

Abstract
We aimed to define the relative contribution of both PKA and Ca2+/calmodulin-dependent protein kinase II (CaMKII) cascades to the phosphorylation of RyR2 and the activity of the channel during β-adrenergic receptor (βAR) stimulation. Rat hearts were perfused with increasing concentrations of the β-agonist isoproterenol in the absence and the presence of CaMKII inhibition. CaMKII was inhibited either by preventing the Ca2+ influx to the cell by low [Ca]o plus nifedipine or by the specific inhibitor KN-93. We immunodetected RyR2 phosphorylated at Ser2809 (PKA and putative CaMKII site) and at Ser2815 (CaMKII site) and measured [3H]-ryanodine binding and fast Ca2+ release kinetics in sarcoplasmic reticulum (SR) vesicles. SR vesicles were isolated in conditions that preserved the phosphorylation levels achieved in the intact heart and were actively and equally loaded with Ca2+. Our results demonstrated that Ser2809 and Ser2815 of RyR2 were dose-dependently phosphorylated under βAR stimulation by PKA and CaMKII, respectively. The isoproterenol-induced increase in the phosphorylation of Ser2815 site was prevented by the PKA inhibitor H-89 and mimicked by forskolin. CaMKII-dependent phosphorylation of RyR2 (but not PKA-dependent phosphorylation) was responsible for the β-induced increase in the channel activity as indicated by the enhancement of the [3H]-ryanodine binding and the velocity of fast SR Ca2+ release. The present results show for the first time a dose-dependent increase in the phosphorylation of Ser2815 of RyR2 through the PKA-dependent activation of CaMKII and a predominant role of CaMKII-dependent phosphorylation of RyR2, over that of PKA-dependent phosphorylation, on SR-Ca2+ release during βAR stimulation.
Keywords: Ryanodine receptor, β-adrenergic stimulation, CaMKII-dependent phosphorylation, Sarcoplasmic reticulum Ca2+ release
1. Introduction
Activation of β-adrenergic receptors (βAR) plays a pivotal role in the modulation of cardiac function in response to stress or exercise. Stimulation of βAR by catecholamines results in the activation of cAMP-dependent protein kinase (PKA) cascade. However, the prevalent concept that cAMP/PKA pathway is solely responsible for βAR-mediated cardiac cellular responses has been challenged in the last few years. Evidence has grown supporting a critical role of Ca2+/calmodulin-dependent protein kinase (CaMKII) in the effects induced by both acute and chronic βAR stimulation [1-6]. Similar to PKA, CaMKII phosphorylates the major components of the cardiac excitation-contraction coupling (ECC), including the L-type Ca2+ channel, the sarcoplasmic reticulum (SR) Ca2+ release channel or type 2 ryanodine receptor (RyR2) and the SR Ca2+-ATPase regulator phospholamban (PLN).
In spite of the extensive studies on the effects of βAR stimulation on the individual components of the ECC, the physiological significance of the β-adrenergic regulation of RyR2 is not yet fully understood. Results from different laboratories aiming to answer this matter remain controversial [6-8]. One of the reasons for these disparate results probably arises from the difficulty inherent to measure the intrinsic function of the phosphorylated RyR2, since it is well established that Ca2+ release through the RyR2 in active cells is strongly dependent on the characteristics of the trigger Ca2+ current (ICa) and on SR Ca2+ load [8].
Three different phosphorylation sites at the RyR2 have been identified up to now in in vitro studies: Ser2809, Ser2815 (or Ser2808 and Ser2814, respectively, depending on the species) and Ser2030. Whereas Ser2815 has been shown to be the target site for CaMKII [9] and Ser2030 for PKA [10], results regarding the kinase involved in the phosphorylation of Ser2809 residue are controversial. Although originally described as a CaMKII site [11], Ser2809 was found to be exclusively phosphorylated by PKA [9,12], stoichiometrically phosphorylated by both PKA and CaMKII [13] or susceptible of being phosphorylated by cGMP-dependent protein kinase (PKG) and insensitive to either activation or inhibition of PKA [14].
During βAR stimulation, RyR2 was found to be phosphorylated at Ser2809 or at Ser2030 sites [9,12,14]. However, the possible CaMKII-dependent phosphorylation of Ser2809 (if any) and of Ser2815 sites of RyR2 and its physiological significance during βAR stimulation, is far from being clear. In an effort to further understand this issue, our research has focused on two aspects: first, to define the relative contribution of PKA and CaMKII to the phosphorylation of Ser2809 evoked by isoproterenol and to study the effects of β-agonists on the phosphorylation of Ser2815 site. Second, to determine the effect of CaMKII-dependent phosphorylation of RyR2 during βAR stimulation on the channel activity and on its capacity to release Ca2+ from the SR. To accomplish this purpose, we measured in SR vesicles, [3H]-ryanodine binding and Ca2+ fluxes with a temporal resolution of ms (stopped flow technique), to better mimic the response of RyR2 channels to the sudden Ca2+ changes that occurred during L-type Ca2+ channel activation. The experiments were performed under careful control of SR Ca2+ load in the different situations explored. This strategy allowed us to dissect the functional role of the PKA and CaMKII pathways of RyR2 phosphorylation during βAR stimulation.
2. Materials and methods
2.1. Ex vivo experiments
2.1.1. Heart perfusions
Experiments were performed in isolated hearts from male Wistar rats (200–300 g body wt) perfused according to the Langendorff technique at constant temperature (37 °C), flow (12–14 ml/min) and heart rate (250 beats/min), as previously described [15]. The composition of the physiological salt solution was (in mM): 128.3 NaCl, 4.7 KCl, 1.35 CaCl2, 20.2 NaHCO3, 0.4 NaH2PO4, 1.1 MgCl2, 11.1 glucose and 0.04 Na2EDTA; this solution was equilibrated with 95% O2–5% CO2 to give a pH of 7.4. The mechanical activity of the heart was assessed by passing into the left ventricle a latex balloon connected to a pressure transducer (AD Instruments MLT9580, CO, USA). The balloon was filled with aqueous solution to achieve a left ventricular end-diastolic pressure of 7–13 mm Hg. The investigation conforms the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-234, revised 1996).
2.1.2. Experimental protocol
After stabilization, hearts were perfused for a 3 min period with either physiological salt solution with 1.35 mM [Ca]o or the same solution with 0.07 mM [Ca]o plus 0.4 μM nifedipine (to inhibit the Ca2+-influx to the cell), in the absence or the presence of different isoproterenol (Iso) concentrations between 0.3 and 300 nM. In additional experiments, hearts were perfused with: (a) 30–300 nM Iso in the absence and the presence of 5 μM of the specific CaMKII-inhibitor, KN-93, and in the absence and the presence of 30 μM of the PKA-inhibitor H-89 and (b) 1 μM forskolin. At the end of the experiments, hearts were freeze-clamped and stored at −80 °C until biochemical assays were performed.
2.1.3. Preparation of SR membrane vesicles for Western blot, [3H]-ryanodine binding and Ca2+ release kinetic assays
SR membrane vesicles for Western blot were prepared following the protocol previously described [1]. Briefly, frozen hearts were homogenized in 6 volumes of ice-cold solution which contained: 5 mM Na2EDTA, 25 mM NaF, 300 mM sucrose, 30 mM KH2PO4 (pH 7.0 at 4 °C) and 1 mM PMSF, 1 mM benzamidine, 0.5 μg/ml leupeptin and 1 μg/ml pepstatin as protease inhibitors. The homogenate was sedimented twice at 14,000×g and 16,000×g for 20 min. The resulting supernatant was centrifuged at 45,000×g for 45 min. The pellet obtained was suspended in 3 volumes of the above buffer containing 600 mM KCl and centrifuged as in the previous step. The final pellet was resuspended in small volumes of 10 mM Na2EDTA, 10 mM NaF, 250 mM sucrose and 30 mM histidine (pH 7.0 at 4 °C). SR vesicles for [3H]-ryanodine binding assay were isolated as above except that Na2EDTA was omitted and NaF was replaced by 1 μM okadaic acid as phosphatase inhibitor, in all the buffers. SR preparations were enriched approximately threefold with respect to homogenates as estimated by Western blot analysis of RyR2 and [3H]-ryanodine binding assay. SR vesicles for Ca2+ release kinetic assays were prepared following the protocol previously described [16] except that the homogenization buffer contained 20 mM MOPS/Tris (pH 6.8), 300 mM sucrose, 1 mM okadaic acid and protease inhibitors as above. Protein was measured by the method of Bradford, using bovine serum albumin as standard.
2.1.4. Electrophoresis and Western Blot analysis
For immunological detection of RyR2 and phosphorylated RyR2, SDS-PAGE was performed according to the method of Laemmli in 6% acrylamide gels [17]. Twelve and 50 μg of SR membrane protein were electrophoresed per gel lane for the in vitro assay and for the assessment of phosphorylation level in the perfused heart, respectively. Separated proteins were transferred to PVDF membranes (Immobilon-P, Millipore) and probed with antibodies RyR2-PS2809 (1:5000; Badrilla, Leeds, UK), RyR2-PS2809 (1:1000) and RyR2-PS2815 (1:1000), both kindly provided by A. Marks and X. Wehrens (Columbia University, USA), and RyR2 antibody (1:2500; Affinity Bioreagents Inc.). Immunoreactivity was visualized by peroxidase-conjugated antibodies using a peroxidase-based chemiluminescence detection kit (ECL, Amersham). The signal intensity of the bands was quantified using ImageJ (NIH). Results of phosphorylation of RyR2 normalized by the total RyR2 content were expressed as percentage of the maximal phosphorylation induced by the β agonist, isoproterenol, which is achieved at 30–300 nM. Phosphorylation of Thr17 of PLN was detected as previously described [1].
2.1.5. In vitro phosphorylation/dephosphorylation assays
Phosphorylation reactions of SR vesicles (0.6 mg/ml) were conducted for 10 min at room temperature in a buffer containing 50 mM histidine (pH 7.0), 5 mM MgSO4, 5 mM NaF and 2.5 μM okadaic acid. For PKA phosphorylation, 1.5 U of the catalytic subunit per μg of SR protein, 1 mM EGTA and 1 μM of the CaMKII inhibitor, KN-93, were added to the assay medium. For phosphorylation by the endogenous CaMKII, 0.5 mM CaCl2, 50 μg/ml calmodulin (CaM) and 1 μM of the PKA inhibitor PKI were added. For the control samples, Ca2+ (1 mM EGTA present), CaM and PKA were omitted from the medium. Phosphorylation reactions were initiated by the addition of 0.2 mM ATP. For dephosphorylation reactions, cardiac SR vesicles (1 mg/ml) were incubated for 30 min at 37 °C in a solution containing (in mM): 50 histidine (pH 7.0), 5 MgSO4 and 1 EGTA with 2–4 U of alkaline phosphatase per μg of SR protein. After incubations, all reactions were stopped by the addition to the medium of SDS sample buffer.
2.1.6. [3H]-ryanodine binding
[3H]-ryanodine binding was performed in SR cardiac membranes (60 μg) assayed in 0.25 ml of incubation medium according to Lokutta et al. [18]. The medium contained 1 mM benzamidine, 1 mM PMSF, 0.5 μg/ml leupeptin, 1 μg/ml pepstatin A as protease inhibitors and 1 μM okadaic acid as phosphatase inhibitor, without addition of Mg2+ or ATP. The necessary CaCl2 to set free [Ca2+] in the range of 0.1 to 100 μM was calculated using the stability constants of Fabiato [19]. [3H]-ryanodine was diluted directly into the incubation medium to achieve a final concentration of 10 nM. Incubation lasted 90 min at 36 °C. Samples were run in duplicate, filtered onto glass fiber filters (Munktell MGB) and washed four times with 5 ml cold water. The filters were placed in vials containing scintillation liquid cocktail and retained radioactivity was measured in a β-counter. The specific binding was defined as the difference between the binding in the absence (total binding) and in the presence (non-specific binding) of 10 μM unlabeled ryanodine.
2.1.7. Active Ca2+ loading of cardiac SR vesicles
SR vesicles were actively loaded with Ca2+ in the presence of an ATP-regenerating system as previously described [16]. Briefly, SR vesicles (1 mg/ml) were added to a solution containing: 0.05 mM CaCl2, 100 mM KCl, 2 mM ATP, 3 mM MgCl2, 10 mM phosphocreatine, 15 U/ml creatine kinase and 20 mM imidazole MOPS (pH 7.2 at 4 °C). To avoid changes in the redox and phosphorylation status of SR vesicles during Ca2+ uptake, we included a redox buffer (3 mM of reduced glutahione/glutathione oxidized (GSSG/GSH) in a ratio 1/100), 1 μM okadaic acid and 0.5 μM staurosporine as phosphatase and kinase inhibitors, respectively. The active Ca2+ loading of these vesicles was determined by measuring the decay in extravesicular free [Ca2+] [16]. Active Ca2+ uptake was completed in 25 min at 30 °C. At this time all SR vesicles were equally loaded with Ca2+ (extravesicular [Ca2+] stabilized at 0.59±0.11 μM Ca2+, n=6).
2.1.8. Ca2+ release kinetics
Ca2+ release kinetics were determined in a SX.18MV fluorescence stopped-flow spectrometer from Applied Photo-physics Ltd. (Leatherhead, U.K.). The increase in extravesicular [Ca2+] was measured by the increase in fluorescence of the Ca2+ indicator Calcium Green-5N (Molecular Probes, Eugene, OR). The fluorescent emission of the dye was measured through a 515 nm cut-off long-pass filter, using an excitation wavelength of 488 nm. Ca2+ release was initiated by rapidly mixing (less than 2 ms) 1 volume of the solution containing Ca2+ loaded SR vesicles with 10 volumes of releasing solution containing (in mM): 100 KCl, 1.6 ATP, 20 imidazole-MOPS, pH 7.2 and 1 μM Calcium Green-5N. After mixing, this solution produced free concentrations of 1.2 mM ATP, 17 μM Mg2+ and pCa 6 [16].
2.1.9. Statistics
For comparison among different groups, ANOVA was used followed by Tukey test. A P value <0.05 was considered statistically significant.
3. Results
3.1. Phosphorylation studies
3.1.1. In vitro phosphorylation/dephosphorylation of Ser2809 and Ser2815 sites of RyR2
The goals of the in vitro phosphorylation/dephosphorylation assays were twofold: (1) to verify the specificity and the ability of the antibodies used to detect the phosphorylation level of RyR2; (2) to elucidate the controversial issue regarding the phosphorylation of Ser2809 site: two independent groups using phospho-specific-antibodies reported that this site was either exclusively phosphorylated by PKA [9,12] or phosphorylated by both PKA and CaMKII [13]. We therefore performed experiments using the antibodies from both laboratories under the same experimental conditions to determine whether discrepancies in the results could be ascribed to the different methodology and source of the antibodies used. Representative immunoblots and overall results are shown in Fig. 1. Treatment of the SR membrane preparation with alkaline phosphatase reduced the immunorecognition by the phospho-antibodies with respect to a non-treated sample. Densitometric analysis with the two phospho-Ser2809 antibodies provided equivalent results: the signal increased when SR membrane vesicles were exposed to the catalytic subunit of PKA and after the addition of Ca2+ and CaM to activate the endogenous CaMKII. The signal with the phospho-Ser2815 antibody only increased in the Ca2+/CaM-treated samples and did not show any significant increase in the PKA-treated samples. In all cases, immunodetection with an antibody against RyR2 showed no difference in the content of RyR2 among the different treatments. Taken together the results indicated that the antibodies were faithfully reporting on the phosphorylation level of RyR2 and confirmed that Ser2809 can be phosphorylated by either PKA or CaMKII and that Ser2815 site can be exclusively phosphorylated by CaMKII.
Fig. 1.
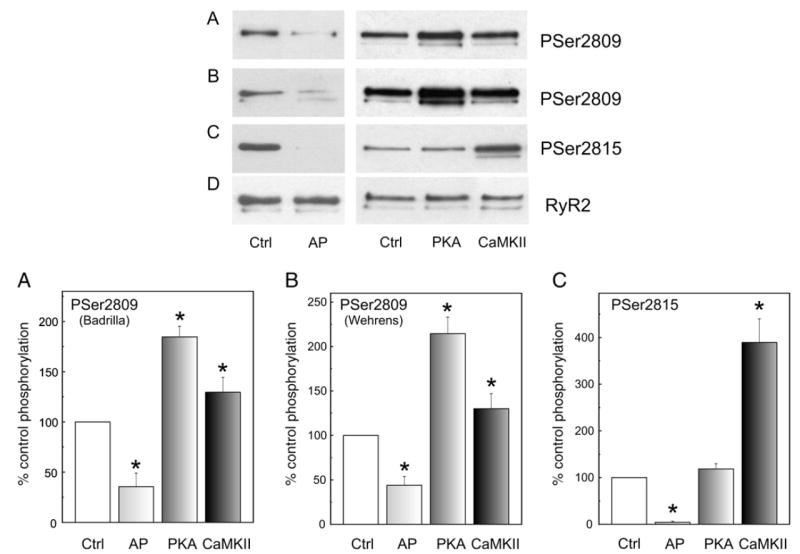
Site-specific phosphorylation of RyR2. Representative immunoblots and overall results of SR membrane vesicles dephosphorylated by treatment with alkaline phosphatase (AP) and phosphorylated in vitro by the catalytic subunit of PKA in the presence of EGTA (PKA) or by the endogenous SR-associated CaMKII in the presence of the PKA inhibitor PKI (CaMKII) as described under Materials and methods. For the control samples, Ca2+ (1 mM EGTA present), calmodulin and the catalytic subunit of PKA were omitted (Ctrl). SR proteins were separated on SDS-PAGE and transferred to PVDF membranes. Blots were probed with anti-PS2809-RyR2 from Badrilla (A), anti-PS2809-RyR2 (B) and anti-PS2815-RyR2 (C) from X. Wehrens and A. Marks laboratories and anti-RyR2 from Affinity Bioreagents (D). Results are expressed as percentage of the phosphorylation obtained in Ctrl. *p<0.05 with respect to Ctrl.
3.1.2. β-adrenergic-dependent phosphorylation of Ser2809 and Ser2815 sites of RyR2 in the intact heart
The concentration-dependence of the phosphorylation of Ser2809 and Ser2815 in response to isoproterenol stimulation was studied in the absence and presence of low extracellular Ca2+ concentration ([Ca]o) and nifedipine to avoid Ca2+-influx to the cell and the consequent activation of CaMKII. Under this latter condition, basal contractile activity as well as the response to isoproterenol were completely suppressed indicating that the maneuver is an effective experimental tool to decrease intracellular Ca2+ (Fig. 2A).
Fig. 2.
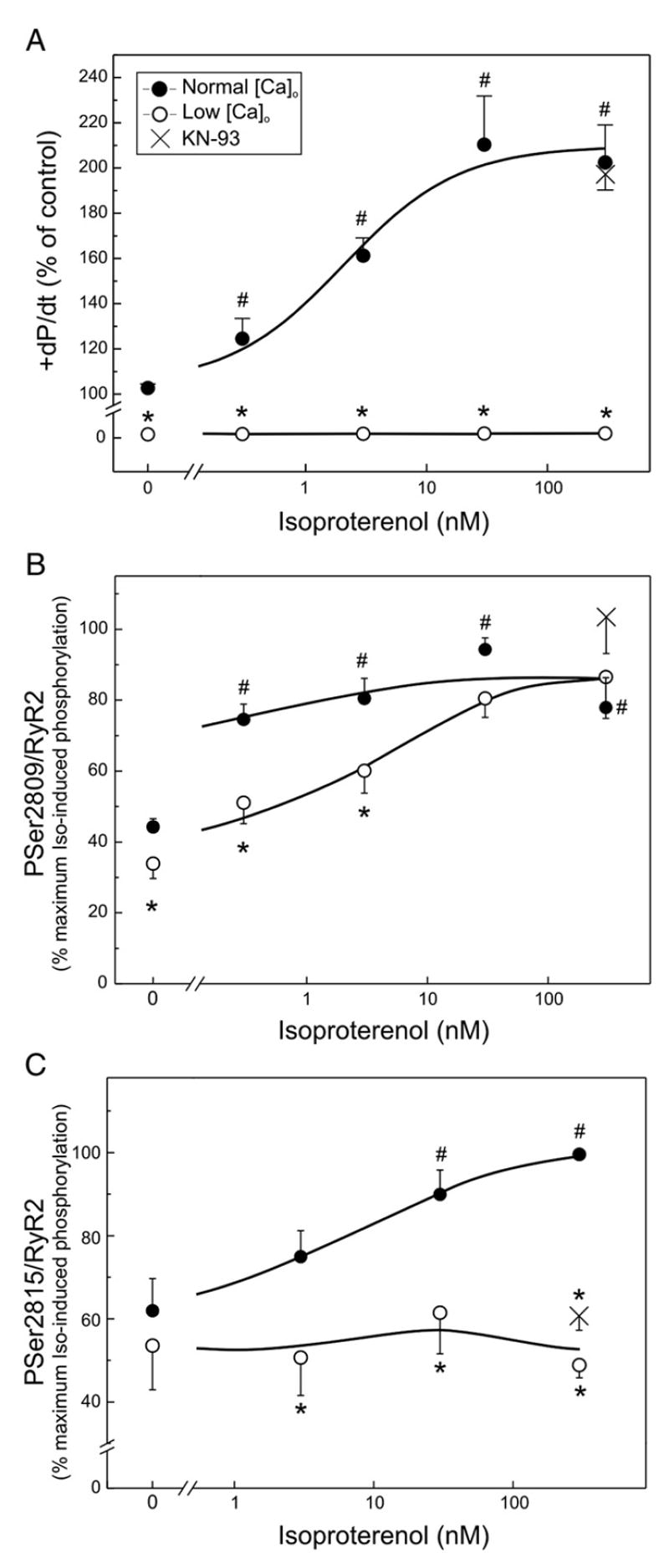
β-adrenergic-dependent effects on left ventricular function and phosphorylation of RyR2 in the perfused rat heart in the absence and the presence of low [Ca]o and KN-93. Maximal rate of rise of pressure (+dP/dt, A), phosphorylation of Ser2809 (B) and of Ser2815 (C) of RyR2 in response to isoproterenol in the absence (closed circles) and presence of low [Ca]o and nifedipine (open circles), to avoid Ca2+ influx to the cells. Isoproterenol increased contractility and the phosphorylation of both Ser2809 and Ser2815 sites of RyR2. Low [Ca]o plus nifedipine (Low [Ca]o) abolished contractility in the absence and presence of isoproterenol (+dP/dt, A) and significantly diminished basal Ser2809 phosphorylation and that produced by the lowest isoproterenol concentrations (0.3 and 3 nM) without significantly affecting the phosphorylation evoked by the highest concentrations of the β-agonist (B). Low [Ca]o plus nifedipine did not decrease basal phosphorylation of Ser2815 but significantly diminished to basal values the phosphorylation of Ser2815 at all isoproterenol concentrations tested (C). 5 μM of the CaMKII inhibitor KN-93 (crosses) did not affect the increase in contractility and phosphorylation of Ser2809 induced by 300 nM isoproterenol, but mimicked the effects of low [Ca]o plus nifedipine in the phosphorylation of Ser2815 induced by 300 nM isoproterenol. Mean±SEM, n=3–10 experiments. #p<0.05 with respect to hearts perfused with normal [Ca]o in the absence of drugs. *p<0.05 with respect to the corresponding situation in the presence of normal [Ca]o.
Fig. 2B shows that basal phosphorylation levels of Ser2809 were high, in agreement with previous findings [10,13,14,20,21]. The figure also shows that 0.3 nM, the lowest isoproterenol concentration studied, produced a significant increase in the phosphorylation of Ser2809 site. Higher isoproterenol concentrations produced a slight increase in the phosphorylation of this residue, which did not attain, however, significant levels when compared with 0.3 nM isoproterenol. Thus, in agreement with previous results [14,22], Ser2809 was virtually maximally phosphorylated at the lowest isoproterenol concentration used. Low [Ca]o plus nifedipine produced a modest albeit significant decrease in the basal phosphorylation of Ser2809 and also in the phosphorylation produced by the lowest isoproterenol concentrations (0.3 and 3 nM). These results would indicate that under basal conditions and at the lower isoproterenol concentrations, PKA and CaMKII pathways both contribute to the phosphorylation of Ser2809 site. In contrast, low [Ca]o plus nifedipine did not affect the phosphorylation evoked by the highest concentrations of the β-agonist, which would indicate that at the higher isoproterenol concentrations, Ser2809 site is phosphorylated only by PKA. Thus, the procedure of avoiding Ca2+ entry to the cell allows us to dissect the relative participation of PKA and CaMKII in the phosphorylation of Ser2809 site under βAR stimulation. The maneuver further reveals that the PKA phosphorylation of Ser2809 increased in a concentration-dependent manner with the concentration of the β-agonist.
Fig. 2C shows that basal phosphorylation values of Ser2815 were high as in the case of Ser2809. However, and in contrast to what was observed with Ser2809 site, basal Ser2815 phosphorylation was not affected by low [Ca]o plus nifedipine. Moreover, phosphorylation of Ser2815 increased with the increase in isoproterenol concentration, attaining significant levels at 30 and 300 nM. At all isoproterenol concentrations the presence of low [Ca]o plus nifedipine decreased the phosphorylation of Ser2815 residue to basal levels. Of interest, the dose–response curve to PKA-dependent phosphorylation of RyR2 (Fig. 2B) was shifted to lower isoproterenol concentrations with respect to the dose–response curve to CaMKII-dependent phosphorylation (Fig. 2C). A similar shift has been previously described for the PKA and CaMKII-dependent PLN phosphorylation [2,23]. Since CaMKII-dependent RyR2 phosphorylation at Ser2815 site was shown to be enhanced in isolated rabbit hearts submitted to increased heart rates [9], and the maneuver of reducing Ca2+ plus nifedipine renders a non-beating heart (see Fig. 2A), we repeated the experiments with 300 nM isoproterenol in the absence and the presence of the CaMKII inhibitor KN-93. In these two conditions contraction frequency was the same, thus ruling out any possible frequency-dependent CaMKII activity change. As shown in Fig. 2C, 5 μM KN-93 was able to mimic the effects of low [Ca]o plus nifedipine on the isoproterenol-induced phosphorylation of Ser2815. The results suggest that Ser2815 site of RyR2 is concentration-dependently phosphorylated by the activation of the CaMKII-dependent pathway of the β-adrenergic stimulation. KN-93 also mimicked the effect of low [Ca]o plus nifedipine on Ser2809 site, failing to reduce the phosphorylation of Ser2809 produced by 300 nM isoproterenol (Fig. 2B).
Recent experiments by Curran et al. [6] indicated that activation of CaMKII by a PKA-independent pathway was responsible for the enhancement of isoproterenol-induced SR Ca2+ leak. We therefore explored whether the increase in the phosphorylation of Ser2815 site of RyR2 observed during βAR stimulation here described was also due to a PKA-independent activation of CaMKII. For this purpose, we inhibited PKA with H-89 during βAR stimulation and we directly activated the adenylate cyclase and therefore PKA, bypassing the βAR, with forskolin. Thirty micromolar H-89 was required to achieve the complete inhibition of the positive inotropic effect of isoproterenol (results not shown). Forskolin was used at 1 μM, a concentration that produced an increase in contractility and relaxation not significantly different from that evoked by isoproterenol (maximal rate of rise of pressure: 174.5±9.3 vs. 181.3±7.1% of control and half relaxation time −10.4±2.8 vs. −12.1±2.3 Δ ms from control, for forskolin and isoproterenol, respectively). Fig. 3 shows typical immunoblots (upper panels) and average data (lower panels) of the phosphorylation results obtained in this experimental series. H-89 blunted the isoproterenol-induced increase in the phosphorylation of Ser2815 of RyR2. Moreover, forskolin evoked a phosphorylation of Ser2815 of RyR2 similar to that produced by isoproterenol (Fig. 3A). Thus, in our conditions, the CaMKII-dependent phosphorylation of Ser2815 of RyR2 is dependent on the increase in PKA activity evoked by βAR stimulation. βAR stimulation also phosphorylated Thr17 residue of PLN, a well known intracellular marker of increased CaMKII activity, through a PKA-dependent pathway (Fig. 3B).
Fig. 3.
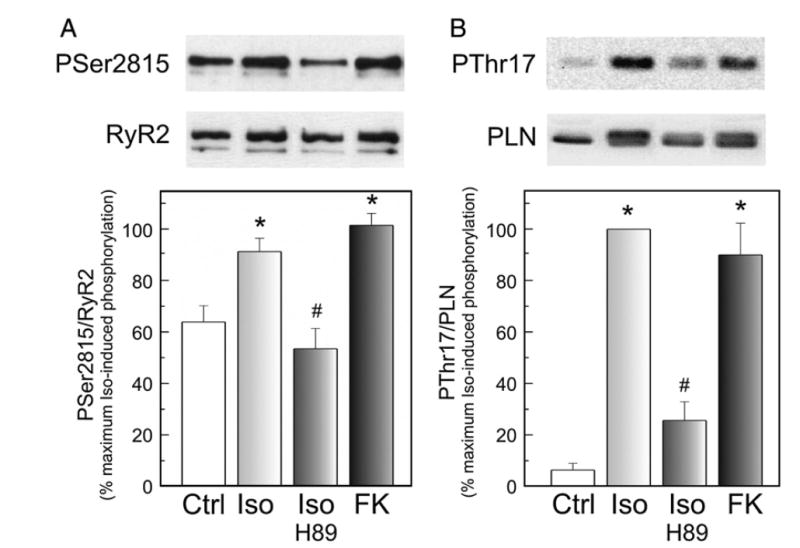
Activation of PKA is required for the CaMKII-dependent phosphorylation of Ser2815 of RyR2 and Thr17 of PLN. Representative immunoblots and overall results of SR vesicles isolated from hearts perfused without (Ctrl) and with 30 nM isoproterenol in the absence (Iso) and the presence of 30 μM of the PKA inhibitor H-89 (Iso-H-89) and with 1 μM of forskolin (FK). Phosphorylation of Ser2815 of RyR2 and Thr17 of PLN is shown as ratio between the signals of the site-specific phospho-antibody and the antibody used to recognized the non-phosphorylated form. *p<0.05 with respect to Ctrl. #p<0.05 with respect to Iso-treated hearts.
3.2. Functional studies
Functional studies were performed in SR membrane vesicles isolated in conditions which preserved the phosphorylation levels achieved under the different experimental situations in the perfused rat heart. We studied the β-adrenergic effect at 300 nM isoproterenol, a concentration at which Ser2809 was exclusively phosphorylated by PKA and Ser2815 by CaMKII (see Figs. 2B and C). Thus at this concentration, the inhibition of CaMKII activity either by preventing the Ca2+ influx to the cell by low [Ca]o plus nifedipine or by the specific inhibitor KN-93, allowed to dissect the relative role of both kinases, PKA and CaMKII, during β-adrenergic stimulation.
3.2.1. [3H]-ryanodine binding
Fig. 4 shows the [Ca2+] dependence of [3H]-ryanodine binding to SR vesicles isolated from the differently treated hearts. Specific binding under control conditions showed a graded dependence of [Ca2+], which was maximal at 10 μM [Ca2+]. Membrane vesicles isolated from isoproterenol perfused hearts, showed an increased [3H]-ryanodine binding, reaching statistical significance at 1, 10 and 100 μM [Ca2+]. This upward shift in the [3H]-ryanodine binding produced by isoproterenol, indicated an increased Ca2+-induced activation of RyR2 compared with control. The isoproterenol-induced stimulation of the binding was abolished when either KN-93 or low [Ca]o plus nifedipine was perfused simultaneously with isoproterenol. Neither KN-93 nor low [Ca]o plus nifedipine perfused in the absence of the β-agonist, modified [3H]-ryanodine binding. At 10 μM [Ca2+], the ryanodine binding was 0.21±0.02 (control), 0.22±0.02 (KN) and 0.20±0.03 (low Ca) pmol/mg protein in agreement with previous findings [18].
Fig. 4.
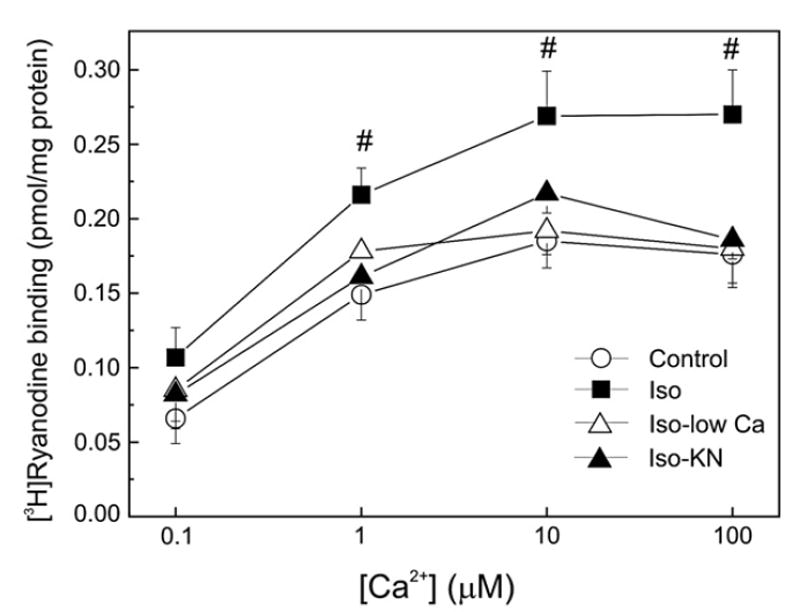
[3H]-ryanodine binding in SR vesicles isolated from treated hearts. Ca2+ dependence of [3H]-ryanodine binding in cardiac SR vesicles isolated from hearts perfused in the absence (Control) and the presence of 300 nM isoproterenol (Iso) and in the presence of Iso, either with low [Ca]o plus nifedipine (Iso-low Ca) or with 5 μM KN-93 (Iso-KN). Isoproterenol increased [3H]-ryanodine binding. This increase did not occur in the presence of KN-93 or low [Ca]o plus nifedipine. Of note, at 300 nM isoproterenol, the decrease in Ca2+ influx to the cell or the inhibition of CaMKII did not affect the phosphorylation of Ser2809 site but significantly decreased the phosphorylation of Ser2815 site (see Fig. 2). Neither KN-93 nor low [Ca]o plus nifedipine modified [3H]-ryanodine binding in the absence of isoproterenol. #p<0.05 with respect to control hearts.
3.2.2. Ca2+ release kinetics
Fig. 5 shows the time course of Ca2+ release from SR vesicles isolated from hearts perfused under the different experimental conditions. In every case the increase in fluorescence followed a double exponential function: A1 [1−exp (−k1t)]+A2 [1−exp (−k2t)], characterized by a fast rate constant k1 and a slower rate constant k2, which differed in magnitude by approximately 20-fold under control conditions. Previous studies in isolated SR vesicles have demonstrated that the faster rate constant of Ca2+ release, k1, correlates with the average open probability of a population of channels in response to a sudden [Ca2+] change, while k2 is most likely due to a time-dependent decrease in channel activity following rapid activation by Ca2+ [16,24,25]. Overall results in Fig. 6A show that k1 increased 5.8-fold in isoproterenol-treated hearts and that this increase was prevented in hearts simultaneously perfused with either low [Ca]o plus nifedipine or the CaMKII inhibitor KN-93. This behavior was similar to the one observed in the phosphorylation of Ser2815 site as shown for comparison in the same figure. The fractional Ca2+ release that occurs with the faster rate constant (A1/A1+A2) did not significantly change among the different experimental conditions (control: 0.28±0.09; 300 nM isoproterenol: 0.23±0.02; 300 nM isoproterenol in low [Ca]o plus nifedipine: 0.21±0.07 and 300 nM isoproterenol in the presence of KN-93, 0.22±0.07). In addition, SR vesicles from isoproterenol-treated hearts showed a significant increase in k2 with respect to control values. Of interest this increase in k2 was not significantly affected by low [Ca]o plus nifedipine or KN-93 and it would therefore represent a PKA-dependent effect of βAR stimulation. These results paralleled the results obtained with the PKA-dependent phosphorylation of Ser2809 site of RyR2 (Fig. 6B).
Fig. 5.
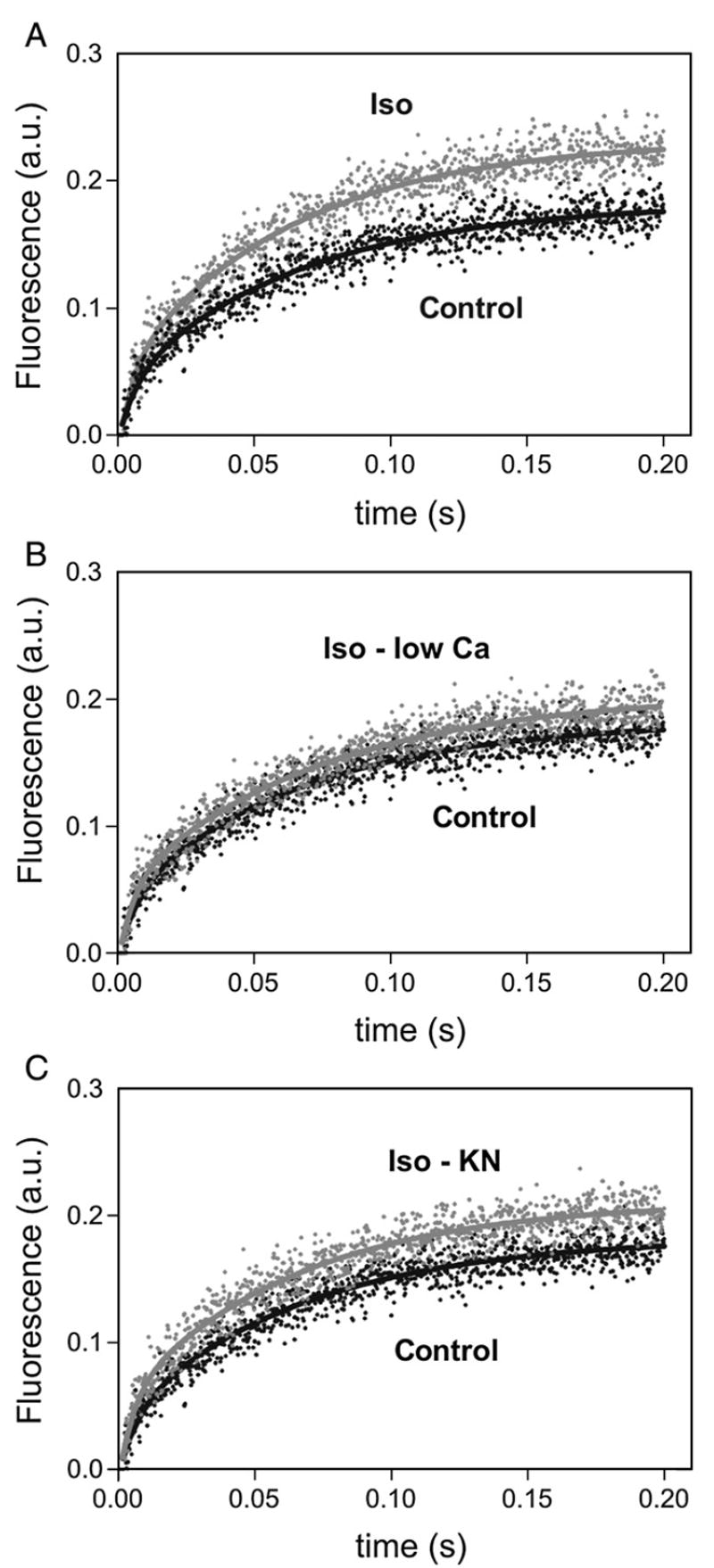
Effect of isoproterenol on Ca2+ release kinetics in isolated hearts. Release kinetic was measured following the change in fluorescence of Calcium Green 5 and was fitted to the double exponential function: A1 [1−exp (−k1t)]+A2 [1−exp (−k2t)]. Representative Ca2+ release records in SR vesicles isolated from hearts submitted to the different experimental protocols. The increase in Ca2+ release kinetics produced by isoproterenol (Iso, A) was significantly decreased bylow [Ca]o plus nifedipine (Iso-low Ca, B) or by CaMKII-inhibition (Iso-KN, C). Records shown are the average of 5 to 7 determinations.
Fig. 6.
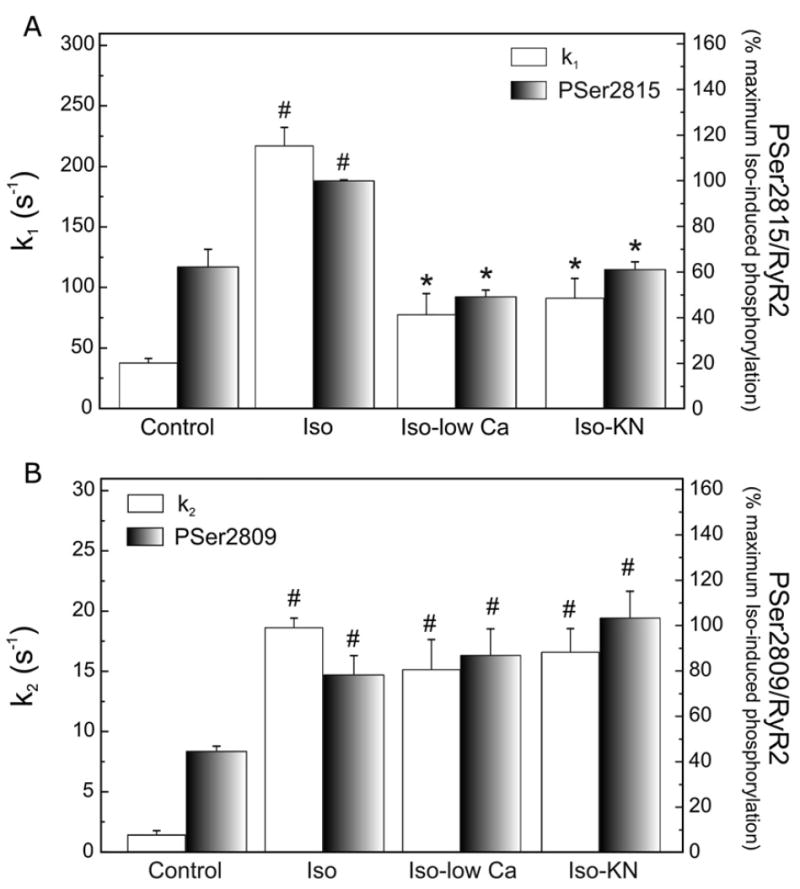
Ca2+ release rate constants (k1 and k2) and phosphorylation of RyR2 sites. The rate constant of the fast-exponential release (k1, A) increased 5.8-fold in isoproterenol-treated hearts (Iso) and this increase was prevented in hearts perfused with low [Ca]o plus nifedipine (Iso-low Ca) or the CaMKII inhibitor KN-93 (Iso-KN). k2 increased with Iso and it did no decrease by inhibition of CaMKII (B). Changes in the phosphorylation of PSer2815 (A) and PSer2809 sites (B) in the same experimental conditions are shown for comparison. #p<0.05 with respect to control hearts; *p<0.05 with respect to Iso.
4. Discussion
The functional role of RyR2 phosphorylation has been a controversial issue over the years. Numerous and contradictory results have been reported regarding the physiological and pathophysiological relevance not only of PKA-dependent phosphorylation but also of CaMKII-dependent phosphorylation of RyR2 [6-14,18,26-35]. In spite of this profusion of work regarding RyR2 phosphorylation and function, the PKA and CaMKII-dependent phosphorylations of RyR2 under βAR stimulation and their functional consequences are not well defined. The main goal of the present experiments was to investigate this issue.
Our results demonstrated that: (1) Ser2809 and Ser2815 of RyR2 are dose-dependently phosphorylated under βAR stimulation by PKA and CaMKII respectively; (2) CaMKII-dependent phosphorylation of Ser2815 during βAR stimulation is secondary to PKA activation; (3) CaMKII contributes to the phosphorylation of Ser2809 of RyR2 under basal conditions and at the lowest isoproterenol concentrations and (4) CaMKII-dependent phosphorylation of RyR2 (but not PKA-dependent phosphorylation), is responsible for the β-induced increase in the channel activity as indicated by [3H]-ryanodine binding and fast SR Ca2+ release kinetic studies. To the best of our knowledge, the present results reveal for the first time the predominant role of CaMKII-dependent phosphorylation of RyR2 on the Ca2+ release response of the RyR2 to a sudden Ca2+ change over that of PKA-dependent phosphorylation during βAR stimulation. Our approach is unique in the sense that we used for our functional assays SR membrane vesicles that preserved the phosphorylation level achieved under the different experimental conditions in the perfused intact heart, but that were equally loaded with Ca2+ immediately before the functional assay.
4.1. The CaMKII pathway of βAR stimulation on RyR2 phosphorylation and activity
The functional effects of βAR stimulation have been naturally linked to the cAMP-PKA intracellular cascade and the associated PKA-dependent phosphorylations. However, βAR stimulation also triggers the activity of Ca2+-dependent kinases and phosphatases, among which is CaMKII. The effects of these secondary cascades, which may modify the classic cAMP-PKA pathway during βAR stimulation, are frequently dismissed. Our results showed that CaMKII phosphorylates RyR2 at Ser2815 site during βAR stimulation in a dose-dependent manner. These results differed from those of Wehrens et al. [9], who failed to detect the isoproterenol-induced increase in Ser2815 phosphorylation. The reason for this discrepancy is difficult to explain. Since their experiments also failed to show a significant increase in the phosphorylation of Thr17 of PLN which is known to increase under βAR stimulation [1,2,23], one might speculate that either CaMKII was not sufficiently activated by isoproterenol in their experimental conditions or that the activity of phosphatases relative to that of CaMKII in their preparations is too high, avoiding the detection of the phosphorylation of Thr17 of PLN and Ser2815 of RyR2.
Ginsburg and Bers [8] studied the effects of the β-agonist isoproterenol under conditions of controlled ICa trigger and SR Ca2+ load. They found that βAR stimulation did cause a significant increase in the maximal rate of SR Ca2+ release. Moreover, Lindegger and Niggli elegantly demonstrated that βAR stimulation may directly increase the SR Ca2+ release [33]. Both studies, however, failed to discriminate whether the stimulatory effect of isoproterenol was due to PKA and/or CaMKII-dependent phosphorylation of the RyR2. The present findings are in line with these previous observations and further show that CaMKII-dependent phosphorylation of RyR2 is responsible for the increased activation of SR Ca2+ channels in this situation. Of note, the functional consequences of β-adrenergic-induced CaMKII-dependent phosphorylation may not be exclusively attributed to the phosphorylation of Ser2815 site, since in vitro results revealed other putative CaMKII-dependent sites as yet not identified [13].
The faster SR Ca2+ release kinetics produced by the CaMKII-dependent phosphorylation of RyR2 cannot be attributed to the β-adrenergic-induced increase in SR Ca2+ load associated with the phosphorylation of PLN [34,35], since SR vesicles isolated from hearts submitted to the different experimental conditions were equally loaded with Ca2+ before the functional assay. Obviously under in vivo conditions, the increased ICa trigger and SR Ca2+ load should contribute to the overall action of βAR stimulation on SR Ca2+ release.
An additional finding of our experiments is that in spite of the increase in the faster rate constant (k1) associated with the CaMKII-dependent phosphorylation of RyR2 during β-adrenergic stimulation, the fractional Ca2+ release that occurs with this constant (A1 / [A1+A2]) did not significantly change among the different experimental conditions. A possible explanation for this finding might be that the decrease in intravesicular Ca2+ in the isoproterenol-treated vesicles leads to an anticipated end of the fast Ca2+ release, resulting in an unvarying fraction of Ca2+ released. Alternatively, CaMKII-dependent phosphorylation of the channel could enhance not only the transition to a high opening probability mode but also diminish the time at which the channel remains in this state. In line with these findings, experiments of Guinsburg and Bers [8] demonstrated in isolated myocytes that isoproterenol did systematically increase the maximal rate of Ca2+ release for a given ICa and SR Ca2+ load without increasing the release amplitude, suggesting that isoproterenol must also hasten the turn-off of Ca2+ release.
Recent experiments indicated that βAR stimulation enhances diastolic Ca2+ leak by an increase in the activity of CaMKII that was independent of PKA activation [6]. The present findings complement those previous experiments indicating that CaMKII also regulates Ca2+-induced systolic Ca2+ release during βAR stimulation. However, our experiments failed to support the conclusion that the increase in CaMKII activity was independent of PKA activation. We showed that CaMKII-dependent phosphorylation of Ser2815 site of RyR2 as well as that of Thr17 site of PLN, was blunted by a concentration of the PKA-inhibitor H-89, at which the mechanical effects of the β-agonist were also blocked. We are aware of the fact that the concentration of H-89 required for the suppression of the isoproterenol-induced increase in contractility in the intact heart was rather high (30 μM) which would limit the value of this drug as a useful tool in dissecting signaling pathways (See also Bracken et al. [36]). Indeed it has been shown that 30 μM H-89 is the IC50 for CaMKII in in vitro conditions. However, this concentration failed to affect CaMKII activity in more intact preparations [37]. Moreover, our experiments also showed that activation of PKA by forskolin was able to mimic the mechanical and CaMKII-dependent phosphorylation effects of isoproterenol, strongly suggesting that the CaMKII-dependent phosphorylation of Ser2815 site of RyR2 and Thr17 residue of PLN is secondary to PKA activation. Further studies are warranted to solve the discrepancy regarding the pathways of activation of CaMKII during βAR stimulation.
4.2. The PKA pathway of βAR stimulation on RyR2 phosphorylation
The phosphorylation of Ser2809 site of RyR2 has been controversial over the years [9,11-13]. In vitro experiments by Witcher et al. first described Ser2809 residue of RyR2 as a CaMKII-dependent site [11]. This notion was challenged by a large piece of evidence provided by Marks’ group, both in in vitro and ex vivo experiments, which indicates that Ser2809 site is solely phosphorylated by PKA [9,12]. Subsequent studies from Colyer’s group [13] indicated that both kinases equally contribute to the phosphorylation of this site. Our experiments showed that Ser2809 could be phosphorylated by both PKA and CaMKII when interrogated by the specific antibodies used by both groups. Furthermore, although CaMKII seems to contribute to the basal phosphorylation of Ser2809, PKA is the main kinase that phosphorylates this site under βAR stimulation. In fact, at maximal isoproterenol concentrations, the strong PKA activation can make up for any loss in CaMKII activation when CaMKII is blocked. Our final major finding is that PKA-dependent phosphorylation of RyR2 during βAR stimulation does not seem to directly contribute to the fast Ca2+ release during the stimulation with the β-agonist. These results are in line with the findings from Li et al. [35] and Guo et al. [30] that suggest that PKA effects on SR Ca2+ release are secondary to the increased in SR Ca2+ load produced by the kinase. The present findings suggest, however, that PKA-dependent phosphorylation of the channel would increase the slow rate constant of Ca2+ release, k2. Studies on Ca2+ release kinetics in canine cardiac SR vesicles using the same protocol of the present experiments, suggested that k2 may most probably represent a time-dependent decrease in RyR2 channel open probability following Ca2+ activation, compatible with an increase in the inactivation/adaptation processes [16]. If these results can be extrapolated to the rat heart, our findings would indicate that the increase in k2 produced by isoproterenol represents a decrease in time-dependent channel inactivation/adaptation. This conclusion differs from that reported by Valdivia et al. [38] who found that PKA accelerates the kinetics of adaptation of RyR2 incorporated in planar lipid bilayers. Alternatively the increase in k2 might represent a PKA-dependent subconductance opening of the channel as described by Wehrens et al. [9]. Unfortunately our results did not allow to discriminate between these two attractive possibilities.
4.3. Implications
The increase in SR Ca2+ release and intracellular Ca2+ transient produced by isoproterenol depends primarily on the increased ICa trigger and SR Ca2+ load. These two effects are regulated by CaMKII-activity [28] and cannot be avoided in the intact heart. It is therefore difficult to dissect whether the increase in the velocity of the fast SR Ca2+ release described here has any significant contribution to the overall increase in intracellular Ca2+ transient and contractility evoked by βAR stimulation. Experiments from Eisner’s group [39-41] indicated that in conditions where the SR Ca2+ content is not controlled, factors that alter the sensitivity of RyR2 to trigger Ca2+, as is the case of CaMKII-dependent phosphorylation, will have only transitory effects on the amplitude of Ca2+ transients. This is because the increase in Ca2+ transient amplitude evoked by the increase in Ca2+ release due to RyR2 sensitization, decreases Ca2+ influx via the L-type Ca2+ channel and increases Ca2+ efflux via NCX. Ultimately this would decrease SR Ca2+ content, counteracting the initial effect of RyR2 sensitization. It is noteworthy, however, that under these conditions, the maintenance of Ca2+ transient amplitude occurs in spite of a lower SR Ca2+content and ICa, which would actually indicate an enhanced ECC gain. Thus, even though the enhancement of ICa and SR Ca2+ uptake through the phosphorylation of PLN appear as the primary mechanisms of the positive inotropic effect of β-agonists [8,34,35], the CaMKII-dependent enhancement of Ca2+-induced SR fast Ca2+ release may also play a significant role in this positive inotropic action. Recent experiments by Curran et al. [6] indicated that CaMKII activation during βAR stimulation enhanced SR Ca2+ leak, which would tend to decrease SR Ca2+ load and reduce the positive inotropic effect of isoproterenol. The CaMKII-dependent stimulation of systolic SR Ca2+ release during βAR stimulation here described may constitute a mechanism able to counteract, at the same RyR2 level, the negative action of SR Ca2+ leak on the positive inotropy of βAR stimulation. In this scenario, it is interesting to mention that a CaMKII-dependent increase in diastolic Ca2+ leak was described in a rabbit heart failure model [31]. In this model, CaMKII blockade restored SR Ca2+ content but has only modest effects on intracellular Ca2+ transient amplitude. Our experiments support the view that a CaMKII-dependent increase in systolic SR Ca2+ release may effectively counteract the deleterious effects of SR Ca2+ leak in this heart failure model.
Acknowledgments
P.F. and C.V. are fellows from Consejo Nacional de Investigaciones Científicas y Técnicas (Argentina). M.S., L.V., A.M. and C.M.W. are established Investigators of Consejo Nacional de Investigaciones Científicas y Técnicas (Argentina). This work has been partially supported by PICT 26117 (FONCyT) and PIP 5300 (CONICET) and by NIH research grant # R03 TW07713 to A. Mattiazzi and FONDECyT 1030449 to P. Donoso.
References
- 1.Mundiña-Weilenmann C, Vittone L, Ortale M, de Cingolani GC, Mattiazzi A. Immunodetection of phosphorylation sites gives new insights into the mechanisms underlying phospholamban phosphorylation in the intact heart. J Biol Chem. 1996;271:33561–7. doi: 10.1074/jbc.271.52.33561. [DOI] [PubMed] [Google Scholar]
- 2.Said M, Mundiña-Weilenmann C, Vittone L, Mattiazzi A. The relative relevance of phosphorylation of the Thr17 residue of phospholamban is different at different levels of β-adrenergic stimulation. Pflugers Arch. 2002;444:801–9. doi: 10.1007/s00424-002-0885-y. [DOI] [PubMed] [Google Scholar]
- 3.Zhu WZ, Wang SQ, Chakir K, Yang D, Zhang T, Brown JH, et al. Linkage of β1-adrenergic stimulation to apoptotic heart cell death through protein kinase A-independent activation of Ca2+/calmodulin kinase II. J Clin Invest. 2003;111:617–25. doi: 10.1172/JCI16326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang W, Zhu W, Wang S, Yang D, Crow MT, Xiao RP, et al. Sustained β1-adrenergic stimulation modulates cardiac contractility by Ca2+/calmodulin kinase signaling pathway. Circ Res. 2004;95:798–806. doi: 10.1161/01.RES.0000145361.50017.aa. [DOI] [PubMed] [Google Scholar]
- 5.Sucharov CC, Mariner PD, Nunley KR, Long C, Leinwand L, Bristow MR. A β1-adrenergic receptor CaM kinase II-dependent pathway mediates cardiac myocyte fetal gene induction. Am J Physiol. 2006;291:H1299–308. doi: 10.1152/ajpheart.00017.2006. [DOI] [PubMed] [Google Scholar]
- 6.Curran J, Hinton MJ, Ríos E, Bers DM, Shannon TR. β-Adrenergic enhancement of sarcoplasmic reticulum calcium leak in cardiac myocytes is mediated by calcium/calmodulin-dependent protein kinase. Circ Res. 2007;100:391–8. doi: 10.1161/01.RES.0000258172.74570.e6. [DOI] [PubMed] [Google Scholar]
- 7.Reiken S, Gaburjakova M, Guatimosim S, Gomez AM, D’Armiento J, Burkhoff D, et al. Protein kinase A phosphorylation of the cardiac calcium release channel (ryanodine receptor) in normal and failing hearts. Role of phosphatases and response to isoproterenol. J Biol Chem. 2003;278:444–53. doi: 10.1074/jbc.M207028200. [DOI] [PubMed] [Google Scholar]
- 8.Ginsburg KS, Bers DM. Modulation of excitation-contraction coupling by isoproterenol in cardiomyocytes with controlled SR Ca2+ load and Ca2+ current trigger. J Physiol. 2004;556:463–80. doi: 10.1113/jphysiol.2003.055384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wehrens XH, Lehnart SE, Reiken SR, Marks AR. Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ Res. 2004;94:e61–70. doi: 10.1161/01.RES.0000125626.33738.E2. [DOI] [PubMed] [Google Scholar]
- 10.Xiao B, Zhong G, Obayashi M, Yang D, Chen K, Walsh MP, et al. Ser-2030, but not Ser-2808, is the major phosphorylation site in cardiac ryanodine receptors responding to protein kinase A activation upon beta-adrenergic stimulation in normal and failing hearts. Biochem J. 2006;396:7–16. doi: 10.1042/BJ20060116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Witcher DR, Kovacs RJ, Schulman H, Cefali DC, Jones LR. Unique phosphorylation site on the cardiac ryanodine receptor regulates calcium channel activity. J Biol Chem. 1991;266:11144–52. [PubMed] [Google Scholar]
- 12.Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, et al. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–76. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 13.Rodriguez P, Bhogal MS, Colyer J. Stoichiometric phosphorylation of cardiac ryanodine receptor on serine 2809 by calmodulin-dependent kinase II and protein kinase A. J Biol Chem. 2003;278:38593–600. doi: 10.1074/jbc.C301180200. [DOI] [PubMed] [Google Scholar]
- 14.Xiao B, Jiang MT, Zhao M, Yang D, Sutherland C, Lai FA, et al. Characterization of a novel PKA phosphorylation site, serine-2030, reveals no PKA hyperphosphorylation of the cardiac ryanodine receptor in canine heart failure. Circ Res. 2005;96:847–55. doi: 10.1161/01.RES.0000163276.26083.e8. [DOI] [PubMed] [Google Scholar]
- 15.Vittone L, Mundiña C, Chiappe de Cingolani G, Mattiazzi A. cAMP and calcium-dependent mechanisms of phospholamban phosphorylation in intact hearts. Am J Physiol. 1990;258:H318–25. doi: 10.1152/ajpheart.1990.258.2.H318. [DOI] [PubMed] [Google Scholar]
- 16.Sánchez G, Hidalgo C, Donoso P. Kinetic studies of calcium-induced calcium release in cardiac sarcoplasmic reticulum vesicles. Biophys J. 2003;84:2319–30. doi: 10.1016/S0006-3495(03)75037-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–5. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 18.Lokuta AJ, Rogers TB, Lederer WJ, Valdivia HH. Modulation of cardiac ryanodine receptors of swine and rabbit by a phosphorylation–dephosphorylation mechanism. J Physiol. 1995;487:609–22. doi: 10.1113/jphysiol.1995.sp020904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fabiato A, Fabiato F. Calculator programs for computing the composition of the solutions containing multiple metals and ligands used for experiments in skinned muscle cells. J Physiol. 1979;75:463–505. [PubMed] [Google Scholar]
- 20.Stange M, Xu L, Balshaw D, Yamaguchi N, Meissner G. Characterization of recombinant skeletal muscle (Ser-2843) and cardiac muscle (Ser-2809) ryanodine receptor phosphorylation mutants. J Biol Chem. 2003;278:51693–702. doi: 10.1074/jbc.M310406200. [DOI] [PubMed] [Google Scholar]
- 21.Carter S, Colyer J, Sitsapesan R. Maximum phosphorylation of the cardiac ryanodine receptor at serine-2809 by protein kinase A produces unique modifications to channel gating and conductance not observed at lower levels of phosphorylation. Circ Res. 2006;98:1506–13. doi: 10.1161/01.RES.0000227506.43292.df. [DOI] [PubMed] [Google Scholar]
- 22.Obayashi M, Xiao B, Stuyvers BD, Davidoff AW, Mei J, Chen SR, et al. Spontaneous diastolic contractions and phosphorylation of the cardiac ryanodine receptor at serine-2808 in congestive heart failure in rat. Cardiovasc Res. 2006;69:140–51. doi: 10.1016/j.cardiores.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 23.Bartel S, Vetter D, Schlegel WP, Wallukat G, Krause EG, Karczewski P. Phosphorylation of phospholamban at threonine-17 in the absence and presence of beta-adrenergic stimulation in neonatal rat cardiomyocytes. J Mol Cell Cardiol. 2000;32:2173–85. doi: 10.1006/jmcc.2000.1243. [DOI] [PubMed] [Google Scholar]
- 24.Miller C. Ion channels in liposomes. Annu Rev Physiol. 1984;46:549–58. doi: 10.1146/annurev.ph.46.030184.003001. [DOI] [PubMed] [Google Scholar]
- 25.Meszaros LG, Wrenn RW, Varadi G. Sarcoplasmic reticulum-associated and protein kinase C-regulated ADP-ribosyl cyclase in cardiac muscle. Biochem Biophys Res Commun. 1997;234:252–6. doi: 10.1006/bbrc.1997.6620. [DOI] [PubMed] [Google Scholar]
- 26.Hain J, Onoue H, Mayrleitner M, Fleischer S, Schindler H. Phosphorylation modulates the function of the calcium release channel of sarcoplasmic reticulum from cardiac muscle. J Biol Chem. 1995;270:2074–81. doi: 10.1074/jbc.270.5.2074. [DOI] [PubMed] [Google Scholar]
- 27.Yang D, Zhu WZ, Xiao B, Brochet DXP, Chen SR, Lakatta EG, et al. Ca2+/calmodulin kinase II-dependent phosphorylation of ryanodine receptors suppresses Ca2+ sparks and Ca2+ waves in cardiac myocytes. Circ Res. 2007;100:399–407. doi: 10.1161/01.RES.0000258022.13090.55. [DOI] [PubMed] [Google Scholar]
- 28.Li L, Satoh H, Ginsburg KS, Bers DM. The effect of Ca2+-calmodulin-dependent protein kinase II on cardiac excitation-contraction coupling in ferret ventricular myocytes. J Physiol. 1997;501:17–31. doi: 10.1111/j.1469-7793.1997.017bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maier LS, Zhang T, Chen L, DeSantiago J, Brown JH, Bers DM. Transgenic CaMKIIdeltaC overexpression uniquely alters cardiac myocyte Ca2+ handling: reduced SR Ca2+ load and activated SR Ca2+ release. Circ Res. 2003;92:904–11. doi: 10.1161/01.RES.0000069685.20258.F1. [DOI] [PubMed] [Google Scholar]
- 30.Guo T, Zhang T, Mestril R, Bers DM. Ca2+/calmodulin-dependent protein kinase II phosphorylation of ryanodine receptor does affect calcium sparks in mouse ventricular myocytes. Circ Res. 2006;99:398–406. doi: 10.1161/01.RES.0000236756.06252.13. [DOI] [PubMed] [Google Scholar]
- 31.Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res. 2005;97:1314–22. doi: 10.1161/01.RES.0000194329.41863.89. [DOI] [PubMed] [Google Scholar]
- 32.Kohlhaas M, Zhang T, Seidler T, Zibrova D, Dybkova N, Steen A, et al. Increased sarcoplasmic reticulum calcium leak but unaltered contractility by acute CaMKII overexpression in isolated rabbit cardiac myocytes. Circ Res. 2006;98:235–44. doi: 10.1161/01.RES.0000200739.90811.9f. [DOI] [PubMed] [Google Scholar]
- 33.Lindegger N, Niggli E. Paradoxical SRCa2+ release in guinea-pig cardiac myocytes after beta-adrenergic stimulation revealed by two-photon photolysis of caged Ca2+ J Physiol. 2005;565:801–13. doi: 10.1113/jphysiol.2005.084376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Han S, Schiefer A, Isenberg G. Ca2+ load of guinea-pig ventricular myocytes determines efficacy of brief Ca2+ currents as trigger for Ca2+ release. J Physiol. 1994;480:411–21. doi: 10.1113/jphysiol.1994.sp020371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li Y, Kranias EG, Mignery GA, Bers DM. Protein kinase A phosphorylation of the ryanodine receptor does not affect calcium sparks in mouse ventricular myocytes. Circ Res. 2002;90:309–16. doi: 10.1161/hh0302.105660. [DOI] [PubMed] [Google Scholar]
- 36.Bracken N, Elkadri M, Hart G, Hussain M. The role of constitutive PKA-mediated phosphorylation in the regulation of basal ICa in isolated rat cardiac myocytes. Br J Pharmacol. 2006;148:1108–15. doi: 10.1038/sj.bjp.0706809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chijiwa T, Mishima A, Hagiwara M, Sano M, Hayashi K, Inoue T, et al. Inhibition of forskolin-induced neurite outgrowth and protein phosphorylation by a newly synthesized selective inhibitor of cyclic AMP-dependent protein kinase, N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide (H-89), of PC12D pheochromocytoma cells. J Biol Chem. 1990;265:5267–72. [PubMed] [Google Scholar]
- 38.Valdivia HH, Kaplan JH, Ellis-Davies GC, Lederer WJ. Rapid adaptation of cardiac ryanodine receptors: modulation by Mg2+ and phosphorylation. Science. 1995;267:1997–2000. doi: 10.1126/science.7701323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trafford AW, Diaz ME, Eisner DA. Stimulation of Ca-induced Ca release only transiently increases the systolic Ca transient: measurements of Ca fluxes and sarcoplasmic reticulum Ca. Cardiovasc Res. 1998;37:710–7. doi: 10.1016/s0008-6363(97)00266-6. [DOI] [PubMed] [Google Scholar]
- 40.Choi HS, Trafford AW, Orchard CH, Eisner DA. The effect of acidosis on systolic Ca2+ and sarcoplasmic reticulum calcium content in isolated rat ventricular myocytes. J Physiol. 2000;529:661–8. doi: 10.1111/j.1469-7793.2000.00661.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eisner DA, Trafford AW. No role for the ryanodine receptor in regulating cardiac contraction? News Physiol Sci. 2000;15:275–9. doi: 10.1152/physiologyonline.2000.15.5.275. [DOI] [PubMed] [Google Scholar]