

Abstract
The isolation of primary strains of human immunodeficiency virus (HIV) is an invaluable tool for assessing properties of viruses replicating in HIV-infected subjects. A common method for obtaining a primary isolate is coculture of peripheral blood mononuclear cells (PBMCs) from HIV-infected subjects with PBMCs from uninfected donors. However, such in vitro expansion may disturb the composition (identities and relative proportions of constituting viral species) of the original viral population. We developed a GeneScan assay to monitor HIV populations by detecting variants that differ in the length of the V1/V2 coding region of the envelope gene. This assay was used to compare proviral DNAs from the PBMCs of eight subjects to the corresponding primary isolates. Major variants found in uncultured PBMCs usually persisted during culturing, while the minor variants frequently disappeared, resulting in a reduction in viral diversity. The outgrowth of the initial (2 to 4 days) viral population appeared to be determined by random events. However, subsequent changes in the population were deterministic, and as a result, the compositions of primary isolates from parallel cultures were often very similar. For two of three subjects studied, the source of HIV-negative PBMCs had little effect on the composition of primary isolates, while for the third subject donor-dependent effects were observed. Overall, our results show that most primary isolates accurately represent the major viruses found in a subject's blood and that rapid population-based genotyping methods are useful for detecting isolates with perturbed viral populations.
A common approach to studying the properties of human immunodeficiency virus (HIV) variants found in infected subjects (in vivo) is expansion of the viral population in tissue culture (in vitro). Peripheral blood mononuclear cells (PBMCs) from the subject often serve as a starting material and are mixed with cells from uninfected donors in a process often called coculture (9). Because HIV exists under specific selective pressures in vivo, including the host's immune response, viral expansion within in vitro cultures, where immune pressure is not relevant, may lead to changes in the composition of viral quasispecies (identities and relative proportions of constituting viruses). This raises the question of whether viral populations obtained by coculture (primary isolates) are representative of viral populations replicating in vivo. Several previous studies investigated the effects of PBMC cocultures on the composition of primary HIV isolates (13, 19, 31). However, they were limited by the small numbers of subjects analyzed and by limited sampling of viral variants. Therefore, we sought to establish a genotyping assay that would allow us to quickly and quantitatively assess the composition and diversity of HIV primary isolates.
Variable loops V1 and V2 of the envelope present a convenient marker for general genotype diversity of HIV populations. The V1/V2 coding region is extremely diverse, even within a single infected individual (10, 22), and multiple variants are often found simultaneously in the blood (11, 22, 27, 28). Sequence variation in V1/V2 has been shown to affect viral coreceptor specificity, cytopathogenicity, ability to form syncytia, replicative fitness, and sensitivity to neutralizing antibodies (20, 22-25). The V1/V2 region is also known to diversify in length. Indeed, a PCR-based GeneScan approach has been used successfully to qualitatively distinguish in vivo variants with different V1/V2 lengths (12, 35). Because there should be some concordance between V1/V2 length and other sequence characteristics among viruses that are evolutionarily related, the diversity in V1/V2 length should serve as a surrogate marker for general sequence diversity in a viral population.
In this study, we developed a quantitative PCR-based GeneScan assay to quantify HIV envelope variants based on the V1/V2 length. We used this assay to closely follow the time course and magnitude of changes occurring in HIV isolates during coculture. We found that random events dominate early during coculture but that the subsequent expansion of viruses in culture occurs deterministically. This leads to outgrowth of similar viral populations in parallel cocultures starting from the same HIV-positive sample, even when different seronegative donors are used to amplify the virus.
MATERIALS AND METHODS
Samples.
PBMC samples were obtained from women at high risk for HIV infection who were enrolled in an HIV-seronegative cohort in Mombasa, Kenya (16). All women consented to research. Blood samples from these women were obtained approximately monthly and tested for HIV infection, which allowed reliable estimation of the time of infection (14, 17). The infected women continued to provide blood samples after seroconversion. None of the subjects analyzed here reported using antiretroviral therapy at any time during follow-up. At each visit, one or two vials of PBMCs collected from 15 to 30 ml of blood were frozen in 10% dimethyl sulfoxide. After being thawed, a typical sample contained 10 to 30 million cells, of which 30 to 60% were viable, as determined by a trypan blue dye exclusion assay. The sample was usually divided for use in coculture and DNA extraction.
Primary isolates and cocultures.
Primary isolates were obtained by standard PBMC coculture techniques (29), with minor variations. Briefly, PBMCs from HIV-negative donors were extracted by the Ficoll gradient technique, activated for 48 to 72 h with 10 U/ml of phytohemagglutinin M (Roche), and then maintained in RPMI medium supplemented with 25 mM HEPES and 10 units/ml of interleukin-2 (Roche). To establish cocultures, the frozen sample of HIV-positive PBMCs was thawed, and 2 × 106 to 10 × 106 cells were mixed with 10 × 106 PBMCs from an uninfected donor in 4 ml of medium. After 2 h, the cells were washed with medium and resuspended at 2 × 106 to 3 × 106 cells/ml. For initial coculture analysis, cells were cultured at a density of 2 × 106 cells/ml for 10 to 28 days, with the addition of 5 × 106 to 10 × 106 stimulated donor PBMCs at days 7 and 14. The growth of virus was monitored by use of a p24 antigen enzyme-linked immunosorbent assay (ELISA) kit (Coulter) according to the manufacturer's instructions. When the coculture was identified as positive for HIV type 1 (HIV-1) by p24 antigen ELISA, supernatants and cells were stored.
For experiments in which viral populations were monitored over time, two-thirds of the medium was replaced every 2 days and additional cells were added to keep the cell density at 3 × 106 to 5 × 106 cells/ml and to gradually increase the volume of the coculture to 15 ml. Cell-free samples of culture supernatant were stored at −80°C for consequent p24 antigen assay and GeneScan analysis. HIV-negative donors of PBMCs used in these experiments were screened for the absence of a 32-bp deletion in the CCR5 gene (29); the forward primer was labeled with 6-carboxyfluorescein (6-FAM), which allowed detection of a PCR product by GeneScan analysis (see below).
DNAs of primary isolates.
DNAs was extracted from 2 to 5 million cultured PBMC by using a QIAamp DNA Blood mini kit (QIAGEN) according to the manufacturer's instructions, eluted in 200 μl of H2O, and frozen in 50-μl aliquots. For primary isolates from subjects QA210, QB374, and QC805, frozen PBMCs collected at the end point of coculture were used for DNA extraction. For primary isolates from subjects QA203, QA584, QB424, QC449, and QD385, only frozen virus was available. Therefore, 1 ml of frozen virus was thawed and used to infect 5 million activated PBMCs from an HIV-negative donor in the presence of 20 μg/ml of DEAE-dextran for 2 to 3 h. After infection, cells were washed once with fresh medium, and DNAs were extracted 24 to 32 h later.
Single-copy PCR and sequencing of viral variants.
The HIV-1 proviral copy number was estimated for the PBMC DNA by using real-time quantitative PCR with pol-specific primers as described previously (2). Nested PCR of the V1-to-V5 region was performed using a single copy of proviral DNA template to minimize sampling bias, using the following primers and conditions. The first-round primers were Env13 (5′-TTGCAATAGAAAAATTCTCCTC-3′) and Env12 (5′-CCTGGTGGGTGCTACTCCTA-3′). The second-round primers were Env15 (5′-CCATGTGTAAAGTTAACCCC-3′) and Env-10 (5′-ATGAGGGACAATTGAGAAGTGTCTAG-3′). Amplification started with 5 min at 94°C, followed by 35 cycles of 1 min at 94°C, 1 min at 50°C for first-round PCR and at 55°C for second-round PCR, and 3 min at 72°C, with a final extension at 72°C for 8 min. At least 24 independent nested PCRs were performed on HIV-1 proviral DNA extracted from every sample. Generally, <50% of all independent PCRs yielded a product (range, 13% to 67%). Excess primers and deoxynucleoside triphosphates were removed from the PCR product by ExoSAP-IT (Amersham Biosciences) or by gel extraction. HIV-1 envelope V1-to-V5 sequences were derived by directly sequencing the products, using ABI automated sequencing machinery. Both template strands of the viral genome were sequenced, and the sequences were assembled using Sequencher software (Gene Codes, Ann Arbor, MI). When multiple mixed peaks were present in sequences, indicating amplification of more than one copy of the viral genome, the sequence was discarded from the analysis. Once assembled, the sequences were reconciled into contigs and checked against both the Los Alamos National Laboratory HIV Sequence Database and GenBank public databases to identify it as a viral sequence.
For phylogenetic analysis, sequences of viral variants were aligned using MacClade software and neighbor-joining trees were built using PAUP* software.
V1/V2 GeneScan assay.
In the GeneScan assay, the V1/V2 coding regions of multiple viruses are amplified by PCR. Length polymorphism in this region results in PCR products of different lengths, which can be separated in a gel and sized using a set of standards with 1-nucleotide resolution. Because one of the primers is labeled with a fluorescent dye, 6-FAM, relative amounts of gel-separated products can be quantified (Fig. 1A). PCR primers were optimized for HIV-1 subtype A because viruses found in the region where the samples were obtained belong primarily to subtype A, with small proportions of subtypes C and D (21). The following primers were used: 519, 5′-6-FAM-GCCATGTGTAAAGTTAACCCCTCTCTG-3′; and 604, 5′-GCCTGTGTAATGGCTGAGGTATTAC-3′. Alignment of the primers with sequences of 36 subtype A viruses from Kenya showed very little variation. The binding site for the forward primer was completely conserved in 27 variants, and a single mismatch was found in 9 variants, usually at the 5′ end of the primer. The binding site for the reverse primer was completely conserved in 12 variants, had a single mismatch within the three 5′-terminal nucleotides in 15 variants, and had two or three mismatches in 9 variants (predominantly in the 5′ half of the primer). The primers were tested on more than 10 cloned subtype A envelopes with sequences of known V1/V2 length, and PCR products of the expected lengths were reliably obtained in all cases (data not shown).
FIG. 1.

GeneScan assay. (A) The top line shows a schematic of the gp120 part of the viral envelope gene, with variable (V) and conserved (C) regions indicated. Arrows show the approximate positions of primers used for PCR. A star at the end of one primer indicates the presence of a fluorescent label. The size of the PCR product depends on the length of the V1/V2 region and generally varies from 250 to 300 bp. As an example, the results of a GeneScan assay of a mixture of two viral strains, M-E2 and M-B1, is shown. PCR amplification of the shorter V1/V2 region of M-E2 virus results in a PCR product of 225 bp, while amplification of the longer V1/V2 region of M-B1 virus results in a PCR product of 276 bp. GeneScan analysis separates the products by size (x axis on the graph) and measures the corresponding fluorescence signal of each PCR product (y axis on the graph). Relative amounts of the PCR products can be calculated from the areas of the two peaks. (B) Proportion of HIV M-E2 clone in a 1:1 mixture with an M-B1 clone, as determined by the GeneScan assay. Bars show averages for five replicates, and error bars are standard deviations. The amount of virus used in each replicate is shown at the bottom. (C) Relationship between expected and observed ratios of M-E2 to M-B1 variants. Circles indicate averages of three GeneScan measurements. The line indicates the best-fitting trend line for the data. (D) Representative results of three independent GeneScan analyses of a PBMC DNA sample from an HIV-infected subject. The lengths of detected PCR products are shown below the graph, and the bars indicate the percentages of corresponding viruses in the total amount of viruses detected in each analysis.
For samples with more than 300 pg/ml of p24 antigen, 2 μl of cell-free virus-containing medium was added to the PCR mix to a final volume of 20 μl and amplified as previously described (32). Briefly, a OneStep reverse transcriptase PCR (RT-PCR) kit (QIAGEN) was used according to the manufacturer's instructions, with some small modifications, including the addition of 0.4% Triton X-100 to lyse virions and release viral genomic RNA. The RT step was performed for 30 min at 30°C, followed by 15 min of RT inactivation at 95°C and 35 cycles of amplification by two-step PCR (15 s at 93°C and 1 min 15 s at 59°C). The PCR product was diluted 2- to 500-fold with water to obtain peak intensities in the linear range and was run on an ABI PRISM 3700 machine. The data were analyzed using GeneScan Analysis v3.1 software (Applied Biosystems).
For samples with less than 300 pg/ml of p24 antigen, 5 μl of virus was used in a 50-μl total PCR volume and amplified as described above. After amplification, PCR products were purified with a QIAGEN PCR purification kit according to the manufacturer's instructions, eluted in 15 μl of H2O, and then analyzed as described above.
For analysis of proviral DNA, total DNA from 2 to 5 million cells was extracted using a QIAamp DNA Blood mini kit (QIAGEN), eluted in 200 μl, and frozen in 50-μl aliquots. The GeneScan analysis for these samples was done at least in triplicate, using 5 μl of DNA and PCR product purification as described above for viral samples with low p24 loads.
The assay's accuracy and sensitivity were validated using mixtures of virions from two HIV-1 clones, M-E2 and M-B1, which were identical in the primer-binding regions but had V1/V2 regions of significantly different lengths. Amplification of variant M-E2 results in a short, 225-bp fragment, while amplification of variant M-B1 produces a 276-bp fragment (Fig. 1A). The absolute amount of each virus was measured by a Gen-Probe HIV-1 RNA assay (6), and the viruses were mixed in a 1:1 ratio. Tenfold serial dilutions were used to add decreasing amounts of virions to the PCR mix, and the relative amounts of the two variants were measured using the GeneScan assay. The measured proportion of variant M-E2 was ∼70%, higher than the expected 50%, indicating a slightly better amplification of the shorter variant (Fig. 1B). The results were reproducible in at least four independent assays when 103 or more virions were used in each reaction mix, but when 100 virions were used, the results became less reproducible, with low peak signals and artifact products of unexpected sizes appearing in the reaction mix (Fig. 1B and data not shown). Thus, all assays reported in this paper were established with inputs of at least 103 virions, unless otherwise specified.
The ability of the assay to measure relative proportions of two PCR products is limited on the high end by the maximum peak height of 7,000 relative light units (RLU), which can be quantitated accurately by the machine, and on the lower end by the level of noise in the fluorescent signal. Because >95% of the noise showed signal intensities below 150 RLU (data not shown), 150 RLU was set as the lower detection limit for the assay. This established a theoretical limit of 1:45 for the ratio of the two viral variants, which can be measured by the assay. Mixes of M-E2 and M-B1 variants at different ratios showed that the assay is linear for a wide range of relative proportions of variants, including the 1:25 ratio (Fig. 1C).
Because the percentages of viral variants in the population are calculated from the total area of all detected peaks, the GeneScan analysis is able to detect low-frequency viruses when analyzed mixtures contain more variants. In GeneScan analyses reported in this paper, the detection limit was typically between 1 and 3%. Results of an analysis in which we were unable to detect viruses that were present in the viral population at or below a frequency of 5% (detection limit, >5%) were discarded, and the analysis was repeated. In addition, for samples with low copy numbers of viral DNA or RNA, we considered any minor viral species as truly existing in the viral population only if it was detected in at least two separate PCR runs.
Calculations and statistical tests.
The diversity of viral populations was measured by normalized Shannon entropy for viral populations, which was calculated as follows:
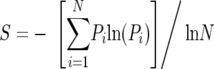 |
where Pi is the proportion of variant i and N is the total number of variants in the sample (5). The weighted average of V1/V2 lengths within a viral population was calculated as follows:
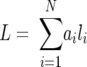 |
where ai is the proportion of V1/V2 variants of length li and N is the total number of variants in the sample. Differences between the compositions of any two populations were summarized by calculating the total change, as follows:
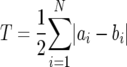 |
where ai and bi are proportions of variant i in the two compared populations. The statistical analysis was done using a paired t test, assuming equal variance of the data.
RESULTS
Development of rapid and quantitative assay to measure viral diversity.
We developed a GeneScan assay to measure viral diversity based on detection and quantification of length polymorphisms in the V1/V2 region of the HIV-1 envelope gene. The assay is based on PCR amplification of the variable region and subsequent detection and quantitation of the PCR products labeled with the fluorescent dye 6-FAM (Fig. 1A; see Materials and Methods). Using two HIV-1 clones, M-E2 and M-B1, with known V1/V2 lengths, we showed that the assay is accurate over a range of viral concentrations and is able to detect rare variants constituting as little as 2 to 3% of the total population (Fig. 1B and C; see Materials and Methods for details). The slightly more efficient amplification of the shorter variant, M-E2, did not present a problem for our purposes because we were interested in an assay that allowed accurate measurement of changes in the relative amounts of different variants in the population (demonstrated in Fig. 1C), not their absolute amounts.
A representative example of GeneScan analysis of DNAs extracted from an HIV-infected subject is shown in Fig. 1D. The results of three independent PCR runs were consistent with each other and showed peaks that were separated by 3-bp intervals. The presence of multiple variants in the population increases the sensitivity of the assay. As a result, the detection limits for the three independent GeneScan runs were 0.7%, 0.8%, and 2.2%, which allowed reliable detection of viral species constituting less than 1% of the total population.
We compared the results of the GeneScan assay to the results of a standard PCR genotyping assay in which single copies of the HIV genome were used to amplify and sequence the V1-to-V5 region. The DNAs from uncultured PBMCs of eight HIV-positive subjects were analyzed by both assays, and the proportions of variants with different V1/V2 lengths were compared (Fig. 2). For most of the samples, the results were quite similar, as the GeneScan assay detected many of the variants found by direct sequencing, often in comparable proportions. For example, in a sample from subject QA584, both assays detected the same major variant as well as two minor variants. In addition, the GeneScan assay detected a fourth variant, whose proportion was likely too small to be detected by sequencing individual clones. Similar results were observed for samples from subjects QA203, QB374, QC449, QC805, and QD385. In two cases, QA210 and QB424, the results of the two assays were less congruent. For samples from subject QA210, a variant that was detected in three of five sequencing reactions appeared as only a minor variant in the GeneScan assay. The reasons for this were not clear and may include differences in specificities of primers used in the two assays. For samples from subject QB424, 5 different variants were detected by sequencing and 11 variants were detected by GeneScan analysis. Only three variants were common between the two assays, indicating the limitations of both assays in assessing the diversity in a sample containing a large number of different variants. In addition, the reliability of the GeneScan assay in this case was likely to be lower than that for other samples due to limited amounts of available DNA, which allowed us to run only two (not three) independent reactions with this sample. To summarize, for the majority of samples, the results of the GeneScan assay agreed well with the results of sequencing.
FIG. 2.

Comparison of V1/V2 genotyping of proviral DNAs isolated from infected subjects by GeneScan analysis and by sequencing. PBMC DNAs from eight HIV-infected subjects (QA203 through QD385) were analyzed by both GeneScan analysis and sequencing of single-copy PCR products of the V1-to-V5 region (the method of analysis is indicated at the top of the figure). Proportions of viral variants with different V1/V2 lengths in each sample are plotted as sectors of different colors. Fractions within sectors correspond to the number of clones with a certain V1/V2 length over the total number of clones sequenced.
Changes in viral population associated with cocultures.
We then used the GeneScan assay to determine the differences between viral diversity in PBMCs and that in primary isolates grown in culture. Thus, we compared the distributions of V1/V2 lengths between DNAs extracted directly from subject PBMCs and DNAs of primary isolates (Fig. 3A). For this analysis, we selected primary isolates cultured from samples from eight HIV-positive subjects infected for at least 2.5 years, with a median time of infection of 4.5 years. The isolates were generated in independently maintained cultures, using HIV-negative PBMCs from different donors. The lengths of coculture were also different and varied from 9 to 24 days, depending on the time it took for the virus to reach high p24 antigen amounts (above 300 pg/ml) in the coculture supernatant (Table 1).
FIG. 3.

Comparison of viral populations in uncultured PBMC DNAs and in primary isolates. (A) Schematic of the analysis procedure. Proviral DNAs extracted from uncultured PBMCs and from cells after coculture were analyzed by the V1/V2 GeneScan assay. (B to I) Comparisons of percentages of viral variants obtained by GeneScan analysis of DNAs extracted from PBMCs before (black bars) and after (white bars) coculture for subjects QA203 through QD385. The nucleotide lengths of detected PCR products are shown under the corresponding bars.
TABLE 1.
Subjects and samples used for analyses of effects of coculture on viral populations
| Subject | Days postinfection | Viral load (104) | Coculture length (days) | Avg diversity (%)a |
|---|---|---|---|---|
| QA203 | 1,234 | 4.5 | 24 | 7.0 |
| QA210 | 2,254 | 41 | 15 | 2.9 |
| QA584 | 2,046 | 48 | 9 | 2.9 |
| QB374 | 2,631 | 320 | 18 | 4.0 |
| QB424 | 919 | 35 | 24 | 3.1 |
| QC449 | 1,240 | 19 | 15 | 3.3 |
| QC805 | 2,071 | 750 | 12 | 0.9 |
| QD385 | 1,309 | 430 | 22 | 2.6 |
Average nucleotide diversity was calculated from pairwise sequence comparisons of single-copy PCR envelope clones, using the C2 to V5 regions.
For four samples (QA203, QC805, QC449, and QA584), only a single variant was detected after coculture (Fig. 3B to E). The variant had the same V1/V2 length as the predominant variant in DNAs from uncultured PBMCs. For sample QD385, the primary isolate contained not only the predominant variant from the blood but also three additional variants (Fig. 3F). One of these variants (228 bp) showed a large expansion during coculture, but it increased its proportion by displacing other minor variants and the major variant (249 bp) remained undisturbed. Sample QA210 showed the most drastic change in the viral population during coculture. The 258-bp variant found in small proportion in uncultured PBMCs greatly expanded and displaced all other variants (Fig. 3G). For sample QB374, the primary isolate contained all three variants detected in uncultured PBMCs, in approximately the same proportions (Fig. 3H). Finally, primary isolate QB424 contained eight different variants, with no single variant appearing as dominant, which accurately reflected the viral population in uncultured PBMCs (Fig. 3I). Overall, the results of this experiment showed that in only one culture (QA210) did a minor variant completely outgrow the major variant, whereas in the majority of cocultures the predominant in vivo variant either remained dominant or was found in a significant proportion in the primary isolate.
To more formally test the effect of coculture, we applied statistical tests to the cumulative data from all eight cocultures. First, we compared the numbers of variants detected by GeneScan analysis before and after coculture and found a statistically significant reduction (t test; P = 0.01) (Fig. 4A). To measure changes in diversity, we calculated the Shannon entropy for populations before and after coculture and also found a significant reduction (t test; P = 0.01) (Fig. 4B). We also considered the possibility that V1/V2 length had an effect on viral replication capacity and that in vitro culture may have selected for viruses with shorter (or longer) V1/V2 regions. The average V1/V2 length weighted by the proportion of each variant was calculated for populations before and after coculture (Fig. 4C). There was no significant change associated with coculture (t test; P = 0.54), suggesting that the length of this region has minor effects on viral replicative fitness in the short-term PBMC coculture.
FIG. 4.

Statistical analyses of viral populations before and after coculture. Populations were compared by the number of viral variants above the detection limit (A), by diversity-measuring Shannon entropy (B), and by the average percentage-weighted length of V1/V2 (C). Lines connect viral populations from the same subject. The P value from paired t-test analyses is shown for each comparison.
V1/V2 length as a marker for sequence diversity.
Measurements of V1/V2 length polymorphism will invariably underestimate the true sequence diversity existing in an HIV population. Our experimental approach was based on the assumption that closely related quasispecies in an HIV-infected subject would often possess the same V1/V2 length. The assumption was based on our finding that within the same patient, the average pairwise genetic distance for the V3-to-V5 region is significantly lower among viruses with equal V1/V2 lengths (2.34%) than among viruses with distinct V1/V2 lengths (4.0%) (P = 0.034; paired t test [data not shown]). To test this assumption directly, we PCR amplified single copies of viral envelope genes and sequenced the region used for GeneScan analysis. The sequences were aligned and neighbor-joining trees were built to reveal the phylogenetic relationships between the clones obtained directly from subject PBMCs and those obtained from postculture isolates. We analyzed sequences from three subjects, namely, QA584, QB374, and QA210 (Fig. 5). In all three cases, V1/V2 length served as a good marker for sequence diversity. For example, for subject QA584, viruses from the primary isolate clustered closely with viruses from blood (shown in boxes) that had the identical V1/V2 length (267 bp) but not with blood variants that had different V1/V2 lengths (228 and 291 bp). Subject QB374 was predicted by GeneScan analysis to contain two dominant variants with V1/V2 lengths of 237 and 240 bp (Fig. 3H). Both variants were found by sequencing, and viruses with different V1/V2 lengths clustered separately, with very strong bootstrap support (≥98%). Nonetheless, viruses with identical V1/V2 lengths do contain additional genetic variation that is revealed by sequence data but not by length alone. The analyses also supported our conclusion that primary isolates are not dominated by a single variant but consist of closely related quasispecies that generally dominate viral populations in blood.
FIG. 5.

Phylogenetic analyses of viruses from primary isolates. The region used in the GeneScan-based genotyping assay was PCR amplified from single copies of viral genomes and sequenced. The neighbor-joining trees were built based on pairwise genetic distances between the sequences. The number at the tip of each branch indicates the length of the expected V1/V2 GeneScan product. Boxes indicate sequences that were obtained directly from noncultured PBMCs, and unboxed numbers correspond to postculture viruses. Subject numbers are shown above each tree. Numbers next to branches indicate bootstrap values.
Subject QA210 was unusual, because GeneScan analysis suggested an outgrowth of a minor blood variant (Fig. 3G). The sequence analysis confirmed the existence of a variant with a V1/V2 length of 258 bp (Fig. 5). This variant was distinct from variants found in blood (bootstrap values, ≥78%). Interestingly, this primary isolate was also distinct from primary isolates from subjects QA584 and QB374 in that it contained just a single viral variant, not a group of related but diverse variants. Thus, the sequencing results confirmed the unusual nature of primary isolate QA210, which was evident from the GeneScan analysis.
Time course and donor dependence of primary isolate outgrowth.
Given the diversity of coculture outcomes that were observed, we examined the consistency of the selection of viral species in primary isolates and the time course of changes in viral populations. Therefore, we established multiple cocultures with PBMCs from three HIV-positive subjects (QA203, QB374, and QA210), which were selected by the presence of multiple variants in the blood (Fig. 3) and by sample availability. For each subject, two cocultures were established with the same HIV-negative donor PBMCs and two cocultures were established with PBMCs from different HIV-negative donors. Thus, PBMCs from each of the three subjects were cultured with PBMCs from three different donors.
Viral growth in cultures was monitored every 2 days by p24 antigen ELISA assay (Fig. 6A). When PBMCs from the same donor (cultures labeled AT-1 and AT-2 or AP-1 and AP-2) were used to establish cocultures, the amount of p24 antigen increased in a similar manner in parallel cultures (the difference in p24 kinetics of QA210 cultures AP-1 and AP-2 was due to technical problems on day 6). However, as previously observed (31, 34), both the rate of viral spread and the peak p24 value varied 10- to 100-fold, depending on the donor of HIV-negative PBMCs (Fig. 6A). Interestingly, no single PBMC donor was responsible for low viral replication. Instead, low replication was limited to specific combinations of PBMCs and viruses, with the same donor allowing robust replication of one virus and poor replication of another (for example, compare the p24 kinetics in cocultures of isolates QA203 and QB374 with cells from donor AS).
FIG. 6.

Changes in viral populations associated with coculture. Samples from three HIV-positive subjects (QA203, QB374, and QA210) were used to establish parallel cocultures with PBMCs from three HIV-negative donors, randomly chosen from four available donors (AR, AS, AP, and AT). Each sample consisted of two vials of frozen PBMCs, one of which was used to establish two cocultures with PBMCs from the same donor and the other of which was used to establish cocultures with PBMCs from two different donors. (A) Kinetics of p24 antigen in culture medium of cocultures. All cocultures were established on the same date, except for cocultures with PBMCs from donor AP, which were done at a different time. (B to D) Compositions of cell-free viral populations in cocultures, as determined by GeneScan analysis. For each time point, the results are shown as a stacked column of boxes of different colors, where each color represents the detected variant according to the legend in the lower right corner and the height corresponds to the proportion of the variant in the total population. (B) GeneScan analysis of cocultures established with a sample taken from subject QA203 on day 1655 postinfection (viral load, 1.9 × 105 copies/ml). (C) GeneScan analysis of cocultures established with a sample taken from subject QB374 on day 2729 postinfection (viral load, 2.7 × 105 copies/ml). (D) GeneScan analysis of cocultures established with a sample taken from subject QA210 on day 2478 postinfection (viral load, 4.5 × 105 copies/ml).
Changes in the viral populations in these cocultures were also monitored every 2 days. The composition of the starting population before culturing (day 0) was determined by analysis of DNAs extracted directly from subject PBMCs. On all following days, the GeneScan assay was performed on cell-free virus in culture medium. Interestingly, we found that many of the variants found in PBMCs were found in culture 2 to 4 days after its initiation, indicating that these variants were not defective but were at least transcriptionally active (Fig. 6B to D, days 2 and 4). Many of them were likely replication competent, because they persisted in culture up to day 6.
In sample QA203, two variants, of 237 bp and 276 bp, dominated the PBMC DNAs (Fig. 5B, day 0). One of these variants (237 bp; blue bars) quickly dominated in all four cocultures. In three of the cocultures, with PBMCs from donors AT, AR, and AS, this variant completely displaced all other variants by 6 to 12 days of culture. In the fourth culture, AT-2, three more variants were present until day 20 and one of those variants was the other dominant in vivo strain (276 bp; red bars). Analysis of a different sample taken from subject QA203 7 months earlier than the sample shown in Fig. 6B showed similar results: the 237-bp variant constituted a significant proportion (∼40%) of the viral population in the subject's PBMC DNA, and two cocultures with cells from donor AP resulted in outgrowth of the same 237-bp variant (data not shown). This indicated that PBMC cocultures allowed isolation of a variant that persistently replicated in the subject for prolonged periods and was not an immunologically suppressed virus or a newly emerging high-fitness variant, as suggested previously (31). The fact that the final populations in all six cocultures were almost identical demonstrated that outgrowth of particular viral variants was very reproducible.
Sample QB374 showed more similar populations in parallel cultures at the early time points (days 2 and 4) than did sample QA203, possibly due to an overall lower level of diversity (Fig. 6C). Similar to cultures with sample QA203, three cultures eventually became homogeneous. In the fourth culture, with PBMCs from donor AS, the 240-bp variant (dark red bars) was found as a minor species in the beginning but eventually recovered and became the most abundant variant in the population by day 20. Thus, all four cultures reproducibly amplified the major in vivo viral strain.
Sample QA210 showed the most diversity in the outcome of the cocultures. Two cocultures with cells from donor AP resulted in primary isolates consisting of two variants, of 258 bp (red bars) and 270 bp (dark green bars), both of which were dominant variants in noncultured PBMCs. This supported the results of experiments with samples QA203 and QB374. However, two other cocultures with this sample gave drastically different results. Coculture with PBMCs from donor AR never became p24 positive, and coculture with cells from donor AT was significantly delayed compared to other cocultures. In the latter coculture, a minor, 261-bp variant (yellow bars) expanded and displaced all other variants, resulting in an isolate that was very different from the isolates from both cocultures with donor AP.
To summarize, 10 of the 11 successful cocultures resulted in primary isolates that either contained or were dominated by a major in vivo variant. The composition of the primary isolate was very reproducible for two of three tested samples, regardless of the donor used to obtain PBMCs or the viral replication rate.
DISCUSSION
The results presented here show that primary isolates are, in general, representative of the major viral variants present in the blood of an infected individual. The viral strain dominant in blood was successfully obtained in at least one coculture for all eight studied subjects (Fig. 3 and 6). The composition of primary isolates was very reproducible in parallel cocultures. For two of three subjects, similar viruses were selected when different donors were used as a source of HIV-negative PBMCs.
Three previous studies addressed the question of viral population changes associated with PBMC cocultures (13, 19, 31) and came to the conclusion that a minor blood variant usually becomes dominant in vitro, in conflict with the results presented here. However, all three studies were limited by analysis of a single patient and also by sequencing of a fairly small number (20 or less) of individual viral clones. The difference in results may also be explained by the use of different assays to monitor viral species. For example, outgrowth of a minor variant could remain undetected in our assay as long as that variant had the same V1/V2 length as the major variant in uncultured PBMCs. While we cannot exclude that this happened in some of our cocultures, the fact that the majority of primary isolates were reproducibly dominated by the major V1/V2 variant found in blood makes such an explanation unlikely. In addition, our sequencing analysis of three primary isolates (Fig. 5) showed directly that V1/V2 length was a good marker for related viral quasispecies. Sequence analysis of full-length envelope genes from these cocultures showed the existence of recombinant viruses that contained similar V1/V2 but different V3/V5 regions (data not shown). However, such recombinants were not frequent and did not indicate selection for a rare V3/V5 variant.
The factors important for outgrowth of a primary isolate which is representative of blood variants remain unknown and may be either patient specific (disease progression and stage) or virus specific (viral subtype or tropism), or they may involve a particular aspect of coculture technique. For example, in two of the previous studies, no additional donor PBMCs were added to the coculture after it was established (19, 31). Since cell-to-cell viral spread is likely to play an important role in viral replication (4, 30), a higher cell density should promote more efficient viral growth and allow a more representative viral isolate to grow out. Low cell density and a limited number of targets favor variants that are less cytopathic, creating selective pressure that does not correspond to in vivo selective pressures and may therefore affect relative viral fitness and benefit a virus that is less fit in vivo (32).
Our analyses of viral populations soon after the start of coculture (days 2 and 4) showed large differences between parallel cultures, even when the starting conditions were identical. For example, on day 2 of culture AT-1 for subject QA203, the 237-bp variant constituted 80% of all virions, but in culture AT-2, the proportion of this variant was below 20%. Similar variation was observed for cultures of QB374 and QA210 samples, indicating that the initial composition of the viral population was determined by stochastic events. The viral population found in the medium after only 2 days of coculture will largely have undergone only a single cycle of replication and should have consisted mostly of virions produced by PBMCs from the original HIV-positive sample. Comparisons of DNAs isolated from identical vials of frozen PBMCs showed only small differences in the distributions of viral species (day 0 data in Fig. 6), suggesting that variation between parallel cultures did not arise due to small numbers of infected PBMCs or due to sample mix-up. Instead, we propose that small differences in survival of PBMCs infected with each variant, their activation and metabolic states, and random interactions with susceptible target cells are among the stochastic factors that influence the composition of the initial viral population in coculture. Once the number of infected cells becomes sufficiently high, viral expansion becomes deterministic and changes in viral populations depend on relative replicative abilities of viral variants.
The ability of a variant to grow in vitro in the absence of immune or drug pressure and in the presence of abundant target cells is often defined as viral replicative fitness (3, 26). Thus, the deterministic changes in relative amounts of viral variants observed in culture are due to competition between variants with different replicative fitness levels. One popular model to explain the in vivo coexistence of multiple viral variants with different replicative fitness levels invokes balancing selection between variants with high replicative fitness and variants that are able to escape the immune system at the cost of their replication ability (1, 8, 15). This model predicts that variants with higher replicative fitness would be suppressed by the immune system in vivo but would quickly outgrow the predominant in vivo immune escape variants in tissue culture. This specifically applies to viral variants with different V1/V2 lengths, because V1/V2 is a major target for antibodies and its length increases over the course of infection, presumably to allow expansion of the “glycan shield” around the viral envelope (28, 33). Therefore, one might predict that viruses with shorter V1/V2 regions may have higher replicative fitness in vitro than viruses with longer V1/V2 regions and might preferentially expand in PBMC cocultures. However, our results showed no significant changes in average V1/V2 length as a result of short-term coculture, indicating that this was not the case. Alternatively, differences in viral replicative fitness between wild-type and escape variants may be relatively minor. Insertions and deletions in the V1/V2 region may either not impose a significant cost on replicative fitness or quickly be compensated by mutations in other regions of the envelope (7, 24). Variants with differential replicative fitness levels may also coexist stably in different compartments or be in a process of constant replacement of some variants with others (11, 18, 22, 28). While we could not distinguish between these possibilities in this study, our finding that the predominant blood variant is usually the most fit variant in tissue culture strongly suggests that replicative fitness is very important for viral replication in vivo.
Primary isolates obtained from subject QA210 were the most variable in composition among isolates from the eight studied subjects. The 2,254-day sample from this subject showed a drastic change during coculture, with the minor, 258-bp blood variant completely displacing all other variants at day 15 of culture (Fig. 3). Sequence analysis of this isolate showed a single viral variant dominating the population. Such low diversity could be due to either a severe bottleneck at the early stages of coculture or a drastic replicative fitness advantage of this particular variant. We could not distinguish between the two possibilities based on our results. However, the cocultures of the 2,478-day sample from this subject produced the following three different outcomes: mixed populations of the 258-bp and 270-bp variants when cultured with PBMCs from donor AP; outgrowth of the minor, 261-bp variant when cultured with PBMCs from donor AT; and unsuccessful coculture with PBMCs from donor AR (Fig. 6). These results are inconsistent with the hypothesis of the existence of a variant suppressed by the immune system, but with high replicative fitness, which dominates the in vitro culture, because such a variant would be expected to dominate in all cocultures. The diversity in coculture outcomes suggests that viruses from this subject are very sensitive to culture conditions and that PBMCs obtained from different donors differentially affect the replication of different variants.
Overall, the work presented in this paper shows that the outcome of PBMC cocultures usually leads to outgrowth of the most common V1/V2 variant found in blood. Under the conditions used here, cocultures with 9 of the 11 analyzed samples (from eight different subjects) preserved the dominant HIV-1 variant found in each subject's PBMCs, and three isolates even maintained a certain degree of in vivo diversity. We observed the outgrowth of a minor variant in samples from only one subject, and this outgrowth appeared to be donor dependent. Samples from two more subjects showed little donor-associated variation and resulted in reproducible primary isolates. In addition, our results demonstrate the usefulness of the GeneScan approach as a fast and reliable method of genotyping viral populations in culture.
Acknowledgments
We thank Catherine Blish, Molly OhAinle, Anne Piantadosi, and Masahiro Yamashita for carefully reading the manuscript and for insightful comments and suggestions. We also thank the FHCRC Shared Genomics Resource for excellent technical assistance.
This work was supported by NIH grants AI34251, AI38518, and R37AI30927. Y.V. was supported in part by amFAR grant 106594-36-RFGN.
Footnotes
Published ahead of print on 25 July 2007.
REFERENCES
- 1.Allen, T. M., M. Altfeld, S. C. Geer, E. T. Kalife, C. Moore, K. M. O'Sullivan, I. Desouza, M. E. Feeney, R. L. Eldridge, E. L. Maier, D. E. Kaufmann, M. P. Lahaie, L. Reyor, G. Tanzi, M. N. Johnston, C. Brander, R. Draenert, J. K. Rockstroh, H. Jessen, E. S. Rosenberg, S. A. Mallal, and B. D. Walker. 2005. Selective escape from CD8+ T-cell responses represents a major driving force of human immunodeficiency virus type 1 (HIV-1) sequence diversity and reveals constraints on HIV-1 evolution. J. Virol. 79:13239-13249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benki, S., R. S. McClelland, S. Emery, J. M. Baeten, B. A. Richardson, L. Lavreys, K. Mandaliya, and J. Overbaugh. 2006. Quantification of genital human immunodeficiency virus type 1 (HIV-1) DNA in specimens from women with low plasma HIV-1 RNA levels typical of HIV-1 nontransmitters. J. Clin. Microbiol. 44:4357-4362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bonhoeffer, S., A. D. Barbour, and R. J. De Boer. 2002. Procedures for reliable estimation of viral fitness from time-series data. Proc. Biol. Sci. 269:1887-1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carr, J. M., H. Hocking, P. Li, and C. J. Burrell. 1999. Rapid and efficient cell-to-cell transmission of human immunodeficiency virus infection from monocyte-derived macrophages to peripheral blood lymphocytes. Virology 265:319-329. [DOI] [PubMed] [Google Scholar]
- 5.Delwart, E. L., H. Pan, H. W. Sheppard, D. Wolpert, A. U. Neumann, B. Korber, and J. I. Mullins. 1997. Slower evolution of human immunodeficiency virus type 1 quasispecies during progression to AIDS. J. Virol. 71:7498-7508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Emery, S., S. Bodrug, B. A. Richardson, C. Giachetti, M. A. Bott, D. Panteleeff, L. L. Jagodzinski, N. L. Michael, R. Nduati, J. Bwayo, J. K. Kreiss, and J. Overbaugh. 2000. Evaluation of performance of the Gen-Probe human immunodeficiency virus type 1 viral load assay using primary subtype A, C, and D isolates from Kenya. J. Clin. Microbiol. 38:2688-2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fernandez, G., B. Clotet, and M. A. Martinez. 2007. Fitness landscape of human immunodeficiency virus type 1 protease quasispecies. J. Virol. 81:2485-2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Friedrich, T. C., E. J. Dodds, L. J. Yant, L. Vojnov, R. Rudersdorf, C. Cullen, D. T. Evans, R. C. Desrosiers, B. R. Mothe, J. Sidney, A. Sette, K. Kunstman, S. Wolinsky, M. Piatak, J. Lifson, A. L. Hughes, N. Wilson, D. H. O'Connor, and D. I. Watkins. 2004. Reversion of CTL escape-variant immunodeficiency viruses in vivo. Nat. Med. 10:275-281. [DOI] [PubMed] [Google Scholar]
- 9.Hollinger, F. B., J. W. Bremer, L. E. Myers, J. W. Gold, and L. McQuay. 1992. Standardization of sensitive human immunodeficiency virus coculture procedures and establishment of a multicenter quality assurance program for the AIDS Clinical Trials Group. J. Clin. Microbiol. 30:1787-1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hughes, E. S., J. E. Bell, and P. Simmonds. 1997. Investigation of population diversity of human immunodeficiency virus type 1 in vivo by nucleotide sequencing and length polymorphism analysis of the V1/V2 hypervariable region of env. J. Gen. Virol. 78:2871-2882. [DOI] [PubMed] [Google Scholar]
- 11.Kitrinos, K. M., N. G. Hoffman, J. A. Nelson, and R. Swanstrom. 2003. Turnover of env variable region 1 and 2 genotypes in subjects with late-stage human immunodeficiency virus type 1 infection. J. Virol. 77:6811-6822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klevytska, A. M., M. R. Mracna, L. Guay, G. Becker-Pergola, M. Furtado, L. Zhang, J. B. Jackson, and S. H. Eshleman. 2002. Analysis of length variation in the V1-V2 region of Env in nonsubtype B HIV type 1 from Uganda. AIDS Res. Hum. Retrovir. 18:791-796. [DOI] [PubMed] [Google Scholar]
- 13.Kusumi, K., B. Conway, S. Cunningham, A. Berson, C. Evans, A. K. Iversen, D. Colvin, M. V. Gallo, S. Coutre, E. G. Shpaer, et al. 1992. Human immunodeficiency virus type 1 envelope gene structure and diversity in vivo and after cocultivation in vitro. J. Virol. 66:875-885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lavreys, L., J. M. Baeten, J. K. Kreiss, B. A. Richardson, B. H. Chohan, W. Hassan, D. D. Panteleeff, K. Mandaliya, J. O. Ndinya-Achola, and J. Overbaugh. 2004. Injectable contraceptive use and genital ulcer disease during the early phase of HIV-1 infection increase plasma virus load in women. J. Infect. Dis. 189:303-311. [DOI] [PubMed] [Google Scholar]
- 15.Li, B., A. D. Gladden, M. Altfeld, J. M. Kaldor, D. A. Cooper, A. D. Kelleher, and T. M. Allen. 2007. Rapid reversion of sequence polymorphisms dominates early human immunodeficiency virus type 1 evolution. J. Virol. 81:193-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martin, D. J., J. G. Sim, G. J. Sole, L. Rymer, S. Shalekoff, A. B. van Niekerk, P. Becker, C. N. Weilbach, J. Iwanik, K. Keddy, et al. 1995. CD4+ lymphocyte count in African patients co-infected with HIV and tuberculosis. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 8:386-391. [PubMed] [Google Scholar]
- 17.Martin, H. L., Jr., P. M. Nyange, B. A. Richardson, L. Lavreys, K. Mandaliya, D. J. Jackson, J. O. Ndinya-Achola, and J. Kreiss. 1998. Hormonal contraception, sexually transmitted diseases, and risk of heterosexual transmission of human immunodeficiency virus type 1. J. Infect. Dis. 178:1053-1059. [DOI] [PubMed] [Google Scholar]
- 18.Masciotra, S., S. M. Owen, D. Rudolph, C. Yang, B. Wang, N. Saksena, T. Spira, S. Dhawan, and R. B. Lal. 2002. Temporal relationship between V1V2 variation, macrophage replication, and coreceptor adaptation during HIV-1 disease progression. AIDS 16:1887-1898. [DOI] [PubMed] [Google Scholar]
- 19.Meyerhans, A., R. Cheynier, J. Albert, M. Seth, S. Kwok, J. Sninsky, L. Morfeldt-Manson, B. Asjo, and S. Wain-Hobson. 1989. Temporal fluctuations in HIV quasispecies in vivo are not reflected by sequential HIV isolations. Cell 58:901-910. [DOI] [PubMed] [Google Scholar]
- 20.Nabatov, A. A., G. Pollakis, T. Linnemann, A. Kliphius, M. I. Chalaby, and W. A. Paxton. 2004. Intrapatient alterations in the human immunodeficiency virus type 1 gp120 V1V2 and V3 regions differentially modulate coreceptor usage, virus inhibition by CC/CXC chemokines, soluble CD4, and the b12 and 2G12 monoclonal antibodies. J. Virol. 78:524-530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neilson, J. R., G. C. John, J. K. Carr, P. Lewis, J. K. Kreiss, S. Jackson, R. W. Nduati, D. Mbori-Ngacha, D. D. Panteleeff, S. Bodrug, C. Giachetti, M. A. Bott, B. A. Richardson, J. Bwayo, J. Ndinya-Achola, and J. Overbaugh. 1999. Subtypes of human immunodeficiency virus type 1 and disease stage among women in Nairobi, Kenya. J. Virol. 73:4393-4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palmer, C., P. Balfe, D. Fox, J. C. May, R. Frederiksson, E. M. Fenyo, and J. A. McKeating. 1996. Functional characterization of the V1V2 region of human immunodeficiency virus type 1. Virology 220:436-449. [DOI] [PubMed] [Google Scholar]
- 23.Pantophlet, R., and D. R. Burton. 2006. GP120: target for neutralizing HIV-1 antibodies. Annu. Rev. Immunol. 24:739-769. [DOI] [PubMed] [Google Scholar]
- 24.Pastore, C., R. Nedellec, A. Ramos, S. Pontow, L. Ratner, and D. E. Mosier. 2006. Human immunodeficiency virus type 1 coreceptor switching: V1/V2 gain-of-fitness mutations compensate for V3 loss-of-fitness mutations. J. Virol. 80:750-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pinter, A., W. J. Honnen, Y. He, M. K. Gorny, S. Zolla-Pazner, and S. C. Kayman. 2004. The V1/V2 domain of gp120 is a global regulator of the sensitivity of primary human immunodeficiency virus type 1 isolates to neutralization by antibodies commonly induced upon infection. J. Virol. 78:5205-5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quinones-Mateu, M. E., and E. J. Arts. 2002. Fitness of drug resistant HIV-1: methodology and clinical implications. Drug Resist. Updat. 5:224-233. [DOI] [PubMed] [Google Scholar]
- 27.Ritola, K., C. D. Pilcher, S. A. Fiscus, N. G. Hoffman, J. A. Nelson, K. M. Kitrinos, C. B. Hicks, J. J. Eron, Jr., and R. Swanstrom. 2004. Multiple V1/V2 Env variants are frequently present during primary infection with human immunodeficiency virus type 1. J. Virol. 78:11208-11218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sagar, M., X. Wu, S. Lee, and J. Overbaugh. 2006. Human immunodeficiency virus type 1 V1-V2 envelope loop sequences expand and add glycosylation sites over the course of infection, and these modifications affect antibody neutralization sensitivity. J. Virol. 80:9586-9598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schuitemaker, H., and N. A. Kootstra. 2005. Isolation, propagation, and titration of human immunodeficiency virus type 1 from peripheral blood of infected individuals. Methods Mol. Biol. 304:17-24. [DOI] [PubMed] [Google Scholar]
- 30.Sourisseau, M., N. Sol-Foulon, F. Porrot, F. Blanchet, and O. Schwartz. 2007. Inefficient human immunodeficiency virus replication in mobile lymphocytes. J. Virol. 81:1000-1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spira, A. I., and D. D. Ho. 1995. Effect of different donor cells on human immunodeficiency virus type 1 replication and selection in vitro. J. Virol. 69:422-429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Voronin, Y., J. Overbaugh, and M. Emerman. 2005. Simian immunodeficiency virus variants that differ in pathogenicity differ in fitness under rapid cell turnover conditions. J. Virol. 79:15091-15098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wei, X., J. M. Decker, S. Wang, H. Hui, J. C. Kappes, X. Wu, J. F. Salazar-Gonzalez, M. G. Salazar, J. M. Kilby, M. S. Saag, N. L. Komarova, M. A. Nowak, B. H. Hahn, P. D. Kwong, and G. M. Shaw. 2003. Antibody neutralization and escape by HIV-1. Nature 422:307-312. [DOI] [PubMed] [Google Scholar]
- 34.Williams, L. M., and M. W. Cloyd. 1991. Polymorphic human gene(s) determines differential susceptibility of CD4 lymphocytes to infection by certain HIV-1 isolates. Virology 184:723-728. [DOI] [PubMed] [Google Scholar]
- 35.Zhang, L., C. Chung, B. S. Hu, T. He, Y. Guo, A. J. Kim, E. Skulsky, X. Jin, A. Hurley, B. Ramratnam, M. Markowitz, and D. D. Ho. 2000. Genetic characterization of rebounding HIV-1 after cessation of highly active antiretroviral therapy. J. Clin. Investig. 106:839-845. [DOI] [PMC free article] [PubMed] [Google Scholar]