineage toward its root are under way (11, 27). Mycothiol, the mycobacterial equivalent of glutathione, utilizes a cysteine thiol to provide the antioxidant protection that the organism needs to survive, particularly during oxidative stress (35), and lower mycothiol levels correlate with enhanced susceptibility to antibiotics, including rifampin and isoniazid (32). The cysteine-biosynthetic pathway, a primary means of assimilating sulfur, has been linked to the survival of the organism during the chronic phase of infection, the basis of which may lie in its resistance to reactive oxygen and nitrogen species (39). The current work explores the reduction of sulfite, an essential step in the biosynthesis of cysteine, in a model organism, Mycobacterium smegmatis.
Mycobacterial assimilation of sulfate begins with its active transport into the cell, whereupon it is chemically activated by the enzyme ATP sulfurylase (cysDN) to form activated sulfate (adenosine 5′-phosphosulfate [APS]) (Fig. 1). APS is then either phosphorylated by APS kinase (cysC) to form the universal sulfuryl group donor 3′-phosphoadenosine 5′-phosphosulfate (PAPS) or reduced by APS reductase (cysH) to form sulfite (12, 38). If sulfate assimilates through the sulfuryl transfer branch of the pathway, the sulfuryl group is transferred from PAPS, via sulfotransferases, to metabolic recipients whose activities are regulated by the modification. If, on the other hand, sulfate is drawn into the reductive branch of the pathway by the action of APS reductase (7, 8), the resulting sulfite is reduced further, in a six-electron reduction, to sulfide by the enzyme sulfite reductase (sirA). Sulfide is then incorporated into cysteine by O-acetyl-l-serine sulfhydrylase (cysK1) (30), and from there the sulfur atom, originally present in sulfate, flows into a myriad of reduced-sulfur-containing metabolites.
FIG. 1.

Segment of sulfur metabolism in mycobacteria. Red arrows and mnemonic terms identify the activities and genes associated with the sulfur reduction operon. Dotted arrows highlight steps that are the primary focus of this study. The cysteine feedback inhibition loop is hypothesized based on similar inhibition in Salmonella enterica serovar Typhimurium (25) and E. coli (21) and complete conservation of the residues involved in the binding of cysteine (28, 29) across numerous organisms, including M. tuberculosis (29). Ac−, without acetyl; CoA, coenzyme A; AcCoA, acetyl coenzyme A; 3Fd(ox), oxidized ferredoxin; 3Fd(red), reduced ferredoxin; Trx(ox), oxidized thioredoxin; Trx(red), reduced thioredoxin.
Nitrite and sulfite reductases have a number of similarities, including conserved catalytic architectures designed to guide an essential siroheme cofactor to function as the active center for a six-electron reduction of substrate to product (12, 13, 22). These enzymes are sufficiently similar that they will often catalyze the reduction of both sulfite and nitrite (12), suggesting that sulfur and nitrogen metabolism may be redundant at the point of sulfite/nitrite reduction and/or in the provision of the siroheme cofactor. Such redundancy is particularly pertinent in the case of M. tuberculosis, which must persist amid high levels of destructive reactive nitrogen species, which it elicits from activated T cells during infection (40).
The genes encoding APS reductase (cysH) and sulfite reductase (sirA) are located in the sulfur reduction operon (see below) in both M. smegmatis and M. tuberculosis, along with a third coding region, Rv2393, which encodes a protein of unknown function. In the current study, the sulfite reduction step in M. smegmatis was explored by deletion mutagenesis, metabolite screening, enzymatic characterization, and complementation.
MATERIALS AND METHODS
Bacterial strains and cultures.
The bacterial strains used in this study are listed in Table 1. The Escherichia coli XL1-Blue and HB101 strains, used for cloning, and the BL21(DE3) Rosetta pLysS strain, used for expression, were propagated in Luria-Bertani (LB) broth or on LB agar at 37°C. M. smegmatis strain mc2155 was grown at 37°C in 7H9 liquid medium (Difco) supplemented with 0.2% (vol/vol) glycerol, 0.2% glucose, and 0.05% Tween 80 or on Middlebrook 7H10 solid medium supplemented as described above. l-Cysteine was obtained from the Sigma Chemical Co. The following antibiotics were used at the concentrations indicated: ampicillin (100 μg/ml) (Fisherbiotech), kanamycin A (25 μg/ml) (Sigma), hygromycin B (50 μg/ml with E. coli and 50 to 150 μg/ml with M. smegmatis) (Roche).
TABLE 1.
Bacterial strains used in this study
| Strain | Description | Source or reference |
|---|---|---|
| E. coli strains | ||
| XL1-Blue | recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac [F′ proAB lacIqZΔM15 Tn10 (Tetr)] | Stratagene |
| BL21(DE3) Rosetta pLysS | F′ ompT hsdSB (rB− mB−) gal dcm (DE3)pLysSRARE2 (Camr) | Novagen |
| JR01 | Rosetta pLysS carrying plasmid pJR1 | This work |
| 302Δa | E. coli cysG Nirs Lac+ CysG− | 31 |
| cysG pER247 strain | Strain 302Δa carrying the plasmid pER247 (pACYC184-lacq-Ptac-P. denitrificans cobA) | 31 |
| RP7 | E. coli cysG pER247 containing plasmid pET23a, M. tuberculosis Rv2393 | This work |
| RP8 | E. coli cysG pER247 containing plasmid pET23a, M. smegmatis Rv2393 | This work |
| HB101 | E. coli K-12 F−Δ(gpt-proA)62 leuB1 glnV44 ara-14 galK2 lacY1 hsdS20 rpsL20 xyl-5 mtl-1 recA13 | 4 |
| M. smegmatis strains | ||
| mc2155 | ept-1 | 43 |
| RPMS001 | mc2155ΔsirA::hyg sacB | This work |
| RPMS002 | mc2155ΔRv2393::hyg sacB | This work |
| RPMS004 | RPMS001 carrying plasmid pRP15 (M. smegmatis sirA complementation vector [Table 2]) | This work |
| RPMS005 | RPMS002 carrying plasmid pRP16 (M. smegmatis che1 complementation vector [Table 2]) | This work |
To obtain the growth curves associated with Fig. 4, M. smegmatis was grown to late exponential phase at 37°C; washed six to eight times in M9 minimal salts without a sulfur source (Na2HPO4 [42 mM], KH2PO4 [24 mM], NaCl [9.0 mM], NH4Cl [19 mM], glucose [0.5%, wt/vol], Tween 80 [0.05%, vol/vol]); suspended in prewarmed M9 medium supplemented with a specific source of sulfur, SO4−2 (1.0 mM), Na2S (1.0 mM), Na2SO3 (1.0 mM), or cysteine (200 μM); and cultured further by shaking at 37°C. Bacterial growth was monitored at 600 nm. To normalize the wild-type and mutant genetic backgrounds in the strains used in Fig. 4C, the wild-type strain was transformed with pMV361, which codes for Hygr and integrates into the genome (44), and pMV261, which confers kanamycin resistance. The mutant strains associated with Fig. 4C were transformed using derivatives of pMV261 that contained either the wild-type sirA gene (pRP15) or Rv2393 (pRP16).
FIG. 4.

Growth of M. smegmatis mutants on various sulfur nutrients. (A) Growth of the ΔsirA mutant on minimal media containing cysteine (□), sulfide (♦), sulfite (○), or sulfate (•) as the sole sulfur source. Generation times for growth on cysteine and sulfide were 7.5 and 6.7 h, respectively; no growth was observed in media containing sulfite or sulfate. (B) Growth of the ΔRv2393 mutant on the same sulfur sources as those mentioned for panel A. The generation time for the mutant was 10 h when the medium contained cysteine or sulfide. Both cells fed sulfate and cells fed sulfite exhibited steady-state growth with a generation time of 19 h. (C) To demonstrate that the growth effects of the deletions were due solely to a lack of expression of the intended coding regions, the growth on sulfate of the deletion strains complemented with plasmids that express the wild-type gene was compared to that of the wild-type strain containing an empty vector. The doubling times of the three strains were extremely similar: for the wild type (○), 8.8 h; for the ΔsirA strain compared with the sirA strain (nirA:comp sirA) (□), 8.6 h; and for the ΔRv2393 strain compared with the Rv2393 strain (Δrv2939:comp rv2393) (♦), 8.5 h. Growth protocols are described in Materials and Methods. Each data point represents the average of results from three experiments; single standard deviation units (not shown) are comparable to the diameters of the symbols representing the averaged values. OD600, optical density at 600 nm.
The cell growth studies presented in Fig. 6 were accomplished by shaking cells overnight at 37°C in the following M9 minimal medium: NaNO3 (20 mM), cysteine (200 μM), Na2HPO4 (42 mM), KH2PO4 (24 mM), NaCl (9.0 mM), glucose (0.5%, wt/vol), Tween 80 (0.05%, vol/vol). The cells were then washed as described above in prewarmed M9 medium lacking a sulfur source. The growth studies were initiated by suspending the washed cells in prewarmed M9 medium with either cysteine or sulfate.
FIG. 6.

Growth of M. smegmatis mutants on nitrate. (A) Growth of the ΔsirA mutant on minimal medium containing nitrate as the sole nitrogen source and either cysteine (○) or sulfate (•) as the sole sulfur source. The generation time for growth on cysteine was 7.1 h, as no growth was observed in medium containing sulfate. (B) Growth of the Δche1 mutant on the same nitrogen and sulfur sources as described for panel A. Generation times for growth on cysteine or sulfate were virtually identical, 7.3 h. Growth protocols are described in Materials and Methods. Each data point represents the average of results from three experiments; single standard deviation units (not shown) are comparable to the diameters of the symbols representing the averaged values. OD600, optical density at 600 nm.
Ferrochelatase complementation.
To assess whether Rv2393 encodes a ferrochelatase, an E. coli cysteine auxotroph that requires ferrochelatase function for growth on SO4−2 (strain 302Δa, a cysG deletion strain containing pER247, a P15 origin plasmid that expresses uroporphyrinogen III methyltransferase) was transformed with ColE1 origin plasmids that express Rv2393 either from M. tuberculosis(pRP17) or M. smegmatis(pRP18) and then tested for the ability to grow on SO4−2 as a sole source of sulfur. Conversion to prototrophy was assessed on minimal agar containing either SO4−2 or cysteine as the sulfur source. Minimal agar contained Na2HPO4 (42 mM), KH2PO4 (24 mM), NaCl (9.0 mM), NH4Cl (19 mM), agar (15 g/liter), CaCl2 (0.10 mM), MgCl2 (1.0 mM), glucose (2.0%), and either MgSO4 (2.0 mM) or cysteine (280 μM). 302Δa carrying pKK (which expresses E. coli CysG) was used as the positive control for growth; the negative-growth-control strain was 302Δa, which carries pER247 and pET23a (the empty Rv2393 expression vector). All plates were incubated at 37°C for 24 to 48 h.
DNA manipulation.
Restriction enzymes (REs) were purchased from New England Biolabs, and digestions were performed according to the manufacturer's recommendations. The purification of DNA from agarose gels and the isolation of plasmid DNA were done using QIAquick gel extraction and QIAprep spin miniprep kits (QIAGEN) according to the manufacturer's protocols. Isolation of M. tuberculosis and M. smegmatis chromosomal DNA was carried out as described previously (5). Standard heat shock protocols were used for the transformation of E. coli strains (34).
Plasmid construction.
All of the plasmids and primers used in this study are listed in Tables 2 and 3, respectively.
TABLE 2.
Plasmids used in this study
| Plasmid | Description | Reference or source |
|---|---|---|
| p0004S | Cosmid containing the Hygr-SacB cassette | 2 |
| phAE159 | Shuttle phasmid, TM4ts::pYUB328 | J. Kriakov and W. R. Jacobs, unpublished |
| pMV261 | Kanr ColE1 oriM aph Phsp60′ | 44 |
| pMV361 | Integrative vector Hygr | 44 |
| pMS2391 | Recombinant cosmid derived from p0004S containing the M. smegmatis sirA AES | This study |
| pMS2393 | Recombinant cosmid derived from p0004S containing the M. smegmatis Rv2393 AES | This study |
| pSKB4-9His-GST | Derived from pGEX-6P-1; contains an N-terminal His tag upstream of a GST tag | 1 |
| pJR1 | pSKB4-9His-GST carrying the M. tuberculosis sirA gene | This study |
| pJR2 | pJR1 with E. coli cysG cloned downstream of sirA | This study |
| pRP15 | pMV261 carrying the M. smegmatis sirA gene | This study |
| pRP16 | pMV261 carrying the M. smegmatis Rv2393 gene | This study |
| pET23a | Cloning vector containing an N-terminal His tag | Novagen |
| pRP17 | pET23a carrying M. tuberculosis Rv2393 | This study |
| pRP18 | pET23a carrying M. smegmatis Rv2393 | This study |
| pKK E. coli cysG | pKK223.3, an overexpression vector derived from pBR322 with the tac promoter, carrying E. coli cysG | 31 |
TABLE 3.
Primers used in this study
| Primera | Sequence (5′-3′) | Amplified sequence |
|---|---|---|
| TBsirA F | GGAATTCCATATGTCCGCGAAGGAGAACCCC | M. tuberculosis sirA |
| TBsirA R | AAGGAAAAAAGCGGCCGCTCATCGCAGGTCGTCCTCCTCGGCCCGGAT | |
| colicysG F | TTTGCGGCCGCAAACGGCTGCCGGTTAATTACTAAGGGGTTTTTAC | E. coli cysG |
| colicysG R | GGCCTAGGTTAATGGTTGGAGAACCAGTTCAGTTTATC | |
| smegsirA F | TACAGCTGATGCTCGAAGACGAGTACTTCAT | M. smegmatis sirA |
| smegsirA R | CCCAAGCTTTCACGTTGCCTACCTCAAATCCGCTTCGTC | |
| smegRv2393 F | CGCAGCTGGTGACGCTCGTCCTGACCGCACAC | M. smegmatis Rv2393 |
| smegRv2393 R | CCCAAGCTTTCAGCGCATGGCCCTGGCCCGTGCACT | |
| 1 | TTTTTTTTCCATAAATTGGGTAGATGCCGAAGGGCCGGCCCGGTGTGGG | 3′-end-flanking region of M. smegmatis sirA |
| 1′ | TTTTTTTTCCATTTCTTGGGCCGACGGCGTCGAGGCGCTTCCAGATCTC | |
| 2 | TTTTTTTTCCATAGATTGGGGCCCCGAGGAGGGTTTCCAGGTGCATCTG | 5′-end-flanking region of M. smegmatis sirA |
| 2′ | TTTTTTTTCCATCTTTTGGGCCCGCATTCGGTCTTGGACAGCCCGGCCC | |
| 3 | TTTTTTTTCCATAAATTGGGTGCGCAACTTCGTGAAACAACGCGAGGAC | 3′-end-flanking region of M. smegmatis Rv2393 |
| 3′ | TTTTTTTTCCATTTCTTGGAGATTCGGTTCGTTCTGCTCACAGAAC | |
| 4 | TTTTTTTTCCATAGATTGGTCGACCGCTACGAAGAGGCGATCGAGG | 5′-end-flanking region of M. smegmatis Rv2393 |
| 4′ | TTTTTTTTCCATCTTTTGGTATCGGTTTCGTGTTCCAGCACTATGCGGC |
F and R refer to forward and reverse primers.
sirA expression vector.
The expression of catalytically competent SirA requires the coexpression of CysG, which produces the quantities of the siroheme cofactor needed for stoichiometric incorporation into SirA. The construction of the coexpression plasmid pJR2 was accomplished in two steps. First, a 1.7-kb fragment containing sirA was PCR amplified from M. tuberculosis genomic DNA using primers TBsirA F and TBsirA R and inserted into the NdeI and NotI sites of pSKB4 (1), yielding pJR1. In the second step, E. coli cysG was PCR amplified (primers colicysG F and colicysG R) and cloned into pJR1 linearized with NotI and BsaAI. The resulting plasmid, pJR2, was used to transform the E. coli BL21(DE3) Rosetta pLysS strain for expression and purification (see below). This strain was named JR01.
Complementation plasmids.
M. smegmatis sirA and Rv2393 were PCR amplified from genomic DNA using the primer pairs smegsirA F/smegsirA R and smegRv2393 F/smegRv2393 R, respectively. In both cases, the forward and reverse primers introduced PvuII and HindIII restriction sites, respectively, which were used to subclone the PCR products into pMV261 (cleaved with the same enzymes), producing plasmids pRP15 (sirA) and pRP16 (Rv2393). pMV261 contains the mycobacterial hsp60 promoter, which facilitates constitutive expression of M. smegmatis SirA and M. smegmatis Rv2393 (44). All constructs were sequenced (AECOM DNA sequencing facility) to confirm the fidelity of the clones.
Construction of M. smegmatis gene deletion mutants.
The M. smegmatis gene deletion mutants were constructed in three stages: recombinant cosmids containing the DNA sequences needed for allelic exchange (allelic-exchange substrates [AESs]) were prepared, high-titer mycobacteriophages needed to isolate genomic deletion events (which occur at low levels) were obtained, and mycobacterial DNA was transduced and selected for allelic exchange. The protocols used to create mycobacterial gene deletion mutants have been described previously (5).
Cosmid construction.
DNA flanking the 5′ and 3′ regions of sirA was PCR amplified, using primer pairs 1/1′ and 2/2′ (see Fig. 3A and Table 3). The flanking regions were subcloned directionally, using the Van911 RE, on either side of the hygromycin resistance-sacB gene cassette found in cosmid p0004S. The resulting recombinant cosmid, pMS2391, contained the AES needed to construct the bacteriophage. Using an identical protocol, primer pairs 3/3′ and 4/4′ were used to generate pMS2393, which contains the Rv2393 AES.
FIG. 3.

Insertion of a hygromycin resistance cassette into sirA and Rv2393 in M. smegmatis mc2155. (A) Primer sets used for the construction of Southern blot probes and AESs. (B) Southern blot of genomic DNAs from wild-type (wt) and ΔsirA strains. BanII-digested genomic DNA was probed using a PCR fragment generated using primers 1 and 1′. The blot revealed that insertion of the resistance cassette caused the expected shift from 1.1 to 2.5 kb. (C) Southern blot of genomic DNAs from the wild-type (wt) and ΔRv2393 strains. MscI-digested genomic DNA was probed using a PCR fragment generated using primers 4 and 4′. Insertion of the resistance cassette resulted in the expected shift from 6.3 to 5.0 kb.
Mycobacteriophage construction.
The AES cosmids and purified phagemid DNA (phAE159) were digested separately with PacI, ligated together, and in vitro packaged (using the GIGApackIII Gold packaging extract [Stratagene]) to produce the transducing λ bacteriophage. E. coli HB101 was then transduced with the phage, and transductants were selected on medium containing hygromycin (150 μg/ml). Phasmid construction was confirmed by PacI digestion prior to electroporation of the phasmid DNA into M. smegmatis. Electroporated cells were plated on 7H10 medium and incubated at the permissive temperature, 30°C, for 3 days. All transducing phages were plaque purified, and high-titer phage lysate (1010 to 1011 PFU/ml) was prepared. M. smegmatis was then transduced by mixing late-log-phase cells with phage lysate (1:1, vol/vol) overnight at 37°C. Transductants were plated onto 7H10 medium plates containing hygromycin (75 μg/ml) and cysteine (40 μg/ml) and incubated at the nonpermissive temperature, 37°C, for 5 days. M. smegmatis deletion mutants were screened by Southern blot analysis (see below).
Southern blotting.
Five micrograms of M. smegmatis genomic DNA was digested completely with either the BanII or the MscI RE and separated by electrophoresis on a 0.7% DNA agarose gel. The gel was immersed in 0.25 M HCl for 5 min, rinsed in H2O, and soaked in 0.40 M NaOH for 10 min. DNA fragments were transferred onto a nylon membrane (Amersham) overnight by the capillary action of a 0.40 M NaOH transfer solution. The membrane was cross-linked (Stratagene) to allow immobilization of DNA fragments on the membrane and then incubated for 10 min in 5 ml of Rapid-hyb buffer (Amersham). Changes in DNA fragment size caused by the deletions were probed using 32P-labeled probes that spanned the 5′ end of sirA and 3′ end of Rv2393. The probes were generated by PCR using the 1/1′ and 4/4′ primer pairs (Table 3 and see Fig. 3A).
Expression and purification of M. tuberculosis SirA.
LB medium was inoculated with an overnight culture of JR01 at an A600 of 0.01, and the cells were grown at 37°C until the optical density at 600 nm reached ∼0.7, at which point SirA expression was induced by adding isopropyl-β-d-thiogalactopyranoside (IPTG) to a final concentration of 0.80 mM. The incubation temperature was then shifted to 17°C, and cells were harvested by centrifugation 17 h later. The cell pellet was suspended in 4.2 ml/g cell paste of lysis buffer (KPO4 [50 mM, pH 8.0], KCl [0.4 M], phenylmethylsulfonyl fluoride [290 μM], pepstatin A [1.5 μM], lysozyme [0.10 mg/ml]), and the solution was stirred for 1 h at 4°C prior to sonication on ice (Branson sonifier). Cellular debris was removed by centrifugation (20 min at 31,000 × g), and the supernatant was loaded onto a 5-ml-chelating-Sepharose fast-flow column charged with Ni2+ and equilibrated with buffer A (KPO4 [50 mM, pH 7.3], KCl [0.4 M]). The column was washed with 10 bed volumes of buffer B (KPO4 [50 mM, pH 7.3], KCl [0.4 M], imidazole [10 mM]). The His-glutathione S-transferase (GST)-tagged fusion protein was then eluted from the Ni2+-chelated Sepharose column with 10 bed volumes of buffer C (KPO4 [50 mM, pH 7.3], KCl [0.4 M], imidazole [250 mM], KCl [0.40 M]). The N-terminal His-GST tag was removed by digestion with PreScission protease (GE Healthcare) during overnight dialysis against HEPES-K+ (25 mM, pH 7.5), KCl (100 mM), glycerol (5%), and β-mercaptoethanol (5.0 mM) at 4°C. To remove the tag and any uncut fusion protein, the dialyzed proteolysate was passed back over the Ni2+ affinity column and washed extensively with buffer B to remove the SirA, which exhibited an affinity for the resin. Typically, 10 mg of protein (>95% pure, as judged by eye from Coomassie blue-stained sodium dodecyl sulfate-polyacrylamide gels) was obtained per liter of culture.
Sulfite reductase assay.
M. tuberculosis SirA was assayed for sulfite and nitrite reductase activity under anaerobic conditions activity using methyl viologen (MV) as an electron donor, which was reduced with Zn metal immediately prior to use (42). The assay mix contained O-acetyl-serine (5.0 mM), sodium sulfite (13 μM), reduced MV (250 μM), O-acetyl-serine sulfhydrylase (1.5 μM), SirA (1.3 μM), and 50 mM HEPES-K+ (pH 8.0) (25 ± 3°C). The assay was carried out under anaerobic conditions; argon gas scrubbed with reduced MV and ascorbic acid was used to degas all buffers and to maintain all samples under positive pressure in a vacuum manifold. A custom optical cuvette that allowed maintenance of positive pressure while the mixture was stirred was used to optically monitor reactions. The assay was initiated by the addition of the sodium salts of sulfite or nitrite. The enzymatic consumption of either sulfite or nitrite was monitored by following the decrease in absorbance at 684 nm (ɛ = 4.8 × 103 M−1 cm−1) caused by the oxidation of MV (24).
RESULTS
Sulfur reduction operon—an overview.
The M. smegmatis and M. tuberculosis sulfur reduction operons are quite similar (Fig. 1A; Table 4). At their 5′ termini, they begin with sirA (sulfite reductase) followed by cysH (APS reductase) and Rv2393 (a hypothetical protein). In both organisms, each of the three coding regions appear to be translationally coupled (i.e., the stop codon of the 5′ gene overlaps the start codon of its downstream partner). The homologous coding regions in the two strains are similar in both length and sequence (Table 4; Fig. 2) (45). The metabolically linked, sulfate transport operons, which include an ABC transporter (encoded by cysTWA) and a sulfate binding protein (encoded by subI), are antiparallel to the sulfur reduction operon in both cases but differ in that they are separated by ∼5 kb in M. tuberculosis and only 10 bp in M. smegmatis.
TABLE 4.
Primary sequence similarity of SirA from M. smegmatis and M. tuberculosisa
| Enzyme | Coding region | Identity (%) | Similarity (%) |
|---|---|---|---|
| Sulfite reductase | sirA | 82 | 89 |
| APS reductase | cysH | 70 | 78 |
| Hypothetical protein | Rv2393 | 58 | 70 |
CLUSTAL W comparison metrics for homologous proteins encoded by the M. smegmatis and M. tuberculosis sulfur reduction operons (45).
FIG. 2.

Sulfur reduction operons of M. smegmatis and M. tuberculosis. (A) Schematic representation of the M. smegmatis (M. smeg) and M. tuberculosis (M. tb) sirA operons. The double hatch mark in the M. tuberculosis diagram indicates a 5-kb intergenic region between the operons.
Construction of sirA and Rv2393 deletion mutants.
To begin to assess the role of sirA and Rv2393 in vivo, each gene in M. smegmatis was deleted using allelic exchange. First, allelic-exchange cosmids containing a selectable gene cassette (γδ res hyg sacB res γδ) flanked by ∼900-bp regions of DNA that spanned the 3′ and 5′ edges of sirA or Rv2393 were constructed (see Materials and Methods). The AESs were then incorporated into the conditionally replicating phasmid phAE159, in vitro packaged into λ phage heads, and transduced into E. coli HB101. Recombinant shuttle cosmids were purified from Hygr E. coli transductants and converted into mycobacteriophage-packaged DNA molecules by transfecting them into M. smegmatis cells. These cells were plated for phage plaques at the permissive temperature of 30°C. High-titer transducing lysates were obtained by propagation of the mycobacteriophage in M. smegmatis. The lysates were used to transduce M. smegmatis at the nonpermissive temperature (37°C) and plated on selective medium. Restriction of phage replication results in a double-crossover event between the homologous DNA arms flanking the disrupted gene (5). Fifteen transductants were obtained in the case of sirA and six in the case of Rv2393. Six isolates from each set were tested by Southern blot analysis, all of which had undergone the expected allelic replacement. A single representative from each set is shown in Fig. 3B. The observed shifts in DNA fragment size caused by insertion of the Hygr-SacB gene cassette into the genomic copies of sirA and Rv2393 are in excellent agreement with the expected values—1.4 and 1.3 kb, respectively.
sirA is essential for growth in minimal media.
Upon entering the macrophage, M. tuberculosis maintains the expression of SirA, a sulfite reductase that catalyzes the six-electron reduction of sulfite (37). Frequently, both sulfite and nitrite can be reduced by either nitrite or sulfite reductases, which are classified on the basis of their substrate preference (12). Thus, a strain lacking sirA might obtain the sulfide needed for cysteine biosynthesis from the reduction of sulfite by other reductases, which would render sirA nonessential and perhaps diminish its efficacy as an antimicrobial target. To establish whether the organism depends essentially entirely on SirA for the provision of sulfide, the M. smegmatis sirA deletion mutant was tested for its ability to grow, relative to the wild type, on sulfur metabolites that straddle either side of the point of action of sirA in the cysteine-biosynthetic pathway. When the mutant and wild type were fed downstream metabolites (i.e., S−2 or cysteine), their growth rates were virtually indistinguishable; however, when fed an upstream metabolite (SO4−2 or SO3−2), the mutant showed no detectible growth over a 50-h incubation, while the wild type grew with a doubling time of 7.1 (±0.4) h (Fig. 4A). sirA is clearly essential for growth on SO4−2 or SO3−2 and central to the sulfur-reducing metabolism of the organism.
SirA—the enzyme.
The kinetic parameters and efficiency of the enzyme toward sulfite reduction have not yet been determined. For this reason, the enzyme from M. tuberculosis was expressed in E. coli and purified to homogeneity (see Materials and Methods), and the kinetic constants were determined. Obtaining pure SirA with the expected levels of siroheme cofactor (22) required coexpression of E. coli CysG, a trifunctional protein that catalyzes the last three steps in siroheme biosynthesis (see Materials and Methods). The reduction reaction was monitored continuously, under anaerobic conditions, by following the change in absorbance at 684 nm associated with the oxidation of the electron donor, MV (24). Sulfide produced by SirA was removed rapidly by serine sulfhydrylase (from E. coli), which converts O-acetyl-l-serine and sulfide to cysteine (46). The removal of the product ensures that product inhibition will not contribute significantly to the reaction, which simplifies extracting kinetic constants from the reaction progress curve (1). Under such conditions, slopes taken over sufficiently small regions (∼3%) of the progress curve provide initial rate measurements over an essentially continuously varying range of substrate concentrations. The double-reciprocal plot of the SirA-catalyzed reduction of sulfite is shown in Fig. 5. The kinetic constants obtained by fitting the data are as follows:  was equal to 27 (±1) μM, the kcat was equal to 0.17 (±0.01) electron consumed s−1 (sulfite reduced to sulfide) or 1.0 electron consumed s−1, and the catalytic efficiency (kcat/Km) was equal to 3.7 × 104/M−1 s−1. It should be noted that, consistently with previous work (38), no enzymatic turnover was observed when sulfite was replaced by nitrite at concentrations as high as 3.0 mM.
was equal to 27 (±1) μM, the kcat was equal to 0.17 (±0.01) electron consumed s−1 (sulfite reduced to sulfide) or 1.0 electron consumed s−1, and the catalytic efficiency (kcat/Km) was equal to 3.7 × 104/M−1 s−1. It should be noted that, consistently with previous work (38), no enzymatic turnover was observed when sulfite was replaced by nitrite at concentrations as high as 3.0 mM.
FIG. 5.

Initial rate determination of the Michaelis constants for the SirA-catalyzed reduction of sulfite. Sulfite reduction was monitored continuously (under stringent, anaerobic conditions) via the change in absorbance at 684 nm associated with the oxidation of the electron donor, MV (see Materials and Methods). The assay conditions were as follows: sulfite (13 μM), reduced MV (250 μM), O-acetyl-l-serine (30 μM), SirA (1.3 μM), O-acetyl-l-serine sulfhydrylase (1.2 μM), and HEPES-K+ (50 mM, pH 8.0) (25 ± 3°C). The reaction rate is given in terms of sulfite reduced per unit time (μM/s−1). The reduction of one equivalent of sulfite requires the oxidation of six equivalents of MV. v, initial velocity.
Growth phenotype of the Rv2393 deletion mutant.
Rv2393 is situated at the C terminus of the sulfur reduction operon. This locale suggests that Rv2393, whose function is unknown, may be important for cysteine biosynthesis. To explore the ways in which Rv2939 might function in the cysteine-biosynthetic pathway of M. smegmatis, an Rv2393 deletion strain was constructed (see Materials and Methods). The deletion removes the entire gene with the exception of short, 140- and 53-nucleotide stretches of sequence at the 5′ and 3′ edges of the coding region, respectively (Fig. 3A). Growth of the deletion strain on sulfide or cysteine is indistinguishable from that of the wild type (Fig. 4B and C), suggesting that Rv2393 acts upstream of the enzymes that incorporate sulfide into cysteine (O-acetylserine sulfhydrylase [cysK1] and serine transacetylase [cysE]). In contrast, the doubling time of the mutant on sulfite or sulfate was approximately twofold slower than that of the wild type (Fig. 4B and C). Thus, the point of action of Rv2393 coincides with that of SirA: the reduction of sulfite.
The primary structure of Rv2393 clusters with an orthologous group of proteins from ancient phylogenies (COG2381.1), many of which are type II chelatases—catalysts that insert divalent cations (Co2+, Fe2+, Mg2+, or Ni2+) into modified tetrapyrroles to produce redox-sensitive cofactors, including chlorophylls, vitamin B12, heme, coenzyme F430, and siroheme (6). Sulfite and nitrite reductases, which are often essential for the assimilation of sulfur and nitrogen, require siroheme to accomplish their redox chemistry. The linking of Rv2393 to the chelatase family and to sulfite reductase function provides a rationale for the colocalization of sirA and Rv2393 and the common metabolic point of action of the proteins that they encode, which is that Rv2393 is the ferrochelatase that inserts Fe2+ into sirohydrochlorin to produce the siroheme cofactor that is necessary for SirA function.
To test the hypothesis that Rv2393 is, in fact, a ferrochelatase, plasmid-borne Rv2393 was used to complement an E. coli ΔcysG mutant (302Δa) (Table 1), in which ferrochelatase function is deleted. The genome of the mutant lacks cysG, which encodes siroheme synthase, a trifunctional protein that catalyzes the last three steps in siroheme biosynthesis: (i) methylation of uroporphyrinogen III (at C-2 and C-7) to produce dihydrosirohydrochlorin; (ii) oxidation of dihydrosirohydrochlorin to produce sirohydrochlorin; and (iii) insertion of Fe2+ into sirohydrochlorin to produce siroheme (31). The cysteine-biosynthetic pathway is disrupted in cysG deletion strains at the point of insertion of iron into sirohydrochlorin. The cysteine auxotrophy that results from the inability to produce siroheme provides a nutrient-based selection for methyltransferase and/or chelatase function. It should be noted that the dehydrogenase function needed by these strains is provided by endogenous activity (47); thus, selecting for ferrochelatase function exclusively requires restoration of the methyltransferase activity in 302Δa, which is accomplished by transformation with pER247, a P15 origin plasmid that expresses the Pseudomonas denitrificans CobA protein, a uroporphyrinogen III methyltransferase that does not exhibit chelatase function (3, 14). The 302Δa strain harboring pER247 and pET23a with or without the Rv2393 coding region were plated onto minimal medium plates containing either sulfate or cysteine as the sole source of sulfur (see “Ferrochelatase complementation” in Materials and Methods) and incubated at 37°C for 24 to 48 h. The methyltransferase-competent strain (302Δa/pER247/pET23a) showed no detectible growth on sulfate as a sole source of sulfur. However, when transformed with a pET23A plasmid containing Rv2393, either from M. tuberculosis or M. smegmatis, the growth on the sulfate-containing plates was comparable to that of the cysG-complemented strain. The observed Rv2393-dependent transformation of the strain to cysteine prototrophy strongly supports the idea that Rv2393 encodes a ferrochelatase that is capable of inserting Fe2+ into sirohydrochlorin to produce siroheme. Based on these findings, we have named Rv2393 che1 to indicate both its chelatase function and that it does not appear to be the only chelatase in the M. tuberculosis genome.
Metabolic complementarity in sulfite and nitrite reduction.
Assimilatory nitrite and sulfite reductases, like those found in M. smegmatis and M. tuberculosis, share similar catalytic strategies, molecular architectures, and substrate specificities. These enzymes are named on the basis of their relative catalytic efficiencies (Vmax/Km) for sulfite versus nitrite, and interestingly, despite a greater overall efficiency toward sulfite, sulfite reductases often turn over faster with nitrite [i.e., 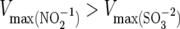 ] (12). Thus, the inability of the ΔsirA strain to grow on sulfite could reflect the inefficiency of the mycobacterial nitrite reductase toward sulfite, which has not been measured, and/or the level at which the enzyme is expressed in the organism when grown on ammonia (as was the case in the present study).
] (12). Thus, the inability of the ΔsirA strain to grow on sulfite could reflect the inefficiency of the mycobacterial nitrite reductase toward sulfite, which has not been measured, and/or the level at which the enzyme is expressed in the organism when grown on ammonia (as was the case in the present study).
Given that sulfite reductase (SirA) is remarkably inefficient toward nitrite (see above), it appears from the M. smegmatis genome that nitrite reductase (nirBD) is the organism's sole means of obtaining ammonia from either nitrate or nitrite (10, 23). Ammonia is required for the biosynthesis of amino acids, and from there, nitrogen is drawn into the urea cycle, glutamate metabolism, pyrimidine biosynthesis, and ultimately every nitrogen-containing compound in the cell (23). To assess the ability of nitrite reductase to reduce sulfite in vivo, the growth of the ΔsirA mutant on sulfate was assessed in a medium selected to optimize the expression of nitrite reductase for the assimilation of nitrogen, M9 medium in which nitrate was substituted for ammonia at an equivalent concentration, 20 mM. Under this condition, all assimilated nitrogen must pass either through nitrite reductase or through other, as-yet-unidentified, reductases in the cell. The substitution of nitrate for ammonia did not result in detectible growth of the organism on sulfate over a 70-h period at 37°C; however, replacement of sulfate by cysteine produced normal growth with nitrite (Fig. 6A). Clearly, the nitrite reductase activity of the cell is sufficient to allow normal growth on metabolites that lie downstream of the point of action of SirA. Thus, nitrogen and sulfur metabolism are well isolated at the point of nitrite and sulfite reduction by what may ultimately prove to be pronounced differences in the substrate specificities of the enzymes that catalyze these reactions.
Unlike the ΔsirA mutation, which causes a no-growth phenotype, the Δche1 mutation results in slow growth of the organism on sulfate or sulfite. Thus, the orthogonality seen in the sulfite reduction step clearly does not extend to the siroheme cofactor, which can be provided to SirA from alternative metabolic sources. Amelioration of slow growth by plasmid-born Che1 suggests that the endogenous siroheme pool(s) is in some way insufficient to achieve the levels of SirA activity needed for normal growth. Because nitrite reductase requires siroheme, it is plausible that the growth of the organism on nitrite, compared to that on ammonia, will up-regulate the cellular levels of siroheme; if so, the growth rate of theΔche1 mutant, when grown on sulfate or sulfite, will increase when the nitrogen source is switched from ammonia to nitrate. This is precisely what was observed (Fig. 6B). Clearly, the perturbation of nitrogen metabolism caused by the ammonia-to-nitrate substitution has impacted sulfur metabolism at the intersection of sulfite reduction and ferrochelatase function.
DISCUSSION
The structure of SirA was determined recently, and the enzyme was shown to reduce sulfite (at a single sulfite concentration, 5.0 mM) under oxygen-depleting conditions (excess sodium dithionite) (38). In the current work, the initial rate of sulfite reduction was studied under strict anaerobic conditions as a function of sulfite concentration, which yielded kinetic constants for the enzyme [kcat = 0.17 (±0.1) electron consumed s−1 (sulfite reduced) and  = 27 (±1) μM] that are in line with previously published values for sulfite reductases from other organisms (12, 24). The value of kcat/Km, 3.4 × 104/M−1 s−1, indicates that SirA catalyzes SO3−2 reduction with modest efficiency, and the inability to detect the SirA-catalyzed reduction of nitrite at concentrations as high as 3.0 mM reveals that the efficiency of the enzyme toward NO2−1 is extremely low and suggests that this reaction is not likely to be physiologically relevant.
= 27 (±1) μM] that are in line with previously published values for sulfite reductases from other organisms (12, 24). The value of kcat/Km, 3.4 × 104/M−1 s−1, indicates that SirA catalyzes SO3−2 reduction with modest efficiency, and the inability to detect the SirA-catalyzed reduction of nitrite at concentrations as high as 3.0 mM reveals that the efficiency of the enzyme toward NO2−1 is extremely low and suggests that this reaction is not likely to be physiologically relevant.
High-density transposon insertion mutagenesis in combination with microarray mapping of the M. tuberculosis genome has indicated that sirA is important for optimal growth of the organism on minimal medium containing sulfate as a sole source of sulfur (36). sirA was deleted in the present work and shown to be essential for the growth of M. smegmatis on sulfate or sulfite, regardless of whether nitrate or ammonia was used as a nitrogen source. These facts are of value not only for their contribution to the genetic and biochemical fundamentals of sulfur reduction in M. smegmatis and related organisms but also for what they reveal about the orthogonality of the sulfur and nitrogen-reducing pathways in these species. On this note, it is interesting that it may be possible to assess, and therefore more accurately annotate, the sulfite-versus-nitrite substrate preferences of ferredoxin-dependent reductases based on primary sequence (38).
The common metabolic point of action of Rv2393 and SirA, the siroheme requirement for SirA function, and the ability of Rv2393 to complement an E. coli ferrochelatase mutant argue strongly that Rv2393, a protein with previously unidentified function, is a ferrochelatase, which we have named che1. The fact that the slow-growth phenotype of the Δche1 mutant is rescued by plasmid-encoded Che1 or by growth on nitrate supports the idea that the organism's growth rate is limited by siroheme access. While nitrogen and sulfur metabolism are well isolated by what appear to be nonoverlapping substrate specificities of the nitrite and sulfite reductases, these pathways do overlap at the level of siroheme production. It is interesting to note that a second CbiX-like open reading frame (Rv0259c) is found quite near the genes that encode nitrite reductase in M. smegmatis. Perhaps the siroheme pool(s) in the mutant grown on rate-limiting sulfur metabolites is simply too small to satisfy the metabolic demands placed on it; alternatively, access of SirA to the pool(s) could be limited by intrinsic specificities that bias delivery of the metalated porphyrin to particular recipients. It is exciting to consider the possibility that the Δche1 strain, whose growth rate is limited by sulfite reduction, presents us with the opportunity to establish a linkage between sulfite reduction and metabolic sources of siroheme.
Acknowledgments
This work was supported by National Institutes of Health grants GM54469 (T.S.L.) and RO1 AI26170 (W.R.J.).
We thank Martin Warren and Evelyne Raux, Department of Biosciences, University of Kent, for kindly providing the E. coli cysG mutant strains and Ann-Francis Miller, Department of Chemistry and Biochemistry, University of Kentucky, for her generous guidance and support in executing the anaerobic, sulfite reductase assays.
Footnotes
Published ahead of print on 20 July 2007.
REFERENCES
- 1.Andreassi, J. L., II, and T. S. Leyh. 2004. Molecular functions of conserved aspects of the GHMP kinase family. Biochemistry 43:14594-14601. [DOI] [PubMed] [Google Scholar]
- 2.Bardarov, S., S. Bardarov, Jr., M. S. Pavelka, Jr., V. Sambandamurthy, M. Larsen, J. Tufariello, J. Chan, G. Hatfull, and W. R. Jacob, Jr. 2002. Specialized transduction: an efficient method for generating marked and unmarked targeted gene disruptions in Mycobacterium tuberculosis, M. bovis BCG and M. smegmatis. Microbiology 148:3007-3017. [DOI] [PubMed] [Google Scholar]
- 3.Blanche, F., L. Debussche, D. Thibaut, J. Crouzet, and B. Cameron. 1989. Purification and characterization of S-adenosyl-l-methionine: uroporphyrinogen III methyltransferase from Pseudomonas denitrificans. J. Bacteriol. 171:4222-4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boyer, H. W., and D. Roulland-Dussoix. 1969. A complementation analysis of the restriction and modification of DNA in Escherichia coli. J. Mol. Biol. 41:459-472. [DOI] [PubMed] [Google Scholar]
- 5.Braunstein, M., S. S. Bardarov, and W. R. Jacobs, Jr. 2002. Genetic methods for deciphering virulence determinants of Mycobacterium tuberculosis. Methods Enzymol. 358:67-99. [DOI] [PubMed] [Google Scholar]
- 6.Brindley, A. A., E. Raux, H. K. Leech, H. L. Schubert, and M. J. Warren. 2003. A story of chelatase evolution: identification and characterization of a small 13-15-kDa “ancestral” cobaltochelatase (CbiXS) in the archaea. J. Biol. Chem. 278:22388-22395. [DOI] [PubMed] [Google Scholar]
- 7.Carroll, K. S., H. Gao, H. Chen, J. A. Leary, and C. R. Bertozzi. 2005. Investigation of the iron-sulfur cluster in Mycobacterium tuberculosis APS reductase: implications for substrate binding and catalysis. Biochemistry 44:14647-14657. [DOI] [PubMed] [Google Scholar]
- 8.Chartron, J., K. S. Carroll, C. Shiau, H. Gao, J. A. Leary, C. R. Bertozzi, and C. D. Stout. 2006. Substrate recognition, protein dynamics, and iron-sulfur cluster in Pseudomonas aeruginosa adenosine 5′-phosphosulfate reductase. J. Mol. Biol. 364:152-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clemens, D. L. 1996. Characterization of the Mycobacterium tuberculosis phagosome. Trends Microbiol. 4:113-118. [DOI] [PubMed] [Google Scholar]
- 10.Cole, S. T., R. Brosch, J. Parkhill, T. Garnier, C. Churcher, D. Harris, S. V. Gordon, K. Eiglmeier, S. Gas, C. E. Barry III, F. Tekaia, K. Badcock, D. Basham, D. Brown, T. Chillingworth, R. Connor, R. Davies, K. Devlin, T. Feltwell, S. Gentles, N. Hamlin, S. Holroyd, T. Hornsby, K. Jagels, A. Krogh, J. McLean, S. Moule, L. Murphy, K. Oliver, J. Osborne, M. A. Quail, M. A. Rajandream, J. Rogers, S. Rutter, K. Seeger, J. Skelton, R. Squares, S. Squares, J. E. Sulston, K. Taylor, S. Whitehead, and B. G. Barrell. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393:537-544. [DOI] [PubMed] [Google Scholar]
- 11.Converse, S. E., J. D. Mougous, M. D. Leavell, J. A. Leary, C. R. Bertozzi, and J. S. Cox. 2003. MmpL8 is required for sulfolipid-1 biosynthesis and Mycobacterium tuberculosis virulence. Proc. Natl. Acad. Sci. USA 100:6121-6126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crane, B. R., and E. D. Getzoff. 1996. The relationship between structure and function for the sulfite reductases. Curr. Opin. Struct. Biol. 6:744-756. [DOI] [PubMed] [Google Scholar]
- 13.Crane, B. R., L. M. Siegel, and E. D. Getzoff. 1997. Structures of the siroheme- and Fe4S4-containing active center of sulfite reductase in different states of oxidation: heme activation via reduction-gated exogenous ligand exchange. Biochemistry 36:12101-12119. [DOI] [PubMed] [Google Scholar]
- 14.Crouzet, J., L. Cauchois, F. Blanche, L. Debussche, D. Thibaut, M. C. Rouyez, S. Rigault, J. F. Mayaux, and B. Cameron. 1990. Nucleotide sequence of a Pseudomonas denitrificans 5.4-kilobase DNA fragment containing five cob genes and identification of structural genes encoding S-adenosyl-l-methionine: uroporphyrinogen III methyltransferase and cobyrinic acid a,c-diamide synthase. J. Bacteriol. 172:5968-5979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferrari, G., H. Langen, M. Naito, and J. Pieters. 1999. A coat protein on phagosomes involved in the intracellular survival of mycobacteria. Cell 97:435-447. [DOI] [PubMed] [Google Scholar]
- 16.Gangadharam, P. R., M. L. Cohn, and G. Middlebrook. 1963. Infectivity, pathogenicity and sulpholipid fraction of some Indian and British strains of tubercle bacilli. Tubercle 44:452-455. [DOI] [PubMed] [Google Scholar]
- 17.Goren, M. B., O. Brokl, and W. B. Schaefer. 1974. Lipids of putative relevance to virulence in Mycobacterium tuberculosis: correlation of virulence with elaboration of sulfatides and strongly acidic lipids. Infect. Immun. 9:142-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goren, M. B., P. D'Arcy Hart, M. R. Young, and J. A. Armstrong. 1976. Prevention of phagosome-lysosome fusion in cultured macrophages by sulfatides of Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 73:2510-2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goren, M. B., J. M. Grange, V. R. Aber, B. W. Allen, and D. A. Mitchison. 1982. Role of lipid content and hydrogen peroxide susceptibility in determining the guinea-pig virulence of Mycobacterium tuberculosis. Br. J. Exp. Pathol. 63:693-700. [PMC free article] [PubMed] [Google Scholar]
- 20.Grange, J. M., V. R. Aber, B. W. Allen, D. A. Mitchison, and M. B. Goren. 1978. The correlation of bacteriophage types of Mycobacterium tuberculosis with guinea-pig virulence and in vitro-indicators of virulence. J. Gen. Microbiol. 108:1-7. [DOI] [PubMed] [Google Scholar]
- 21.Hindson, V. J. 2003. Serine acetyltransferase of Escherichia coli: substrate specificity and feedback control by cysteine. Biochem. J. 375:745-752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Janick, P. A., D. C. Rueger, R. J. Krueger, M. J. Barber, and L. M. Siegel. 1983. Characterization of complexes between Escherichia coli sulfite reductase hemoprotein subunit and its substrates sulfite and nitrite. Biochemistry 22:396-408. [DOI] [PubMed] [Google Scholar]
- 23.Kanehisa, M., S. Goto, M. Hattori, K. F. Aoki-Kinoshita, M. Itoh, S. Kawashima, T. Katayama, M. Araki, and M. Hirakawa. 2006. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 34:D354-D357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krueger, R. J., and L. M. Siegel. 1982. Spinach siroheme enzymes: isolation and characterization of ferredoxin-sulfite reductase and comparison of properties with ferredoxin-nitrite reductase. Biochemistry 21:2892-2904. [DOI] [PubMed] [Google Scholar]
- 25.Leu, L. S., and P. F. Cook. 1994. Kinetic mechanism of serine transacetylase from Salmonella typhimurium. Biochemistry 33:2667-2671. [DOI] [PubMed] [Google Scholar]
- 26.Middlebrook, G., C. M. Coleman, and W. B. Schaefer. 1959. Sulfolipid from virulent tubercle bacilli. Proc. Natl. Acad. Sci. USA 45:1801-1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mougous, J. D., M. D. Leavell, R. H. Senaratne, C. D. Leigh, S. J. Williams, L. W. Riley, J. A. Leary, and C. R. Bertozzi. 2002. Discovery of sulfated metabolites in mycobacteria with a genetic and mass spectrometric approach. Proc. Natl. Acad. Sci. USA 99:17037-17042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Olsen, L. R., B. Huang, M. W. Vetting, and S. L. Roderick. 2004. Structure of serine acetyltransferase in complexes with CoA and its cysteine feedback inhibitor. Biochemistry 43:6013-6019. [DOI] [PubMed] [Google Scholar]
- 29.Pye, V. E., A. P. Tingey, R. L. Robson, and P. C. Moody. 2004. The structure and mechanism of serine acetyltransferase from Escherichia coli. J. Biol. Chem. 279:40729-40736. [DOI] [PubMed] [Google Scholar]
- 30.Rabeh, W. M., and P. F. Cook. 2004. Structure and mechanism of O-acetylserine sulfhydrylase. J. Biol. Chem. 279:26803-26806. [DOI] [PubMed] [Google Scholar]
- 31.Raux, E., H. K. Leech, R. Beck, H. L. Schubert, P. J. Santander, C. A. Roessner, A. I. Scott, J. H. Martens, D. Jahn, C. Thermes, A. Rambach, and M. J. Warren. 2003. Identification and functional analysis of enzymes required for precorrin-2 dehydrogenation and metal ion insertion in the biosynthesis of sirohaem and cobalamin in Bacillus megaterium. Biochem. J. 370:505-516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rawat, M., G. L. Newton, M. Ko, G. J. Martinez, R. C. Fahey, and Y. Av-Gay. 2002. Mycothiol-deficient Mycobacterium smegmatis mutants are hypersensitive to alkylating agents, free radicals, and antibiotics. Antimicrob. Agents Chemother. 46:3348-3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rousseau, C., O. C. Turner, E. Rush, Y. Bordat, T. D. Sirakova, P. E. Kolattukudy, S. Ritter, I. M. Orme, B. Gicquel, and M. Jackson. 2003. Sulfolipid deficiency does not affect the virulence of Mycobacterium tuberculosis H37Rv in mice and guinea pigs. Infect. Immun. 71:4684-4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
- 35.Sareen, D., G. L. Newton, R. C. Fahey, and N. A. Buchmeier. 2003. Mycothiol is essential for growth of Mycobacterium tuberculosis Erdman. J. Bacteriol. 185:6736-6740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sassetti, C. M., D. H. Boyd, and E. J. Rubin. 2003. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48:77-84. [DOI] [PubMed] [Google Scholar]
- 37.Schnappinger, D., S. Ehrt, M. I. Voskuil, Y. Liu, J. A. Mangan, I. M. Monahan, G. Dolganov, B. Efron, P. D. Butcher, C. Nathan, and G. K. Schoolnik. 2003. Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: insights into the phagosomal environment. J. Exp. Med. 198:693-704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schnell, R., T. Sandalova, U. Hellman, Y. Lindqvist, and G. Schneider. 2005. Siroheme- and [Fe4-S4]-dependent NirA from Mycobacterium tuberculosis is a sulfite reductase with a covalent Cys—Tyr bond in the active site. J. Biol. Chem. 280:27319-27328. [DOI] [PubMed] [Google Scholar]
- 39.Senaratne, R. H., A. D. De Silva, S. J. Williams, J. D. Mougous, J. R. Reader, T. Zhang, S. Chan, B. Sidders, D. H. Lee, J. Chan, C. R. Bertozzi, and L. W. Riley. 2006. 5′-Adenosinephosphosulphate reductase (CysH) protects Mycobacterium tuberculosis against free radicals during chronic infection phase in mice. Mol. Microbiol. 59:1744-1753. [DOI] [PubMed] [Google Scholar]
- 40.Shiloh, M. U., and C. F. Nathan. 2000. Reactive nitrogen intermediates and the pathogenesis of Salmonella and mycobacteria. Curr. Opin. Microbiol. 3:35-42. [DOI] [PubMed] [Google Scholar]
- 41.Sirakova, T. D., A. K. Thirumala, V. S. Dubey, H. Sprecher, and P. E. Kolattukudy. 2001. The Mycobacterium tuberculosis pks2 gene encodes the synthase for the hepta- and octamethyl-branched fatty acids required for sulfolipid synthesis. J. Biol. Chem. 276:16833-16839. [DOI] [PubMed] [Google Scholar]
- 42.Skjeldal, L., J. Krane, and T. Ljones. 1989. Proton n.m.r. of ferredoxin from Clostridium pasteurianum. Int. J. Biol. Macromol. 11:322-325. [DOI] [PubMed] [Google Scholar]
- 43.Snapper, S. B., R. E. Melton, S. Mustafa, T. Kieser, and W. R. Jacobs, Jr. 1990. Isolation and characterization of efficient plasmid transformation mutants of Mycobacterium smegmatis. Mol. Microbiol. 4:1911-1919. [DOI] [PubMed] [Google Scholar]
- 44.Stover, C. K., V. F. de la Cruz, T. R. Fuerst, J. E. Burlein, L. A. Benson, L. T. Bennett, G. P. Bansal, J. F. Young, M. H. Lee, G. F. Hatfull, et al. 1991. New use of BCG for recombinant vaccines. Nature 351:456-460. [DOI] [PubMed] [Google Scholar]
- 45.Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wei, J., Q. X. Tang, O. Varlamova, C. Roche, R. Lee, and T. S. Leyh. 2002. Cysteine biosynthetic enzymes are the pieces of a metabolic energy pump. Biochemistry 41:8493-8498. [DOI] [PubMed] [Google Scholar]
- 47.Woodcock, S. C., E. Raux, F. Levillayer, C. Thermes, A. Rambach, and M. J. Warren. 1998. Effect of mutations in the transmethylase and dehydrogenase/chelatase domains of sirohaem synthase (CysG) on sirohaem and cobalamin biosynthesis. Biochem. J. 330:121-129. [DOI] [PMC free article] [PubMed] [Google Scholar]