

Abstract
Adenovirus type 5 encodes two highly structured short RNAs, the virus-associated (VA) RNAI and RNAII. Both are processed by Dicer into small RNAs that are incorporated into the RNA-induced silencing complex (RISC). We show here, by cloning of small RNAs, that approximately 80% of Ago2-containing RISC immunopurified from late-infected cells is associated with VA RNA-derived small RNAs (mivaRNAs). Most surprisingly, VA RNAII, which is expressed at 20-fold lower levels compared to that of VA RNAI, appears to be the preferred substrate for Dicer and accounts for approximately 60% of all small RNAs in RISC. The mivaRNAs are derived from the 3′ strand of the terminal stems of the VA RNAs, with the major fraction of VA RNAII starting at position 138. The small RNAs derived from VA RNAI were more heterogeneous in size, with the two predominant small RNAs starting at positions 137 and 138. Collectively, our results suggest that the mivaRNAs are efficiently used for RISC assembly in late-infected cells. Potentially, they function as miRNAs, regulating translation of cellular mRNAs. In support of this hypothesis, we detected a fraction of the VA RNAII-derived mivaRNAs on polyribosomes.
Small noncoding RNAs of approximately 21 to 23 nucleotides in length regulate a variety of cellular processes, including mRNA degradation, translation, and transcription (for reviews see references 2, 6 and 39). One class of small RNAs, called microRNAs (miRNAs), are processed in the nucleus by an enzyme called Drosha into a hairpin RNA that is transported to the cytoplasm and further processed by Dicer into a short miRNA duplex intermediate. The miRNA strand of the duplex is then incorporated into the RNA-induced silencing complex (RISC) that will bind to a target mRNA. If the complementarity between the mRNA and the miRNA is partial, the assembled RISC can induce translational repression. In a related reaction where the complementarity between the small RNA and the mRNA is extensive, RISC will cause a cut in the mRNA strand that will result in its degradation.
The significance of cellular miRNAs as regulators of processes like cell growth and development is rapidly becoming a well-established field of research (for reviews see references 9 and 36). Recently, it has also become increasingly clear that several vertebrate viruses encode their own small RNAs that may function as miRNAs or small interfering RNAs (siRNAs). Several of these small RNAs have been shown to play key functions both in the infectious and the latent phase of virus infections (reviewed in references 11 and 34). For example, an miRNA encoded by herpes simplex virus type 1 downregulates genes activating apoptosis, thereby providing a maintenance function for the latent stage of a herpesvirus infection (14). Also, simian virus 40 (SV40) produces an siRNA at late times of infection that targets the mRNAs from the viral early transcription unit, and this, as a consequence, causes a reduction in T antigen expression, a mechanism that appears to reduce the efficiency of clearance of SV40-infected cells by cytotoxic T lymphocytes (37). Most remarkably, hepatitis C virus, which does not encode its own miRNA, uses a cellular miRNA, the liver-specific miR-122, to enhance virus replication (16).
In our work, we have focused on human adenovirus type 5 (Ad5), which encodes two 160-nucleotide-long noncoding RNA polymerase III transcripts, the virus-associated (VA) RNAs I and II (reviewed in reference 24). The VA RNAs are highly structured (22), with imperfect stem-loop structures that resemble precursor RNAs used for the production of miRNAs. VA RNAI has a well-characterized function as an RNA that ensures that protein synthesis is sustained at a high level in virus-infected cells by blocking activation of the interferon-induced double-stranded RNA (dsRNA)-binding enzyme PKR (reviewed in reference 13). Several recent studies have also suggested that the VA RNAs are processed to small RNAs that may play an important role in subverting the RNA interference machinery during a lytic infection (3, 4, 33). Both VA RNAI and VA RNAII are processed by Dicer into small RNAs that are incorporated into RISC (3, 33). They can function as siRNAs or miRNAs, depending on the complementarity to the reporter mRNA (33). The VA RNAs accumulate to enormous quantities in late-infected cells (>108 copies per cell) (24) and have been shown to function as competitive substrates squelching Dicer (3). Furthermore, VA RNAI has been suggested to block the nucleus-to-cytoplasm export of pre-miRNAs by competing for binding to the nuclear export receptor exportin-5 (21).
In addition to the effect of the VA RNAs on Dicer, we have also noted that RISC cleavage of an exogenous target RNA is drastically suppressed in late-infected cells (3). Here we have addressed how adenovirus suppresses the activity of the RISC. Our results suggest that the VA RNAs saturate RISC with VA RNA-specific small RNAs. Thus, we find that approximately 80% of all small RNAs in RISC isolated from late-infected cells are derived from the VA RNAs. Most surprisingly, VA RNAII, which is expressed at about 5% of the level of VA RNAI, accounts for around 60% of all small RNAs in Ago2-containing RISC. The function of these VA RNA-derived small RNAs is currently not known. However, we note that a fraction of the VA RNAII-derived small RNAs are associated with polyribosomes, arguing that they may regulate gene expression by functioning as miRNAs.
MATERIALS AND METHODS
Cell culture and transfection.
293 and 293-Ago2 cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% newborn calf serum (NCS), 100 U/ml PEST at 37°C in 5% CO2. All cell culture reagents were purchased from Gibco/BRL. DNA was transfected using the calcium phosphate coprecipitation technique (5), whereas RNA was transfected with Lipofectamine 2000 (Invitrogen), using the protocol supplied by the manufacturer. Plasmid DNA was prepared by using a Maxiprep kit (QIAGEN), whereas dsRNA was purchased from Dharmacon.
Establishment of the 293-Ago2 cell line.
293 cells grown on 6-cm culture plates were transfected with plasmid pIRESneo-FLAG/HA Ago2 (25). Selection for stably transfected cells was initiated 48 h posttransfection by the addition of G418 (400 μg/ml). After 3 weeks, when all 293 cells in an untransfected control plate were dead, the single colonies of 293-Ago2 cells were isolated and transferred to a 24-well plate for further passage. A collection of 20 clones was picked and assayed for Ago2 expression by Western blotting using the FLAG M2 antibody. One clone showing a high (approximately sevenfold overexpression) and stable Ago2 protein expression was selected and used for subsequent experiments.
Virus infection.
Cells were infected with wt900 (38) at a multiplicity of 100 fluorescence-forming units (FFU) per cell (if not otherwise stated) in DMEM containing 2% NCS. After 45 min, the medium was replaced with fresh medium containing 10% NCS. The uninfected control cells were treated identically except that virus was omitted. Twenty-four hours later, the cells were harvested as described below.
Immunopurification of RISC.
Cytoplasmic cell extracts were prepared by the treatment of cells on ice for 20 min with IsoB-NP-40 (10 mM Tris-HCl [pH 7.9], 150 mM NaCl, 1.5 mM MgCl2, 1% NP-40) followed by a centrifugation at 16,000 × g for 10 min at 4°C. The supernatant from one 15-cm culture plate was incubated with 4 μl of anti-FLAG M2 agarose beads (Sigma) with constant rotation overnight at 4°C. The beads were washed three times in NET-1 buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 2.5% Tween 20). Complexes were eluted with NET-1 supplemented with 1% 2-mercaptoethanol and 150 ng/μl 3× FLAG peptide (Sigma) with constant rotation for 4 h at 4°C. Supernatants recovered after centrifugation were used for RNA extraction or Western blotting analysis.
Western blotting analysis.
Protein extracts were separated by 8% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and processed as described previously (27). Primary antibody anti-FLAG M2 (Sigma) was used at a concentration of 10 μg/ml, and anti-Ago2 (8C7) was used at a 1:500 dilution.
Cloning of small RNA.
Seven 15-cm culture plates of 293-Ago2 cells were infected with wt900. Twenty-four hours postinfection, cells were lysed by IsoB-NP-40 treatment, and the Ago2 protein-containing complexes were purified by immunoprecipitation (see description above). RNA was purified by proteinase K treatment, followed by phenol-chloroform extraction and ethanol precipitation. The purification of small RNAs and the cloning strategy were performed as described previously (5, 31).
Sequencing and sequence analysis.
Clones containing small RNA inserts were sequenced using an ABI Prism 3700 machine, and the small RNAs were extracted with PERL script. Sequences were filtered to remove RNAs shorter than 17 nucleotides and then compared, using WU-BLASTN, to the Ad5 genome (GenBank accession number AC_000008), the human subset of miRBase (ftp://ftp.sanger.ac.uk/pub/mirbase/sequences/8.0/mature.fa Feb8-06 release), the human mRNAs (ftp://ftp.wip.ncbi.nlm.nih.gov/genomes/H_sapiens/RNA/rna.fa.gz Mar3-06 release), and the human tRNAs (http://lowelab.ucsc.edu/GtRNAdb/Hsapi/Hg17-tRNAs.fa). The small RNAs were annotated based on the best score or were classified as unknown if no significant match was found. The coordinates of the high-scoring segment pairs were extracted and used to determine which parts of the adenovirus genome and known miRNAs had been cloned.
Cytoplasmic S15 extract preparation.
Cytoplasmic extracts were prepared as previously described (3). Briefly, cells were disrupted by 20 to 30 strokes with a 23-gauge syringe needle. The nuclei were pelleted, and the supernatant was centrifuged at 15,000 × g for 60 min, supplemented with 5% glycerol, frozen in liquid nitrogen, and stored at −80°C. The protein concentration was typically 6 to 8 μg/μl.
RISC assay.
A synthetic 21-base pair siRNA directed against the p99 mRNA (3) was transfected into four 10-cm plates of 293-Ago2 cells (40 nM). Six hours posttransfection, cells were infected with wt900 (at 100 FFU/cell). Twenty-four hours postinfection, S15 cytoplasmic extracts were prepared, and the RISC was isolated by affinity purification (as described above). The beads were resuspended in 50 μl of hypotonic buffer with 5% glycerol. Each 10-μl RISC assay contained 5 μl of S15 cytoplasmic extract or 5 μl of beads and the 32P-labeled p99 reporter mRNA under the assay conditions described previously (3). Reaction mixtures were incubated for 2 h at 30°C, and RNA was isolated and separated on a denaturing 8% polyacrylamide gel.
Northern blotting analysis.
Small RNAs were separated on a denaturing 15% polyacrylamide gel, transferred to a Hybond NX membrane (Amersham), chemically cross-linked, and hybridized as described previously (29). Hybridization probes were generated by 5′ end labeling one strand of an siRNA or by random priming of longer DNA fragments (5).
Polyribosome analysis.
Polyribosomes were prepared and fractionated as previously described (32). To disrupt actively translating ribosomes, cells were placed in fresh medium containing 200 μM puromycin (Sigma) 20 min before harvest. Briefly, cells were lysed in a buffer containing 5 mM Tris-HCl (pH 7.5), 1.5 mM KCl, 2.5 mM MgCl2, 0.5% deoxycholate, 0.5% Triton X-100, 100 μg/ml cycloheximide, 200 μg/ml yeast tRNA, and 120 U/μl RNasin, and the supernatant was separated on a 10 to 50% sucrose gradient in a model SW40 rotor. To disrupt polysomes and other metal-stabilized complexes, a lysis buffer containing 30 mM EDTA and a reduced concentration of MgCl2 (0.2 mM) was used. Ten fractions were collected, RNA was extracted, and VA RNA-derived small RNAs (miva)RII-138 and β-actin mRNA were visualized by Northern blotting.
RESULTS
Incorporation of small RNAs into RISC is suppressed in late-adenovirus-infected cells.
The observation that RISC activity directed toward a target RNA is suppressed in late-infected cells (3) could be explained by several mechanisms. An attractive model is that large amounts of VA RNA-specific small RNAs are produced during infection and outcompete other small RNAs, thereby saturating RISC with VA RNA-derived small RNAs. In such a model, RISC would still be active but potentially redirected toward VA RNA-specific sequences (3). To test this hypothesis, we established a 293 cell line stably expressing an amino-terminal FLAG/HA-tagged Ago2 protein (25). The 293-Ago2 cell line was transfected with a synthetic siRNA directed toward a reporter mRNA (3) and infected with adenovirus 6 hours posttransfection. Following a 24-h incubation, the cells were harvested, and cytoplasmic S15 extracts were prepared. Ago2-containing complexes were captured with an anti-FLAG M2 agarose resin. RISC activity was assayed by cleavage of a 32P-labeled capped target mRNA. As shown in Fig. 1A, the mRNA cleavage activity of the immunopurified RISC was significantly reduced in virus-infected cells (Fig. 1A, compare lanes 3 and 4), a result that agrees with our previous findings, which were produced from cytoplasmic S15 extracts (3). Furthermore, the reduction in RISC activity (Fig. 1A) was accompanied by a similar decrease in siRNA content in RISC (Fig. 1B, lanes 3 and 4). To demonstrate that the reduced activity and siRNA content in purified RISC were not due to a less efficient purification of Ago2 RNPs from infected cells, we probed Western blots with the Ago2 antibody 8C7 (Fig. 1C, lanes 3 and 4). Taken together, the results show that the suppression of RISC activity in late-adenovirus-infected cells toward an exogenous target RNA results from a reduced efficiency of siRNA assembly into functional RISC.
FIG. 1.

Incorporation of exogenously added siRNA into RISC is suppressed in late-adenovirus-infected cells. 293-Ago2 cells were transfected with a synthetic siRNA. Six hours later, the cells were infected with wt900. S15 cytoplasmic extracts were prepared 24 h postinfection, and Ago2 containing RISC was isolated by FLAG affinity purification. (A) RISC activity was assayed by incubating the immunopurified Ago2 RNP complexes with a uniformly labeled target mRNA. The siRNA guides RISC to cleave this target mRNA, producing a 252-nucleotide 5′ cleavage product and a 232-nucleotide 3′ cleavage product. Lane M, DNA molecular size marker. (B) Total RNA was isolated from the same volume of immunopurified RISC and analyzed by Northern blotting and probed with a 5′ end-labeled sense strand siRNA. Lane C, the siRNA antisense strand was loaded as a size marker. (C) The same volume of immunopurified RISC was analyzed by Western blotting and probed with Ago2 antibody 8C7.
To determine whether the incorporation of a cellular miRNA into Ago2-containing complexes was similarly suppressed in late-infected cells, we analyzed the content of miR-16 in purified RISC. For this experiment, 293-Ago2 cells were infected with adenovirus, and cells were harvested 24 h postinfection. Small RNAs were prepared from total cytoplasmic extracts and compared to the small RNA content in purified Ago2-containing RNPs. As shown in Fig. 2A, an adenovirus infection did not have any adverse effects on total miR-16 accumulation (Fig. 2A, lanes 3 and 4). In contrast, the miR-16 content in purified RISC was significantly reduced in virus-infected cells compared to that in uninfected control cells (Fig. 2A, lanes 1 and 2). Probing Western blots with the 8C7 Ago2 antibody demonstrated that the analyzed fractions of total cytoplasmic extracts and purified RISC contained equal amounts of Ago2 protein (Fig. 2B), demonstrating that the reduced miR-16 content in purified RISC was not due to a selective loss of Ago2 RNPs in late-infected cells. Taken together, these results show that an adenovirus infection also results in a reduced efficiency of miRNA assembly into RISC at late times of infection.
FIG. 2.
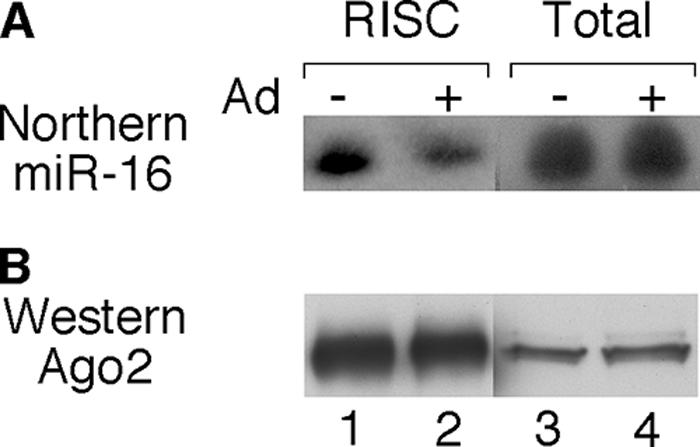
Reduced efficiency of RISC assembly of miR-16 in adenovirus-infected cells. 293-Ago2 cells were infected with wt900, and S15 extracts were prepared 24 h postinfection. Ago2-containing RISC was isolated by FLAG affinity purification. (A) RNA was isolated from equal fractions of total extracts and immunopurified RISC and was analyzed by Northern blotting probed by a 5′ end-labeled oligonucleotide hybridizing to miR-16. (B) Equal fractions of total extracts or immunopurified RISC was analyzed by Western blotting and probed with Ago2 antibody 8C7.
Both VA RNAI- and VA RNAII-derived small RNAs are stably associated with RISC.
To determine whether the previously observed small RNAs derived from the VA RNAs (3, 4, 33) were stably associated with RISC, we affinity purified Ago2-containing complexes from infected 293-Ago2 cells and assayed RISC activity against reporter RNAs containing sequences complementary to the 5′ or 3′ halves of VA RNAI or VA RNAII, respectively. In agreement with our previous results, using cytoplasmic S15 extracts (3), the size of the cleavage products suggests that Dicer cleaves only the terminal stem of VA RNAI (Fig. 3B) and VA RNAII (Fig. 3C). Furthermore, the size of the cleavage product suggests that all VA RNAI- and nearly all VA RNAII-derived small RNAs incorporated into RISC are derived from the 3′ strand of the respective terminal stem. The same asymmetry in RISC assembly for VA RNAI has previously been observed by others (4, 33).
FIG. 3.

VA RNA-derived small RNAs are stably associated with RISC. A schematic diagram of RISC substrate RNAs is shown in panel A. The VA RNA genes were separated near the apical loop into two halves and cloned into a reporter mRNA, generating target regions complementary to the 5′ and 3′ halves of both VA RNAs (see reference 3). Immunopurified RISC from uninfected 293-Ago2 cells (Mock) or cells infected with wt900 was assayed for RISC activity against the synthetic reverse VA transcripts with target regions complementary to the 5′ or 3′ half of VA RNAI (B) or VA RNAII (C). Arrows indicate the span of the VA RNA target regions in respective transcript. The positions of 5′ and 3′ cleavage products generated by Dicer cleavage at the terminal stem in VA RNAI and VA RNAII are indicated by a dot. Bands labeled with * indicate the 3′ end cleavage product of the substrate RNA, which usually is degraded in S15 cytoplasmic extracts but is often seen in reactions using immunopurified RISC.
Cloning of RISC-associated small RNAs from adenovirus-infected cells.
To directly test whether RISC becomes saturated with VA RNA-specific small RNAs in vivo, we cloned and sequenced approximately 1,200 small RNAs from the 17- to 26-nucleotide-long RNA population from uninfected and adenovirus-infected 293-Ago2 cells. To determine whether the incorporation of VA RNA-derived small RNAs into RISC was unbiased with respect to the total small RNA pool, we cloned both the total cytoplasmic small RNA pool and the small RNAs stably associated with immunopurified Ago2-containing RISC. Each small RNA was assigned to an annotation class based on the BLASTN query (Fig. 4). The average length of the cloned small RNAs was 20.0 nucleotides, and by comparing the sequences to annotated, known miRNAs, we could conclude that the 5′ end was generally intact (86%), whereas the 3′ end was frequently degraded by a few nucleotides (62%). On average, 1.5 nucleotides at the 3′ end were missing in our samples. The seed sequence (20), however, was intact in 95% of the samples.
FIG. 4.

Changes in the small RNA pool of the cytoplasm and in immunopurified RISC after an adenovirus infection. Small RNAs (17 to 26 nucleotides) were cloned, sequenced, and annotated from uninfected (A) and adenovirus-infected (B) cytoplasmic extracts. The content of immunopurified (IP) RISC from uninfected (C) and infected (D) cells was also sampled by the same method. The “?” represents small RNAs of unknown origin. The distribution of individual small RNA classes is shown in Table S1 in the supplemental material.
Small RNAs derived from VA RNAII are preferentially assembled into RISC in adenovirus-infected cells.
In striking contrast to what we had expected, VA RNAII appears to be the major source for virus-derived small RNAs incorporated into RISC. We cloned approximately twice the amount of small RNAs derived from VA RNAII as that derived from VA RNAI from the total pool of small RNAs in infected cells (Fig. 4B). Since VA RNAII accounts for only 5% of the total VA RNA pool of infected cells (see below), this result suggests that VA RNAII is preferentially processed by Dicer. This disproportionate overrepresentation of VA RNAII was even more pronounced in the immunopurified RISC, where VA RNAII-derived sequences accounted for approximately 60% of the total small RNA population (Fig. 4D). In total, the VA RNAs accounted for approximately 80% of all small RNAs in immunopurified RISC. The selective loading of VA RNA sequences into RISC consequently occurs at the expense of other small RNAs, including cellular miRNAs. Thus, in uninfected cells, known miRNAs account for approximately 55% of the cloned small RNAs from immunopurified RISC (Fig. 4C). In adenovirus-infected cells, this figure has been reduced to approximately 10% (Fig. 4D).
Characterization of the VA RNA-derived small RNAs.
An analysis of the cDNA clones revealed that essentially all VA RNAI- and VA RNAII-derived small RNAs originated from the terminal stem. A tiny fraction (0.5%) of the cloned small RNAs originated from the 5′ strand of the apical stem of VA RNAI. However, these small RNAs were not detected in the population cloned from immunopurified RISC (Fig. 5A). Furthermore, both VA RNAI and VA RNAII showed a strand bias, in which the 3′ strand of the terminal stem was selectively incorporated into RISC. For VA RNAII, a small fraction (2.5%) of the small RNAs was derived from the 5′ strand (Fig. 5B). The small RNAs show a certain heterogeneity, both in terms of the starting nucleotide and at the 3′ terminus. As discussed above, the variation at the 3′ end could, in many cases, result from a shortening of the small RNAs during the cloning process. However, the majority of the differences in the 5′ nucleotide most likely represent a slight variation in Dicer cleavage. Since such a variation may produce small RNAs with different biological functions, we have named them accordingly dependent on the start nucleotide in the RNA. For example, the major small RNA from VA RNAII starts at nucleotide 138 and therefore is named mivaRII-138 (Fig. 5B). Similarly, the two major small RNAs from VA RNAI are named as mivaRI-137 and mivaRI-138 (Fig. 5A).
FIG. 5.

VA RNA-derived small RNAs present in RISC. Schematic picture showing the exact positioning and relative amounts of VA RNAI-derived (A) and VA RNAII-derived (B) small RNAs cloned from immunopurified RISC. Variations in length dependent on partial 3′ end degradations are indicated by shades of gray. The most abundant forms are named after their starting positions (e.g., mivaRI-137, mivaRI-138, and mivaRII-138). The VA RNA structures are from reference 22.
VA RNA expression in 293-Ago2 cells.
The majority of the mivaRNAs comes from the 3′ half of the terminal stem of VA RNAII, both in the total cytoplasmic pool of small RNAs and in the immunopurified RISC (Fig. 4). This result was completely unexpected, since VA RNAI has been shown to be the major VA RNA expressed during a lytic adenovirus infection (8). To verify that the VA RNAs were expressed at the expected ratio in the 293-Ago2 cell line, under our experimental conditions, we quantitated VA RNA expression by Northern blotting. Although VA RNAI and VA RNAII are identical in length, VA RNAII appears to have a more compact structure and migrates significantly faster than VA RNAI on a denaturing polyacrylamide gel (Fig. 6A, lanes 2 and 3 and 5 and 6). This difference in migration rate made it possible to simultaneously visualize both RNAs using a 32P-labeled probed spanning the whole VA RNA region (Fig. 6A, lanes 8 and 9). The result confirms the previous conclusions that VA RNAI is the major VA RNA expressed late in infection. At a multiplicity of infection of 10 FFU/cell, VA RNAII accounts for approximately 10% of the total pool of VA RNAs. In contrast, VA RNAII levels are reduced to only about 5% at the higher multiplicity (100 FFU/cell). This finding is in agreement with previous results showing that the VA RNAII promoter is much weaker than the VA RNAI promoter and loses activity under limiting conditions (8). It should be noted that all our experiments were done using a multiplicity of infection of 100 FFU/cell.
FIG. 6.

VA RNAII is preferentially associated with RISC. (A) Five micrograms of total RNA prepared 24 h postinfection from uninfected or wt900-infected (10 or 100 FFU/cell) 293-Ago2 cells was separated on an 8% denaturing polyacrylamide gel and transferred to a Hybond membrane. 32P-labeled DNA probes detecting VA RNAI (lanes 1 to 3), VA RNAII (lanes 4 to 6) or both VA RNAs (lanes 7 to 9) were used as probes to detect the full-length VA RNAs, as indicated by the arrows. The relative ratio of VA RNAI and VA RNAII expression in wt900-infected 293-Ago2 cells was based on the quantitative results shown in lanes 8 and 9. Lane MOI, multiplicity of infection (FFU/cell). (B) S15 cytoplasmic extracts prepared from uninfected 293-Ago2 cells (lanes 1 and 2) or wt900-infected cells (lanes 3 and 4) were assayed for RISC activity, using target transcripts detecting RISC loaded with the 3′ strand of VA RNAI or of VA RNAII. Arrows show the position of the cleavage products. The quantitative results of target RNA cleavage are mean values based on three independent experiments.
The cloning of small RNAs from RISC is not biased toward VA RNAII.
The overrepresentation of VA RNAII in the small RNA cDNA library could hypothetically also result from a cloning artifact, where the VA RNAII small RNA population might be favored. This hypothesis predicts that the proportion of the VA RNAI-specific RISC should be greater than the proportion observed for VA RNAI-derived clones. To test this hypothesis, we quantitated, in a parallel experiment, the amount of RISC activity in S15 extracts directed toward VA RNAI and VA RNAII. As shown in Fig. 6B, the VA RNAII-derived mivaRNAs yield approximately sevenfold higher RISC activity than that of VA RNAI mivaRNAs. This should be compared to an approximately twofold overrepresentation of VA RNAII-derived mivaRNAs in the cloned small RNA population (Fig. 4B). Taken together, these results clearly argue against the hypothesis that the VA RNAII-derived mivaRNAs are preferentially cloned. However, it should be noted that we do not have kinetic information establishing whether the VA RNAI- and VA RNAII-derived mivaRNAs differ in their ability to induce catalytically active RISC. Thus, we can only conclude that the RISC activity profile as determined in our experimental setup clearly indicates that the overrepresentation VA RNAII-derived mivaRNA in immunopurified RISC does not support the hypothesis that these small RNAs are preferentially cloned.
The asymmetric assembly of the VA RNAII terminal stem into RISC is not mimicked by a synthetic siRNA.
It has previously been noted that the relative stability of the first four base pairs of the siRNA is critical for strand selection during RISC assembly (18, 35). Potentially, the asymmetry in the assembly of the 3′ strand of VA RNAII into RISC may result from the position of a bulge three nucleotides from the 5′ end of mivaRII-138 (Fig. 5B and 7A). To test this hypothesis, 293-Ago2 cells were (i) infected with wild-type virus, (ii) transfected with a plasmid expressing VA RNAII, or (iii) transfected with a synthetic mivaRII-138 dsRNA (Fig. 7A). Cytoplasmic extracts were prepared, and the RISC activity was directed toward the 5′ and 3′ halves of VA RNAII, measured by in vitro cleavage of a capped substrate RNA (3). As shown in Fig. 7B, transfection of the mivaRII-138 dsRNA resulted in an almost symmetrical incorporation of the 5′ and 3′ strands of the dsRNA into RISC (Fig. 7B, lanes 7 and 8). In contrast, in adenovirus-infected cells (Fig. 7B, lanes 3 and 4) and in cells transfected with a plasmid expressing VA RNAII (Fig. 7B, lanes 9 and 10), the 3′ strand was almost exclusively assembled into RISC. MivaRII-138 contains bulged nucleotides (Fig. 7A). However, this does not seem to severely impair dsRNA formation. Thus, more than 70% of the mivaRII-138 transfected into cells was double stranded (data not shown). Collectively, our results suggest that the asymmetrical RISC assembly favoring the 3′ strand of VA RNAII is enhanced if mivaRII-138 is generated by Dicer cleavage of the native VA RNA molecule.
FIG. 7.

Asymmetry of VA RNAII RISC assembly. (A) The sequence of the synthetic mivaRII-138 dsRNA. (B) S15 cytoplasmic extracts from uninfected 293-Ago2 cells (control), cells infected by wt900 (Ad), nonspecific siRNA-transfected cells (siGFP), synthetic mivaRII-138 dsRNA-transfected cells (mivaRII-138), or VA RNAII plasmid-transfected cells (Plasmid) were assayed for RISC activity, using target RNAs detecting RISC loaded with the 5′ or the 3′ strand of VA RNAII. Arrows indicate the span of the VA RNAII target region. The positions of the 5′ and 3′ cleavage products generated by Dicer cleavage at the terminal stem in VA RNAII are indicated by a dot. Lane M, DNA molecular size marker.
VA RNAII small RNAs are associated with dense polysome-like RNPs.
Previous studies have shown that cellular miRNAs are associated with polyribosomes and control host cell gene expression at the level of translation (19, 26, 28). To test whether the mivaRNAs are similarly associated with polyribosomes, we fractionated cytoplasmic extracts prepared from infected cells on a sucrose gradient and analyzed the presence of mivaRII-138 in the polyribosome fractions by Northern blotting. As shown in Fig. 8B, most of mivaRII-138 was detected in the top fraction of the gradient (Fig. 8, lane 11). This is probably not so unexpected, since mivaRNAII-138 is produced in massive amounts in late-infected cells (see Discussion). However, a significant proportion of mivaRII-138 sedimented in the polyribosome region of the gradient (Fig. 8B, lanes 15 to 18). This signal was specific since it was not detected in uninfected cells (Fig. 8B, lanes 1 to 10) or in cells infected with the VA RNAII mutant virus dl328 (data not shown). To support the theory that some of the faster migrating complexes were polysomes, we treated cells with puromycin, which disrupts translating ribosomes by acting as an acceptor for the growing peptide chain. As shown in Fig. 8B, puromycin treatment resulted in a dramatic increase in mivaRII-138 in the top fraction of the gradient (Fig. 8B, lane 11) and a significant shift of small RNAs toward smaller structures in the gradient (Fig. 8B, lanes 12 to 17). This result indicates that a fraction of mivaRII-138 is indeed associated with polysomes in late-infected cells. However, it is also clear that a large fraction of mivaRII-138 exists in dense puromycin-resistant complexes. Treatment of extracts with EDTA, which breaks down ribosomes and other metal-stabilized noncovalent complexes, completely abolished the rapidly sedimenting mivaRII-138 complexes, suggesting that mivaRII-138 associates with dense RNP complexes of currently unknown composition (Fig. 8B).
FIG. 8.

A fraction of the VA RNAII small RNAs is associated with polysomes. Adenovirus-infected (wt900) or uninfected (Mock) 293 cells were incubated in the absence or presence of puromycin or EDTA. (A) The absorbance profile is shown with the position of 80S monosomes indicated. The polysomal distributions of mivaRII-138 (B) and β-actin mRNA (C) were analyzed by Northern blotting and quantified by PhosphorImager scanning.
It is well established that host cell translation is effectively shut off in late adenovirus-infected cells, although essentially normal levels of cellular mRNAs are present in the cytoplasm at late times of infection (7). In agreement with this, we find that the bulk of the β-actin mRNA population appears to be “free” or associated with smaller polysomes in adenovirus-infected cells compared to mock-infected cells (Fig. 8C). This finding agrees with previous work showing that cellular protein synthesis is blocked at the level of translation initiation (10) and probably also translation elongation (17). It is noteworthy that a large fraction of the β-actin mRNA appears also to sediment as puromycin-resistant but EDTA-sensitive RNP complexes in both infected and uninfected cells (Fig. 8C).
DISCUSSION
This study shows that during an adenovirus infection, VA RNAII appears to be a more favorable substrate for Dicer cleavage than VA RNAI and a preferred small RNA for RISC assembly. Thus, although 20-fold more VA RNAI accumulates in late-infected cells (Fig. 6A), approximately 2-fold more VA RNAII-derived mivaRNAs were present in the total pool of small RNAs (Fig. 4B). This translates into a 40-fold higher substrate preference for Dicer to cleave VA RNAII than VA RNAI. We estimate that under our experimental conditions, approximately 1.5% of VA RNAII is cleaved by Dicer, resulting in the production of approximately 75,000 copies of mivaRII-138 in late-infected cells. Furthermore, in immunopurified Ago2-containing RISC, the VA RNAII excess seemed even more pronounced (Fig. 4D), suggesting that there might be a slight preference for VA RNAII also at the level of RISC assembly. The decrease in RISC-associated cellular miRNAs appears to arise from the fact that the mivaRNAs are produced in large quantities and are efficiently incorporated into RISC, thereby competing cellular miRNAs. However, a potential caveat with an overexpression strategy in a stable cell line is that if Ago2 is the limiting factor in the cell, production of an expanded pool of Ago2 may create an excess of RISC rather than showing that the mivaRNAs are efficient competitors for Ago2. This is a potential problem in all overexpression approaches, which we are aware of and will address in our future work. However, we note that the RISC assembly of a synthetic siRNA is significantly impaired (Fig. 1) and that incorporation of a cellular miRNA is less efficient in adenovirus-infected cells (Fig. 2), suggesting that the mivaRNAs indeed compete for the formation of new RISC complexes in late-infected cells.
A complementarity between the mRNA and positions two to seven of a miRNA, the “seed sequence” (20), is critical for translational repression, while the 3′ region of the miRNA has a modulating effect under certain circumstances (12). Since the adenovirus genome is symmetrically transcribed, essentially every base pair is expressed as cytoplasmic mRNA (either as a rightward or leftward transcribed mRNA). Thus, complementary hexamers to the mivaRI-137, mivaRI-138, or mivaRII-138 seed sequence occur as much as 50 times in the Ad5 genome (see Fig. S1 in the supplemental material). However, a more careful analysis indicates that there has been a selection against having hexamers complementary to the VA RNA seed sequences expressed in mature viral mRNA. None of the mivaRNA complementary sequence interactions are sufficiently long enough to be expected to induce mRNA degradation. Also, since miRNAs appear to bind to the 3′ untranslated region of a mRNA (discussed in reference 6), there is only one hypothetical mivaRNA interaction that targets cytoplasmic mRNA: mivaRII-138 targeting the L2 family of mRNAs (see Fig. S1 in the supplemental material).
Asymmetry is the hallmark of miRNA assembly (reviewed in reference 15). Thus, typically only one strand of the double-stranded miRNA intermediate is assembled into RISC. Similarly, mivaRNA assembly is also highly asymmetric, with essentially only the 3′ strand of both VA RNAs incorporated into RISC (Fig. 5). Most likely the mivaRNAs are viral equivalents of cellular miRNAs. Although the mechanism by which miRNAs regulate translation is currently debated (initiation or elongation), several studies have shown that they are stably associated with polyribosomes (for a discussion see reference 30). Accordingly, we found that mivaRII-138 is associated with polyribosomes (Fig. 8), a result that is in line with the hypothesis that it functions as an miRNA and regulates gene expression at the level of translation. The list of putative cellular target mRNAs for the mivaRNAs is long and includes genes involved in transcription regulation, cell cycle control, cell signaling, etc. However, we have not so far experimentally demonstrated that the mivaRNAs function as translational repressors.
Our finding that the adenovirus type 5 VA RNAs are processed into small RNAs that are expressed at high levels may be related to the fact that this virus efficiently establishes persistent infections. In fact, as much as 20% of the adult population and 50% of healthy children may be persistently infected by adenovirus (1). It is also known that adenovirus devotes a significant portion of its genome to the production of gene products whose functions appear to combat the host immune system (reviewed in reference 23). For example, VA RNAI has been shown to play an important role in restricting the host cell immune response against an adenovirus-infected cell by blocking activation of the interferon-induced enzyme PKR (reviewed in reference 13). An intriguing possibility is that the mivaRNAs aid in the establishment of these benign long-term infections by modulating activation of the interferon response or other aspects of the immune system.
The majority of all adenoviruses encode two VA RNA genes. However, a substantial fraction (9 out of 47 sequenced) has only one VA RNA gene. It has been speculated that VA RNAII may enhance virus growth in a tissue-specific fashion. Thus, it has been noted that adenoviruses that infect the gastroenteric tract encode one VA RNA gene, whereas adenoviruses that cause respiratory tract infections and keratoconjunctivitis encode two VA RNA genes (22). It is possible that VA RNAII provides a unique function that is superfluous in gastroenteric infections. From this point, it is interesting to note that the enteric types Ad40 and Ad41, which contain only one VA RNA gene (22), do not appear to establish persistent infections (1). Thus, one may speculate that the hijacking of RISC by mivaRII-138 may be related to an important function of VA RNAII in the establishment of persistent infections.
The difficult but key challenge for our future work is to identify natural target mRNAs that are regulated by the mivaRNAs. What will the targets be? Will they be related, and which cellular processes will be involved? Moreover, does the fact that VA RNAII-derived small RNAs predominate in RISC reflect a more significant biological function of these small RNAs in virus multiplication and/or persistent infections? This report represents the beginning of what hopefully will be an exciting scientific story.
Supplementary Material
Acknowledgments
We thank M. Gunnar Andersson for much help during the initiation phase of this project. We also thank Thomas Tuschl for the pIRESneo-FLAG/HA Ago2 plasmid, Gideon Dreyfuss for the 8C7 Ago2 antibody, and Catharina Svensson for critical comments on the manuscript.
This work was supported by the Swedish Cancer Society and the Swedish Research Council through a grant to the Uppsala RNA Research Centre (UCCR).
Footnotes
Published ahead of print on 25 July 2007.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Allard, A., B. Albinsson, and G. Wadell. 1992. Detection of adenoviruses in stools from healthy persons and patients with diarrhea by two-step polymerase chain reaction. J. Med. Virol. 37:149-157. [DOI] [PubMed] [Google Scholar]
- 2.Ambros, V. 2004. The functions of animal microRNAs. Nature 431:350-355. [DOI] [PubMed] [Google Scholar]
- 3.Andersson, M. G., P. C. Haasnoot, N. Xu, S. Berenjian, B. Berkhout, and G. Akusjärvi. 2005. Suppression of RNA interference by adenovirus virus-associated RNA. J. Virol. 79:9556-9565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aparicio, O., N. Razquin, M. Zaratiegui, I. Narvaiza, and P. Fortes. 2006. Adenovirus virus-associated RNA is processed to functional interfering RNAs involved in virus production J. Virol. 80:1376-1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ausubel, F. M., R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl. 2006. Current protocols in molecular biology. John Wiley & Sons, Inc., Boston, MA.
- 6.Bartel, D. P. 2004. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281-297. [DOI] [PubMed] [Google Scholar]
- 7.Beltz, G. A., and S. J. Flint. 1979. Inhibition of HeLa cell protein synthesis during adenovirus infection. Restriction of cellular messenger RNA sequences to the nucleus. J. Mol. Biol. 131:353-373. [DOI] [PubMed] [Google Scholar]
- 8.Bhat, R. A., and B. Thimmappaya. 1984. Adenovirus mutants with DNA sequence perturbations in the intragenic promoter of VAI RNA gene allow the enhanced transcription of VAII RNA gene in HeLa cells. Nucleic Acids Res. 12:7377-7388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carthew, R. W. 2006. Gene regulation by microRNAs. Curr. Opin. Genet. Dev. 16:203-208. [DOI] [PubMed] [Google Scholar]
- 10.Cuesta, R., Q. Xi, and R. J. Schneider. 2001. Preferential translation of adenovirus mRNAs in infected cells. Cold Spring Harbor Symp. Quant. Biol. 66:259-267. [DOI] [PubMed] [Google Scholar]
- 11.Cullen, B. R. 2006. Viruses and microRNAs. Nat. Genet. 38:25-30. [DOI] [PubMed] [Google Scholar]
- 12.Doench, J. G., and P. A. Sharp. 2004. Specificity of microRNA target selection in translational repression. Genes Dev. 18:504-511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gale, M., Jr., and M. G. Katze. 1998. Molecular mechanisms of interferon resistance mediated by viral-directed inhibition of PKR, the interferon-induced protein kinase. Pharmacol. Ther. 78:29-46. [DOI] [PubMed] [Google Scholar]
- 14.Gupta, A., J. J. Gartner, P. Sethupathy, A. G. Hatzigeorgiou, and N. W. Fraser. 2006. Anti-apoptotic function of a microRNA encoded by the HSV-1 latency-associated transcript. Nature 442:82-85. [DOI] [PubMed] [Google Scholar]
- 15.Hutvagner, G. 2005. Small RNA asymmetry in RNAi: function in RISC assembly and gene regulation. FEBS Lett. 579:5850-5857. [DOI] [PubMed] [Google Scholar]
- 16.Jopling, C. L., M. Yi, A. M. Lancaster, S. M. Lemon, and P. Sarnow. 2005. Modulation of hepatitis C virus RNA abundance by a liver-specific microRNA. Science 309:1577-1581. [DOI] [PubMed] [Google Scholar]
- 17.Katze, M. G., D. DeCorato, and R. M. Krug. 1986. Cellular mRNA translation is blocked at both initiation and elongation after infection by influenza virus or adenovirus. J. Virol. 60:1027-1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khvorova, A., A. Reynolds, and S. D. Jayasena. 2003. Functional siRNAs and miRNAs exhibit strand bias. Cell 115:209-216. [DOI] [PubMed] [Google Scholar]
- 19.Kim, J., A. Krichevsky, Y. Grad, G. D. Hayes, K. S. Kosik, G. M. Church, and G. Ruvkun. 2004. Identification of many microRNAs that copurify with polyribosomes in mammalian neurons. Proc. Natl. Acad. Sci. USA 101:360-365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lewis, B. P., C. B. Burge, and D. P. Bartel. 2005. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets Cell 120:15-20. [DOI] [PubMed] [Google Scholar]
- 21.Lu, S., and B. R. Cullen. 2004. Adenovirus VA1 noncoding RNA can inhibit small interfering RNA and microRNA biogenesis. J. Virol. 78:12868-12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma, Y., and M. B. Mathews. 1996. Structure, function, and evolution of adenovirus-associated RNA: a phylogenetic approach. J. Virol. 70:5083-5099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mahr, J. A., and L. R. Gooding. 1999. Immune evasion by adenoviruses. Immunol. Rev. 168:121-130. [DOI] [PubMed] [Google Scholar]
- 24.Mathews, M. B. 1995. Structure, function, and evolution of adenovirus virus-associated RNAs. Curr. Top. Microbiol. Immunol. 199:173-187. [DOI] [PubMed] [Google Scholar]
- 25.Meister, G., M. Landthaler, A. Patkaniowska, Y. Dorsett, G. Teng, and T. Tuschl. 2004. Human Argonaute2 mediates RNA cleavage targeted by miRNAs and siRNAs. Mol. Cell 15:185-197. [DOI] [PubMed] [Google Scholar]
- 26.Nelson, P. T., A. G. Hatzigeorgiou, and Z. Mourelatos. 2004. miRNP:mRNA association in polyribosomes in a human neuronal cell line. RNA 10:387-394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Öhrmalm, C., and G. Akusjärvi. 2006. Cellular splicing and transcription regulatory protein p32 represses adenovirus major late transcription and causes hyperphosphorylation of RNA polymerase II. J. Virol. 80:5010-5020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Olsen, P. H., and V. Ambros. 1999. The lin-4 regulatory RNA controls developmental timing in Caenorhabditis elegans by blocking LIN-14 protein synthesis after the initiation of translation. Dev. Biol. 216:671-680. [DOI] [PubMed] [Google Scholar]
- 29.Pall, G. S., C. Codony-Servat, J. Byrne, L. Ritchie, and A. Hamilton. 2007. Carbodiimide-mediated cross-linking of RNA to nylon membranes improves the detection of siRNA, miRNA and piRNA by northern blot. Nucleic Acids Res. 35:e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Petersen, C. P., M. E. Bordeleau, J. Pelletier, and P. A. Sharp. 2006. Short RNAs repress translation after initiation in mammalian cells Mol. Cell 21:533-542. [DOI] [PubMed] [Google Scholar]
- 31.Pfeffer, S., A. Sewer, M. Lagos-Quintana, R. Sheridan, C. Sander, F. A. Grasser, L. F. van Dyk, C. K. Ho, S. Shuman, M. Chien, J. J. Russo, J. Ju, G. Randall, B. D. Lindenbach, C. M. Rice, V. Simon, D. D. Ho, M. Zavolan, and T. Tuschl. 2005. Identification of microRNAs of the herpesvirus family Nat. Methods 2:269-276. [DOI] [PubMed] [Google Scholar]
- 32.Pillai, R. S., S. N. Bhattacharyya, C. G. Artus, T. Zoller, N. Cougot, E. Basyuk, E. Bertrand, and W. Filipowicz. 2005. Inhibition of translational initiation by Let-7 microRNA in human cells. Science 309:1573-1576. [DOI] [PubMed] [Google Scholar]
- 33.Sano, M., Y. Kato, and K. Taira. 2006. Sequence-specific interference by small RNAs derived from adenovirus VAI RNA. FEBS Lett. 580:1553-1564. [DOI] [PubMed] [Google Scholar]
- 34.Sarnow, P., C. L. Jopling, K. L. Norman, S. Schutz, and K. A. Wehner. 2006. MicroRNAs: expression, avoidance and subversion by vertebrate viruses. Nat. Rev. Microbiol. 4:651-659. [DOI] [PubMed] [Google Scholar]
- 35.Schwarz, D. S., G. Hutvagner, T. Du, Z. Xu, N. Aronin, and P. D. Zamore. 2003. Asymmetry in the assembly of the RNAi enzyme complex. Cell 115:199-208. [DOI] [PubMed] [Google Scholar]
- 36.Shivdasani, R. A. 2006. MicroRNAs: regulators of gene expression and cell differentiation. Blood 108:3646-3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sullivan, C. S., A. T. Grundhoff, S. Tevethia, J. M. Pipas, and D. Ganem. 2005. SV40-encoded microRNAs regulate viral gene expression and reduce susceptibility to cytotoxic T cells. Nature 435:682-686. [DOI] [PubMed] [Google Scholar]
- 38.Ulfendahl, P. J., S. Linder, J. P. Kreivi, K. Nordqvist, C. Svensson, H. Hultberg, and G. Akusjärvi. 1987. A novel adenovirus-2 E1A mRNA encoding a protein with transcription activation properties. EMBO J. 6:2037-2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Valencia-Sanchez, M. A., J. Liu, G. J. Hannon, and R. Parker. 2006. Control of translation and mRNA degradation by miRNAs and siRNAs. Genes Dev. 20:515-524. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.