

Abstract
Ocular infection with herpes simplex virus (HSV) sets off an array of events that succeed in clearing virus from the cornea but leaves the tissue with a CD4+ T-cell-orchestrated chronic inflammatory lesion that impairs vision. We demonstrate that Toll-like receptor (TLR) signaling forms a part of the recognition system that induces the syndrome that eventually culminates in immunopathology. Accordingly, in a comparison of the outcomes of infection in wild-type (WT) mice and those lacking TLR function, it was apparent that the absence of TLR2 and, to a lesser extent, TLR9 resulted in significantly diminished lesions. Similarly, mice lacking the adapter molecule MyD88 were resistant to lesion development, but such animals were also unable to control infection, with most succumbing to lethal encephalitis. The susceptibility of TLR4−/− animals was also evaluated. These animals developed lesions, which were more severe, more rapidly than did WT animals. We discuss the possible mechanisms by which early recognition of HSV constituents impacts the subsequent development of immunopathological lesions.
Viruses induce several immediate host-reactive events that impact the long-term consequences of infection. For example, ocular infection of mice with herpes simplex virus (HSV) causes a multitude of cytokine, chemokine, and cellular changes to occur (9). Manipulating the expression of many of these affects the severity of the subsequent vision impairing consequences, such as neovascularization and chronic inflammation (8, 14, 20, 22). The latter reaction is immunopathological and is set off initially by infection but sustained well after replication has ceased and viral antigens are removed (9). These stromal keratitis (SK) reactions occur in response to HSV in humans and represent the commonest cause of infectious blindness in developed countries (30).
We know that viruses and their components can trigger multiple innate immune protective and inflammatory responses (35). These include interferon induction, the production of multiple cytokines, and activation of complement as well as innate cellular defenses (31, 35). Recognition mechanisms that initiate and control innate reactions to viruses remain poorly understood, but accumulating evidence indicates that membrane-bound cell surface or intracellular Toll-like receptors (TLRs) form part of the surveillance system (10). After the initial observation with respiratory syncytial virus (25), several viruses were shown to express ligand activities for one or more TLRs (10). Infection could also result in the release and induction of host molecules, such as heat shock proteins, that may themselves activate TLRs and so contribute to the innate response (5, 12). The observation that TLRs participate during HSV infection came from several lines of investigation. Initially, purified HSV DNA, such as synthetic CpG DNA, the ligand for TLR9 (23), was shown to induce ocular neovascularization, an essential step in the pathogenesis of SK (42). HSV DNA was later confirmed to possess TLR9 ligand activity (28), and studies with knockout mice revealed a role for TLR9 in HSV-induced type I interferon production (23). Other investigations demonstrated that HSV also possesses TLR2 activity (24), with mice lacking TLR2 showing more resistance to lethal HSV encephalitis than wild-type (WT) animals (24). This perhaps unexpected observation that brain inflammation and death were reduced in the absence of TLR2 (but virus levels were similar in WT and TLR2−/− mice) indicated that encephalitis, the usual cause of death with HSV infection, was the consequence of the virus acting as a TLR2 ligand, driving an inflammatory reaction in the brain (24). In addition to inflammatory processes, herpetic encephalitis, especially that in humans, may represent a direct destructive effect of the virus (19), and even in mice, neurovirulence appears to be associated with the success of virus gaining access to and replicating in the central nervous system (CNS) (33). Thus, both viral and host components may contribute to disease pathogenesis.
For the present report, we measured the influence of TLR2 and other potential TLR ligand effects of HSV in a corneal scarification disease model where lesion expression is widely agreed to represent a host immunopathological response (9). In this model, animals lacking lymphocytes fail to develop SK lesions and, in fact, die of herpetic encephalitis, with an abundance of virus in the brain (32). We examined the sequences of events that occurred following infection of different TLR knockout mice compared to those after infection of WT animals. Our results demonstrate that TLR2−/−, MyD88−/−, and to a lesser extent TLR9−/−, but not TLR4−/−, mice showed significantly reduced SK lesions upon HSV infection. Similarly, TLR2−/− and MyD88−/− mice also showed reduced neovascularization compared to other groups. The latter strains also showed marked reductions in early nonlymphoid inflammatory infiltrates as well as lower levels of proinflammatory cytokines than those in WT or TLR4−/− animals. In addition, whereas both TLR2−/− and TLR9−/− animals resisted the lethal effects of HSV, most MyD88−/− animals infected with the same dose succumbed to lethal disease. The TLR4−/− animals developed lesions, which were more severe, more rapidly than did WT animals. We discuss the possible mechanisms by which early recognition of HSV constituents impacts the subsequent development of immunopathological lesions.
MATERIALS AND METHODS
Animals.
TLR2−/−, TLR4−/−, TLR9−/−, TLR2/4d−/−, and MyD88−/− mice were the kind gift of S. Akira (Osaka University, Osaka) and were backbred for more than 10 generations onto the C57BL/6 background. Backbreeding was confirmed by examining a 110-marker panel that distinguishes between C57BL/6 and 129 mice (Charles River Laboratories). These microsatellite markers are spread across the 19 autosomes and the X chromosome at approximately 15-centimorgan intervals. C57BL/6 mice were obtained from Harlan Sprague-Dawley (Indianapolis, IN). All experimental procedures followed the guidelines of the Association for Research in Vision and Ophthalmology resolution on the use of animals in research.
Corneal HSV-1 infection and clinical observation.
HSV type 1 (HSV-1) strain RE was used in all procedures. Details of corneal infections of mice under deep anesthesia were described in a previous publication (3). Animals were infected with a range of virus doses, although in most of the reported experiments mice were infected with 5 × 103 PFU/eye. The experiments were repeated multiple times, especially the comparison of TLR2−/− animals with control animals. Lesion scores and the extent of angiogenesis were measured at various times postinfection (p.i.), as described in detail previously (4, 13).
Histopathology.
For histopathologic analysis, eyes from different groups of mice were extirpated at the indicated time points p.i. and snap-frozen in OCT compound (Miles, Elkart, IN). Six-micrometer-thick sections were cut and air dried in a desiccation box. Staining was performed with hematoxylin and eosin (Richard Allen Scientific, Kalamazoo, MI).
Flow cytometry.
For flow cytometry measurement of the infiltrating cells, four corneas or two trigeminal ganglia (TG) were collected per time point. Corneas or TG were digested in liberase (Roche Diagnostics Co., Indianapolis, IN) for 45 min at 37°C. Single-cell suspensions were prepared as described elsewhere (15). The Fc receptors were blocked with unconjugated anti-CD16/32 (BD Pharmingen, San Diego, CA) for 30 min. Samples were incubated with fluorescein isothiocyanate (FITC)-labeled anti-Gr-1, anti-CD11b, anti-CD11c (BD Pharmingen), anti-F4/80 (eBioscience), or anti-CD4 (BD Pharmingen) antibody for 30 min. For intracellular staining for cyclooxygenase-2 (COX-2), goat anti-COX-2 and donkey anti-goat immunoglobulin G (IgG) (Santa Cruz Biotechnology, Santa Cruz, CA) were used as primary and secondary antibodies, respectively. For detection of induced Hsp70, FITC-conjugated Hsp70 (Hsp72) antibody was obtained from Stressgen (Ann Arbor, MI). FITC-conjugated rat IgG was used as an isotype control for Hsp70. Intracellular staining for COX-2 and Hsp70 was performed using a fixation/permeabilization solution kit with BD GolgiPlug (BD Bioscience). All samples were collected on a FACScan flow cytometer (BD Biosciences, San Diego, CA), and data were analyzed by using Cell Quest 3.1 software (BD Biosciences).
Cytokine and chemokine ELISAs with corneal lysates.
For preparation of corneal lysates, six corneas per indicated time point were pooled from each group of mice and were processed as described previously (3). For draining lymph nodes (DLNs), cervical lymph nodes from each mouse were collected and processed. Levels of interleukin-6 (IL-6), IL-1β, macrophage inflammatory protein 2 (MIP-2), vascular endothelial growth factor (VEGF), and IL-10 in corneal lysates and of IL-10, IL-6, and gamma interferon (IFN-γ) in DLN lysates were determined by a standard sandwich enzyme-linked immunosorbent assay (ELISA) protocol. Anti-IL-6, anti-IL-10, and anti-IFN-γ capture and detection antibodies were purchased from BD Pharmingen. Anti-IL-1β, anti-MIP-2, and anti-VEGF capture and biotinylated detection antibodies were purchased from R&D Systems (Minneapolis, MN). The color reaction was developed using ABTS [2,2′-azinobis(3-ethylbenzthiazolinesulfonic acid); Sigma-Aldrich, St. Louis, MO] and measured with an ELISA reader (Spectramax 340; Molecular Devices, Sunnyvale, CA) at 405 nm. Quantification was performed with Spectramax ELISA reader software, version 1.2.
Reverse transcription and real-time PCR.
Total RNAs from naïve and infected C57BL/6 corneas and the DC2.4 cell line were isolated at different days p.i. by using an RNeasy MinElute cleanup kit (QIAGEN) and were reverse transcribed by using murine leukemia virus reverse transcriptase (Life Technologies, Bethesda, MD), with oligo(dT) as the primer (Invitrogen, San Diego, CA). All cDNAs were divided into aliquots and stored at −20°C until further use. All murine dendritic cells (DCs) express TLR2, -4, and -9, as does the DC2.4 cell line (a DC line; obtained from K. L. Rock). PCR was performed in a PTC-100 programmable thermal controller (MJ Research, Cambridge, MA), using Hot Start PCR master mix (Promega, Madison, WI). The primers used were murine GAPDH forward (CATCCTGCACCACCAACTGCTTAG) and reverse (GCCTGCTTCACCACCTTCTTGATG), murine TLR2 forward (AAGAGGAAGCCCAAGAAAGC) and reverse (CGATGGAATCGATGATGTTG), murine TLR4 forward (ACCTGGCTGGTTTACACGTC) and reverse (CTGCCAGAGACATTGCAGAA), and murine TLR9 forward (ACTGAGCACCCCTGCTTCTA) and reverse (AGATTAGTCAGCGGCAGGAA).
Real-time PCR was performed using an iQ5 real-time PCR detection system (Bio-Rad). PCR was performed using SYBR green PCR master mix (Applied Biosystems) according to the manufacturer's protocol. PCR amplification of a housekeeping gene, the murine hypoxanthine phosphoribosyltransferase gene, was done for each sample as a control for sample loading and to allow normalization between samples. The primers used were murine HPRT forward (GACCGGTCCCGTCATGC) and reverse (TCATAACCTGGTTCATCATCGC), murine β-defensin 2 forward (TATGCTGCCTCCTTTTCTCA) and reverse (GACTTCCATGTGCTTCCTTC), and murine β-defensin 3 forward (CTCCTGGTGCTGCTGTCTCCAC) and reverse (AACTCCACAACTGCCAATCTGA).
Virus recovery and titrations.
Eye swabs were taken from infected corneas (four eyes/group), using sterile swabs, at the indicated time points. Similarly, brain tissue and TG were taken from the infected animals at the indicated time points (n = 3). The tissues were disrupted by homogenization in 1-ml ground glass grinders (Wheaton) and centrifuged, and the supernatant was used to assay viral titers. Titration was performed by a standard plaque assay as described previously (3). Titers were calculated as log10 PFU/ml per a standard protocol (37).
Statistical analysis.
Statistical significance was determined by Student's t test. P values of <0.05 were regarded as representing significant differences between the groups.
RESULTS
Expression of TLRs and TLR ligands in naïve and infected corneas.
Since HSV replication in the eye occurs almost exclusively in corneal epithelial cells (9), corneal buttons taken from uninfected eyes were examined by reverse transcription-PCR for the presence of TLR2, -4, and -9 mRNAs. All could be demonstrated (Fig. 1A). Whereas previous reports had shown that both TLR2- and TLR9-activating ligands are expressed by HSV (16), endogenous TLR ligands might also be induced and released from dead or dying cells following infection (5, 12). As shown in Fig. 1B, the TLR4 ligand, inducible Hsp70 (5), could be demonstrated in corneal tissues in samples analyzed at 24 h post-HSV infection. Similarly, as shown in Fig. 1C, both β-defensin 2 and β-defensin 3 mRNA expression was increased in the infected compared to naïve corneas, and β-defensin 3 has been shown to trigger TLR4 (6). Hence, TLR4 as well as previously demonstrated TLR2 and TLR9 ligand effects could be involved in the recognition events following ocular HSV infection.
FIG. 1.

Induced TLR ligands in HSV-infected corneas. (A) Corneal buttons from uninfected mice were harvested, and isolated total mRNAs were tested by reverse transcription-PCR for TLR2, TLR4, and TLR9. All three could be demonstrated in the naïve cornea. The DC2.4 cell line was used as a positive control. (B) Single-cell suspensions of corneal cells were prepared from four corneas at 24 h p.i. Intracellular staining was done with FITC-labeled anti-Hsp70 antibody, as mentioned in Materials and Methods. The number of Hsp70-expressing cells was determined by fluorescence-activated cell sorting. Rat IgG was used as the isotype control. The number in each upper right corner represents the percentage of Hsp70 cells/1 × 105 corneal cells at 24 h p.i. (C) C57BL/6 mice were infected with 2 × 104 PFU of HSV-1 RE. At the indicated time points, four corneas from infected mice were collected and total mRNAs were extracted. Real-time PCR was performed to compare the expression of mouse β-defensin 2 (mBD2) and mouse β-defensin 3 (mBD-3) mRNAs in the infected corneas at different times p.i. as described in Materials and Methods. The bar diagram demonstrates the relative increases (n-fold) in the expression of mouse β-defensin 2 and mouse β-defensin 3 compared to those in naïve corneas.
Disease expression following HSV infection in control and knockout mice.
To determine the role of TLRs in HSV pathogenesis in the eye, TLR knockout mice that were fully backbred onto the C57BL/6 background were ocularly infected with different doses of HSV-1 RE and evaluated over a 15- to 20-day period to quantify the extent of corneal neovascularization as well as the severity of SK. At infection doses of 2 × 105 PFU/eye and above, all groups developed lesions and neovascularization responses that were similar in magnitude. However, significantly different responses in the various groups became evident at infection doses of 2 × 104 PFU/eye and below. Figure 2 shows the results of one of two experiments in which the animals were infected with 5 × 103 PFU virus. As evident at both days 8 and 15 p.i., both the TLR2−/− and MyD88−/− animals showed significantly diminished lesions as well as neovascularization compared to WT animals. For instance, whereas almost 90% of control WT animals developed SK lesions with severity scores of 3 or greater and neovascularization scores of 8 or greater, for TLR2−/− and MyD88−/− animals only around 30% of animals had such severity scores. Moreover, at the peak response time (day 15 p.i.), the mean severity scores for TLR2−/− and MyD88−/− mice were 1.5 ± 0.37 and 1.8 ± 0.4, respectively, which were significantly (P ≤ 0.005) lower than that observed for WT mice (3.8 ± 0.26). On average, the mean severity of SK in TLR2−/− mice was reduced 60% compared to that of controls at day 8 p.i. and was further reduced 65% by day 15 p.i. These experiments indicate that animals unable to respond to the TLR2 ligand activity of HSV showed a resistance phenotype. However, curiously, both TLR2−/− and MyD88−/− animals often showed quite severe herpetic periocular disease, a lesion seldom seen in the WT animals at the low dose of virus used.
FIG. 2.
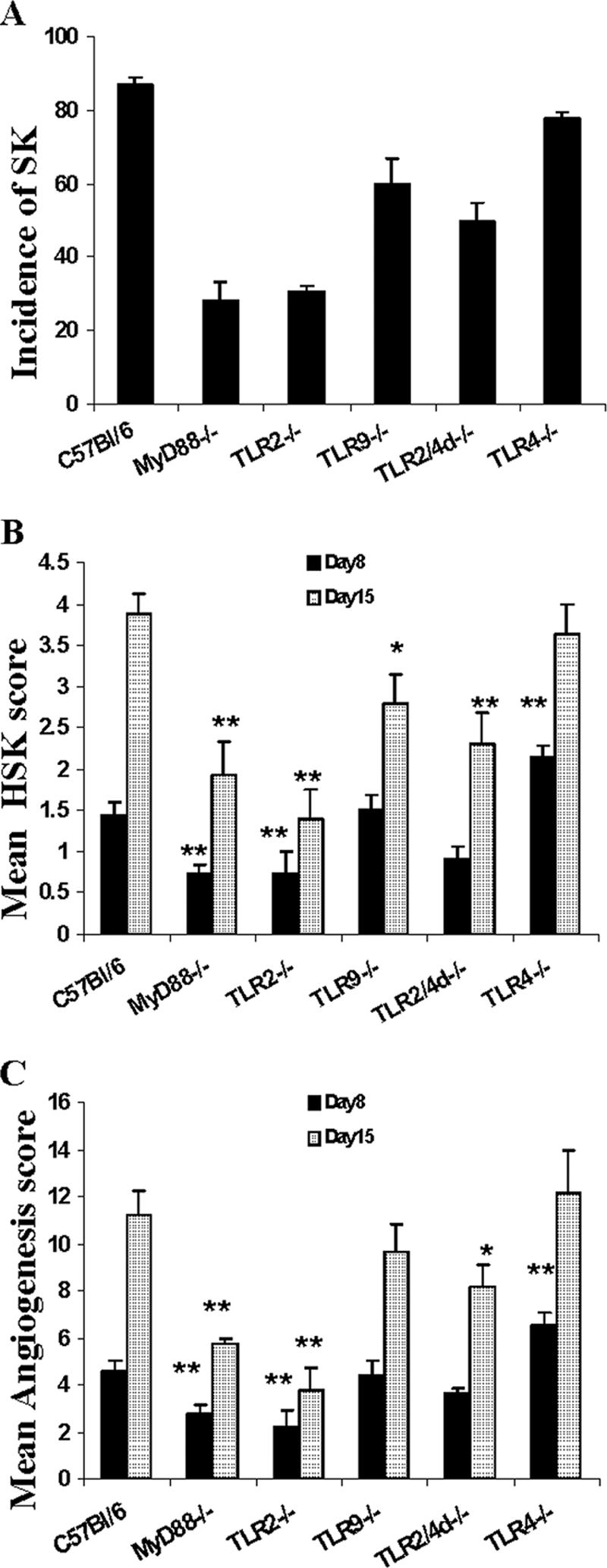
Reduced SK lesions and neovascularization in TLR2−/−, TLR2/4d−/−, and MyD88−/− mice compared to those in TLR9−/−, TLR4−/−, and WT mice. (A) Bar diagram demonstrating the percentage severity of SK in each group of mice (n = 16) infected with 5 × 103 PFU HSV-1 RE at day 15 p.i. TLR2−/−, TLR2/4d−/−, and MyD88−/− mice showed a reduced incidence (herpetic stromal keratitis score, ≥3) of disease compared to TLR9−/−, TLR4−/−, and WT animals. (B) Mean herpetic stromal keratitis lesion scores at day 8 and day 15 p.i. of WT, TLR4−/−, TLR9−/−, TLR2/4d−/−, TLR2−/−, and MyD88−/− mice infected with a 5 × 103-PFU infection dose. **, P ≤ 0.001; *, P ≤ 0.05 (statistically significant differences compared to WT controls). (C) Mean angiogenesis lesion scores at day 8 and day 15 p.i. of WT, TLR4−/−, TLR9−/−, TLR2/4d−/−, TLR2−/−, and MyD88−/− mice infected with a 5 × 103-PFU infection dose. **, P ≤ 0.001; *, P ≤ 0.05 (statistically significant differences compared to WT control animals).
Animals lacking TLR9 also showed more resistance to HSV-induced SK and neovascularization than did WT animals, although the differences were modest and were significant only for the 15-day scores (2.7 ± 0.38 compared to 3.8 ± 0.26; P = 0.03) (Fig. 2). Most of the TLR9−/− animals also developed herpetic periocular disease, but the lesions were less intense than those noted in TLR2−/− and MyD88−/− animals.
The pattern of response for TLR4−/− animals revealed a modestly more susceptible phenotype. Thus, by day 8 p.i., 30% of TLR4−/− animals showed level 3 lesions (mean, 2.3 ± 0.13), whereas almost all WT animals lacked lesions at this time (mean, 1.2 ± 0.15; P = 0.004). The neovascularization score reflected a similar heightened susceptibility of TLR4−/− animals (mean, 7 ± 0.5) compared to WT animals (mean, 4.5 ± 0.4; P = 0.009) (Fig. 2). Curiously, animals lacking both TLR2 and -4 responsiveness showed a resistance phenotype, although this was lower in magnitude than that of TLR2 or MyD88 knockout animals.
MyD88 is an essential downstream adapter of TLR signaling, particularly for TLR2 and TLR9 responses (36). TLR4 also signals via MyD88 but can activate an alternate pathway of MyD88-independent gene expression (18). Interestingly, although the MyD88−/− animals showed a resistance phenotype for disease expression similar to that of the TLR2−/− mice, the MyD88−/− animals were far more susceptible to the lethal effects of HSV infection. As shown in Table 1, even at the 5 × 103-PFU dose, up to 70% of the infected MyD88−/− animals were dying by day 15 p.i., although almost 60% of them showed no signs of clinical disease. The animals were presumed to be dying from viral encephalitis, and replicating virus could be demonstrated in the homogenates of brain tissues harvested from moribund MyD88−/− animals (Table 2). Thus, eliminating signaling through multiple TLRs by knocking out MyD88 reduces local inflammation but may allow enhanced viral replication and/or spread, while eliminating single TLRs, i.e., TLR2 or TLR9, ameliorates local immunopathology, but these receptors may be redundant for control of virus replication/spread.
TABLE 1.
Mortality in various groups by day 15 p.i.
| Mouse group | % Mortality
|
|
|---|---|---|
| 2 × 104 PFU/eye | 5 × 103 PFU/eye | |
| WT | 30 | <20 |
| TLR4−/− | 45 | <20 |
| TLR9−/− | <15 | |
| TLR2−/− | <15 | |
| MyD88−/− | 100 | 70 |
TABLE 2.
Virus titers in TG and brains at different time points p.i.a
| Mouse group | Virus titer (log10 PFU/ml)
|
|||
|---|---|---|---|---|
| Day 4 p.i.
|
Day 7 p.i.b
|
|||
| TG | Brain | TG | Brain | |
| WT | 3.2 ± 0.04 | 1.7 ± 0.29 | UD | UD |
| TLR9−/− | 3.7 ± 0.08 | 2.3 ± 0.2 | UD | UD |
| TLR2−/− | 3.2 ± 0.03 | 2.3 ± 0.07 | UD | UD |
| MyD88−/− | 3.7 ± 0.06 | 2.2 ± 0.20 | UD | 3.8 ± 0.30c |
Mice were infected with 3 × 105 PFU of HSV-1 RE and killed on day 4 or day 7 to measure viral titers, using a standard plaque assay, in brain and TG homogenates (n = 3).
UD, undetected, i.e., below the sensitivity of the assay (<10 PFU/ml).
Virus was also detected in brain tissues collected from dying MyD88−/− mice (days 10 to 15).
As shown in Table 1, for infections at both 2 × 104 PFU/eye and 5 × 103 PFU/eye, neither TLR2−/− nor TLR9−/− animals were more susceptible to lethal infection than were WT animals. In addition to titers in brains and TG, virus titers were also measured in eye swabs collected from infected corneas. As shown in Table 3, in contrast to the case for the CNS, virus titers did not increase in the absence of TLRs or MyD88 in the corneas, and all of the groups could clear the virus efficiently by day 7 p.i. Our data confirm the previous finding by others that MyD88-independent antiviral mechanisms can effectively compensate for the absence of MyD88 in a localized HSV-1 infection (23).
TABLE 3.
Virus titers in corneasa
| Mouse group | Virus titer (log10 PFU/ml) at day 3 p.i. |
|---|---|
| WT | 3.2 ± 0.18 |
| TLR4−/− | 3.7 ± 0.21 |
| TLR9−/− | 3.3 ± 0.17 |
| TLR2−/− | 3.1 ± 0.08 |
| MyD88−/− | 3.2 ± 0.10 |
Mice were infected with 3 × 105 PFU of HSV-1 RE. Virus titers were estimated by standard plaque assay from swabs taken from infected corneas of mice (n = 4) at different time points. Virus was not detected, i.e., was below the sensitivity of the assay (<10 PFU/ml), for all corneas taken at day 7 p.i.
Cellular differences between control and knockout animals.
Soon after HSV infection of the corneal epithelium, cytokine expression and cellular changes occur (9). One prominent event is infiltration in the underlying stroma by mainly neutrophils (38). The stimuli that induce such responses remain to be identified, but they appear to include chemokines such as MIP-2 and macrophage chemoattractant protein 1, induced directly or indirectly by HSV infection in the corneal epithelium (9, 21). Additionally, angiogenic molecules such as VEGF and IL-6 are induced, both of which play a role at causing neovascularization of the cornea (3, 41). To compare the initial inflammatory responses in knockout versus WT control mice, tissue sections were examined. As evident in Fig. 3, diminished cell infiltrates at 48 h compared to those for WT controls were apparent in TLR2−/−, MyD88−/−, and TLR9−/− sections. In the WT sections, the inflammatory infiltrates had fully accessed the central cornea, but in both TLR2−/− and TLR9−/− animals, the infiltrates were largely confined to the limbal region. Of these two TLR knockouts, the infiltration of cells in TLR2−/− animals was far less than that in TLR9−/− animals. Of interest, the inflammatory events in TLR4−/− animals appeared to be slightly elevated compared to those in WT animals. This more severe reaction was also more evident by flow cytometric analysis of Gr1+ and CD11b+ cells in the infected corneas (Fig. 3).
FIG. 3.

Compromised inflammatory cell influx into the corneas of TLR2−/−, TLR9−/−, TLR2/4d−/−, and MyD88−/− mice compared to TLR4−/− and WT mice at 48 h p.i. Mice were infected with 5 × 103 PFU HSV-1 RE. Mice were terminated at 48 h p.i., and eyes were processed for cryo-sections. Hematoxylin and eosin staining was carried out on 6-μm sections. Dot plots associated with the sections represent the Gr1+ CD11b+ cells in the WT and TLR4−/− corneas (Gr1+ on x axis and CD11b+ on y axis). The numbers in the dot plots denote the percentages of inflammatory cells expressing both the Gr1+ and CD11b+ markers.
It has been documented by past studies that after ocular infection with HSV, replicating virus could be detected in the TG for 2 to 7 days p.i. (27). The presence of virus in the TG induces a chronic inflammatory reaction involving multiple cell types that is responsible for controlling the virus load as well as restricting further spread of the virus (27). Therefore, we examined the cells from the TG of different groups at 48 h and 96 h p.i. by flow cytometry. Diminished inflammatory lesions were observed in the infected TG of the TLR2−/− and MyD88−/− animals than in those of WT animals. As shown in Fig. 4, infiltration of Gr1+, CD11b+, CD11c+, and F4/80+ cells was reduced in the TLR2−/− and MyD88−/− animals. Interestingly, similar to the case for the cornea, an increased inflammatory cell number was noticed for the TLR4−/− animals compared to that for the WT controls.
FIG. 4.

Compromised cell infiltration into the TG of TLR2−/− and MyD88−/− mice compared to that into the TG of TLR4−/− and WT mice. Single-cell suspensions of TG cells were prepared from four TG at 48 h and 96 h p.i. for each group of mice. The cells were stained with FITC-labeled anti-Gr-1, -CD11c, and -F4/80 and with phycoerythrin-labeled CD11b antibody. The number of positive cells was determined by fluorescence-activated cell sorting. The bars represent the numbers of cells/4 × 105 cells from the TG at 48 h and 96 h p.i. *, P ≤ 0.05; **, P ≤ 0.01 (statistically significant difference compared to WT animals).
Cytokine and other proinflammatory protein differences between control and knockout animals.
A likely explanation for the reduced influx of inflammatory cells and eventual resistance to disease expression in TLR2−/− (and MyD88−/−) mice could be a reduced proinflammatory cytokine/chemokine response to infection. Lysates of corneas collected at 48 h and 96 h p.i. were used to measure the levels of candidate molecules shown in previous studies to correlate with the outcome of ocular HSV infection (9). These were IL-6, MIP-2, IL-1β, and VEGF as well as the enzyme COX-2. As shown in Fig. 5, compared to the WT, three- to fourfold reductions in the levels of these proinflammatory mediators were evident in the lysates of TLR2−/− and MyD88−/− corneas at 48 h and 96 h p.i. IL-6 and IL-1β, produced from virus-infected cells, were shown to stimulate noninfected resident corneal cells and other inflammatory cells in a paracrine manner to secrete VEGF, a potent angiogenic factor (9). Interestingly, marked reductions in the levels of IL-1β, IL-6, and VEGF were noticed in the TLR2−/− and MyD88−/− mice. The TLR9−/− animals had a moderate decrease in inflammatory mediators, but this was evident only at the 48-h time point. In contrast, TLR4−/− animals had slightly increased levels (elevated 20 to 30%) of inflammatory mediators compared to the WT controls at all time points examined.
FIG. 5.

Reduced levels of cytokines and angiogenic factors in the corneas of TLR2−/− and MyD88−/− mice compared to those in the corneas of TLR4−/− and WT mice. At the indicated time points, six corneas/group were processed to measure the IL-6, IL-1β, MIP-2, and VEGF levels. Levels of IL-6, IL-1β, MIP-2, and VEGF were estimated from the supernatants of corneal lysates of WT, TLR2−/−, TLR2/4d−/−, TLR4−/−, TLR9−/−, and MyD88−/− animals infected with 5 × 103 PFU HSV-1 RE by an antibody capture ELISA as outlined in Materials and Methods. *, P ≤ 0.05; ** P ≤ 0.01 (statistically significant difference compared to WT animals).
In two separate experiments, the level of IL-10 induction in TLR4−/− mice was compared to that in WT animals for both corneal extracts and the cervical DLNs on day 2 and day 5 after infection. As evident in Fig. 6A, levels of IL-10 in the corneas of TLR4−/− mice were lower than those observed for WT animals at both time points, perhaps accounting for the more severe reactions seen in TLR4−/− animals. Although significant differences in the levels of IL-10 in DLNs were not observed at these time points (Fig. 6B), the levels of the proinflammatory Th1 cytokines IL-6 and IFN-γ were significantly higher (P ≤ 0.05) in TLR4−/− animals than in WT animals (Fig. 6C and D). Interestingly, no difference was observed in the IL-10 levels in the DLNs of TLR2−/− animals, although the levels of IL-6 and IFN-γ were lower in TLR2−/− animals than in WT animals (data not shown).
FIG. 6.

Increased expression of proinflammatory cytokines and reduced levels of IL-10 in TLR4−/− animals compared to those in WT animals. At the indicated time points, six corneas/group and cervical DLNs from individual mice (n = 4) were processed to measure the IL-10, IL-6, and IFN-γ levels of WT and TLR4−/− animals infected with 5 × 103 PFU HSV-1 RE. Cytokines were measured by antibody capture ELISA as outlined in Materials and Methods. *, P ≤ 0.05 (statistically significant differences compared to WT animals).
Previous studies showed that HSV infection of the cornea upregulates the expression of COX-2, a critical enzyme for the induction of potential inflammatory mediators such as prostaglandins (2, 11). Moreover, the administration of a selective COX-2 inhibitor diminished virus-induced SK lesions and neovascularization (7). Recent in vitro findings demonstrated that TLR2 signaling could induce COX-2 in peritoneal macrophages in vitro (17, 34, 40). Therefore, we examined the role of TLR2 in HSV-induced COX-2. Intracellular staining revealed a >50% reduction in the percentage of COX-2-expressing cells in HSV-infected TLR2−/− mouse corneas compared to those for the WT and TLR4−/− mice (Fig. 7A). In addition, reduced numbers of COX-2+ Gr1+ cells were evident in the TLR2−/− corneas compared to those in WT corneas (Fig. 7B). Interestingly, although there was no difference in the percentage of COX-2-expressing cells in TLR4−/− animals compared to that in WT animals, such animals showed a higher proportion of COX-2+ Gr1+ cells than did WT animals (Fig. 7A and B). These results may mean that less induction of prostaglandins as well as chemokines could be a possible explanation for the resistant and susceptible phenotypes of TLR2−/− and TLR4−/− mice, respectively.
FIG. 7.

Reduced expression of COX-2 in TLR2−/− animals compared to that in WT animals. Single-cell suspensions of corneal cells were prepared from two corneas (n = 8) at day 3 and day 6 p.i. Intracellular COX-2 staining was carried out as described in Materials and Methods. In panels A and B, bar diagrams demonstrate the total number of COX-2+ cells and the total number of COX-2+ Gr1+ cells/1 × 105 corneal cells, respectively. *, P ≤ 0.05 (statistically significant differences compared to WT animals).
DISCUSSION
Ocular infection with HSV sets off an array of events that succeed in clearing virus from the cornea but leave the tissue with a CD4+ T-cell-orchestrated chronic inflammatory lesion that impairs vision. We demonstrate that TLR signaling forms a part of the recognition system that sets off a syndrome that eventually culminates in immunopathology. Accordingly, in a comparison of the outcomes of infection in WT mice and those lacking TLR function, it was apparent that the absence of TLR2, and to a lesser extent TLR9, resulted in significantly diminished lesions. Similarly, mice lacking the adapter molecule MyD88 were resistant to lesion development, but such animals were also unable to control infection, and most succumbed to lethal encephalitis. Animals lacking TLR4 developed lesions more rapidly, and these became more severe than those in WT animals. It could be that modulating TLRs in an appropriate way could represent an approach to control the severity of SK, an important cause of vision impairment.
Past studies demonstrated that HSV expresses at least two TLR ligand activities (24). These were TLR2 ligand activity, perhaps a property of one of the envelope major glycoproteins (16), and the TLR9 ligand activity of its DNA (28). In this report, we also show that HSV infection can cause cells to endogenously express at least two molecules that act as TLR ligands. These are BD3, a TLR4 ligand (6), and inducible Hsp70, a ligand for TLR4 and perhaps also for TLR2 (5). Our investigations were designed to determine if the absence of TLR2, -4, and -9 impacted the outcome of the immunopathological reaction in the eye that results from HSV infection.
The most dramatic effects were observed when TLR2−/− mice were infected with a low dose of a virulent strain of HSV. Compared to WT animals, TLR2−/− mice showed considerable resistance to disease expression. Fewer eyes showed clinical lesions, and those that did had reduced levels of neovascularization and stromal inflammatory reactions at both the early and peak times of the response. Reduced inflammatory reactions were also evident in the TG, a site where virus locates soon after ocular infection and persists when acute infection resolves (9). Curiously, however, TLR2−/− animals usually expressed notable periocular disease, even when the eye itself exhibited minimal or no lesions. We interpret these data to mean that the TLR2 ligand activity of HSV may be a major mechanism by which the virus, usually confined to the epithelium of the infected cornea (9), induces early events necessary for the development of the inflammatory disease. As shown in previous studies, these include the induction of cytokines such as IL-6, IL-1β, and TNF-α (9), the chemokines MIP-1α and MIP-2 (9), enzymes such as COX-2, associated with the production of inflammatory molecules (9), and molecules responsible for neovascularization of the usually avascular eye, such as VEGF and MMP9 (9). In line with this, when levels of some of these molecules were measured early after infection, all were reduced in TLR2−/− compared to WT eyes. Evidence that cells from TLR2-negative cells make reduced inflammatory cytokine responses to HSV was previously reported by others (1, 24). Moreover, such reduced inflammatory reactions to HSV in the brain were advocated to explain why TLR2−/− mice were less susceptible to HSV encephalitis than were WT controls (24). Similarly, for SK, the resistance phenotype of TLR2−/− animals is likely explained by the reduced induction of key inflammatory mediators.
The observation that periocular disease was more severe in TLR2−/− mice was less easy to explain, especially since viral clearance kinetics in the eye itself between TLR2−/− and WT animals showed no significant differences. Similarly, Kurt-Jones et al. observed no major differences in viral clearance in the brain between control and resistant TLR2−/− animals (24). Since the periocular lesions appeared to be caused by the virus itself, rather than being an immunopathological reaction, conceivably the diminished stimulation of local antiviral molecules, such as interferons and perhaps others involved in the acute inflammatory response, may explain the outcome for TLR2−/− mice. We are further evaluating this issue.
Since HSV DNA is well known to act as a TLR9 ligand (23, 28), we anticipated that TLR9−/− mice would also react differently to the ocular infection than did WT animals. Such TLR9−/− mice were in fact more refractory to lesion expression than were WT animals, but this was only a modest difference. This may be explained by the possibility that the TLR9 ligand activity is inadequately produced under in vivo infection conditions to drive the neovascular and inflammatory responses or that any effect of TLR9 is largely overridden by the proinflammatory effect of the TLR2 ligand activity of the virus. We hope to obtain double-knockout mice to help resolve these issues. Alternatively, there could be a reduced Th1 immune response in the absence of TLR9, which could help to reduce the lesion severity. We are currently investigating the generation of HSV-specific immune responses in the absence of specific TLRs.
Previous studies of herpetic encephalitis had not revealed a response pattern for TLR4−/− animals. However, following ocular infection, such animals were significantly, although not dramatically, more susceptible to both neovascularization and SK lesion development. Lesions also occurred earlier than those in WT animals, and a higher percentage of infected eyes developed lesions. Periocular disease, however, was unusual, as it is in WT animals. Although we currently lack a definitive mechanistic explanation for these observations, several ideas are being pursued. These include the possibilities that the TLR4 ligand activities might serve to induce either the production of anti-inflammatory cytokines (26) or the induction of some type of regulatory T-cell response (39) and that the enhanced reactions occurred because of a heightened anti-HSV immune response in TLR4−/− animals. Currently, we have preliminary support for at least two of these ideas. First, IL-10 levels in corneal extracts of TLR4−/− mice were lower than those present in WT control extracts (Fig. 6). Additionally, the magnitudes of T-cell responses in TLR4−/− animals were marginally elevated over those in WT animals (data not shown). We have yet to evaluate any differential induction of regulatory T-cell responses between TLR4−/− and control animals, but such studies are under way.
Our studies also included minimal investigations on the outcome of infection in animals lacking both TLR2 and TLR4. Such animals also showed the resistance phenotype, but the magnitude was less than that observed for TLR2−/− and MyD88−/− animals. This could mean that the TLR2 ligand effect of HSV is the dominant activity but that this is partially blunted by the apparent anti-inflammatory effect of TLR4 ligand stimulation. Perhaps the use of TLR4 ligands has some place in the future therapy of SK.
Finally, our studies evaluated HSV-induced ocular responses in MyD88−/− mice. Such animals showed approximately the same phenotype as did the TLR2−/− mice, which may mean that TLR2 ligand-mediated effects are the most significant proinflammatory agonists in HSV infections. MyD88−/− mice, however, showed one major response difference compared to TLR2−/− and other TLR−/− animals. This was their marked susceptibility to herpetic encephalitis, with animals usually dying by 15 days p.i. Others have also observed that HSV may prove lethal to MyD88−/− animals infected intranasally (29). The explanation for this heightened susceptibility to encephalitis remains unclear. Accordingly, although virus was cleared normally from the eye and TG in MyD88−/− mice, this was not true for the brain, where virus persisted and was still present in dying animals. We anticipate that the susceptibility of MyD88−/− animals might be explained by a deficiency in some critical innate and/or adaptive immune response that functions in the CNS. In some other infection systems, MyD88−/− animals also show a reduced ability to generate an effective adaptive immune response (43). In our preliminary studies, we noted that antigen-presenting-cell function in MyD88−/− mice is impaired, which might have an effect on the generation of an effective immune response.
In conclusion, our results indicate that the TLR ligand activity of HSV, primarily that of TLR2, plays a role in the induction of an immunopathological response in the cornea and TG. Other TLR ligands, such as TLR4, may serve to counteract the severity of the inflammatory response. It is conceivable that manipulation of the TLR ligand response could provide a means to modulate SK lesions.
Acknowledgments
This study was supported by National Institutes of Health grant EY05093.
We thank Sharvan Sehrawat for his help in the experiments and in examining eyes. We thank Jason Burchett for his help in this work.
Footnotes
Published ahead of print on 8 August 2007.
REFERENCES
- 1.Aravalli, R. N., S. Hu, T. N. Rowen, J. M. Palmquist, and J. R. Lokensgard. 2005. Cutting edge: TLR2-mediated proinflammatory cytokine and chemokine production by microglial cells in response to herpes simplex virus. J. Immunol. 175:4189-4193. [DOI] [PubMed] [Google Scholar]
- 2.Arslan, A., and H. H. Zingg. 1996. Regulation of COX-2 gene expression in rat uterus in vivo and in vitro. Prostaglandins 52:463-481. [DOI] [PubMed] [Google Scholar]
- 3.Banerjee, K., P. S. Biswas, B. Kim, S. Lee, and B. T. Rouse. 2004. CXCR2−/− mice show enhanced susceptibility to herpetic stromal keratitis: a role for IL-6-induced neovascularization. J. Immunol. 172:1237-1245. [DOI] [PubMed] [Google Scholar]
- 4.Banerjee, K., S. Deshpande, M. Zheng, U. Kumaraguru, S. P. Schoenberger, and B. T. Rouse. 2002. Herpetic stromal keratitis in the absence of viral antigen recognition. Cell. Immunol. 219:108-118. [DOI] [PubMed] [Google Scholar]
- 5.Beg, A. A. 2002. Endogenous ligands of Toll-like receptors: implications for regulating inflammatory and immune responses. Trends Immunol. 23:509-512. [DOI] [PubMed] [Google Scholar]
- 6.Biragyn, A., P. A. Ruffini, C. A. Leifer, E. Klyushnenkova, A. Shakhov, O. Chertov, A. K. Shirakawa, J. M. Farber, D. M. Segal, J. J. Oppenheim, and L. W. Kwak. 2002. Toll-like receptor 4-dependent activation of dendritic cells by beta-defensin 2. Science 298:1025-1029. [DOI] [PubMed] [Google Scholar]
- 7.Biswas, P. S., K. Banerjee, B. Kim, P. R. Kinchington, and B. T. Rouse. 2005. Role of inflammatory cytokine-induced cyclooxygenase 2 in the ocular immunopathologic disease herpetic stromal keratitis. J. Virol. 79:10589-10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Biswas, P. S., K. Banerjee, M. Zheng, and B. T. Rouse. 2004. Counteracting corneal immunoinflammatory lesion with interleukin-1 receptor antagonist protein. J. Leukoc. Biol. 76:868-875. [DOI] [PubMed] [Google Scholar]
- 9.Biswas, P. S., and B. T. Rouse. 2005. Early events in HSV keratitis—setting the stage for a blinding disease. Microbes Infect. 7:799-810. [DOI] [PubMed] [Google Scholar]
- 10.Boehme, K. W., and T. Compton. 2004. Innate sensing of viruses by Toll-like receptors. J. Virol. 78:7867-7873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bowman, C. C., and K. L. Bost. 2004. Cyclooxygenase-2-mediated prostaglandin E2 production in mesenteric lymph nodes and in cultured macrophages and dendritic cells after infection with Salmonella. J. Immunol. 172:2469-2475. [DOI] [PubMed] [Google Scholar]
- 12.Burch, A. D., and S. K. Weller. 2004. Nuclear sequestration of cellular chaperone and proteasomal machinery during herpes simplex virus type 1 infection. J. Virol. 78:7175-7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dana, M. R., S. N. Zhu, and J. Yamada. 1998. Topical modulation of interleukin-1 activity in corneal neovascularization. Cornea 17:403-409. [DOI] [PubMed] [Google Scholar]
- 14.Deshpande, S., K. Banerjee, P. S. Biswas, and B. T. Rouse. 2004. Herpetic eye disease: immunopathogenesis and therapeutic measures. Expert Rev. Mol. Med. 2004:1-14. [DOI] [PubMed] [Google Scholar]
- 15.Deshpande, S., M. Zheng, S. Lee, K. Banerjee, S. Gangappa, U. Kumaraguru, and B. T. Rouse. 2001. Bystander activation involving T lymphocytes in herpetic stromal keratitis. J. Immunol. 167:2902-2910. [DOI] [PubMed] [Google Scholar]
- 16.Finberg, R. W., D. M. Knipe, and E. A. Kurt-Jones. 2005. Herpes simplex virus and Toll-like receptors. Viral Immunol. 18:457-465. [DOI] [PubMed] [Google Scholar]
- 17.Gil, M. L., and D. Gozalbo. 2006. TLR2, but not TLR4, triggers cytokine production by murine cells in response to Candida albicans yeasts and hyphae. Microbes Infect. 8:2299-2304. [DOI] [PubMed] [Google Scholar]
- 18.Kaisho, T., K. Hoshino, T. Iwabe, O. Takeuchi, T. Yasui, and S. Akira. 2002. Endotoxin can induce MyD88-deficient dendritic cells to support T(h)2 cell differentiation. Int. Immunol. 14:695-700. [DOI] [PubMed] [Google Scholar]
- 19.Kennedy, P. G., and A. Chaudhuri. 2002. Herpes simplex encephalitis. J. Neurol. Neurosurg. Psych. 73:237-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim, B., S. Lee, S. Suvas, and B. T. Rouse. 2005. Application of plasmid DNA encoding IL-18 diminishes development of herpetic stromal keratitis by antiangiogenic effects. J. Immunol. 175:509-516. [DOI] [PubMed] [Google Scholar]
- 21.Kim, B., P. P. Sarangi, Y. Lee, S. D. Kaistha, S. Lee, and B. T. Rouse. 2006. Depletion of MCP-1 increases development of herpetic stromal keratitis by innate immune modulation. J. Leukoc. Biol. 80:1405-1415. [DOI] [PubMed] [Google Scholar]
- 22.Kim, B., Q. Tang, P. S. Biswas, J. Xu, R. M. Schiffelers, F. Y. Xie, A. M. Ansari, P. V. Scaria, M. C. Woodle, P. Lu, and B. T. Rouse. 2004. Inhibition of ocular angiogenesis by siRNA targeting vascular endothelial growth factor pathway genes: therapeutic strategy for herpetic stromal keratitis. Am. J. Pathol. 165:2177-2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krug, A., G. D. Luker, W. Barchet, D. A. Leib, S. Akira, and M. Colonna. 2004. Herpes simplex virus type 1 activates murine natural interferon-producing cells through Toll-like receptor 9. Blood 103:1433-1437. [DOI] [PubMed] [Google Scholar]
- 24.Kurt-Jones, E. A., M. Chan, S. Zhou, J. Wang, G. Reed, R. Bronson, M. M. Arnold, D. M. Knipe, and R. W. Finberg. 2004. Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc. Natl. Acad. Sci. USA 101:1315-1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kurt-Jones, E. A., L. Popova, L. Kwinn, L. M. Haynes, L. P. Jones, R. A. Tripp, E. E. Walsh, M. W. Freeman, D. T. Golenbock, L. J. Anderson, and R. W. Finberg. 2000. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat. Immunol. 1:398-401. [DOI] [PubMed] [Google Scholar]
- 26.Kwan, W. H., C. Boix, N. Gougelet, W. H. Fridman, and C. G. Mueller. 2007. LPS induces rapid IL-10 release by M-CSF-conditioned tolerogenic dendritic cell precursors. J. Leukoc. Biol. 82:133-141. [DOI] [PubMed] [Google Scholar]
- 27.Liu, T., Q. Tang, and R. L. Hendricks. 1996. Inflammatory infiltration of the trigeminal ganglion after herpes simplex virus type 1 corneal infection. J. Virol. 70:264-271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lund, J., A. Sato, S. Akira, R. Medzhitov, and A. Iwasaki. 2003. Toll-like receptor 9-mediated recognition of herpes simplex virus-2 by plasmacytoid dendritic cells. J. Exp. Med. 198:513-520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mansur, D. S., E. G. Kroon, M. L. Nogueira, R. M. Arantes, S. C. Rodrigues, S. Akira, R. T. Gazzinelli, and M. A. Campos. 2005. Lethal encephalitis in myeloid differentiation factor 88-deficient mice infected with herpes simplex virus 1. Am. J. Pathol. 166:1419-1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McGill, J., and G. M. Scott. 1985. Viral keratitis. Br. Med. Bull. 41:351-356. [DOI] [PubMed] [Google Scholar]
- 31.Melchjorsen, J., L. N. Sorensen, and S. R. Paludan. 2003. Expression and function of chemokines during viral infections: from molecular mechanisms to in vivo function. J. Leukoc. Biol. 74:331-343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Minagawa, H., S. Sakuma, S. Mohri, R. Mori, and T. Watanabe. 1988. Herpes simplex virus type 1 infection in mice with severe combined immunodeficiency (SCID). Arch. Virol. 103:73-82. [DOI] [PubMed] [Google Scholar]
- 33.Mitchell, B. M., and J. G. Stevens. 1996. Neuroinvasive properties of herpes simplex virus type 1 glycoprotein variants are controlled by the immune response. J. Immunol. 156:246-255. [PubMed] [Google Scholar]
- 34.Mitsunari, M., S. Yoshida, T. Shoji, S. Tsukihara, T. Iwabe, T. Harada, and N. Terakawa. 2006. Macrophage-activating lipopeptide-2 induces cyclooxygenase-2 and prostaglandin E(2) via Toll-like receptor 2 in human placental trophoblast cells. J. Reprod. Immunol. 72:46-59. [DOI] [PubMed] [Google Scholar]
- 35.Mogensen, T. H., and S. R. Paludan. 2005. Reading the viral signature by Toll-like receptors and other pattern recognition receptors. J. Mol. Med. 83:180-192. [DOI] [PubMed] [Google Scholar]
- 36.O'Neill, L. A. 2004. Immunology. After the Toll rush. Science 303:1481-1482. [DOI] [PubMed] [Google Scholar]
- 37.Spear, P. G., and B. Roizman. 1972. Proteins specified by herpes simplex virus. V. Purification and structural proteins of the herpesvirion. J. Virol. 9:143-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thomas, J., S. Gangappa, S. Kanangat, and B. T. Rouse. 1997. On the essential involvement of neutrophils in the immunopathologic disease: herpetic stromal keratitis. J. Immunol. 158:1383-1391. [PubMed] [Google Scholar]
- 39.van Eden, W., G. Wick, S. Albani, and I. Cohen. 21 June 2007. Stress, heat shock proteins, and autoimmunity: how immune responses to heat shock proteins are to be used for the control of chronic inflammatory diseases. Ann. N. Y. Acad. Sci. [Epub ahead of print.] http://www.annalsnyas.org/cgi/rapidpdf/annals.1391.020v1. [DOI] [PubMed]
- 40.Villamon, E., P. Roig, M. L. Gil, and D. Gozalbo. 2005. Toll-like receptor 2 mediates prostaglandin E(2) production in murine peritoneal macrophages and splenocytes in response to Candida albicans. Res. Microbiol. 156:115-118. [DOI] [PubMed] [Google Scholar]
- 41.Zheng, M., S. Deshpande, S. Lee, N. Ferrara, and B. T. Rouse. 2001. Contribution of vascular endothelial growth factor in the neovascularization process during the pathogenesis of herpetic stromal keratitis. J. Virol. 75:9828-9835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zheng, M., D. M. Klinman, M. Gierynska, and B. T. Rouse. 2002. DNA containing CpG motifs induces angiogenesis. Proc. Natl. Acad. Sci. USA 99:8944-8949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou, S., E. A. Kurt-Jones, L. Mandell, A. Cerny, M. Chan, D. T. Golenbock, and R. W. Finberg. 2005. MyD88 is critical for the development of innate and adaptive immunity during acute lymphocytic choriomeningitis virus infection. Eur. J. Immunol. 35:822-830. [DOI] [PubMed] [Google Scholar]