

Abstract
This study evaluated the contributions of carboxyl ester lipase (CEL) and pancreatic triglyceride lipase (PTL) in lipid nutrient absorption. Results showed PTL deficiency has minimal effect on triacylglycerol (TAG) absorption under low fat dietary conditions. Interestingly, PTL−/− mice displayed significantly reduced TAG absorption compared with wild type mice under high fat/high cholesterol dietary conditions (80.1 ± 3.7 versus 91.5 ± 0.7%, p < 0.05). Net TAG absorption was reduced further to 61.1 ± 3.8% in mice lacking both PTL and CEL. Cholesterol absorption was 41% lower in PTL−/− mice compared with control mice (p < 0.05), but this difference was not exaggerated in PTL−/−,CEL−/− mice. Retinyl palmitate absorption was reduced by 45 and 60% in PTL−/− mice (p < 0.05) and PTL−/−,CEL−/− mice (p < 0.01), respectively. After 15 weeks of feeding, the high fat/high cholesterol diet, wild type, and CEL−/− mice gained ∼24 g of body weight. However, body weight gain was 6.2 and 8.6 g less (p < 0.01) in PTL−/− and PTL−/−,CEL−/− mice, respectively, despite their consumption of comparable amounts of the high fat/high cholesterol diet. The decrease body weight gain in PTL−/− and PTL−/−,CEL−/− mice was attributed to their absorption of fewer calories from the high fat/high cholesterol diet, thereby resulting in less fat mass accumulation than that observed in wild type and CEL−/− mice. Thus, this study documents that PTL and CEL serve complementary functions, working together to mediate the absorption of a major portion of dietary fat and fat-soluble vitamin esters. The reduced lipid absorption efficiency due to PTL and CEL inactivation also resulted in protection against diet-induced obesity.
Despite the fundamental importance that digestion, absorption, and subsequent trafficking of dietary fat, fat soluble vitamins, and cholesterol play in health and disease, details of the molecular events involved in these processes remain poorly described. Dietary lipids, such as triacylglycerol (TAG),4 phospholipids, cholesteryl esters, and retinyl esters must be cleaved by intestinal hydrolases before absorption. Thus, lipid hydrolases derived from the pancreas are central in the digestion and absorption of these dietary components. The most abundant lipolytic enzymes secreted by the pancreas include pancreatic triglyceride lipase (PTL) and carboxyl ester lipase (CEL, formerly named cholesterol esterase). The principal enzyme involved in the hydrolysis of dietary TAG, thereby mediating its absorption, has generally been accepted as PTL. In contrast, the major role of CEL in the digestive tract is generally ascribed to digestion of cholesteryl esters and retinyl esters before their absorption.
Recent studies with genetically modified mouse models designed to critically examine the role of pancreatic lipolytic enzymes in lipid digestion and transport revealed some unexpected findings. First, TAG absorption was altered very little, but free cholesterol absorption was reduced substantially in PTL−/− mice (1). Second, whereas CEL−/− mice displayed dramatically reduced cholesteryl ester hydrolysis and absorption as expected (2), retinyl ester digestion and absorption were similar between CEL−/− and CEL−/− mice (3). The decrease in cholesterol absorption observed in the PTL−/− mice was attributed to PTL deficiency delaying dietary fat absorption to the distal small intestine (ileum) where intestinal cholesterol uptake is less efficient (4, 5). However, the nearly normal absorption of TAG and retinyl esters in PTL−/− and CEL−/− mice suggested that more than one enzyme participates in the digestion of these lipids in vivo.
In vitro studies examining lipolytic activities in pancreatic extracts from PTL−/− and CEL−/− mice provided insights to the identity of enzymes responsible for TAG and retinyl ester hydrolysis. In these studies pancreatic extracts from PTL−/− mice were shown to contain robust TAG hydrolytic activity, albeit at lower levels than those observed in pancreatic extracts from wild type mice. Importantly, the TAG hydrolytic activity in PTL−/− pancreas was dependent on the presence of taurocholate, whereas taurodeoxycholate was found to be ineffective (1). This selectivity of trihydroxylated bile salt for TAG hydrolytic activity in PTL−/− pancreas is consistent with the trihdroxy- but not dihydroxy-bile salt dependence of CEL (6), thus implying that CEL may have a previously unrecognized role as a supplementary enzyme for TAG digestion in vivo. Likewise, significant colipase-dependent retinyl ester hydrolytic activity was demonstrated in pancreatic extracts from both CEL+/+ and CEL−/− mice (7). This colipase dependence suggests that PTL may also serve as a retinyl ester hydrolase in the digestive tract.
In the current study we crossed PTL−/− mice with CEL−/− mice to obtain mice lacking both enzymes to test the hypothesis that PTL and CEL serve a mutually compensatory role in assuring that sufficient lipolytic enzymes are available to catalyze absorption of dietary lipid nutrients. We also took advantage of the availability of mice lacking one or both of these lipolytic enzymes in the digestive tract to assess their importance in dictating susceptibility to diet-induced obesity.
EXPERIMENTAL PROCEDURES
Animals and Diets
—Wild type C57BL/6 mice were obtained from The Jackson Laboratory (Bar Harbor, ME). The PTL−/− mice and CEL−/− mice in homogenous C57BL/6 background were generated in our laboratory as described previously (1, 2). The PTL−/−,CEL−/− mice were produced by cross-breeding PTL−/− mice with CEL−/− mice. The double knock-out mice were fertile and did not display any obvious abnormalities. The genotype of all genetically modified mice was confirmed by polymerase chain reaction amplification of tail DNA as described (1, 2). All animals were maintained in a temperature and humidity-controlled room with a 12-h light/dark cycle. The mice were maintained on two types of diet obtained from Harlan Teklad (Madison, WI). The low fat control diet was a rodent chow (LM-485), whereas the high fat/high cholesterol Western-type diet (TD88137) contained 21% fat, 0.15% cholesterol and 48.5% carbohydrate by weight. The Western-type diet was introduced at 8−9 weeks of age and continued for the duration of experiments as indicated. All animal protocols used in this study were approved by the Institutional Animal Care and Use Committee at the University of Cincinnati. Only male mice were used to generate data in this study to avoid estrous cycle variations that may introduce confounding factors in data interpretation.
Determination of Net Lipid Absorption in Mice
—Triacylglycerol absorption in mice was assessed using sucrose polybehenate as a nonabsorbable marker as described previously (8). Briefly, a diet mimicking the Harlan Teklad Western-type diet was prepared. The composition of the diet included 42.2% nonfat dry milk, 36.6% sucrose, and 21.2% fat (wt/wt). The fat component consisted of 94.25% anhydrous milk fat, 0.75% cholesterol, and 5% sucrose polybehenate (wt/wt). Mice were fed this powdered preparation for 4 days, and fecal samples were collected daily. Individual fecal samples of ∼10 mg as well as the original powdered diet were dried and analyzed. Esterified fatty acids were chemically hydrolyzed by the addition of 0.5 N NaOH in methanol followed by heating to 80 °C in a water bath for 5 min. Derivatization of the fatty acids to their respective methyl esters was accomplished by addition of boron trifluo-ride in methanol and reheating to 80 °C. Finally, hexane and a saturated solution of NaCl were added followed by rigorous vortexing. The aqueous and organic phases were allowed to separate, and the organic fraction was transferred to a vial containing sodium sulfate to dry the sample. Analysis of the fatty acid methyl esters was accomplished by gas chromatography. Fat absorption was calculated based on the ratio of fatty acids to behenic acid in the diet versus that in the feces.
Net cholesterol absorption was determined in mice consuming the Western-type diet for 4 weeks using the dual isotope procedure described previously (2). Mice were housed individually and fed ad libitum before receiving a bolus of 100 μlof olive oil containing 2 μCi of [14C]cholesterol (GE Healthcare), 0.2 mg of cholesterol (Sigma), and 0.5 μCi of [3H]sitostanol (American Radiolabeled Chemicals, St. Louis, MO) by stomach gavage. Mice continued to have free access to food and water. Feces were collected after 24 and 48 h. The feces were homogenized in water and then extracted with twice the volume of chloroform/methanol (2:1, v/v). An aliquot of the organic phase from each sample was used for scintillation counting to determine the amount of radioactive sterols present. Recovery of nonabsorbed cholesterol in feces was corrected based on the amount of [3H]sitostanol measured in each sample. Absorption of [14C]cholesterol was determined as a percentage of the administered dose as described (2).
Determination of Lipid Nutrient Absorption Rates
—The rates of TAG and retinyl palmitate absorption were determined based on methods described previously (1, 3). Briefly, wild type, PTL−/−, CEL−/−, and PTL−/−,CEL−/− mice fed the Western-type diet for 31 weeks were fasted for 14 h with access to water. The mice were then administered Poloxamer 407 (Pluronic F-127 from BASF Corporation, Parsippany, NJ) at a dose of 1 g/kg of body weight by intraperitoneal injection to block lipolysis and clearance of postprandial triacylglycerol-rich lipoproteins (9). A test meal containing 1 μCi of [3H]retinyl palmitate and 1.5 μCi of [14C]tripalmitin (American Radiolabeled Chemicals) in 100 μl of olive oil was administered by stomach gavage 15 min after Poloxamer 407 injection. In selected experiments 1 μCi of [3H]retinyl palmitate and 1.5 μCi of [14C]cholesteryl oleate (American Radiolabeled Chemicals) were introduced into each mouse in phospholipid vesicles prepared by sonicating the radiolabeled lipids in water containing 35 μg/ml of phosphatidylcholine (2). Plasma samples were obtained from the animals via tail bleeding at 0, 1, 3, 6, and 9 h after delivery of the test meal. The appearance of 3H and 14C in plasma was measured via scintillation counting and used to determine the rate of absorption of retinyl palmitate and TAG or cholesteryl esters, respectively.
Food Intake
—Mice consuming the high fat/high cholesterol Western-type diet were housed individually. A weighed amount of food was presented to mice in the manner that it is normally supplied, and the food hoppers were weighed every 2 days for 8 consecutive days. Mice were returned to their usual housing environment including cohabitating with their cohorts for 1 month. The procedure was then repeated, and the average daily food intake was determined for each mouse from these 16 days of analysis.
Body Composition Assessment
—Body composition was analyzed by quantitative nuclear magnetic resonance (NMR) (10) using an EcoMRI apparatus (Echo Medical Systems, Houston, TX), which allows the determination of fat and lean mass of live mice. Analysis was applied to 11-, 16-, and 25-week-old mice consuming the low fat chow diet or 16- and 25-week old mice fed the high fat/high cholesterol diet beginning at 9 weeks of age.
Lipid, Glucose, and Insulin Determinations
—Mice consuming chow or the Western-type diet for ∼40 weeks were fasted for 14 h. Glucose levels were determined using an Accu-Check Glucometer (Roche Diagnostics) from a drop of blood obtained through the tail vein. Plasma triglyceride and cholesterol levels were measured with commercially available colorimetric assay kits made by ThermoElectron Corp. (Fisher). Lipids were also extracted from the liver by adding 4 ml of chloroform/methanol (2:1, v/v) to 100 mg of liver homogenates in 2 ml of phosphate-buffered saline containing a protease inhibitor mixture (Roche Diagnostics) and 0.02 μCi of [3H]triolein. After phase separation, 1 ml of the organic phase was removed, dried under nitrogen, and resuspended in ethanol. Triglyceride and cholesterol contents were determined by colorimetric assay kits as described above. The efficiency of lipid extraction was normalized based on recovery of the radiolabel.
Fasting plasma insulin levels were determined from age-matched mice consuming either the chow or the Western-type diet for 11 weeks. Plasma was collected via tail bleeding after an overnight fast. Insulin in the plasma was measured using a fluorescence-based enzyme-linked immunosorbent assay from Linco Research (Millipore, Billerica, MA) according to the manufacturer's protocol.
Pancreatic Expression of PTL-related Protein-2 (PTLRP2)
— Pancreati were removed from mice promptly after exsanguination. Approximately 50 mg of pancreas was placed in RNA-Later® (Ambion, Austin, TX). Total RNA was isolated using TR1 Reagent (Molecular Research Center, Cincinnati, OH), and 0.5 μg was used for reverse transcriptase-PCR amplification using the iScript One-Step reverse transcription-PCR kit (Bio-Rad). Relative levels of PTLRP2 were quantified via real time PCR using cyclophilin as a reference gene and SYBR green for detection on a Bio-Rad iCycler apparatus. The forward primer for PTLRP2 is 5′-GGATGATTCAGCGGCCTTCG-3′, and the reverse primer sequence is 5′-TCTCTGGGCTGTACCCCATC-3′. The primers used for amplification of cyclophilin were designed as described (11).
Statistical Analysis
—Data were evaluated using 2-tailed Student's t test of independent groups with the assumption of unequal variances. Results are presented as the mean ± S.E. unless indicated otherwise in the figure legend. Results from genetically modified mice were compared with wild type mice, with statistical significant difference recognized at p < 0.05.
RESULTS
Triacylglycerol and Cholesterol Absorption in Mice
—We have previously reported a reduced rate of TAG absorption in PTL−/− mice, but overall fat absorption efficiency was similar between PTL+/+ and PTL−/− mice when they were maintained on a low fat chow diet (1). Likewise, dietary fat absorption was similar between chow-fed CEL+/+ and CEL−/− mice (3, 12). The current study examined TAG absorption in PTL−/−,CEL−/−, and PTL−/−,CEL−/− double knock-out mice when the animals were maintained under high fat/high cholesterol dietary conditions utilizing the nonabsorbable lipid-based tracer sucrose polybehenate as a marker of nonabsorbed lipid excreted in feces (8). Results showed no difference in the amount of TAG absorbed from a test diet between wild type and CEL−/− mice even under high fat/high cholesterol dietary conditions (Fig. 1). However, in contrast to previous results using fecal fat content assessment to show no difference in TAG absorption between wild type and PTL−/− mice (1), use of the more sensitive tracer fat recovery method in the current study revealed a modest but statistically significant reduction in TAG absorption in PTL−/− mice when the animals were maintained on this Western-type diet (91.5 ± 0.7% for wild type versus 80.1 ± 3.6% for PTL−/− mice). Intriguingly, the lack of both PTL and CEL resulted in a dramatic reduction in fat absorption (61.1 ± 3.8%, p < 0.001 versus wild type mice). Analysis of individual fatty acids in the feces by gas chromatography indicated that saturation and chain length were not factors affecting the decreased absorption in the double knock-out mice, as the absorption was reduced regardless of the fatty acid species measured. As reported previously (1, 2), cholesterol absorption was significantly reduced in PTL−/− mice but not in CEL−/− mice when they were maintained on the high fat/high cholesterol diet (Fig. 2). Inactivation of the CEL gene did not cause additional reduction of cholesterol absorption in PTL−/− mice as cholesterol absorption was similar between PTL−/− and PTL−/−,CEL−/− mice (Fig. 2).
FIGURE 1. Fat absorption efficiency in mice consuming a high fat/high cholesterol Western-type diet.
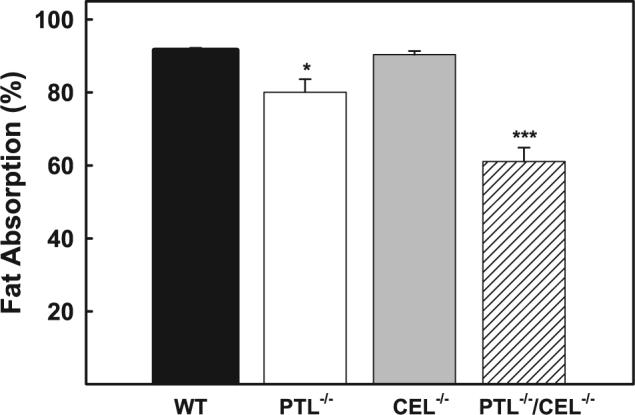
Mice maintained on a high fat/high cholesterol diet were fed a similar high fat/high cholesterol diet for 4 days that also contained the non-absorbable marker sucrose polybehenate as 5% of the dietary fat. Fecal samples were collected daily. Fatty acids present in the original diet as well as the feces were derivatized to their respective methyl esters and quantified by gas chromatography. Fat absorption was calculated from the ratios of behenic acid to other fatty acids in the diet versus the feces. Two experiments were averaged for n = 6 wild type (WT), n =7 PTL−/−, n = 8 CEL−/− and n = 7 PTL−/−,CEL−/− mice and shown as the means ± S.E. for each genotype. *, p < 0.05; ***, p < 0.001 (differences from wild type mice).
FIGURE 2. Cholesterol absorption efficiency in mice consuming a high fat/high cholesterol diet.

Mice maintained on the high fat/high cholesterol diet for 4 weeks received 2 μCi of [14C]cholesterol, 0.2 mg of cholesterol, and 0.5 μCi of [3H]sitostanol in 100 μl of olive oil by stomach gavage. Fecal matter was collected after 24 and 48 h, homogenized in water, and then extracted with chloroform/methanol (2:1, v/v). Scintillation counting was used to determine the amount of radioactive sterols present in the organic phase. Recovery of nonabsorbed cholesterol in feces was corrected based on the amount of [3H]sitostanol in each sample. Absorption of cholesterol is presented as a percentage of the administered dose and is representative of two experiments. Shown is the means ± S.E. for n = 9 wild type (WT), n = 7 PTL−/−, n = 10 CEL−/−, and n = 9 PTL−/−,CEL−/− mice. *, p < 0.05; **, p < 0.01 (differences from wild type mice).
Rate of Lipid Nutrient Absorption in Mice
—The rates of dietary TAG, long chain retinyl ester, and cholesteryl ester absorption by wild type, PTL−/−, CEL−/−, and PTL−/−,CEL−/− mice were compared by determining the accumulation of radiolabeled lipids in plasma after Poloxamer 407 injection followed by oral administration of radiolabeled lipids by gavage. As shown in Fig. 3A, the rate of appearance of radioactivity in plasma after [14C]tripalmitin administration was much slower in PTL−/− mice than in wild type mice, with 47% less 14C radioactivity detected in the circulation of PTL−/− mice after 9 h (Fig. 3A). A small but significantly more pronounced decrease in the rate of [14C]tripalmitin absorption was observed in the [PTL−/−, CEL−/− mice (Fig. 3A). At the 9-h time point, 52% less [14C]tripalmitin absorption occurred in the double knock-out mice compared with that in wild type mice (Fig. 3A).
FIGURE 3. Rate of lipid absorption in mice.
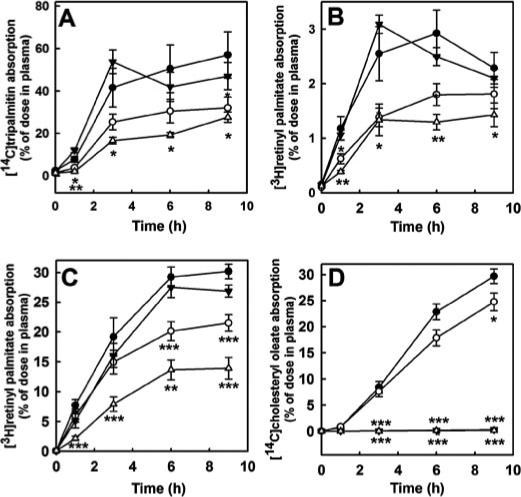
Mice consuming a high fat/ high cholesterol diet were fasted for 14 h, then given Poloxamer 407 via intraperitoneal injection (1 g/kg of body weight). After 15 min mice were given [14C]triolein and [3H]retinyl palmitate in olive oil (A and B) or [14C]cholesteryl oleate and [3H]retinyl palmitate in phospholipid vesicles (C and D). The appearance of radiolabel derived from triacylglycerol (A), retinyl ester (B and C), or cholesteryl ester (D) in the circulation at the indicated times after delivery of the bolus meal was determined by scintillation counting. The data are shown as the means ± S.E. for n = 8 wild type (•), 7 PTL−/− (○), 11 CEL−/− (▼ ), and 8 PTL−/−,CEL−/− (▽) mice for A and B, and n = 9 wild type (•), 7 PTL−/−(○), 7 CEL(−/−) (▼ ), and 9 PTL−/−,CEL−/− (▽) mice for C and D.*, p < 0.05; **, p < 0.01; ***, p < 0.001 (differences from wild type mice).
The rate of retinyl ester absorption was assessed initially by feeding [3H]retinyl palmitate mixed in olive oil to mice fed the high fat/high cholesterol diet. Results of these experiments showed the rate of retinyl ester absorption was similar between wild type and CEL−/− mice (Fig. 3B). In contrast, the rate and amount of retinyl ester absorption over a 9-h period were significantly reduced in PTL−/− mice (Fig. 3B). The rate and amount of retinyl ester absorption were further decreased in PTL−/−,CEL−/− mice (Fig. 3B). Interestingly, the differences in rate and efficiency of retinyl ester absorption between wild type, PTL−/−, CEL−/−, and PTL−/−,CEL−/− mice paralleled their differences in rate and efficiency of TAG absorption (Fig. 3A), suggesting that TAG digestion may be required for efficient retinyl ester absorption. Alternatively, their difference in retinyl ester absorption may be directly related to the effectiveness of PTL and CEL in retinyl ester hydrolysis before the absorption of the [3H]retinol by enterocytes in the brush border. These possibilities were explored by using phospholipid vesicles instead of olive oil as the vehicle to deliver [3H]retinyl palmitate to mice. The latter study was also carried out in mice maintained on a low fat chow diet to avoid the confounding variable of differences in digestion of dietary TAG altering retinyl ester digestion and absorption rates. The delivery of [3H]retinyl palmitate in this manner resulted in greater than 30% of the radiolabel appearing in plasma after9hin wild type mice (Fig. 3C) compared with ∼5% when olive oil was used as the vehicle to deliver retinyl ester to high fat/high cholesterol-fed mice (Fig. 3B). Importantly, the trends in rate and amount of retinyl ester absorption between wild type, PTL−/−, CEL−/−, and PTL−/−,CEL−/− mice remained the same, with wild type and CEL−/− mice being the most efficient and the PTL−/−,CEL−/− mice the least efficient regardless of the vehicle used for retinyl ester delivery (Fig. 3C). Taken together, these results indicated that whereas the presence of TAG in the digestive tract is one variable that dictates retinyl ester absorption, PTL is the major enzyme in the intestinal lumen responsible for retinyl ester digestion and absorption in vivo and that CEL may also participate in this process especially under conditions where PTL is limiting.
The decreased retinyl ester absorption observed in PTL−/−
mice is consistent with the in vitro data demonstrating the ability of PTL to hydrolyze retinyl esters in addition to its well established role in TAG hydrolysis (7). In view of numerous studies using dietary retinyl esters as tracers for cholesteryl ester metabolism in vivo (13, 14), we also tested the ability of PTL−/− mice to absorb cholesteryl ester. Results depicted in Fig. 3D show similar levels of [14C]cholesteryl oleate absorption by wild type and PTL−/− mice. In contrast, [14C]cholesteryl oleate absorption was dramatically reduced in the CEL−/− mice as expected. Because cholesteryl ester absorption was nearly eliminated in CEL−/− mice, no further reduction of [14C]cholesteryl oleate absorption was observed in the PTL−/−,CEL−/− mice (Fig. 3D).
Diet-induced Hypercholesterolemia and Insulin Resistance in Mice
—The difference in dietary lipid absorption efficiency between wild type mice and mice with various lipolytic enzyme deficiencies resulted in their differences in sensitivity to chronic feeding of a high fat/high cholesterol diet. Whereas wild type, PTL−/−, CEL−/−, and PTL−/−,CEL−/− mice displayed similar fasting plasma TAG, cholesterol, and glucose levels when fed the low fat chow diet and these values were increased when fed a Western-type high fat/high cholesterol diet regardless of their lipolytic enzyme genotypes, plasma cholesterol levels were 36 and 45% less in PTL−/− and PTL−/−,CEL−/− mice than those observed in wild type and CEL−/− mice after feeding the Western-type diet for 40 weeks (Table 1). Consumption of the Western-type diet did not alter fasting plasma TAG levels in these animals regardless of their genotypes. The normal TAG levels observed in these animals after feeding the Western-type diet is consistent with rapid clearance of dietary TAG from circulation after absorption through the intestine. These data also indicated that PTL and CEL participate only in the lipid absorption process in the digestive tract, and these enzymes are not involved in plasma TAG metabolism. The reduced severity of hypercholesterolemia and liver weight gain in response to Western diet feeding in PTL−/− and PTL−/−,CEL−/− mice compared with wild type and CEL−/− mice is also consistent with the reduced lipid absorption in these animals (Table 1).
TABLE 1. Characteristics of mice fed chow or Western-type diet.
Samples were obtained from mice consuming the low fat chow or the high fat/high cholesterol Western-type diet for 40 weeks after an overnight fast, except for the insulin levels, which were determined from 5 wild type, 4 PTL−/−, 4 CEL−/−, and 5 PTL−/−, CEL−/− mice after 11 weeks. All data are reported as mean ± S.E. ND, not determined.
| Genotype |
Wild type |
PTL−/− |
CEL−/− |
PTL−/−, CEL−/− |
||||
|---|---|---|---|---|---|---|---|---|
| Chow n = 10 | Western n = 9 | Chow n = 9 | Western n = 9 | Chow n = 9 | Western n = 8 | Chow n = 10 | Western n = 9 | |
| Plasma | ||||||||
| Triglyceride (mg/dl) | 53.5 ± 7.0 | 59.2 ± 1.9 | 59.2 ± 5.1 | 62.4 ± 4.0 | 48.2 ± 4.3 | 67.6 ± 3.6 | 41.4 ± 2.6 | 59.1 ± 3.1 |
| Cholesterol (mg/dl) | 38.9 ± 5.6 | 434 ± 51 | 42.5 ± 2.6 | 277 ± 31a | 43.5 ± 1.4 | 401 ± 21 | 53.3 ± 4.7 | 238 ± 22a |
| Glucose (mg/dl) | 107 ± 4 | 148 ± 4 | 110 ± 4 | 143 ± 5 | 112 ± 4 | 147 ± 3 | 103 ± 3 | 148 ± 8 |
| Insulin (pg/dl) |
488 ± 83 |
907 ± 69 |
484 ± 139 |
642 ± 63b |
293 ± 33 |
736 ± 25 |
491 ± 44 |
506 ± 93a |
| Liver | ||||||||
| Weight (g) | 1.28 ± 0.02 | 5.78 ± 0.29 | 1.45 ± 0.04 | 4.23 ± 0.21a | 1.27 ± 0.04 | 5.38 ± 0.13 | 1.15 ± 0.11 | 4.14 ± 0.35a |
| Triglyceride (μg/mg) | 10.9 ± 0.8 | 15.9 ± 1.3 | 8.3 ± 0.9 | 16.9 ± 0.9 | 9.6 ± 0.6 | 16.4 ± 0.9 | 9.9 ± 0.9 | 17.4 ± 1.2 |
| Cholesterol (μg/mg) |
2.9 ± 0.2 |
5.9 ± 0.5 |
2.3 ± 0.2 |
6.1 ± 0.7 |
2.7 ± 0.6 |
6.4 ± 0.7 |
2.5 ± 0.2 |
6.6 ± 1.0 |
| Pancreas | ||||||||
| PTLRP2 mRNA (AU)c | ND | 1.00 ± 0.42 | ND | 0.71 ± 0.22 | ND | 0.47 ± 1.9 | ND | 0.64 ± 0.10 |
Significant difference from wild type mice at p < 0.01.
Significant difference from wild type mice at p < 0.05.
AU, arbitrary unit based on wild type level of 1.0.
In addition to differences in their severity of hypercholesterolemia, the PTL−/− and PTL−/−,CEL−/− mice also displayed significantly lower fasting insulin levels than wild type and CEL−/− mice upon feeding the Western-type diet for 11 weeks (Table 1). The elevated insulin levels in wild type and CEL−/− mice required to maintain similar fasting glucose levels as PTL−/− and PTL−/−,CEL−/− mice suggested that the latter two strains are more insulin-sensitive.
Diet-induced Obesity in Mice
—The difference in nutrient absorption efficiency and sensitivity to diet-induced insulin resistance between wild type mice and mice with various lipolytic enzyme deficiencies suggested potential differences in their susceptibility to diet-induced obesity. Accordingly, we monitored the body weight of mice of each genotype used in this study starting at 9 weeks of age. The body weights of mice consuming a chow diet were not different over a 17-week period of study (data not shown). The similarity in body weight gain may reflect the low fat content of chow, and thus, equal amounts of calories were absorbed by these animals. Therefore, another cohort of mice was challenged with a high fat/high cholesterol diet beginning at 9 weeks of age. Although the body weights of the gene-disrupted mice were similar to those of wild type mice at the initiation of the feeding study, the body weight of the PTL−/−,CEL−/− mice consuming high fat/high cholesterol diet was found to be significantly less than that observed for wild type mice fed the same diet after only 0.5 weeks (Fig. 4A). After consuming the high fat/high cholesterol diet for 10.5 weeks, PTL−/− mice also weighed significantly less than wild type mice, and their weight remained lower for the duration of the 16-week study period. The differences in body weight between high fat/high cholesterol-fed wild type, PTL−/−,CEL−/−, and PTL−/−,CEL−/− mice was due to their differences in rate of body weight gain in response to the high fat diet (Fig. 4B). The differences cannot be attributed to variation in the amount of food consumed by these animals (Table 2), thus strongly suggesting that differences in calories absorbed is responsible for their differences in weight gain. Consistent with this hypothesis is the observation that total body weight and weight gain in response to the high fat/high cholesterol diet were similar between wild type and CEL−/− mice (Fig. 4A), which displayed comparable TAG absorption efficiency (Fig. 1).
FIGURE 4. Body weight and weight gain in response to high fat/high cholesterol diet in mice.
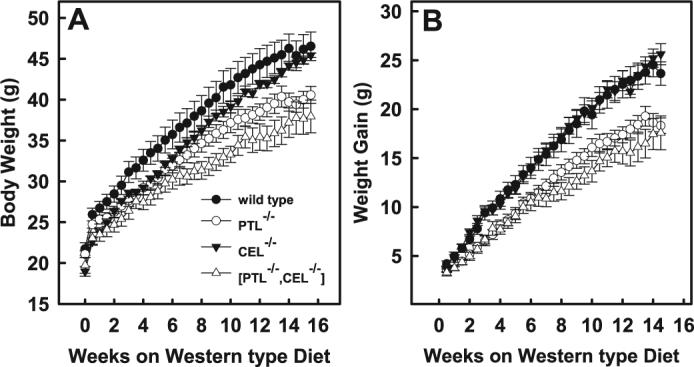
Male mice consuming a chow diet were transferred to a high fat/high cholesterol diet at 9 weeks of age. Body weight was monitored twice per week. Total body weight (A) as well as the rate of body weight gain (B) is plotted as the means ± S.E. from n = 10 wild type (•), 10 PTL−/− (○), 9CEL−/− (▼), and 9 PTL−/−,CEL−/− (▽) mice. The body weight of PTL−/− mice was statistically different from wild type counterparts at 3−4.5, 9.5, and 10.5−13.5 weeks at p < 0.05 and 14−15.5 weeks at p < 0.01. The body weight of CEL−/− mice was statistically different from wild type counterparts at 0, 1.5, 2, and 4 weeks at p < 0.05 and 0.5−1 week at p < 0.01. The body weight of PTL−/−,CEL−/− mice was statistically different from wild type counterparts at 0.5−3.5, 5−6.5, and 14.5 weeks at p < 0.05 and 4, 4.5, 7−14, and 15−15.5 weeks at p < 0.01. The body weight gain of PTL−/− mice was statistically different from wild type counterparts at 1.5−2.5, 4, 5−9.5, 10.5, and 13.5 weeks at p < 0.05 and 3, 3.5, 4.5, 11−13, and 14−14.5 weeks at p < 0.01. The body weight gain of PTL−/−,CEL−/− mice was statistically different from wild type counterparts at 1−2, 5−6.5, 10, and 14.5 weeks at p < 0.05 and 2.5−3, 4, 4.5, 7−9.5, and 10.5−14 weeks at p < 0.01. The body weight gain of CEL−/− mice was not different from wild type controls for the duration of this study.
TABLE 2. Daily food consumption in mice.
Mice consuming a high fat/high cholesterol Western-type diet for 17 weeks were housed individually. Food hoppers were weighed every second day for 8 days. Mice were returned to their normal housing situations involving cohabitation with littermates for 1 month, and then the procedure for monitoring food intake was repeated. Data from 16 days of monitoring was averaged for n = 6 wild type, n = 6 PTL−/−, n = 7 CEL−/−, and n = 6 PTL−/−, CEL−/− mice and shown as the mean ± S.D. for each genotype. No statistically relevant differences in food intake between mice with various genotypes were detected.
| Genotype | Wild type | PTL−/− | CEL−/− | PTL−/−,CEL−/− |
|---|---|---|---|---|
| Food intake per day (g) | 3.10 ± 0.42 | 3.60 ± 0.35 | 3.29 ± 0.47 | 3.60 ± 0.53 |
Additional studies were also carried out using a quantitative NMR method to assess body composition of wild type, PTL−/−, CEL−/−, and PTL−/−,CEL−/− mice consuming either chow or Western-type diets. The body composition of all 4 groups of animals, as determined by percent of fat and lean body mass, was similar at 11, 16, and 25 weeks of age when consuming the low fat chow diet. In general, the chow-fed mice carried <15% fat mass and >85% lean body mass from 9 to 25 weeks of age (data not shown). When challenged with the high fat/high cholesterol diet, there were dramatic alterations in body composition compared with mice consuming the low fat chow diet. The alteration in body composition reflected changes in body weight in response to the high fat/high cholesterol diet. At 16.5 weeks of age, at which time the mice had been consuming the high fat/high cholesterol diet for 7.5 weeks, wild type mice carried 31.2% of their body mass as fat compared with 8.5% fat mass in age-matched chow-fed wild type mice (Fig. 5). At 25 weeks of age, when the animals had been consuming the high fat/high cholesterol diet for 16 weeks, body composition measurements showed that ∼40% of the body mass in wild type mice was attributed to fat mass (Fig. 5). In contrast, only 10.5% of body mass was due to fat mass in age-matched chow-fed wild type mice.
FIGURE 5. Assessment of body composition by NMR.
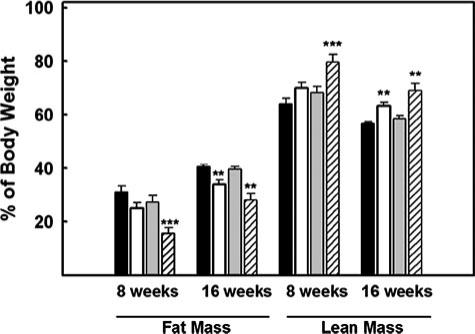
Quantitative nuclear magnetic resonance was used to measure fat and lean body mass of live mice that had been consuming chow or a Western-type diet. The method was applied to mice that were 16 and 25 weeks old and consuming the Western-type diet beginning at 9 weeks of age. Data shown are the means ± S.E. from n = 10 wild type (black bars), 10 PTL−/− (white bars), 9 CEL−/− (gray bars), and 9 PTL−/−,CEL−/− mice (hatched bars). **, p < 0.01; ***, p < 0.001 (differences from wild type mice analyzed at the same time point and consuming the same diet).
Consistent with results obtained from body weight determinations, resistance to diet-induced acquisition of fat mass was readily apparent in the PTL−/−,CEL−/− mice at 16.5 weeks of age (Fig. 5). Fat mass in the double knock-out mice was 15.5% less (p < 0.001), and lean body mass was 15.7% more (p < 0.001) than that observed in similarly fed age-matched wild type mice (Fig. 5). At 25 weeks of age, the PTL−/− mice on the high fat/ high cholesterol diet displayed 6.5% less fat mass (p < 0.01) and 6.7% more lean body mass (p < 0.001) than similarly fed age-matched wild type mice. The difference in diet-induced gain of fat mass was further exaggerated in the double knock-out mice lacking in both PTL and CEL, with 12.5% less fat mass (p < 0.01) and 12.3% more lean body mass (p < 0.01) than wild type mice.
DISCUSSION
Digestion and absorption of long-chain TAG are highly efficient processes with >90% of dietary TAG from each meal being hydrolyzed and absorbed by the intestine. Because of the fundamental importance of efficient dietary lipid absorption, redundancy exists in the repertoire of enzymes capable of hydrolyzing lipids in the gut, thereby ensuring availability of these nutrients. The purpose of this study was to determine how PTL and CEL contribute to dietary lipid absorption in vivo. Results showed that PTL and CEL both participate in lipid absorption and may serve a mutually compensatory role. Importantly, inactivation of either the PTL or CEL gene alone had minimal effects on TAG absorption, whereas disruption of both genes resulted in significant reduction of TAG absorption. Likewise, both PTL and CEL appear to participate in dietary retinyl ester hydrolysis and absorption. The decrease in retinyl palmitate absorption observed in PTL−/− mice but not in CEL−/− mice may imply a more prominent role for PTL in retinyl ester digestion. Nevertheless, CEL is also capable of catalyzing retinyl ester hydrolysis and mediating retinol absorption, as mice with both PTL and CEL deficiency absorbed significantly less retinyl ester than PTL-deficient mice. The difference in retinyl ester absorption efficiency between PTL−/− and PTL−/−,CEL−/− mice indicates a more avid retinyl ester hydrolytic activity of PTL than CEL in vivo, which was not previously appreciated from in vitro enzyme assays. Alternatively, this difference may reflect the abundance of PTL in the digestive tract compared with CEL and that total PTL deficiency results in insufficient enzyme activity in the intestinal lumen for retinyl ester hydrolysis.
An important observation made in the current study is that although PTL and CEL deficiency dramatically reduced dietary TAG and retinyl ester absorption, a substantial amount (∼50 − 60%) of these lipids was still absorbed in PTL−/−,CEL−/− mice. Therefore, at least one other lipolytic enzyme must be present in the intestinal lumen to catalyze TAG and retinyl ester hydrolysis in the absence of PTL and CEL. A likely candidate is PTLRP2, another enzyme secreted by the pancreas with avid in vitro TAG hydrolytic activity (15). The pancreas expresses PTLRP2 before birth, and its expression level persists at a lower level into adulthood (16). In contrast, the pancreas does not express PTL until near the suckling/weaning transition period (17). It is noteworthy that suckling PTLRP2−/− mice have steatorrhea and fat malabsorption with major increases of undigested and partially digested TAG in the feces (18). In addition, weight gain of suckling PTLRP2−/− mice is reduced compared with that of wild type mice, which is consistent with their diminished ability to acquire calories from dietary fat. Those data clearly documented that in the absence of PTL, PTLRP2 is capable of catalyzing TAG digestion and mediating fat absorption in vivo. In the current study we showed no significant difference in the expression level of PTLRP2 between wild type, PTL−/−, CEL−/−, and PTL−/−,CEL−/− mice (Table 1). Thus, the low level of PTLRP2 expression in the digestive tract may be responsible for the luminal lipolytic activity that facilitates TAG digestion and absorption in the PTL−/−,CEL−/− mice. The results showing dramatic decrease of fat absorption efficiency in colipase knock-out mice (19) but marginal decrease observed in PTL−/− mice together with the requirement of colipase for both PTL and PTLRP2 activity are supportive of this possibility. Mice with defective expression of PTL, CEL, and PTLRP2 are now being generated to test this hypothesis. Additionally, a phospholipase B residing in brush border membranes was also shown to hydrolyze retinyl esters in vitro (20). Thus, this enzyme may also participate in retinyl ester hydrolysis and absorption, particularly in the [PTL−/−, CEL−/−] mice.
Our previous study showing that PTL deficiency reduces cholesterol absorption efficiency was confirmed and extended in the current study, with results showing additional reduction of cholesterol absorption efficiency in mice lacking both PTL and CEL. The reduced cholesterol absorption efficiency observed in these animals is consistent with our previous hypothesis that efficient TAG digestion in the proximal intestine is necessary for cholesterol partitioning to micelles for subsequent absorption by enterocytes. The deficiency in PTL alone or in combination with CEL delays TAG hydrolysis to the distal intestine (1) where the cholesterol is less efficiently taken up (4, 5). The lower fasting cholesterol levels observed in PTL−/− and PTL−/−,CEL−/− mice consuming the Western-type diet (Table 1) is consistent with the reduced cholesterol absorption efficiency in these animals.
In contrast to the digestion and absorption of TAG, retinyl ester, and unesterified cholesterol, intestinal absorption of cholesteryl esters was dramatically reduced in CEL−/− mice but was unaffected in PTL−/− mice. Thus, PTL failed to compensate for the lack of CEL in cholesteryl ester digestion in the intestinal lumen. Inactivation of the PTL gene in CEL-deficient mice also did not cause additional inhibition of cholesteryl ester absorption compared with that observed in CEL−/− mice. Taken together these results indicated that CEL is the only enzyme in the digestive tract responsible for cholesteryl ester hydrolysis. The difference between cholesteryl ester and retinyl ester absorption in these mouse models illustrated their unique metabolic fate, suggesting that studies monitoring dietary retinyl ester metabolism as a surrogate for cholesteryl ester metabolism should take into consideration whether they are substrates for these and possibly other lipolytic enzymes.
Finally, the results of this study also revealed that the reduction of dietary TAG absorption due to PTL and CEL inactivation significantly reduced susceptibility to high fat/ high cholesterol diet-induced obesity. This observation has direct clinical implications. Currently, the only strategy to reduce obesity through inhibition of dietary lipid absorption is via tetrahydrolipstatin (Orlistat) therapy (21). Tetrahydrolipstatin inhibits all lipolytic enzyme activities in the digestive tract (22, 23); therefore, its use needs to be titrated carefully to avoid the unpleasant adverse effects of fecal spotting (24, 25). In this study we showed that inactivation of both PTL and CEL without the inhibition of other lipolytic enzymes in the digestive tract is sufficient to provide resistance to diet-induced obesity. Thus, the development of pharmacological strategies aimed at specific inhibition of PTL and CEL may be warranted to suppress diet-induced obesity without the adverse consequences of fat-soluble vitamin deficiency or steatorrhea that culminate upon inhibition of all lipolytic enzymes in the digestive tract.
Acknowledgments—
The excellent technical assistance of Joshua Bas-ford, Beth Coburn, Andy Thomas, and Todd Greer are gratefully appreciated. The fat absorption studies were performed at the University of Cincinnati Mouse Metabolic Phenotype Center, supported by National Institutes of Health Grant U24 DK59630.
Footnotes
The abbreviations used are: TAG, triacylglycerol; CEL, carboxyl ester lipase; PTL, pancreatic triglyceride lipase; PTLRP, PTL-related protein.
This work was supported by National Institutes of Health Grant DK067416. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Huggins KW, Camarota LM, Howles PN, Hui DY. J. Biol. Chem. 2003;278:42899–42905. doi: 10.1074/jbc.M303422200. [DOI] [PubMed] [Google Scholar]
- 2.Howles PN, Carter CP, Hui DY. J. Biol. Chem. 1996;271:7196–7202. doi: 10.1074/jbc.271.12.7196. [DOI] [PubMed] [Google Scholar]
- 3.Weng W, Li L, van Bennekum AM, Potter SH, Harrison EH, Blaner WS, Breslow JL, Fisher EA. Biochemistry. 1999;38:4143–4149. doi: 10.1021/bi981679a. [DOI] [PubMed] [Google Scholar]
- 4.Borgstrom B. J. Clin. Investig. 1960;39:809–815. doi: 10.1172/JCI104100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sylven C, Nordstrom C. Scand. J. Gastroenterol. 1970;5:57–63. [PubMed] [Google Scholar]
- 6.Hui DY. Biochim. Biophys. Acta. 1996;1303:1–14. doi: 10.1016/0005-2760(96)00085-9. [DOI] [PubMed] [Google Scholar]
- 7.van Bennekum AM, Fisher EA, Blaner WS, Harrison EH. Biochemistry. 2000;39:4900–4906. doi: 10.1021/bi9927235. [DOI] [PubMed] [Google Scholar]
- 8.Jandacek RJ, Heubi JE, Tso P. Gastroenterology. 2004;127:139–144. doi: 10.1053/j.gastro.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 9.Palmer WK, Emeson EE, Johnston TP. Atherosclerosis. 1998;136:115–123. doi: 10.1016/s0021-9150(97)00193-7. [DOI] [PubMed] [Google Scholar]
- 10.Taicher GZ, Tinsley FC, Reiderman A, Heiman ML. Anal. Bioanal. Chem. 2003;377:990–1002. doi: 10.1007/s00216-003-2224-3. [DOI] [PubMed] [Google Scholar]
- 11.Agellon LB, Drover VAB, Cheema SK, Gbaguidi GF, Walsh A. J. Biol. Chem. 2002;277:20131–20134. doi: 10.1074/jbc.C200105200. [DOI] [PubMed] [Google Scholar]
- 12.Kirby RJ, Zheng S, Tso P, Howles PN, Hui DY. J. Biol. Chem. 2002;277:4104–4109. doi: 10.1074/jbc.M107549200. [DOI] [PubMed] [Google Scholar]
- 13.Cohn JS, Johnson EJ, Millar JS, Cohn SD, Milne RW, Marcel YL, Russell RM, Schaefer EJ. J. Lipid Res. 1993;34:2033–2040. [PubMed] [Google Scholar]
- 14.Le NA, Coates PM, Gallagher PR, Cortner JA. Metabolism. 1997;46:584–594. doi: 10.1016/s0026-0495(97)90198-0. [DOI] [PubMed] [Google Scholar]
- 15.Lowe ME. J. Lipid Res. 2002;43:2007–2016. doi: 10.1194/jlr.r200012-jlr200. [DOI] [PubMed] [Google Scholar]
- 16.Lowe ME. Biochimie (Paris) 2000;82:997–1004. doi: 10.1016/s0300-9084(00)01184-6. [DOI] [PubMed] [Google Scholar]
- 17.Payne RM, Sims HF, Jennens ML, Lowe ME. Am. J. Physiol. 1994;266:G914–G921. doi: 10.1152/ajpgi.1994.266.5.G914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lowe ME, Kaplan MH, Jackson-Grusby L, D'Agostino D, Grusby MJ. J. Biol. Chem. 1998;273:31215–31221. doi: 10.1074/jbc.273.47.31215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.D'Agostino D, Cordle RA, Kullman J, Erlanson-Albertsson C, Muglia LJ, Lowe ME. J. Biol. Chem. 2002;277:7170–7177. doi: 10.1074/jbc.M108328200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rigtrup KM, Kakkad B, Ong DE. Biochemistry. 1994;33:2661–2666. doi: 10.1021/bi00175a039. [DOI] [PubMed] [Google Scholar]
- 21.Padwal RS, Majumdar SR. Lancet. 2007;369:71–77. doi: 10.1016/S0140-6736(07)60033-6. [DOI] [PubMed] [Google Scholar]
- 22.Borgstrom B. Biochim. Biophys. Acta. 1988;962:308–316. doi: 10.1016/0005-2760(88)90260-3. [DOI] [PubMed] [Google Scholar]
- 23.Fernandez E, Borgstrom B. Biochim. Biophys. Acta. 1989;1001:249–255. doi: 10.1016/0005-2760(89)90107-0. [DOI] [PubMed] [Google Scholar]
- 24.Fox M, Thumshirn M, Menne D, Stutz B, Fried M, Schwizer W. Aliment. Pharmacol. Ther. 2004;19:311–321. doi: 10.1111/j.1365-2036.2004.01848.x. [DOI] [PubMed] [Google Scholar]
- 25.Armand M. Curr. Opin. Clin. Nutr. Metab. Care. 2007;10:156–164. doi: 10.1097/MCO.0b013e3280177687. [DOI] [PubMed] [Google Scholar]