

Abstract
Gemfibrozil, a lipid-lowering drug, inhibited cytokine-induced production of NO and the expression of inducible nitric-oxide synthase (iNOS) in human U373MG astroglial cells and primary astrocytes. Similar to gemfibrozil, clofibrate, another fibrate drug, also inhibited the expression of iNOS. Inhibition of human iNOS promoter-driven luciferase activity by gemfibrozil in cytokine-stimulated U373MG astroglial cells suggests that this compound inhibits the transcription of iNOS. Since gemfibrozil is known to activate peroxisome proliferator-activated receptor-α (PPAR-α), we investigated the role of PPAR-α in gemfibrozil-mediated inhibition of iNOS. Gemfibrozil induced peroxisome proliferator-responsive element (PPRE)-dependent luciferase activity, which was inhibited by the expression of ΔhPPAR-α, the dominant-negative mutant of human PPAR-α. However, ΔhPPAR-α was unable to abrogate gemfibrozil-mediated inhibition of iNOS suggesting that gemfibrozil inhibits iNOS independent of PPAR-α. The human iNOS promoter contains consensus sequences for the binding of transcription factors, including interferon-γ (IFN-γ) regulatory factor-1 (IRF-1) binding to interferon-stimulated responsive element (ISRE), signal transducer and activator of transcription (STAT) binding to γ-activation site (GAS), nuclear factor-κB (NF-κB), activator protein-1 (AP-1), and CCAAT/enhancer-binding protein β (C/EBPβ); therefore, we investigated the effect of gemfibrozil on the activation of these transcription factors. The combination of interleukin (IL)-1β and IFN-γ induced the activation of NF-κB, AP-1, C/EBPβ, and GAS but not that of ISRE, suggesting that IRF-1 may not be involved in cytokine-induced expression of iNOS in human astrocytes. Interestingly, gemfibrozil strongly inhibited the activation of NF-κB, AP-1, and C/EBPβ but not that of GAS in cytokine-stimulated astroglial cells. These results suggest that gemfibrozil inhibits the induction of iNOS probably by inhibiting the activation of NF-κB, AP-1, and C/EBPβ and that gemfibrozil, a prescribed drug for humans, may further find its therapeutic use in neuroinflammatory diseases.
It is now increasingly clear that glial cells (astrocytes and microglia) in the central nervous system (CNS)1 induce the expression of inducible nitric-oxide synthase (iNOS) and the production of NO in response to proinflammatory cytokines, including interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α), and interferon-γ (IFN-γ) (1-4). Although the NO produced by iNOS has bactericidal and tumoricidal properties, it also plays an important role in pathophysiologies of inflammatory neurological diseases including demyelinating disorders (e.g. multiple sclerosis, experimental allergic encephalopathy), neurodegenerative disorder like Alzheimer’s disease, and in ischemic and traumatic brain injuries associated with the activation of glial cells and the production of proinflammatory cytokines (5-8). NO derived from activated glial cells is assumed to contribute to oligodendrocyte degeneration in demyelinating diseases and neuronal death during ischemia and trauma (5, 6). Therefore, characterization of intracellular pathways required to transduce the signal from the cell surface to the nucleus for the induction of iNOS is an active area of investigation, since compounds capable of antagonizing signaling steps for the induction of iNOS may have therapeutic effect in NO-mediated pathophysiological conditions.
Peroxisome proliferator-activated receptors (PPARs), members of the nuclear hormone receptor superfamily, have been implicated in a variety of human diseases (9). Three isotypes have been described to date, PPAR-α, PPAR-β, and PPAR-γ (9). Activation of PPAR-α mainly leads to the induction of a variety of genes such as those coding for the enzymes for β- and ω-oxidation of fatty acids (10). Gemfibrozil, an activator of PPAR-α, has been often prescribed in patients to lower the level of triglycerides (11, 12). This drug decreases the risk of coronary heart disease by increasing the level of high density lipoprotein cholesterol and decreasing the level of low density lipoprotein cholesterol (11, 12). Activation of PPAR-α is also capable of modifying the stress response by activation of heat shock factor 1 (HSF-1) and induction of HSP70 (13, 14). Recently it has been shown that activation of HSP70 inhibits the expression of iNOS in astrocytes (15), suggesting that the expression of iNOS may also be regulated by activators of PPAR-α. Therefore, we investigated the effect of gemfibrozil on the expression of iNOS in cytokine-stimulated human U373MG astroglial cells and primary astrocytes. In the current work, we present evidence that gemfibrozil markedly inhibited the expression of iNOS and the production of NO in human astrocytes independent of PPAR-α. In addition, reporter gene assays reveal that gemfibrozil specifically inhibited cytokine-induced activation of NF-κB, AP-1, and C/EBPβ but not that of GAS. These results raise the possibility that gemfibrozil, a common lipid-lowering drug, may be of therapeutic value in human neuroinflammatory diseases.
MATERIALS AND METHODS
Reagents
Fetal bovine serum and DMEM/F-12 were from Invitrogen. Human recombinant IFN-γ, IL-1β, and TNF-α were purchased from R & D Systems. l-NG-Monomethylarginine (l-NMA) and d-NG-monomethylarginine (d-NMA) were obtained from Biomol. Gemfibrozil and clofibrate were obtained from Sigma. Antibodies against human macrophage iNOS were obtained from Calbiochem. 125I-Labeled protein A and [α-32P]dCTP (3000 Ci/mmol) were purchased from PerkinElmer Life Sciences. Peroxisome proliferator-responsive element (PPRE)-dependent reporter construct (tk-PPREx3-Luc) and dominant-negative mutant of CCAAT/enhancer-binding protein β (ΔC/EBPβ) were kindly provided by Dr. Ronald M. Evans of The Salk Institute and Dr. Steve Smale of the University of California at Los Angeles, respectively.
Preparation of Human Astrocytes
Human CNS tissue was obtained from the Human Embryology Laboratory, University of Washington, Seattle. The CNS tissue from each specimen was processed separately and independently, as were subsequent cell cultures; there was no pooling of CNS tissue from distinct specimens. All the experimental protocols were reviewed and approved by the Institutional Review Board (IRB number 224-01-FB) of the University of Nebraska Medical Center. These cells were grown in a serum-free, defined medium (B16) enriched with 5 ng of basic fibroblast growth factor per ml for optimal growth of astrocytes and for the suppression of fibroblast growth (16). By immunofluorescence assay, these cultures homogeneously expressed glial fibrillary acidic protein (GFAP). Cells were trypsinized, subcultured, and stimulated with cytokines in serum-free DMEM/F-12 medium to induce the production of NO. The human U373MG astrocytoma cell line, purchased from the American Type Culture Collection (ATCC), was also maintained and stimulated under similar conditions.
Assay for NO Synthesis
Synthesis of NO was determined by assay of culture supernatant for nitrite, a stable reaction product of NO with molecular oxygen, using Griess reagent (1-4). Briefly, 400 μl of culture supernatant was allowed to react with 200 μl of Griess reagent and incubated at room temperature for 15 min. The optical density of the assay samples was measured spectrophotometrically at 570 nm. Fresh culture medium served as the blank in all experiments. Nitrite concentrations were calculated from a standard curve derived from the reaction of NaNO2 in the assay. Protein was measured by the procedure of Bradford (17).
Immunoblot Analysis for iNOS
Immunoblot analysis for iNOS was carried out as described earlier (2-4). Briefly, cells were detached by scraping, washed with Hanks’ buffer, and homogenized in 50 mm Tris-HCl (pH 7.4) containing protease inhibitors (1 mm phenylmethylsulfonyl fluoride, 5 μg/ml aprotinin, 5 μg/ml antipain, 5 μg/ml pepstatin A, and 5 μg/ml leupeptin). After electrophoresis the proteins were transferred onto a nitrocellulose membrane, and the iNOS band was visualized by immunoblotting with antibodies against human iNOS and 125I-labeled protein A (2-4).
RNA Isolation and Northern Blot Analysis
Cells were taken out of the culture dishes directly by adding Ultraspec-II RNA reagent (Biotecx Laboratories, Inc.), and total RNA was isolated using Ultraspec-II RNA reagent (Biotecx Laboratories Inc.) according to the manufacturer’s protocol. For Northern blot analyses, 20 μg of total RNA was electrophoresed on 1.2% denaturing formaldehyde-agarose gels, electrotransferred to Hybond-Nylon Membrane (Amersham Biosciences) and hybridized at 68 °C with 32P-labeled cDNA probe using Express Hyb hybridization solution (Clontech) as described by the manufacturer. The cDNA probe was made by polymerase chain reaction amplification using two primers (forward primer, 5′-CTC CTT CAA AGA GGC AAA AAT A-3′; reverse primer, 5′-CAC TTC CTC CAG GAT GTT GT-3′) (2-4, 18). After hybridization filters were washed two or three times in solution I (2× SSC, 0.05% SDS) for 1 h at room temperature followed by solution II (0.1× SSC, 0.1% SDS) at 50 °C for another hour. The membranes were then dried and exposed to x-ray films (Eastman Kodak Co.). The same amount of RNA was hybridized with probe for glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
Assay of iNOS Promoter-driven Reporter Activity
Construction of phiNOS(7.2)Luc, the 7.2-kb human iNOS promoter-luciferase construct, has been described previously (19). Cells plated at 50–60% confluence in six-well plates were cotransfected with 1 μg of phiNOS(7.2)Luc and 50 ng of pRL-TK (a plasmid encoding Renilla luciferase, used as transfection efficiency control; Promega) by LipofectAMINE Plus (Invitrogen) following manufacturer’s protocol (3, 4). Twentyfour h after transfection, cells were treated with different stimuli for 12 h. Firefly and Renilla luciferase activities were obtained by analyzing total cell extract according to standard instructions provided in the Dual Luciferase Kit (Promega) in a TD-20/20 Luminometer (Turner Designs). Relative luciferase activity of cell extracts was typically represented as the ratio of firefly luciferase value/Renilla luciferase value × 10−3.
Assay of Transcriptional Activities of Different Proinflammatory Transcription Factors
Cells plated at 50–60% confluence in six-well plates were cotransfected with 1 μg of either pNF-κB-Luc (NF-κB-dependent reporter construct), pAP-1-Luc (AP-1-dependent reporter construct), pC/EBPβ-Luc (C/EBPβ-dependent reporter construct), pGAS-Luc (GAS-dependent reporter construct), or pISRE-Luc (ISRE-dependent reporter construct) and 50 ng of pRL-TK using LipofectAMINE Plus. Construction of pC/EBPβ-Luc has been described earlier (4). This C/EBPβ-sensitive promoter contains four consensus C/EBPβ-binding sites. Other reporter constructs (pNF-κB-Luc, pAP-1-Luc, pGAS-Luc, and pISRE-Luc) were obtained from Stratagene. After 24 h of transfection, cells were treated with different stimuli for 6 h. Firefly and Renilla luciferase activities were obtained as described above.
Statistics
Statistical comparisons were made using one-way analysis of variance followed by Student’s t test.
RESULTS
Gemfibrozil Inhibits the Expression of iNOS in Cytokine-stimulated Human U373MG Astroglial Cells
Cells were cultured in serum-free media in the presence of IL-1β and IFN-γ. It is evident from Table I that IL-1β and IFN-γ alone were poor inducers of NO production. However, marked induction of NO production was observed by the combination of IL-1β and IFN-γ. This combination of cytokines was used to induce the production of NO in subsequent studies. The inhibition of cytokine-induced production of NO by arginase (an enzyme that degrades the substrate, l-arginine, of NOS) and l-NMA (a competitive inhibitor of NOS) but not by d-NMA (a negative control of l-NMA) suggests that the combination of IL-1β and IFN-γ induces the production of NO in U373MG astroglial cells through NOS-mediated arginine metabolism (Table I). Next we examined the effect of gemfibrozil, an activator of PPAR-α (20), on the cytokine-induced nitrite production in U373MG glial cells. Gemfibrozil itself was neither stimulatory nor much inhibitory to NO production in control cells. However, gemfibrozil, when added 2 h before the addition of cytokines, markedly inhibited cytokine-induced production of NO (Table I).
Table I.
Induction of NO production in human U373MG astroglial cells
U373MG glial cells preincubated in serum-free DMEM/F-12 for 1 h with arginase, l-NMA or d-NMA received the combination of IL-1β and IFN-γ. After 24 h of incubation, nitrite concentrations in the supernatants were measured as described under “Materials and Methods.” Data are expressed as the mean ± S.D. of three different experiments. The concentrations of different compounds were as follows: IL-1β, 10 ng/ml; IFN-γ, 10 units/ml; arginase, 100 units/ml; l-NMA, 0.1 mm; D-NMA, 0.1 mm; gemfibrozil, 200 μm.
| Treatments | Nitrite production |
|---|---|
| μg/mg protein/24 h | |
| Control | 6.2 ± 0.8 |
| IL-1β only | 18.6 ± 1.9 |
| IFN-γ only | 15.9 ± 0.9 |
| IL-1β + IFN-γ | 196.3 ± 26.6 |
| IL-1β + IFN-γ + arginase | 21.2 ± 1.8a |
| IL-1β + IFN-γ + l-NMA | 19.1 ± 2.1a |
| IL-1β + IFN-γ + d-NMA | 194.7 ± 21.3 |
| Gemfibrozil only | 5.9 ± 1.1 |
| IL-1β + IFN-γ + gemfibrozil | 20.8 ± 2.8a |
p < 0.001 versus IL-1β + IFN-γ.
To determine whether inhibition of cytokine-induced NO production by gemfibrozil was simply due to delayed induction, we measured NO concentrations in cytokine-stimulated cultures maintained up to 48 h. When cells were stimulated in the absence of gemfibrozil, NO was detected in culture supernatants after 8 h and increased progressively thereafter for 48 h, the duration of the experiment (Fig. 1). However, when 200 μm gemfibrozil was added 2 h before the addition of the combination of IL-1β and IFN-γ, production of NO was significantly inhibited (Fig. 1). In our studies, maximal suppression of NO production was observed when gemfibrozil was added 2 h before the addition of cytokines (data not shown). When gemfibrozil was added after the addition of cytokines, the extent of inhibition progressively decreased (data not shown). It is evident from Fig. 2A that gemfibrozil dose-dependently inhibited cytokine-induced production of NO. Although at 50 μm concentration, gemfibrozil was not a potent inhibitor of cytokine-induced NO production, it inhibited the induction of NO production by more than 80% at 200 μm concentration. To understand the mechanism of inhibition, we examined the effect of gemfibrozil on protein and mRNA level of iNOS in cytokine-stimulated cells. Consistent with the effect of gemfibrozil on cytokine-induced production of NO, gemfibrozil dose-dependently inhibited cytokine-induced expression of iNOS protein (Fig. 2B) and mRNA (Fig. 2C).
Fig. 1. Time course of cytokine-induced NO production and its suppression by gemfibrozil in human U373MG astroglial cells.
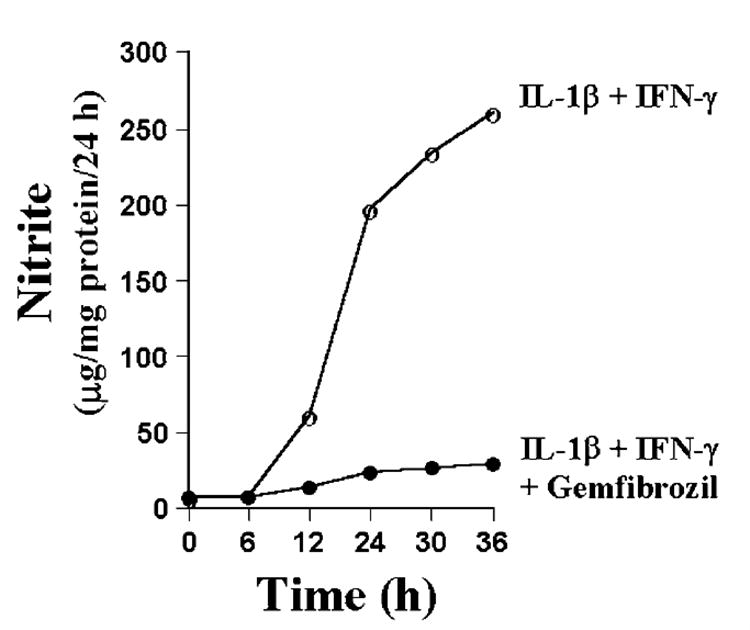
Cells preincubated with 200 μm gemfibrozil for 2 h in serum-free DMEM/F-12 received the combination of IL-1β (10 ng/ml) and IFN-γ (10 units/ml). At different hours of stimulation, the concentration of nitrite was measured in supernatants using the “Griess reagent” as described under “Materials and Methods.” Data are expressed as the mean of two separate experiments.
Fig. 2. Gemfibrozil dose-dependently inhibits the expression of iNOS in cytokine-stimulated human U373MG astroglial cells.
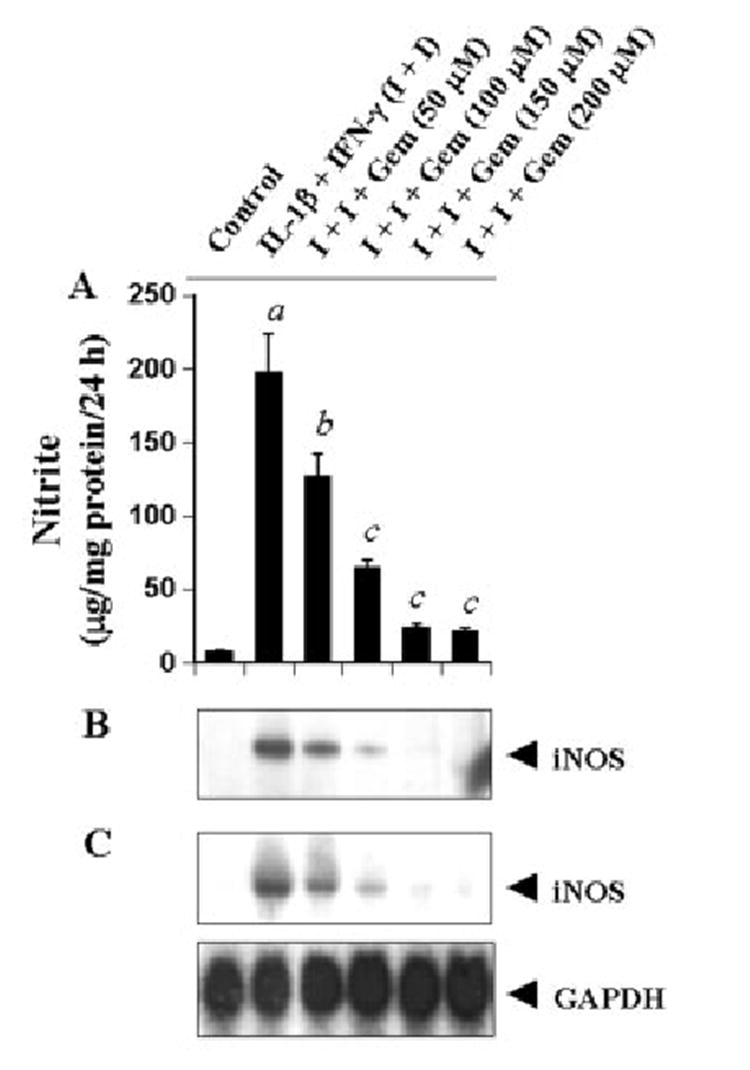
Cells preincubated with different concentrations of gemfibrozil for 2 h in serum-free DMEM/F-12 received the combination of IL-1β (10 ng/ml) and IFN-γ (10 units/ml). A, after 24 h of stimulation, the concentration of nitrite was measured in the supernatants. Data are mean ± S.D. of three different experiments. a, p < 0.001 versus control; b, p < 0.005 versus IL-1β+IFN-γ; c, p < 0.001 versus IL-1β+IFN-γ. B, cell homogenates were immunoblotted with antibodies against mouse macrophage iNOS as described under “Materials and Methods.” C, after 6 h of stimulation, total RNA was isolated, and Northern blot analysis for iNOS mRNA was carried out as described under “Materials and Methods.”
Next we investigated the possibility whether gemfibrozil inhibited cytokine-induced expression of iNOS mRNA by decreasing the stability of iNOS mRNA. Human U373MG astroglial cells were stimulated with the combination of IL-1β and IFN-γ under serum-free condition. After 6 h of stimulation, cells were treated with actinomycin D (an inhibitor of RNA synthesis) in the presence or absence of 200 μm of gemfibrozil. At different h of treatment with actinomycin D, the level of iNOS mRNA was analyzed by Northern blot. It is apparent from figure 3, A and B, that the relative rate of degradation of iNOS mRNA (iNOS/GAPDH) in the presence or absence of gemfibrozil at different time periods remained almost same suggesting that gemfibrozil-mediated inhibition of iNOS mRNA is not due to any alteration of the stability of iNOS mRNA.
Fig. 3. Effect of gemfibrozil on the stability of iNOS mRNA in human U373MG astroglial cells.
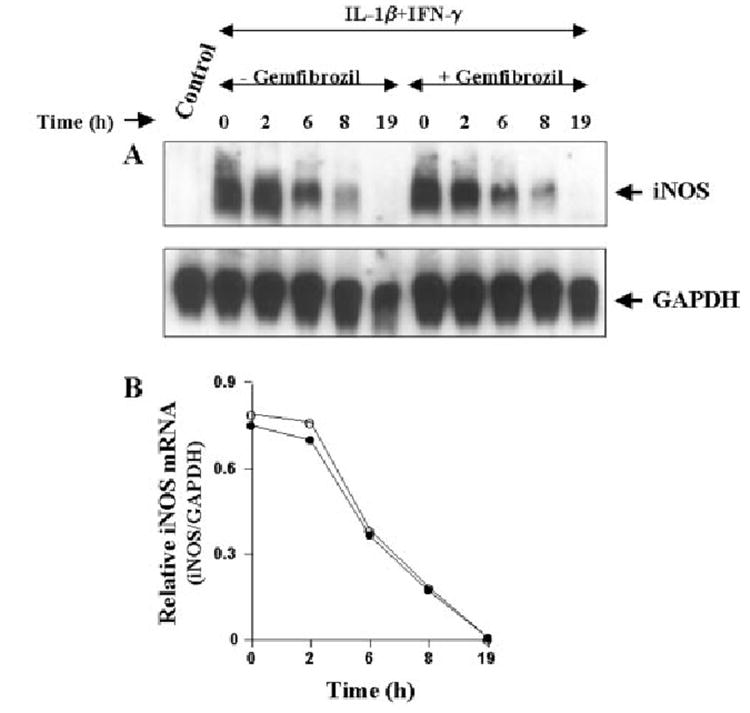
Cells were stimulated with the combination of IL-1β (10 ng/ml) and IFN-γ (10 units/ml) under serum-free condition. After 6 h of stimulation by cytokines, cells were treated with 3 μg/ml actinomycin D in the presence or absence of gemfibrozil, and cells were analyzed for iNOS RNA by Northern blot (A) at different hours of treatment. B, the relative level of iNOS mRNA (iNOS/GAPDH) at different hours of treatment was determined after densitometric scanning of iNOS and GAPDH bands by a Fluor Chem 8800 Imaging System (Alpha Innotech Corp.). The data are expressed as the average of two separate experiments.
Inhibition of Cytokine-induced Expression of iNOS by Clofibrate in Human U373MG Astroglial Cells
To investigate whether other fibrate drugs are also capable of inhibiting cytokine-induced production of NO and expression of iNOS in astrocytes, we examined the effect of clofibrate. Clofibrate is also a hypolipidemic drug that activates PPAR-α and induces proliferation of peroxisomes in rats and mice (9, 20, 21). Similar to gemfibrozil, clofibrate itself was neither stimulatory nor much inhibitory to NO production; however, it dose-dependently inhibited the production of NO (Fig. 4A) and the expression of iNOS protein (Fig. 4B) in cytokine-stimulated U373MG astroglial cells. These studies suggest that fibrate drugs, in general, are inhibitory to cytokine-induced expression of iNOS in human astroglial cells.
Fig. 4. Clofibrate dose-dependently inhibits the expression of iNOS in cytokine-stimulated human U373MG astroglial cells.
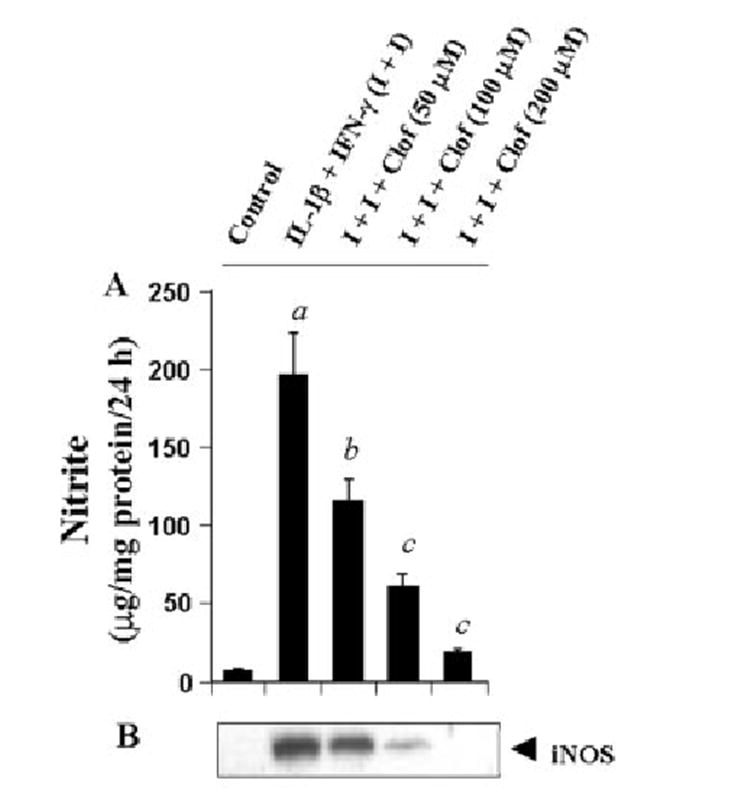
Cells preincubated with different concentrations of clofibrate for 2 h in serum-free DMEM/F-12 received the combination of IL-1β (10 ng/ml) and IFN-γ (10 units/ml). A, after 24 h of stimulation, the concentration of nitrite was measured in the supernatants. Data are mean ± S.D. of three different experiments. a, p < 0.001 versus control; b, p < 0.005 versus IL-1β + IFN-γ; c, p < 0.001 versus IL-1β + IFN-γ. B, cell homogenates were immunoblotted with antibodies against mouse macrophage iNOS.
Inhibition of Cytokine-induced Production of NO by Gemfibrozil in Human Primary Astrocytes
Human primary astrocytes have been shown to induce the expression of iNOS in the presence of different proinflammatory cytokines (22-24). Since gemfibrozil potently inhibited the expression of iNOS in human U373MG astroglial cells, we examined the effect of gemfibrozil on cytokine-induced expression of iNOS in human primary astrocytes. Different cytokines alone were poor inducers of NO production (Table II). However, the combination of IL-1β and IFN-γ markedly induced the production of NO. The addition of TNF-α to the combination of IL-1β and IFN-γ did not further increase the production of NO (Table II). Although gemfibrozil itself had no effect on NO production in control cells, preincubation of human primary astrocytes with 200 μm of gemfibrozil for 2 h markedly inhibited cytokine-induced production of NO (Table II).
Table II. Gemfibrozil inhibits the induction of NO production in human primary astrocytes.
Human primary astrocytes preincubated in serum-free DMEM/F-12 for 2 h with 200 μm gemfibrozil, received IL-1β, IFN-γ, and TNF-α alone or in different combinations. After 24 h of incubation, nitrite concentrations in the supernatants. Data are expressed as the mean ± S.D. of three different experiments. The concentrations of different cytokines were as follows: IL-1β, 10 ng/ml; IFN-γ, 10 units/ml; TNF-α, 10 ng/ml.
| Treatments | Nitrite production |
|---|---|
| μg/mg protein/24 h | |
| Control | 5.3 ± 0.7 |
| IL-1β | 24.5 ± 2.5 |
| IFN-γ | 6.1 ± 0.7 |
| TNF-α | 5.8 ± 0.8 |
| Gemfibrozil | 5.1 ± 0.6 |
| IL-1β + IFN-γ | 215.3 ± 22.3 |
| IL-1β + IFN-γ + TNF-α | 219.2 ± 28.5 |
| IL-1β + IFN-γ + gemfibrozil | 24.8 ± 2.8a |
| IL-1β + IFN-γ + TNF-α + gemfibrozil | 25.7 ± 3.1b |
p < 0.001 versus IL-1β + IFN-γ.
p < 0.001 versus IL-1β + IFN-γ + TNF-α.
Gemfibrozil Inhibits Human iNOS Promoter-driven Luciferase Activity in Cytokine-stimulated Human U373MG Astroglial Cells
To understand the effect of gemfibrozil on the transcription of iNOS gene, U373MG glial cells were transfected with phiNOS(7.2)Luc, a construct containing the human iNOS promoter fused to the luciferase gene (19), and activation of this promoter was measured after stimulating the cells with cytokines in the presence or absence of gemfibrozil. The combination of IL-1β and IFN-γ induced iNOS promoter-driven luciferase activity by about 3.9-fold (Fig. 5). Consistent with the effect of gemfibrozil on the expression of iNOS, gemfibrozil itself had no effect on iNOS promoter-driven luciferase activity but it dose-dependently inhibited iNOS promoter-driven luciferase activity in cytokine-stimulated cells (Fig. 5), suggesting that gemfibrozil inhibits cytokine-induced production of NO and the expression of iNOS mRNA by inhibiting the activation of iNOS promoter.
Fig. 5. Gemfibrozil inhibits human iNOS promoter-derived luciferase activity in cytokine-stimulated human U373MG astroglial cells.
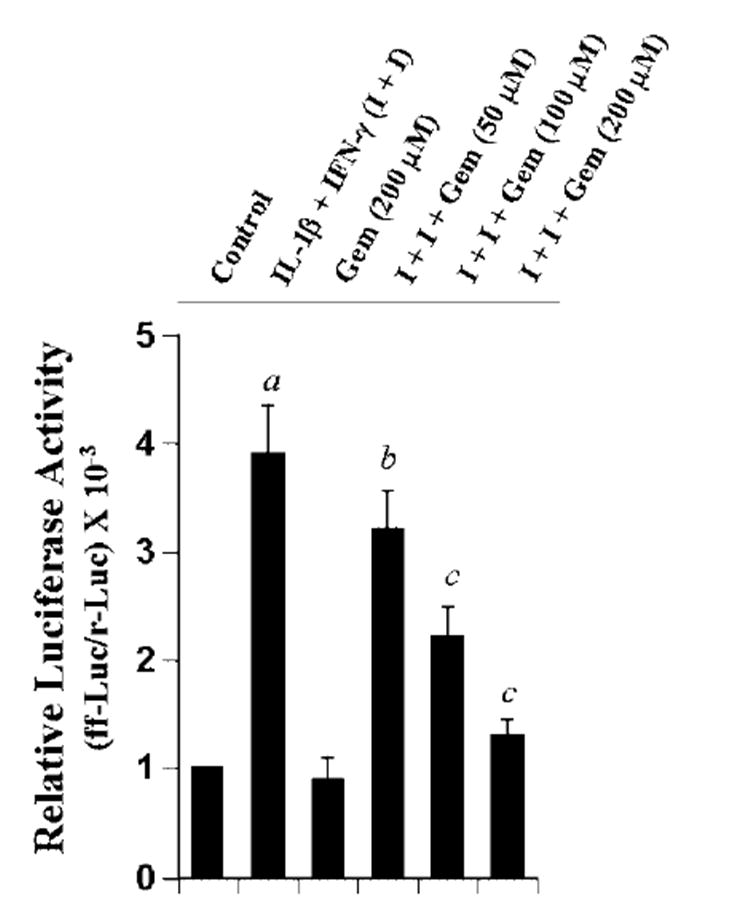
Cells plated at 50–60% confluence in six-well plates were cotransfected with 1 μg of phiNOS(7.2)Luc (a construct containing the human iNOS promoter fused to the luciferase gene) and 50 ng of pRL-TK (a plasmid encoding Renilla luciferase, used as a transfection efficiency control) using the LipofectAMINE Plus (Invitrogen). Twenty-four hours after transfection, cells received different concentrations of gemfibrozil. After 2 h of incubation, cells were stimulated with the combination of IL-1β (10 ng/ml) and IFN-γ (10 units/ml) for 12 h under serum-free condition. Firefly (ff-Luc) and Renilla (r-Luc) luciferase activities were obtained by analyzing the total cell extract as described under “Materials and Methods.” Data are mean ± S.D. of three different experiments. a, p < 0.001 versus control; b, p < 0.05 versus IL-1β + IFN-γ; c, p < 0.001 versus IL-1β + IFN-γ.
Role of PPAR-α in Gemfibrozil-mediated Inhibition of iNOS in Human U373MG Astroglial Cells
Since gemfibrozil is a known activator of PPAR-α (9, 20, 21), a member of the nuclear hormone receptor superfamily, we examined whether gemfibrozil inhibited the induction of iNOS through the activation of PPAR-α. PPARs bind to a consensus sequence known as PPRE (9). Therefore, to study the activation of PPAR, cells were transfected with tk-PPREx3-Luc, a PPRE-dependent luciferase construct, and luciferase activity was measured. As shown in Fig. 6A, gemfibrozil alone was able to induce PPRE-dependent luciferase activity in a dose-dependent manner and the maximum induction (~4-fold) was observed at 100 μm or higher concentration of gemfibrozil. In contrast, the combination of IL-1β and IFN-γ capable of inducing iNOS inhibited PPRE-dependent luciferase activity (Fig. 6A). However, gemfibrozil treatment blocked the inhibitory effect of cytokines on the activation of PPRE and stimulated PPRE-dependent luciferase activity over basal level even in the presence of cytokines (Fig. 6A). To analyze the role of PPAR-α in gemfibrozil-induced activation of PPRE, we used ΔhPPAR-α, the dominant-negative mutant of human PPAR-α (25). Marked abrogation (p < 0.001) of gemfibrozil-induced activation of PPRE (Fig. 6B) by the expression of ΔhPPAR-α but not that of the empty vector suggests that gemfibrozil induced PPRE-dependent reporter activity through the activation of PPAR-α in human astroglial cells.
Fig. 6. The dominant-negative mutant of human PPAR-α (ΔhPPAR-α) inhibits gemfibrozil-induced PPRE-dependent luciferase activity in human U373MG astroglial cells.
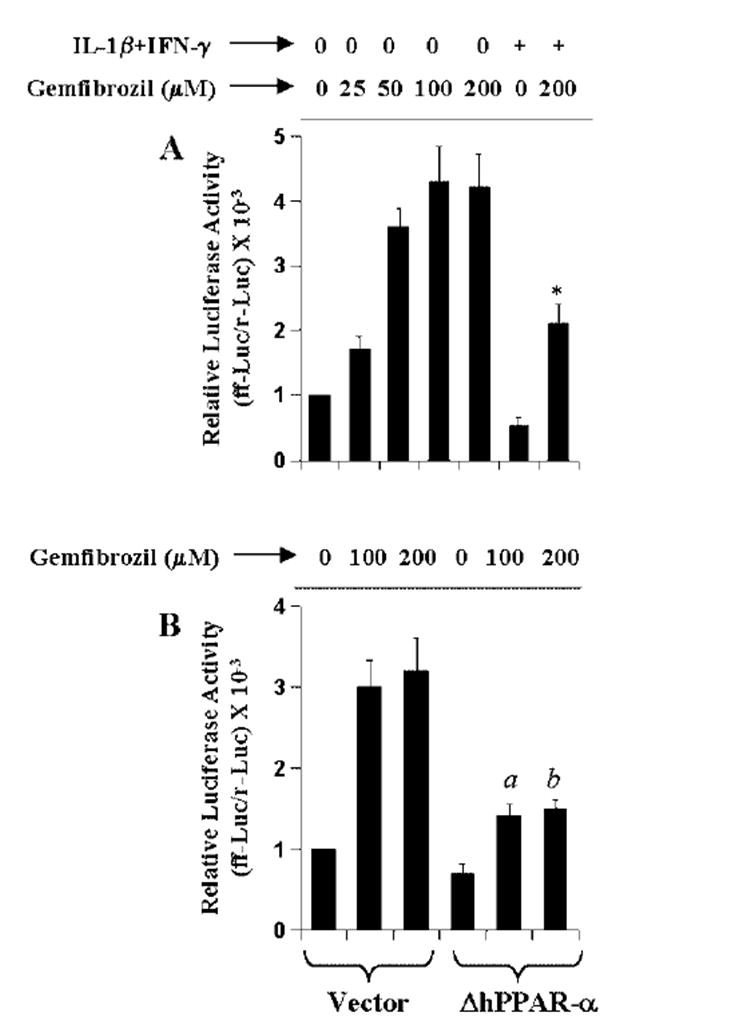
A, cells plated at 50–60% confluence in six-well plates were cotransfected with 1 μg of tk-PPREx3-Luc, a PPRE-dependent luciferase reporter construct, and 50 ng of pRL-TK using LipofectAMINE Plus. Twenty-four hours after transfection, cells were treated with different concentrations of gemfibrozil and/or the combination of IL-1β (10 ng/ml) and IFN-γ (10 units/ ml). After 6 h of incubation, firefly (ff-Luc) and Renilla (r-Luc) luciferase activities were assayed. Data are mean ± S.D. of three different experiments. *, p < 0.001 versus 200 μm gemfibrozil. B, cells were cotransfected with 0.5 μg of either ΔhPPAR-α or an empty vector and 1 μg of tk-PPREx3-Luc. All transfections also included 50 ng/μg of pRL-TK. Twenty-four hours after transfection, cells were treated with different concentrations of gemfibrozil. After 6 h of incubation, firefly (ff-Luc) and Renilla (r-Luc) luciferase activities were assayed. Data are mean ± S.D. of three different experiments. a, p < 0.001 versus 100 μm gemfibrozil; b, p < 0.001 versus 200 μm gemfibrozil.
Next we examined whether ΔhPPAR-α can block the inhibitory effect of gemfibrozil on the induction of iNOS. Cells were cotransfected with phiNOS(7.2)Luc, ΔhPPAR-α, and pRL-TK (a plasmid encoding Renilla luciferase, used as transfection efficiency control). After 24 h of transfection, cells were incubated with gemfibrozil for 2 h followed by stimulation with cytokines. Consistent, to the inhibitory effect of gemfibrozil on the activation of iNOS promoter (Fig. 5), this drug inhibited iNOS promoter-driven luciferase activity in empty vector-transfected cells (Fig. 7). However, despite the ability of ΔhPPAR-α to block gemfibrozil-mediated activation of PPRE (Fig. 6B), the expression of ΔhPPAR-α did not block the inhibitory effect of gemfibrozil on iNOS promoter-driven luciferase activity in cytokine-stimulated cells (Fig. 7). These results suggest that PPAR-α is not involved in gemfibrozil-mediated inhibition of iNOS in human astroglial cells.
Fig. 7. ΔhPPAR-α does not block gemfibrozil-mediated inhibition of iNOS promoter activation in cytokine-stimulated human U373MG astroglial cells .
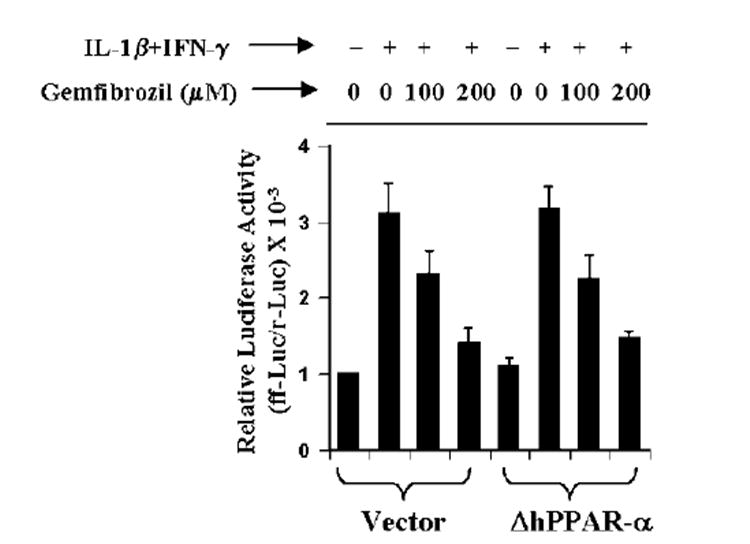
Cells were cotransfected with 0.5 μg of either ΔhPPAR-α or an empty vector, 0.5 μg of phiNOS(7.2)Luc, and 50 ng of pRL-TK. Twenty-four hours after transfection, cells were incubated with gemfibrozil for 2 h followed by stimulation with the combination of IL-1β and IFN-γ. After 12 h of incubation, firefly (ff-Luc) and Renilla (r-Luc) luciferase activities were assayed. Data are mean ± S.D. of three different experiments.
Effect of Gemfibrozil on the Activation of Proinflammatory Transcription Factors in Human U373MG Astroglial Cells
Different proinflammatory transcription factors are known to be involved in the transcription of iNOS (2-4, 26-30). Analysis of human iNOS promoter shows that it has consensus sequences for binding of several transcription factors such as NF-κB, AP-1, C/EBPβ, IRF-1 binding to ISRE, and STAT binding to GAS (29-31). Although several reports have established the involvement of NF-κB, AP-1, and STAT in the induction of iNOS in human cells (29, 32, 33), the role of C/EBPβ in the induction of human iNOS has not been established. Overexpression of dominant-negative molecules provides an effective tool with which to investigate the in vivo functions of different transcription factors and signaling molecules. Therefore, we used the dominant-negative mutant of C/EBPβ (ΔC/EBPβ) (34) to inhibit the activation of C/EBPβ. It is apparent from Fig. 8 that the expression of ΔC/EBPβ but not that of the empty vector inhibited iNOS promoter-driven luciferase activity significantly (p < 0.005) in cytokine-stimulated human U373MG astroglial cells, suggesting the involvement of C/EBPβ in the induction of iNOS in human astroglial cells.
Fig. 8. ΔC/EBPβ inhibits iNOS promoter-driven luciferase activity in cytokine-stimulated human U373MG astroglial cells.
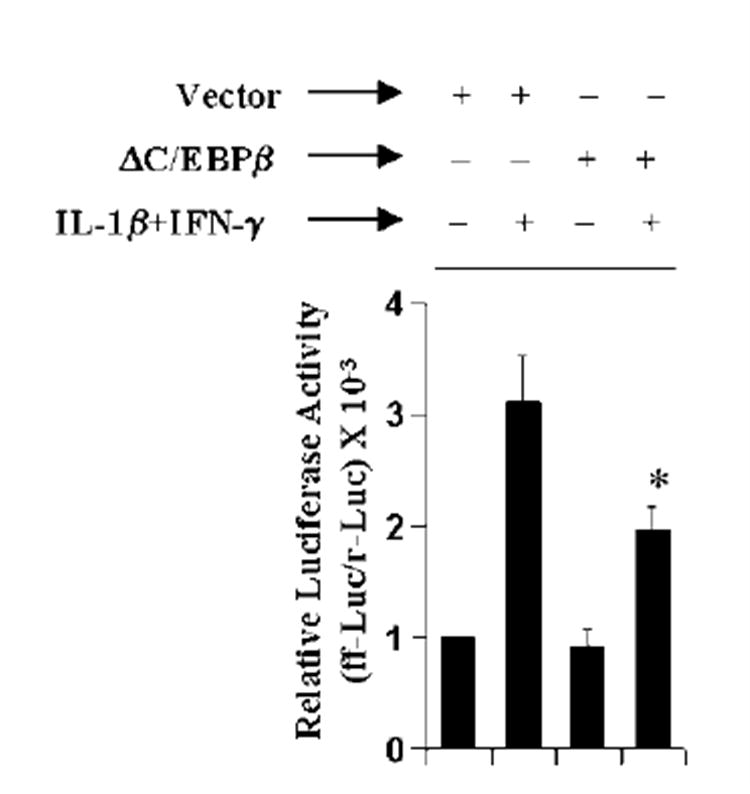
Cells were cotransfected with 0.5 μg of either ΔC/EBPβ or an empty vector, 0.5 μg of phiNOS(7.2)Luc, and 50 ng of pRL-TK. Twenty-four hours after transfection, cells were stimulated with the combination of IL-1β and IFN-γ. After 12 h of incubation, firefly (ff-Luc) and Renilla (r-Luc) luciferase activities were assayed. Data are mean ± S.D. of three different experiments. *, p < 0.005 versus cytokine treatment of empty vector-transfected cells.
Since gemfibrozil inhibited cytokine-induced activation of human iNOS promoter (Fig. 5), we decided to investigate the effect of gemfibrozil on the activation of these proinflammatory transcription factors in cytokine-stimulated human U373MG astroglial cells. Activation of these transcription factors was monitored by transcriptional activity using the expression of luciferase from respective reporter constructs. It is evident from Fig. 9, A-D, that the combination of IL-1β and IFN-γ induced NF-κB-, AP-1-, and C/EBPβ-dependent luciferase activities by 3–5-fold and that of the GAS-dependent one by about 10-fold within 6 h of incubation. However, ISRE-dependent luciferase activity was not induced in the presence of same cytokines under the same condition (Fig. 9E). The combination of IL-1β and IFN-γ was also unable to induce the activation of ISRE within 12 or 18 h of incubation (data not shown). Gemfibrozil itself did not induce the activation of any of these transcription factors (Fig. 9). However, this fibrate drug markedly inhibited cytokine-induced activation of NF-κB, AP-1, and C/EBPβ (Fig. 9, A-C). This inhibitory effect was dose-dependent, and the maximum inhibition was found at 150–200 μm concentration of gemfibrozil. However, activation of C/EBPβ was much more sensitive than that of NF-κB and AP-1 to gemfibrozil (Fig. 9). In contrast, gemfibrozil had no inhibitory effect on GAS-dependent luciferase activities in cytokine-stimulated U373MG astroglial cells (Fig. 9D). At 150 or 200 μm gemfibrozil, only 10–12% inhibition of GAS-dependent luciferase activity was observed. Similar to the effect of ΔhPPAR-α on the inhibitory effect of gemfibrozil on the induction of iNOS, ΔhPPAR-α was also unable to reverse the inhibitory effect of gemfibrozil on cytokine-induced activation of NF-κB, AP-1, and C/EBPβ (data not shown).
Fig. 9. Effect of gemfibrozil on NF-κB- (A), AP-1- (B), C/EBPβ- (C), GAS- (D) and ISRE (E)-dependent luciferase activities in cytokine-stimulated human U373MG astroglial cells.
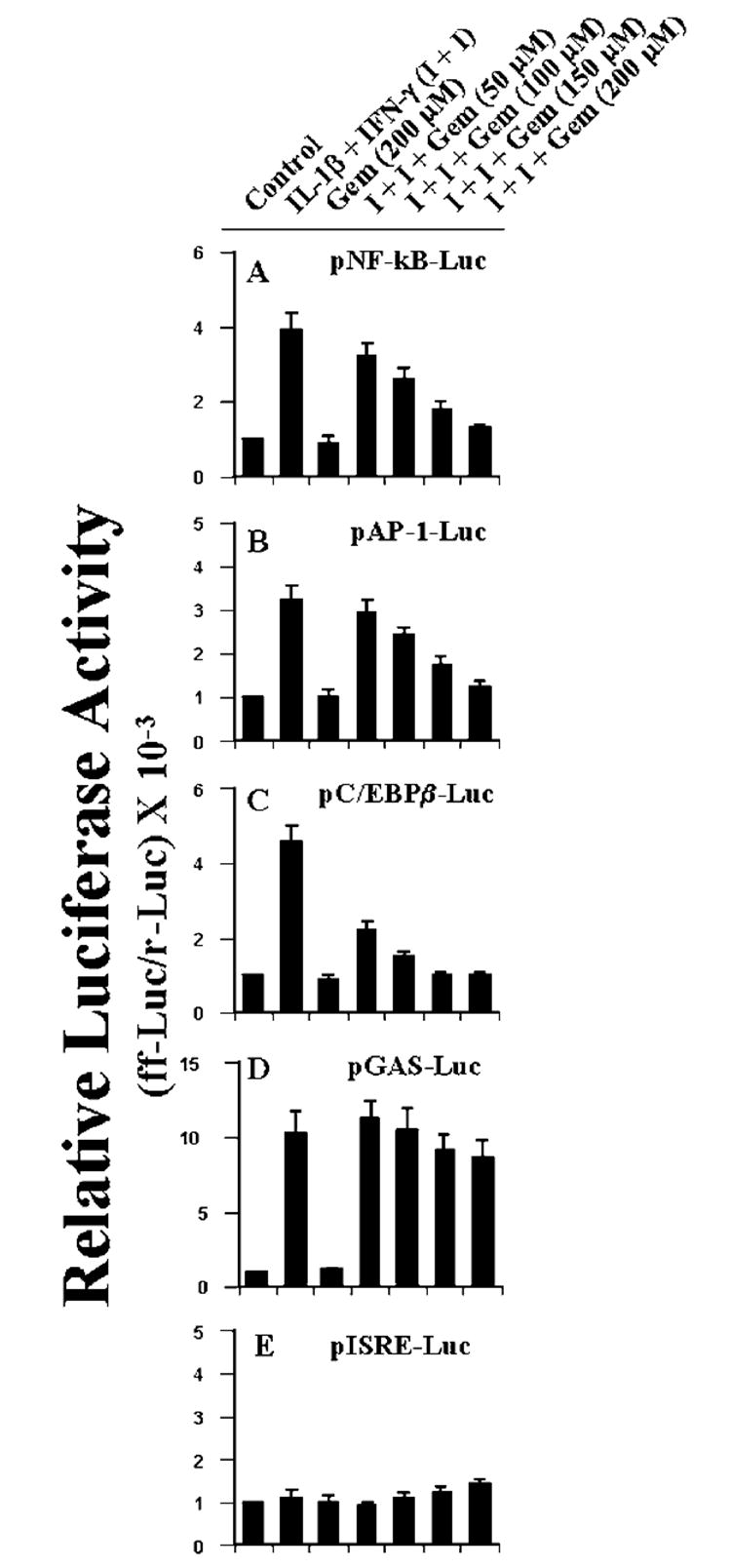
Cells plated at 50–60% confluence in six-well plates were cotransfected with 1 μg of either pNF-κB-Luc, pAP-1-Luc, p C/EBPβ-Luc, pGAS-Luc, or pISRE-Luc and 50 ng of pRL-TK. After 24 h of transfection, cells were incubated with different concentrations of gemfibrozil for 2 h and then stimulated with the combination of IL-1β and IFN-γ for 6 h under serum-free condition. Firefly (ff-Luc) and Renilla (r-Luc) luciferase activities were obtained by analyzing the total cell extract. Data are mean ± S.D. of three different experiments.
Effect of Gemfibrozil on Cell Viability
Human U373MG astrocytoma cells were incubated with different concentrations (50, 100, 150, and 200 μm) of gemfibrozil under serum-free condition as mentioned above, and their viability was determined by the MTT assay. Gemfibrozil used at different concentrations did not decrease the viability of the cells (data not shown). Therefore, inhibition of the expression of iNOS by gemfibrozil was not due to any change in viability of the cells.
DISCUSSION
The signaling events transduced by proinflammatory cytokines for the induction of iNOS are poorly understood. A complete understanding of the cellular signaling mechanisms involved in the induction of iNOS should identify novel targets for the therapeutic intervention in NO-mediated neuroinflammatory diseases. The studies reported in this manuscript clearly demonstrates that gemfibrozil, an activator of PPAR-α, reduces the induction of iNOS in human astrocytes. Since astrocytes express PPAR-α (35), and NO produced from iNOS has been implicated in the pathogenesis of demyelinating and neurodegenerative diseases (5-7), our results provide a potentially important mechanism whereby activators of PPAR-α may ameliorate neural injury. However, gemfibrozil inhibits the induction of iNOS in human astroglial cells independent of PPAR-α. This conclusion is based on the following observations. First, gemfibrozil induced PPRE-dependent luciferase activity, suggesting that gemfibrozil can activate PPAR in human astroglial cells. Second, gemfibrozil inhibited cytokine-induced activation of iNOS promoter suggesting that gemfibrozil inhibits the transcription of iNOS. Third, the expression of ΔhPPAR-α, a dominant-negative mutant of human PPAR-α, blocked gemfibrozil-mediated activation of PPRE suggesting that gemfibrozil activates PPRE through PPAR-α. Fourth, ΔhPPAR-α was unable to block the inhibitory effect of gemfibrozil on the induction of iNOS promoter activation, suggesting that gemfibrozil does not require PPAR-α to inhibit the induction of iNOS in human astroglial cells.
In contrast to the marked induction of iNOS mRNA by the cytokine combination (Fig. 2), there was only a 3.9-fold induction of human iNOS promoter in human astroglial cells (Fig. 5), suggesting that in addition to transcriptional mechanisms posttranscriptional events could also play a significant role in regulating the expression of iNOS gene. Several authors have performed nuclear run-on assays to analyze the induction rate of the human iNOS promoter in different human cell lines (30, 36-38). These authors have shown that the endogenous iNOS promoter displays a significant basal activity and that the induction rate of only 2–10-fold by cytokines is much lower as seen for the induction of the iNOS mRNA expression. Taken together, these results suggest that human iNOS gene is regulated at the level of transcription as well as posttranscription. However, in our experiment, gemfibrozil does not alter the relative rate of degradation of human iNOS mRNA in astroglial cells (Fig. 3), suggesting that gemfibrozil may not couple to posttranscriptional pathways required for the regulation of iNOS expression and that gemfibrozil inhibits the expression of iNOS mRNA mainly at the level of transcription.
Proinflammatory cytokines (TNF-α, IL-1β, or IFN-γ) bind to their respective receptors and induce iNOS expression via activation of NF-κB (2-4, 22, 26-28). The presence of multiple consensus sequences in the promoter region of iNOS for the binding of NF-κB and the inhibition of iNOS expression with the inhibition of NF-κB activation (2-4, 22, 26-28) establishes an essential role of NF-κB activation in the induction of iNOS. Although TNF-α or IL-1β alone is capable of inducing the activation of NF-κB, these cytokines alone were not sufficient to induce the expression of iNOS in human cell lines (39). The fact that a combination of cytokines is required to induce the expression of iNOS suggests that activation of additional transcription factors is also necessary for the expression of iNOS. Consistently, apart from the consensus sequence for binding of NF-κB, the human iNOS promoter contains consensus sequences for the binding of transcription factors including AP-1, C/EBPβ, IRF-1 binding to ISRE, and STAT binding to GAS (29-31). The bulk of the work regarding the involvement of these transcription factors in the transcriptional regulation of the iNOS gene involved the murine system.
Although the role of these transcription factors in the transcription of human iNOS has not been well established, several evidences point to their possible involvement in the induction of iNOS in human cells. Kleinert et al. (32) have shown that the cytokine mixture induces the tyrosine phosphorylation of JAK-2 in human DLD-1 cells. This activated JAK-2 further induces the DNA binding activity of STAT1α (32). Tyrophostin B42, a specific inhibitor of JAK-2 (33), inhibits the phosphorylation of JAK-2, the activation of STAT1α, and the induction of iNOS (32), suggesting that the JAK-2-STAT1α pathway is an important activator of iNOS transcription. Moss and colleagues (29) have recently shown that activation of both NF-κB and AP-1 is an important step for the transcription of iNOS in human cells. Mutation in NF-κB- as well as AP-1-binding site of the iNOS promoter reduces the transcriptional activity of iNOS promoter (29). Furthermore, they (29) have shown that MAP kinase pathways (ERK and p38) regulate the expression of iNOS in human lung epithelial (A549) cells through the modulation of NF-κB and AP-1. Recently, we have found that activation of C/EBPβ is also necessary for the induction of iNOS (4). Overexpression of ΔC/EBPβ, a truncated alternate C/EBPβ translational product, LIP, which acts as a dominant-negative inhibitor of C/EBPβ activity (34), inhibits the production of NO and the expression of iNOS in mouse microglial cells (4). Consistently, here we show that ΔC/EBPβ also inhibits cytokine-induced activation of iNOS promoter in human astroglial cells (Fig. 8).
Here we have found that the combination of IL-1β and IFN-γ markedly induced the activation of NF-κB, AP-1, C/EBPβ, and GAS but not that of ISRE in human U373MG astroglial cells. Interestingly, gemfibrozil suppressed cytokine-induced activation of NF-κB, AP-1, and C/EBPβ but not that of GAS. Since STAT binds to GAS (40) and JAK is known to phosphorylate and activate STAT (40), our results suggest that gemfibrozil may not inhibit the JAK-STAT pathway in human astrocytes. On the other hand, IRF-1 binds to ISRE (41). IRF-1 has been found to be involved in the induction of iNOS by IFN-γ in mouse macrophages (42). Consistently, IFN-γ cannot induce iNOS in macrophages isolated from IRF-1(−/−) mice (43). Although the promoter of human iNOS gene contains ISRE (30, 31, 44), the combination of IL-1β and IFN-γ did not modulate the activation of ISRE in any significant way, suggesting that IRF-1 is unlikely to act as an important regulator of cytokine-induced expression of iNOS in human astrocytes.
Fibrate drugs like gemfibrozil, clofibrate, and fenofibrate induce the proliferation of peroxisomes in rats and mice (45, 46). Continuous administration of fibrate drugs to the rats and mice for 40–50 weeks also leads to the formation of hepatic tumor (45-47). However, induction of hepatic tumor promotion by fibrate drugs has not been demonstrated in human, other primates, and guinea pig (46, 48), species that have lost their ability to synthesize ascorbate due to inherent loss of the gulonolactone oxidase gene. Braun et al. (48) have recently reported that the evolutionary loss of the gulonolactone oxidase gene may contribute to the missing carcinogenic effect of peroxisome proliferators in humans, since ascorbate synthesis is accompanied by H2O2 production, and consequently its induction can be potentially harmful. Furthermore, recent studies have also revealed that humans have considerably lower levels of PPAR-α in liver than rodents, and this difference may, in part, explain the species differences in the carcinogenic response to peroxisome proliferators (46). In addition, gemfibrozil does not require PPAR-α to inhibit the activation of proinflammatory transcription factors and the induction of iNOS in human astroglial cells. Taken together, these observations suggest that gemfibrozil as an anti-neuroinflammatory drug may not cause human health problems.
NO, a short-lived and diffusible free radical, plays many roles as a signaling and effector molecule in diverse biological systems; it is a neuronal messenger and is involved in vasodilation as well as in antimicrobial and antitumor activities (49). On the other hand, NO has also been implicated in several CNS disorders, including inflammatory, infectious, traumatic, and degenerative diseases (5-8, 50). There is considerable evidence for the transcriptional induction of iNOS (the high output isoform of NOS) in the CNS that is associated with autoimmune reactions, acute infection, and traumatic brain injury (5-8, 50). Once NO is formed, it spontaneously reacts with O2− to form peroxynitrite (ONOO−), the most reactive derivative of NO known so far (51). Both NO and peroxynitrite are potentially toxic molecules to neurons and oligodendrocytes that may mediate toxicity through the formation of iron-NO complexes of iron-containing enzyme systems (52), oxidation of protein sulfhydryl groups (51), nitration of proteins, and nitrosylation of nucleic acids and DNA strand breaks (53).
In the CNS, iNOS is expressed mainly by activated astrocytes and microglia, the two glial cell types involved in intracerebral immune regulation. Astrocytes are the major glial cell population in the central nervous system; therefore, induction of iNOS in astrocytes may be an important source of NO in CNS inflammatory disorders associated with neuronal and oligodendrocytes death. Therefore, gemfibrozil being capable of attenuating the activation of proinflammatory transcription factors and the expression of iNOS in human astrocytes may find therapeutic application in neuroinflammatory and neurodegenerative disorders.
Acknowledgments
We thank Tom Dunn and associates of the University of Nebraska Medical Center for help in preparing this manuscript.
The abbreviations used are
- CNS
central nervous system
- iNOS
inducible nitric-oxide synthase
- IL
interleukin
- TNF
tumor necrosis factor
- INF
interferon
- IRF-1
IFN-γ regulatory factor-1
- PPAR
peroxisome proliferator-activated receptor
- STAT
signal transducer and activator of transcription
- NF-κB
nuclear factor-κB
- AP-1
activator protein-1
- C/EBPβ
CCAAT/enhancer-binding protein β
- GAS
γ-activation site
- DMEM
Dulbecco’s modified Eagle’s medium
- l-NMA
l-NG-Monomethylarginine
- d-NMA
d-NG-monomethylarginine
- PPRE
peroxisome proliferator-responsive element
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- JAK
Janus kinase
Footnotes
This work was supported by National Institutes of Health Grant NS39940 (to K. P.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
References
- 1.Feinstein DL, Galea E, Roberts S, Berquist H, Wang H, Reis DJ. J Neurochem. 1994;62:315–321. doi: 10.1046/j.1471-4159.1994.62010315.x. [DOI] [PubMed] [Google Scholar]
- 2.Pahan K, Sheikh FG, Namboodiri AMS, Singh I. J Clin Invest. 1997;100:2671–2679. doi: 10.1172/JCI119812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pahan K, Sheikh FG, Liu X, Hilger S, McKinney M, Petro TM. J Biol Chem. 2001;276:7899–7905. doi: 10.1074/jbc.M008262200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jana M, Liu X, Koka S, Ghosh S, Petro TM, Pahan K. J Biol Chem. 2001;276:44527–44533. doi: 10.1074/jbc.M106771200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mitrovic B, Ignarro LJ, Montestruque S, Smoll A, Merril JE. Neuroscience. 1994;61:575–585. doi: 10.1016/0306-4522(94)90435-9. [DOI] [PubMed] [Google Scholar]
- 6.Merrill JE, Ignarro LJ, Sherman MP, Melinek J, Lane TE. J Immunol. 1993;151:2132–2141. [PubMed] [Google Scholar]
- 7.Koprowski H, Zheng YM, Heber-Katz E, Fraser N, Rorke L, Fu ZF, Hanlon C, Dietzshold B. Proc Natl Acad Sci USA. 1993;90:3024–3027. doi: 10.1073/pnas.90.7.3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Akama KT, Albanese C, Pestell RG, Van Eldik LJ. Proc Natl Acad Sci USA. 1998;95:5795–5800. doi: 10.1073/pnas.95.10.5795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lemberger T, Desvergne B, Wahli W. Annu Rev Cell Dev Biol. 1996;12:335–363. doi: 10.1146/annurev.cellbio.12.1.335. [DOI] [PubMed] [Google Scholar]
- 10.Dreyer C, Krey G, Keller H, Givel F, Helftenbein G, Wahli W. Cell. 1992;68:879–887. doi: 10.1016/0092-8674(92)90031-7. [DOI] [PubMed] [Google Scholar]
- 11.Hsu HC, Lee YT, Yeh HT, Chen MF. J Lab Clin Med. 2001;137:414–421. doi: 10.1067/mlc.2001.114991. [DOI] [PubMed] [Google Scholar]
- 12.Bloomfield RH, Davenport J, Babikian V, Brass LM, Collins D, Wexler L, Wagner S, Papademetriou V, Rutan G, Robins SJ. Circulation. 2001;103:2828–2833. doi: 10.1161/01.cir.103.23.2828. [DOI] [PubMed] [Google Scholar]
- 13.Amici C, Sistonen L, Santoro MG, Morimoto RI. Proc Natl Acad Sci USA. 1992;89:6227–6231. doi: 10.1073/pnas.89.14.6227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elia G, Amici C, Rossi A, Santoro MG. Cancer Res. 1996;56:210–217. [PubMed] [Google Scholar]
- 15.Feinstein DL, Galea E, Aquino DA, Li GC, Xu H, Reis DJ. J Biol Chem. 1996;271:17724–17732. doi: 10.1074/jbc.271.30.17724. [DOI] [PubMed] [Google Scholar]
- 16.McCarthy M, Wood C, Fedoseyeva L, Whittemore S. J Neurovirol. 1995;1:275–285. doi: 10.3109/13550289509114024. [DOI] [PubMed] [Google Scholar]
- 17.Bradford MM. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 18.Pahan K, Namboodiri AMS, Sheikh FG, Smith BT, Singh I. J Biol Chem. 1997;272:7786–7791. doi: 10.1074/jbc.272.12.7786. [DOI] [PubMed] [Google Scholar]
- 19.Taylor BS, de Vera ME, Ganster RW, Wang Q, Shapiro RA, Morris SM, Billiar TR, Geller DA. J Biol Chem. 1998;273:15148–15156. doi: 10.1074/jbc.273.24.15148. [DOI] [PubMed] [Google Scholar]
- 20.Kliewer SA, Forman BM, Blumberg B, Ong ES, Borgmeyer U, Mangelsdorf DJ, Umesono K, Evans RM. Proc Natl Acad Sci USA. 1994;91:7355–7359. doi: 10.1073/pnas.91.15.7355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Illingworth DR, Bacon S. Arteriosclerosis. 1989;9:I121–I134. [PubMed] [Google Scholar]
- 22.Pahan K, Liu X, McKinney M, Wood C, Sheikh FG, Raymond JR. J Neurochem. 2000;74:2288–2295. doi: 10.1046/j.1471-4159.2000.0742288.x. [DOI] [PubMed] [Google Scholar]
- 23.Pahan K, Liu X, Wood C, Raymond JR. FEBS Lett. 2000;472:203–207. doi: 10.1016/s0014-5793(00)01465-4. [DOI] [PubMed] [Google Scholar]
- 24.Zhao ML, Liu JSH, He DK, Dickson DW, Lee SC. Brain Res. 1998;813:402–405. doi: 10.1016/s0006-8993(98)01023-3. [DOI] [PubMed] [Google Scholar]
- 25.Roberts RA, James NH, Woodyatt NJ, Macdonald N, Tugwood JD. Carcinogenesis. 1998;19:43–48. doi: 10.1093/carcin/19.1.43. [DOI] [PubMed] [Google Scholar]
- 26.Xie Q-W, Kashiwabara Y, Nathan C. J Biol Chem. 1994;269:4705–4708. [PubMed] [Google Scholar]
- 27.Pahan K, Sheikh FG, Namboodiri AMS, Singh I. J Biol Chem. 1998;273:12219–12226. doi: 10.1074/jbc.273.20.12219. [DOI] [PubMed] [Google Scholar]
- 28.Pahan K, Raymond JR, Singh I. J Biol Chem. 1999;274:7528–7536. doi: 10.1074/jbc.274.11.7528. [DOI] [PubMed] [Google Scholar]
- 29.Kristof AS, Marks-Konczalik J, Moss J. J Biol Chem. 2001;276:8445–8452. doi: 10.1074/jbc.M009563200. [DOI] [PubMed] [Google Scholar]
- 30.Linn SC, Morelli PJ, Edry I, Cottongim SE, Szabo C, Salzman AL. Am J Physiol. 1997;272:G1499–G1508. doi: 10.1152/ajpgi.1997.272.6.G1499. [DOI] [PubMed] [Google Scholar]
- 31.Spitsin SV, Koprowski H, Michaels FH. Mol Med. 1996;2:226–235. [PMC free article] [PubMed] [Google Scholar]
- 32.Kleinert H, Wallerath T, Fritz G, Ihrig-Biedert I, Rodriguez-Pascual F, Geller DA, Forstermann U. Br J Pharmacol. 1998;125:193–201. doi: 10.1038/sj.bjp.0702039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bright JJ, Du C, Sriram S. J Immunol. 1999;162:6255–6262. [PubMed] [Google Scholar]
- 34.Descombes P, Schibler U. Cell. 1991;67:569–579. doi: 10.1016/0092-8674(91)90531-3. [DOI] [PubMed] [Google Scholar]
- 35.Cullingford TE, Bhakoo K, Peuchen S, Dolphin CT, Patel R, Clark JB. J Neurochem. 1998;70:1366–1375. doi: 10.1046/j.1471-4159.1998.70041366.x. [DOI] [PubMed] [Google Scholar]
- 36.Salzman AL, Linn SC, Szabo C. Int J Mol Med. 2000;6:209–216. doi: 10.3892/ijmm.6.2.209. [DOI] [PubMed] [Google Scholar]
- 37.Laubach VE, Zhang CX, Russell SW, Murphy WJ, Sherman PA. Biochim Biophys Acta. 1997;1351:287–295. doi: 10.1016/s0167-4781(96)06909-6. [DOI] [PubMed] [Google Scholar]
- 38.de Vera ME, Shapiro RA, Nussler AK, Mudgett JS, Simmons RL, Morris SM, Jr, Billiar TR, Geller DA. Proc Natl Acad Sci USA. 1996;93:1054–1059. doi: 10.1073/pnas.93.3.1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Taylor BS, Geller DA. Shock. 2000;13:413–424. doi: 10.1097/00024382-200006000-00001. [DOI] [PubMed] [Google Scholar]
- 40.Darnell JE, Jr, Kerr IM, Stark GR. Science. 1994;264:1415–1421. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 41.Coccia EM, Stellacci E, Marziali G, Weiss G, Battistini A. Int Immunol. 2000;12:977–985. doi: 10.1093/intimm/12.7.977. [DOI] [PubMed] [Google Scholar]
- 42.Saura M, Zaragoza C, Bao C, McMillan A, Lowenstein CJ. J Mol Biol. 1999;289:459–471. doi: 10.1006/jmbi.1999.2752. [DOI] [PubMed] [Google Scholar]
- 43.Fujimura M, Tominaga T, Kato I, Takasawa S, Kawase M, Taniguchi T, Okamoto H, Yoshimoto T. Brain Res. 1997;759:247–250. doi: 10.1016/s0006-8993(97)00264-3. [DOI] [PubMed] [Google Scholar]
- 44.Marks-Konczalik J, Chu SC, Moss J. J Biol Chem. 1998;273:22201–22208. doi: 10.1074/jbc.273.35.22201. [DOI] [PubMed] [Google Scholar]
- 45.Reddy JK, Mannaerts GP. Annu Rev Nutr. 1994;14:343–370. doi: 10.1146/annurev.nu.14.070194.002015. [DOI] [PubMed] [Google Scholar]
- 46.Gonzalez FJ, Peters JM, Cattley RC. J Natl Cancer Inst. 1998;90:1702–1709. doi: 10.1093/jnci/90.22.1702. [DOI] [PubMed] [Google Scholar]
- 47.Yeldandi AV, Rao MS, Reddy JK. Mutat Res. 2000;448:159–177. doi: 10.1016/s0027-5107(99)00234-1. [DOI] [PubMed] [Google Scholar]
- 48.Braun L, Mile V, Schaff Z, Csala M, Kardon T, Mandl J, Banhegyi G. FEBS Lett. 1999;458:359–362. doi: 10.1016/s0014-5793(99)01184-9. [DOI] [PubMed] [Google Scholar]
- 49.Nathan C. FASEB J. 1992;6:3051–3064. [PubMed] [Google Scholar]
- 50.Samdani AF, Dawson TM, Dawson VL. Stroke. 1997;28:1283–1288. doi: 10.1161/01.str.28.6.1283. [DOI] [PubMed] [Google Scholar]
- 51.Radi R, Beckman JS, Bush KM, Freeman BA. J Biol Chem. 1991;266:4244–4250. [PubMed] [Google Scholar]
- 52.Drapier J-C, Hibbs JB. J Immunol. 1988;140:2829–2838. [PubMed] [Google Scholar]
- 53.Wink DA, Kasprazak KS, Maragos CM, Elespuru RK, Misra M, Dunams TM, Cebula TA, Koch WA, Andrews AW, Allen JS. Science. 1991;254:1001–1003. doi: 10.1126/science.1948068. [DOI] [PubMed] [Google Scholar]