

Abstract
Parkinson’s disease is a debilitating neurodegenerative movement disorder characterized by damage to the nigrostriatal dopaminergic system. Current therapies are symptomatic only and may be accompanied by serious side effects. There is therefore a continual search for novel compounds for the treatment of Parkinson’s disease symptoms, as well as to reduce or halt disease progression. Nicotine administration has been reported to improve motor deficits that arise with nigrostriatal damage in parkinsonian animals and in Parkinson’s disease. In addition, nicotine protects against nigrostriatal damage in experimental models, findings that have led to the suggestion that the reduced incidence of Parkinson’s disease in smokers may be due to the nicotine in tobacco. Altogether, these observations suggest that nicotine treatment may be beneficial in Parkinson’s disease. Nicotine interacts with multiple nicotinic receptor (nAChR) subtypes in the peripheral and central nervous system, as well as in skeletal muscle. Work to identify the subtypes affected in Parkinson’s disease is therefore critical for the development of targeted therapies. Results show that striatal α6β2-containing nAChRs are particularly susceptible to nigrostriatal damage, with a decline in receptor levels that closely parallels losses in striatal dopamine. In contrast, α4β2-containing nAChRs are decreased to a much smaller extent under the same conditions. These observations suggest that development of nAChR agonists or antagonists targeted to α6β2-containing nAChRs may represent a particularly relevant target for Parkinson’s disease therapeutics.
Keywords: α-ConotoxinMII, Nicotine, Nicotinic, Parkinson’s disease, Nigrostriatal, Striatum
1. Parkinson’s disease and the nicotinic cholinergic system
The pathological hallmarks of Parkinson’s disease are the presence of intracellular Lewy bodies and an extensive degeneration of the nigrostriatal dopaminergic system. [1–4]. There is a ≥70% decline in striatal dopamine and ≥50% loss of nigral dopaminergic neurons with the onset of clinical symptoms, which include bradykinesia, rigidity, and tremor [1–4].
Although Parkinson’s disease has primarily been considered a dopaminergic disorder, it is becoming increasingly clear that multiple CNS systems are involved in its pathogenesis [5–7]. Braak and coworkers have also identified Lewy bodies in numerous non-dopaminergic brain regions including the locus coeruleus, raphe nuclei, thalamus, amygdala, olfactory nuclei, pedunculopontine nucleus, and cerebral cortex [5, 6]. These observations are in agreement with much earlier studies, which indicated that multiple CNS neuronal systems are affected in Parkinson’s disease [8, 9]. Significant declines have been observed in molecular markers of many neurotransmitters and neuromodulators, such as enkephalin, somatostatin, cholecystokinin, and substance P, as well as changes in the noradrenergic, glutamatergic, serotonergic, and cholinergic systems [8, 9].
This review focuses on the involvement of the nicotinic cholinergic system in Parkinson’s disease for the following reasons. First, its close association and functional interaction with the nigrostriatal dopaminergic system suggests that nicotine or nicotinic agonists have potential in treatment of the motor symptoms of the disease [10, 11]. In addition, the finding that stimulation of the nicotinic cholinergic system ameliorates cognitive declines [12] suggests that nicotinic acetylcholine receptor (nAChR) drugs may be useful in treating some of the nondopaminergic deficits associated with Parkinson’s disease, such as dementia that develops in 50% of Parkinson’s disease patients with disease progression [1–4]. Lastly, there is very compelling epidemiological evidence demonstrating a negative association between smoking and Parkinson’s disease that has been attributed to the nicotine in tobacco, at least in part [11, 13–17].
Altogether these observations suggest that the nicotinic cholinergic system represents a promising direction for much-needed therapies for Parkinson’s disease. Moreover, the identification of unique nAChR subtypes in the nigrostriatal system (as discussed later) may allow for the development of drugs that selectively target this debilitating neurological disorder.
2. Nicotine and nicotinic agonists for neuroprotection
A wealth of epidemiological studies has demonstrated an inverse correlation between smoking and development of Parkinson’s disease. The decreased incidence of Parkinson’s disease in smokers has consistently been demonstrated in both prospective and retrospective studies, is dose- and time-dependent, and does not appear to be due to increased mortality [11, 13–17]. Although the component in cigarette smoke that confers this apparent neuroprotective action remains to be identified, numerous studies using both culture systems and experimental animal models suggest that nicotine may play an important role, as detailed below.
2.1. Nicotine-mediated protection against toxic insults in culture models
Extensive evidence using culture systems has shown that nicotine protects against toxicity mediated by β-amyloid, glutamate, ethanol, trophic factor deprivation, arachidonic acid and others [11, 18–20]. Of more direct relevance are studies using cultured ventral mesencephalic dopaminergic neurons which show that nicotine pre-treatment attenuates toxicity-induced by MPP+, the metabolite of MPTP that selectively destroys dopaminergic terminals [21]. There is thus a firm basis for the hypothesis that nicotine has neuroprotective properties using in vitro systems.
2.2. Nicotine-mediated protection against toxic insults in experimental animal models
There is also a large body of literature demonstrating nicotine-mediated protection against dopaminergic damage in parkinsonian animal models [22]. In rats, nicotine pre-treatment protects against nigrostriatal degeneration induced by the dopaminergic neurotoxin 6-hydroxydopamine or by mechanical lesions [23–28]. A neuroprotective effect of nicotine is also observed in striatum of mice in which nigrostriatal damage is induced by the selective dopaminergic neurotoxin MPTP [24, 29–39]. It should be noted, however, that results are somewhat inconsistent in this model with protection in some but not all studies. Although the nature of this variability is uncertain, it may be due to the relatively large lesion generated with MPTP. This possibility stems from findings that neuroprotection is observed in mice treated with methamphetamine and paraquat, both of which result in milder damage to the nigrostriatal dopaminergic system [24, 40].
Nicotine-induced protection against striatal damage is also observed in MPTP-treated nonhuman primates, a model that may more closely resemble the human disease [41–43]. A chronic nicotine treatment regimen improved a variety of neurochemical markers in MPTP-lesioned nicotine treated animals, including tyrosine hydroxylase, the dopamine and vesicular monoamine transporters, dopamine levels and nAChR expression [42, 43]. In addition, nicotine treatment normalized lesion-induced overactivity of the nigrostriatal pathway and preserved synaptic plasticity lost with nigrostriatal damage [41].
These combined results from different experimental animal models support the idea that the reduced incidence of Parkinson’s disease in smokers may relate to the nicotine in tobacco.
3. Nicotine and nicotinic agonists for the symptomatic treatment of Parkinson’s disease
Results from animal studies also lend support to the idea that nicotine and/or nAChR agonists may prove useful for treatment of Parkinson’s disease symptoms. NAChR stimulation is well known to modulate locomotor activity in unlesioned animals [44], as well as ameliorate motor behaviors in animals with nigrostriatal damage. In rats with a unilateral 6-hydroxydopamine lesion, nicotine or nicotinic agonist treatment reduces apomorphine-induced turning behavior [45, 46]. Treatment with nicotine or the nicotinic agonist SIB-1508Y also improves the antiparkinsonian action of levodopa in MPTP-lesioned monkeys [47, 48]. Interestingly, nAChR activation also ameliorated deficits associated with nigrostriatal damage in tasks that evaluated aspects of attentional and executive cognitive functions [49].
The effect of smoking, and nicotine or nicotinic agonist treatment on Parkinson’s disease symptoms has also been investigated (Table 1). Smoking (possibly acting via nicotine) reduced the tremor, rigidity, bradykinesia and gait disturbances observed in Parkinson’s disease, although effects were transient and short lasting [50]. In another report, improvement was noted in two elderly patients with Parkinson’s disease treated with the nicotine gum and patch for several months [51]. Variable results have been obtained in more recent small-scale clinical trials. Kelton and co-workers [52] observed improvements in areas of cognitive performance and motor measures after treatment with intravenous nicotine and the nicotine patch, with effects sustained for up to 1 month after the drug. However, Vieregge et al. observed no improvements in patients administered the patch for 3 weeks [53], although the lack of efficacy may have related to the fact that Parkinsonism was only assessed 3 weeks after patch cessation. On the other hand, Lemay et al. [54] also obtained no improvement in motor or cognitive deficits with immediate testing of Parkinson’s disease symptoms after 3 to 4 weeks of treatment. Several studies also investigated the effect of acute exposure (1 day) to the nicotine patch or gum, with improvement in one but a worsening of motor performance in Parkinson’s disease patients in two other studies [55, 56]. There was also no observable effect of the nicotinic agonist SIB-1508Y on parkinsonism after 4 weeks of treatment [57]. Possible reasons for these varying effects on Parkinson’s disease symptoms among the different studies may include differences in the severity of parkinsonism, the end-points under study, the study design (open-label versus blinded) and/or the dose, timing and mode of administration of nicotine.
Table 1.
Summary of the studies testing nicotine or a nicotinic agonist in Parkinson’s disease patients
| Study | Test agent | Type of study | # subjects | Duration | Final dose/day | Anti-parkinsonian effect | |
|---|---|---|---|---|---|---|---|
| Dose titration | Dose maintenance | ||||||
| Ishikawa & Miyatake, 1993 | Smoking and nicotine gum | Open-label | 6 | Chronic smoker | NA | Yes | |
| Fagerstrom et al., 1994 | Nicotine gum and patch | Open-label | 2 | ≥ 7 mo | 15 mg patch + gum | Yes | |
| Clemens et al., 1995 | Nicotine gum | Double-blinded placebo-controlled | 48 | ND | 1 day | 3 × 2 mg | No |
| Ebersbach et al., 1999 | Nicotine patch | Double-blinded crossover | 16 | ND | 12 hours | 7 mg# | No |
| Kelton et al., 2000 | Nicotine iv and patch | Open-label | 15 | 2 wk | 1 wk | 14 mg | Yes |
| Vieregge et al., 2001 | Nicotine patch | Double-blinded placebo-controlled | 32 | 1 wk | 2 wk | 14 mg | No |
| Mitsuoka et al., 2002 | Nicotine gum | Open-label | 8 | ND | 1 day | NA | Yes |
| Lemay et al., 2004 | Nicotine patch | Open-label | 22 | 22 days | 3 days | 21 mg | No |
| Shoulson et al., 2006 | SIB-1508Y | Double-blinded placebo controlled | 77 | 2 wk | 2 wk | 10 mg | No |
Used the 35 mg patch, of which 14 mg is absorbed over 24 h, or 7 mg over 12 h. ND – not done; NA, not available.
Similarly variable results have been obtained in nonhuman primates. In our results in nonhuman primates, nicotine administration via the drinking water did not modify Parkinsonism over an 8-week period [58], although Domino et al. [47] did obtain small effects on motor activity using a different short term injection treatment protocol. On the other hand, although nicotine treatment had no effect in the short term in our monkey studies, longer term administration via the drinking water (12 months) did improve striatal dopaminergic measures decreased with nigrostriatal damage [41–43]. These latter findings may suggest that nicotine treatment primarily has a protective and/or restorative effect with chronic use, but no acute symptomatic effects on Parkinsonism, at least at the doses tested to date.
4. Striatal cholinergic system
In order to understand how nAChR activation is involved in neuroprotection and/or symptomatic improvements in Parkinson’s disease, knowledge of the anatomical relationship between the striatal cholinergic and dopaminergic systems is critical. Two major neuronal cell types are present in the striatum. These include the GABAergic medium spiny projection neurons that comprise the greater majority (~95%) of neurons, as well as a much smaller population of interneurons (5%) of which the majority are GABAergic and about a third cholinergic. These larger cholinergic interneurons form a dense network in striatum that closely overlaps with dopaminergic terminals [10]. Thus high levels of acetylcholine, choline acetyltransferase and acetylcholinesterase [59] coincide with expression of dopamine, tyrosine hydroxylase and other dopaminergic markers [60]. Under physiological conditions, the cholinergic interneurons are tonically active [10]. They release acetylcholine, which subsequently interacts with nAChRs on striatal cell bodies and nerve terminals, including dopaminergic projections from the substantia nigra and glutamatergic afferents from the cortex [61–64]. Stimulation of nAChRs at these sites results in dopamine release mediated through various nAChR subtypes [65–67], as discussed in the following section.
5. Nicotinic receptors in the nigrostriatal system
Identification of the nAChRs that modulate dopaminergic function and characterization of changes in their expression with nigrostriatal damage is essential for the development of Parkinson’s disease therapies using nAChR ligands. NAChRs are pentameric ligand gated ion channels comprised of different combinations of α and β subunits to form heteromeric receptors, and select α subunits to form homomeric receptors [67–71]. There is a requirement for α subunits in the receptor complex, as these possess the recognition site for acetylcholine. The β subunit does not bind acetylcholine, however it modulates the interaction of the ligand with the α subunit, thereby affecting the physiological and pharmacological properties of the receptor [67–71]. Multiple nAChR subtypes (Fig. 1) have been identified in the nigrostriatal system using a variety of complementary experimental approaches.
Fig. 1.
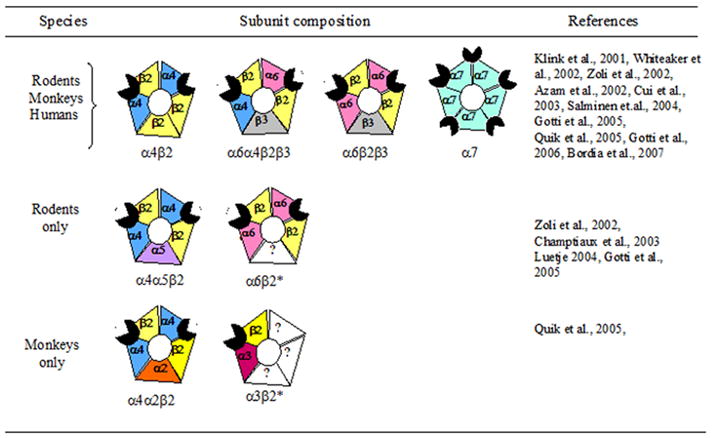
Putative subunit composition of nAChR subtypes in rodent, monkey and human striatum. Note expression of the α4β2, α6α4β2β3, α6β2β3 and α7 nAChR subtypes across the three species. In contrast, the α4α5β2 and α6(nonα4)β2* nAChR subtypes appear to be expressed only in rodent striatum, while the α4α2β2 and α3β2* subtypes are expressed in monkey striatum. The * indicates the possible presence of other nAChR subunits in the receptor complex.
5.1. Rodents
Robust nAChR expression has been identified in the rodent nigrostriatal system using radiolabeled epibatidine, a ligand that labels multiple receptor subtypes. The use of other ligands such as radiolabeled nicotine, α-bungarotoxin and α-conotoxinMII has helped identify nAChR subtypes, including those containing the α4, α6 and α7 subunits [67, 69, 71]. These findings from receptor binding studies are in agreement with the results of in situ hybridization and RT-PCR work, which demonstrate the presence of α2 through α7 and β2 through β4 nAChR subunit transcripts in the striatum and/or substantia nigra [72–77]. The results of immunoprecipitation studies using subunit-selective nAChR antibodies suggest that the greater majority of these transcripts are translated into protein to form receptors [64, 78]. These show that all but the α2, α3 and β4 subunits are present in the striatum and/or substantia nigra of mice and rats. These data, coupled with the results of functional studies (3H-dopamine release from striatal synaptosomes) using wild-type and nAChR null mutant mice, indicate that the major nAChR subtypes in rodent striatum are α6α4β2β3, α6β2β3 and α4β2* (*indicates the possible presence of other subunits in the receptor complex) nAChRs (Fig. 1) [63, 64, 79, 80]. In contrast, α7 nAChRs are present at a much lower density, particularly in rats [81].
5.2. Monkeys
Multiple nAChRs have also been identified in striatum of nonhuman primates. nAChR subunit mRNAs in the primate nigrostriatal system include the α2, α4 through α7, and β2 through β4 transcripts [82, 83]. Receptor binding studies with 125I-epibatidine demonstrate that receptors are expressed in both the striatum and substantia nigra [84–86]. Subsequent studies with more selective ligands, such as 125I-α-conotoxinMII indicated the presence of α6β2* nAChR subtypes, in the nigrostriatal system [84, 87], while the use of 125I-epibatidine in the presence of unlabelled α-conotoxinMII suggested the presence of α4β2* receptors. Immunoprecipitation studies with receptor subunit-directed antibodies show that all subunits except α5 and β4 are expressed in the primate striatum and substantia nigra. The major nAChR subtypes appear similar to those in the rodent and include α6α4β2β3, α6β2β3 and α4β2 (Fig. 1), with only very low α7 nAChR expression [87, 88]. There are some variations in the minor subtypes, with expression of α4α2β2 and α3β2* nAChRs in nonhuman primates, but not rodent striatum [87].
5.3. Humans
Although studies in human striatum are more limited, the nAChR subtypes appear to be similar to those in the rodent and monkey nigrostriatal systems [89, 90]. Evidence for this stems from the results of in situ hybridization and RT-PCR studies which demonstrate the presence of multiple nAChR transcripts including α3, α4, α7, and β2 mRNAs, with the others not yet investigated [91–94]. Radioligand binding studies show that 125I-epibatidine, 3H-nicotine, 125I-A85380, 125I-α-bungarotoxin and 125I-α-conotoxinMII all bind to human striatum suggesting the presence of nAChRs containing the α4, α3 and/or α6, α7, and β2 [89, 90, 95–104]. The α4, α6, β2 and β3 nAChR subunits have also been identified in human striatum using immunoprecipitation studies with subunit specific human nAChR antibodies [105]. These combined data suggest the presence of α4β2*, α6β2* and α7 nAChRs in human striatum (Fig. 1).
6. Alterations in striatal nAChRs with nigrostriatal damage
A key question is whether nAChRs are altered with nigrostriatal damage in experimental models and in Parkinson’s disease. Such knowledge is critical for the development of effective targeted therapies in Parkinson’s disease. To approach this, work has been done in animal models with nigrostriatal damage, as well as with tissue from Parkinson’s disease brains.
6.1. Rodents
The most commonly used parkinsonian rat model involves unilateral 6-hydroxydopamine injection into the striatum, substantia nigra or medial forebrain bundle. Nigrostriatal damage decreases high affinity nicotine binding sites that are thought to represent α4β2* nAChRs, while α7 nAChRs are unaffected [106, 107]. Nigrostriatal damage also leads to dramatic declines in binding of 125I-α-conotoxinMII [108], a ligand that identifies α6β2* in rodent striatum [80, 109]. The loss of α6β2* receptors correlates well with declines in the striatal dopamine transporter, a dopaminergic neuronal marker [64, 78, 108], suggesting that this receptor subtype is located primarily on striatal dopaminergic nerve terminals. The results of immunoprecipitation studies using nAChR subunit-targeted antibodies support these findings and show that the largest reductions are in α6 and β3 subunit proteins, followed by smaller declines in α4, α5 and β2 subunits [64, 78]. There was no change in α7 nAChRs, while the α2, α3 and β4 subunit proteins are not expressed in rodent striatum.
These combined results indicate that there is a loss of both α4β2* and α6β2* nAChRs with nigrostriatal damage in rodents. The larger decrease in the latter population most likely occurs because α6β2* nAChRs are localized primarily on striatal dopaminergic terminals, while α4β2* receptors are expressed on both dopaminergic terminals and other striatal neurons.
6.2. Monkeys
These data in rodents correspond well with those in nonhuman primates with MPTP-induced nigrostriatal damage (Table 2). A moderate lesion led to declines predominately in α6β2* nAChRs that closely paralleled reductions in the dopamine transporter [84]. With severe nigrostriatal damage (≥90%), decreases were also evident in α4β2* nAChR although they were of smaller magnitude compared to those in α6β2* nAChRs [85, 110]. Immunoprecipitation studies with antibodies to human nAChR subunits showed that the major reductions with severe nigrostriatal damage were in the α6 and β3 subunits, followed by decreases in α4, β2, and α3 subunits [87]. There was no change in α7 nAChRs, while the α5 and β4 subunit proteins are not expressed in monkey striatum [87].
Table 2.
Summary of the declines in different nAChR subtypes in monkey striatum with nigrostriatal damage
| nAChR subtype | Present on striatal dopamine terminals | Severity of lesion
|
|
|---|---|---|---|
| Moderate | Severe | ||
| α6α4β2β3 | Yes | ↓↓ | ↓↓ |
| α6β2β3 | Yes | - | ↓ |
| α4β2 | Yes | - | ↓ |
| α4β2* | No | - | - |
| α7 | No | - | - |
The α3β2* nAChR subtype is also expressed in monkey striatum and decreased with lesioning. However, the effect of moderate versus severe nigrostriatal damage remains to be determined [87]. The α4β2* receptors may contain either α4β2 or α4α2β2 subunits. Moderate lesion, ≥ 50% decline in the dopamine transporter; severe lesion, ≥ 90% decline in the dopamine transporter; ↓ indicates a decline; - indicates no change.
In summary, α6β2* nAChRs appear particularly vulnerable to nigrostriatal damage as they are selectively decreased with a moderate lesion. In contrast, α4β2* subtypes are affected only with severe degeneration, while α7 nAChRs are unchanged (Table 2).
6.3. Humans
Results in brains of Parkinson’s disease cases were similar to those in parkinsonian animal models. Declines were observed in radiolabeled epibatidine, which targets most nAChR subtypes and 125I-A85380, which identifies β2-containing nAChRs [89, 90, 111]. Receptor studies with 3H-nicotine also demonstrate ~50% decline in striatum, as well as in other areas including substantia nigra, cortex and hippocampus [98–101]. These latter findings support the contention that there is a generalized loss of neuronal integrity in Parkinson’s disease [5, 6]. As in the rodent and monkey studies, declines in α6β2* nAChRs in Parkinson’s disease brains are significantly greater than in α4β2* receptors in some striatal regions, as evaluated using receptor binding assays [103, 104]. This observation is supported by results from immunoprecipitation studies which showed that the α6 subunit is decreased by ≥80%, whereas the α4 and β2 subunits were decreased by ~50% [105].
Thus, both α4β2* and α6β2* nAChRs were decreased in Parkinson’s disease brains, with somewhat greater declines in this latter receptor population. There appeared to be little change in the α7 nAChR population, although declines have been observed in some studies [90, 111].
7. Receptor studies with the α-conotoxinMII analog E11A
7.1. Identification of multiple α6β2* nAChR subtypes in control striatum across species
As already mentioned, the α6β2* nAChR population may be of particular relevance to nigrostriatal function and to Parkinson’s disease [71]. This idea is based on results showing that its localization is fairly restricted in the CNS to the dopaminergic nigrostriatal and a few other catecholaminergic systems. In addition, declines in α6β2* nAChRs closely parallel the loss of the striatal dopamine transporter. Previous rodents studies have shown that mouse striatum expresses two α6β2* nAChR subtypes, those containing the α6α4β2β3 and α6β2β3 subunits [63, 79]. Similarly, our studies in monkeys suggest the presence of at least two striatal α6β2* nAChRs [87, 112]. In a continued effort to more precisely determine the subunit composition of these receptors, experiments were recently performed with α-conotoxinMII analogs to determine whether these might discriminate between α6β2* subtypes. One such analog α-conotoxinMII E11A, in which the glutamic acid at position 11 is replaced with alanine [113], inhibited 125I-α-conotoxinMII binding to striatum from mice, monkeys and humans in a biphasic manner (Fig. 2) [88]. These data indicate that α-conotoxinMII E11A distinguishes between a very high (fM) and high affinity (pM) α6β2* subtype. Subsequent receptor studies showed that only the very high affinity site was lost in striatum of α4 null mutant mice (Fig. 2) [88]. Altogether, these data suggest that the very high affinity site is the α6α4β2β3 receptor and the high affinity site the α6β2β3 subtype.
Fig. 2.
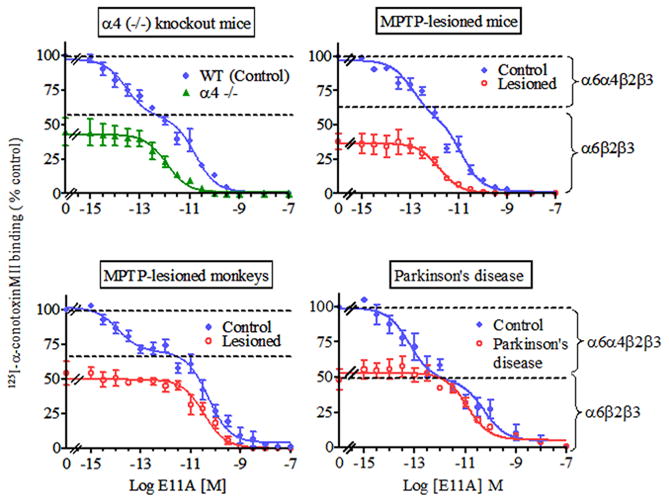
Preferential decline in the striatal α6α4β2β3 nAChR subtype in animal models (mouse and monkey) of nigrostriatal damage and Parkinson’s disease. 125I-α-conotoxinMII (0.5 nM) competition assays were performed using varying concentrations of the α-conotoxinMII analog E11A. In the upper left panel, biphasic inhibition curves (data fit best to a two site model) were obtained with E11A in the striatum of wild type mice suggesting the presence of at least two E11A-sensitive 125I-α-conotoxinMII binding sites. In contrast monophasic inhibition (data fit best to a one site model) was obtained using striatal sections from α4 nAChR null mutant mice. Since these mice do not express the α4 subunit, the remaining high affinity-binding site is most likely the α6β2β3 nAChR, while the very high affinity-binding site that is lost may represent the α6α4β2β3 subtype. The three remaining panels depict the effect of nigrostriatal damage on the nAChR subtypes in mouse, monkey and human striatum. Biphasic inhibition curves (data fit best to two site model) were obtained with E11A under control conditions. These data suggest that E11A discriminates between at least two α6β2-containing nAChR populations, a very high α6α4β2β3 and a high affinity α6β2β3 nAChR subtype. Similar competition analyses using sections from lesioned animals and Parkinson’s disease striatum yielded monophasic curves, suggesting the loss of the very high affinity E11A-sensitive α6α4β2β3 nAChR subtype. Symbols represent means ± S.E.M. of 4–8 mice, 4–6 monkeys and 5 control and 4 Parkinson’s disease cases. Reproduced in modified form with permission from reference [88]
7.2. Preferential loss of the α6α4β2β3 nAChR subtype with nigrostriatal damage in mice, monkeys and humans
We subsequently investigated the effect of nigrostriatal lesioning on these two striatal α6β2* populations in two animal models and in Parkinson’s disease (Fig. 2). There was a preferential loss of the very high affinity E11A binding site (α6α4β2β3) in striatum of MPTP-treated mice, MPTP-treated monkeys and Parkinson’s disease cases [88]. This selective loss of the α6α4β2β3 binding site correlated well with reductions in striatal dopamine transporter [88]. In monkeys with moderate nigrostriatal damage (~50% reduction in dopamine transporter) there was a preferential reduction in the α6α4β2β3 subtype (Fig. 2 and 3), while the α6β2β3 and α4β2 nAChR populations were reduced primarily with severe lesions (≥ 90%) (Fig. 3; Table 2) [88]. The α6α4β2β3 subtype may thus provide a unique marker for dopamine nerve terminals that are particularly sensitive to nigrostriatal degeneration. This possibility is consistent with previous work indicating that dopaminergic afferents from the nigra to the striatum are variably susceptible to nigrostriatal insults [114, 115]. Studies to understand the molecular basis for this enhanced vulnerability have linked calbindin to nigrostriatal dopamine neuron survival [116–118] while neuromelanin has been negatively associated [119–122]. However, a definitive connection of either of these markers with nigrostriatal damage remains to be established. On the other hand, there was a fairly clear-cut relationship between the loss of the α6α4β2β3 nAChR and nigrostriatal damage, possibly indicating that this subtype identifies selectively vulnerable dopaminergic terminals. Studies to investigate this possibility may also provide insight about the mechanisms responsible for the nigrostriatal neurodegeneration.
Fig. 3.
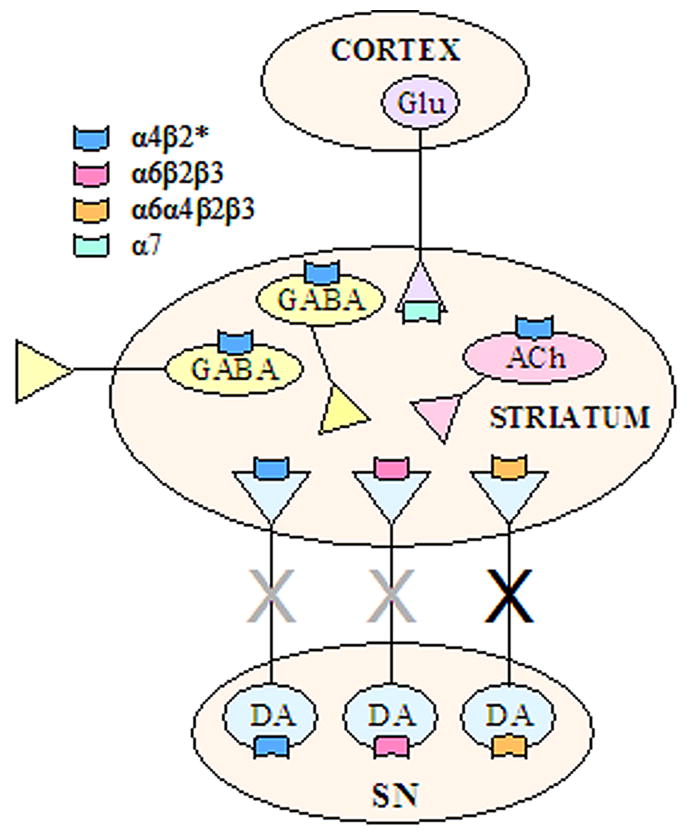
Schematic representation of nAChR localization within the nigrostriatal pathway based on receptor binding, antibody immunoprecipitation and functional studies. The nigrostriatal dopaminergic terminals express three subtypes, the α4β2*, α6β2β3 and α6α4β2β3 nAChR populations. The results of lesion studies in animal models suggest that α6α4β2β3 subtype is localized to a select population of dopaminergic terminals that are particularly vulnerable to nigrostriatal insults (black “X”). The α4β2* and α6β2β3 subtypes are most likely present on terminals other than those expressing α6α4β2β3 nAChRs since they are reduced only with more severe dopaminergic lesions (grey “X”). It is not known whether these latter two subtypes are on the same or separate dopaminergic terminals. The precise postsynaptic localization of the α4β2* nAChR subtype in striatum is presently uncertain, but they may be present on cholinergic and/or GABAergic neurons. α7 nAChRs are located on glutamatergic afferent terminals from the cortex. ACh, acetylcholine; DA, dopamine; Glu, glutamate; SN, substantia nigra. The * indicates the possible presence of other nAChR subunits in the receptor complex.
8. Concluding remarks
Accumulating evidence suggests that nigrostriatal nAChRs represent potential targets for the treatment of Parkinson’s disease for symptomatic improvements and/or for long-term neuroprotection. Several nAChR populations have been identified in the nigrostriatal system. These include α4β2* and α6β2β3 subtypes that are decreased primarily with severe nigrostriatal degeneration, and the α6α4β2β3 subtype that is significantly reduced even with only moderate damage (Fig. 3). These latter findings suggest that the α6α4β2β3 nAChR subtype represents a marker for neurons particularly vulnerable to nigrostriatal damage. In addition, these data suggest that this receptor subtype may represent a particularly relevant target for Parkinson’s disease therapeutics as stimulation of the remaining α6α4β2β3 nAChRs may enhance function lost with nigrostriatal damage. This possibility stems from results showing that chronic nicotine treatment improved striatal integrity and function. On the other hand, nicotine is well known to desensitize or block nAChRs raising the question whether nicotine may induce its beneficial effects by blocking aberrant nAChR-mediated activity. This latter interpretation would suggest that an α6α4β2β3 nAChR antagonist (and not an agonist) may be the drug of choice to counteract the effects of nigrostriatal injury and/or provide symptomatic improvements. Continued studies to elucidate the role of α6β2* nAChRs in nigrostriatal function should help resolve what type of α6β2* nAChR ligand would prove most beneficial against the effects of nigrostriatal damage in neurological disorders such as Parkinson’s disease.
Acknowledgments
This work was supported by NIH grants NS42091 and NS47162.
Abbreviations
- nAChR
nicotinic acetylcholine receptor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Samii A, Nutt JG, Ransom BR. Parkinson’s disease. Lancet. 2004;363:1783–93. doi: 10.1016/S0140-6736(04)16305-8. [DOI] [PubMed] [Google Scholar]
- 2.Olanow CW. The scientific basis for the current treatment of Parkinson’s disease. Annu Rev Med. 2004;55:41–60. doi: 10.1146/annurev.med.55.091902.104422. [DOI] [PubMed] [Google Scholar]
- 3.Savitt JM, Dawson VL, Dawson TM. Diagnosis and treatment of Parkinson disease: molecules to medicine. J Clin Invest. 2006;116:1744–54. doi: 10.1172/JCI29178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lang AE, Obeso JA. Challenges in Parkinson’s disease: restoration of the nigrostriatal dopamine system is not enough. Lancet Neurol. 2004;3:309–16. doi: 10.1016/S1474-4422(04)00740-9. [DOI] [PubMed] [Google Scholar]
- 5.Braak H, Del Tredici K, Bratzke H, Hamm-Clement J, Sandmann-Keil D, Rub U. Staging of the intracerebral inclusion body pathology associated with idiopathic Parkinson’s disease (preclinical and clinical stages) J Neurol. 2002;249(Suppl 3):III/1–5. doi: 10.1007/s00415-002-1301-4. [DOI] [PubMed] [Google Scholar]
- 6.Braak H, Muller CM, Rub U, Ackermann H, Bratzke H, de Vos RA, Del Tredici K. Pathology associated with sporadic Parkinson’s disease--where does it end? J Neural Transm. 2006;(Suppl):89–97. doi: 10.1007/978-3-211-45295-0_15. [DOI] [PubMed] [Google Scholar]
- 7.Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 8.Marsden CD. Basal ganglia disease. Lancet. 1982;2:1141–7. doi: 10.1016/s0140-6736(82)92797-0. [DOI] [PubMed] [Google Scholar]
- 9.Agid Y, Javoy-Agid F. Peptides and Parkinson’s disease. Trends in Neuroscience. 1985;6:30–35. [Google Scholar]
- 10.Zhou FM, Wilson CJ, Dani JA. Cholinergic interneuron characteristics and nicotinic properties in the striatum. J Neurobiol. 2002;53:590–605. doi: 10.1002/neu.10150. [DOI] [PubMed] [Google Scholar]
- 11.Quik M. Smoking, nicotine and Parkinson’s disease. Trends Neurosci. 2004;27:561–8. doi: 10.1016/j.tins.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 12.Levin ED, McClernon FJ, Rezvani AH. Nicotinic effects on cognitive function: behavioral characterization, pharmacological specification, and anatomic localization. Psychopharmacology (Berl) 2006;184:523–39. doi: 10.1007/s00213-005-0164-7. [DOI] [PubMed] [Google Scholar]
- 13.Morens DM, Grandinetti A, Reed D, White LR, Ross GW. Cigarette smoking and protection from Parkinson’s disease: false association or etiologic clue? Neurology. 1995;45:1041–51. doi: 10.1212/wnl.45.6.1041. [DOI] [PubMed] [Google Scholar]
- 14.Baron JA. Beneficial effects of nicotine and cigarette smoking: the real, the possible and the spurious. Br Med Bull. 1996;52:58–73. doi: 10.1093/oxfordjournals.bmb.a011533. [DOI] [PubMed] [Google Scholar]
- 15.Checkoway H, Nelson LM. Epidemiologic approaches to the study of Parkinson’s disease etiology. Epidemiology. 1999;10:327–36. [PubMed] [Google Scholar]
- 16.Gorell JM, Rybicki BA, Johnson CC, Peterson EL. Smoking and Parkinson’s disease: a dose-response relationship. Neurology. 1999;52:115–9. doi: 10.1212/wnl.52.1.115. [DOI] [PubMed] [Google Scholar]
- 17.Allam MF, Campbell MJ, Hofman A, Del Castillo AS, Fernandez-Crehuet Navajas R. Smoking and Parkinson’s disease: systematic review of prospective studies. Mov Disord. 2004;19:614–21. doi: 10.1002/mds.20029. [DOI] [PubMed] [Google Scholar]
- 18.O’Neill MJ, Murray TK, Lakics V, Visanji NP, Duty S. The role of neuronal nicotinic acetylcholine receptors in acute and chronic neurodegeneration. Curr Drug Target CNS Neurol Disord. 2002;1:399–411. doi: 10.2174/1568007023339166. [DOI] [PubMed] [Google Scholar]
- 19.Mudo G, Belluardo N, Fuxe K. Nicotinic receptor agonists as neuroprotective/neurotrophic drugs. Progress in molecular mechanisms. J Neural Transm. 2007;114:135–47. doi: 10.1007/s00702-006-0561-z. [DOI] [PubMed] [Google Scholar]
- 20.Zanardi A, Leo G, Biagini G, Zoli M. Nicotine and neurodegeneration in ageing. Toxicol Lett. 2002;127:207–15. doi: 10.1016/s0378-4274(01)00502-1. [DOI] [PubMed] [Google Scholar]
- 21.Jeyarasasingam G, Tompkins L, Quik M. Stimulation of non-α7 nicotinic receptors partially protects dopaminergic neurons from 1-methyl-4-phenylpyridinium-induced toxicity in culture. Neuroscience. 2002;109:275–85. doi: 10.1016/s0306-4522(01)00488-2. [DOI] [PubMed] [Google Scholar]
- 22.Quik M, O’Neill M, Perez XA. Nicotine neuroprotection against nigrostriatal damage: importance of the animal model. Trends Pharmacol Sci. 2007 doi: 10.1016/j.tips.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 23.Janson AM, Fuxe K, Agnati LF, Kitayama I, Harfstrand A, Andersson K, Goldstein M. Chronic nicotine treatment counteracts the disappearance of tyrosine-hydroxylase-immunoreactive nerve cell bodies, dendrites and terminals in the mesostriatal dopamine system of the male rat after partial hemitransection. Brain Res. 1988;455:332–45. doi: 10.1016/0006-8993(88)90092-3. [DOI] [PubMed] [Google Scholar]
- 24.Ryan RE, Ross SA, Drago J, Loiacono RE. Dose-related neuroprotective effects of chronic nicotine in 6-hydroxydopamine treated rats, and loss of neuroprotection in α4 nicotinic receptor subunit knockout mice. Br J Pharmacol. 2001;132:1650–6. doi: 10.1038/sj.bjp.0703989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Soto-Otero R, Mendez-Alvarez E. Hermida-Ameijeiras A, Lopez-Real AM, Labandeira-Garcia JL, Effects of (-)-nicotine and (-)-cotinine on 6-hydroxydopamine-induced oxidative stress and neurotoxicity: relevance for Parkinson’s disease. Biochem Pharmacol. 2002;64:125–35. doi: 10.1016/s0006-2952(02)01070-5. [DOI] [PubMed] [Google Scholar]
- 26.Abin-Carriquiry JA, McGregor-Armas R, Costa G, Urbanavicius J, Dajas F. Presynaptic involvement in the nicotine prevention of the dopamine loss provoked by 6-OHDA administration in the substantia nigra. Neurotox Res. 2002;4:133–9. doi: 10.1080/10298420290015863. [DOI] [PubMed] [Google Scholar]
- 27.Costa G, Abin-Carriquiry JA, Dajas F. Nicotine prevents striatal dopamine loss produced by 6-hydroxydopamine lesion in the substantia nigra. Brain Res. 2001;888:336–342. doi: 10.1016/s0006-8993(00)03087-0. [DOI] [PubMed] [Google Scholar]
- 28.Visanji NP, O’Neill MJ, Duty S. Nicotine, but neither the α4β2 ligand RJR2403 nor an α7 nAChR subtype selective agonist, protects against a partial 6-hydroxydopamine lesion of the rat median forebrain bundle. Neuropharmacology. 2006;51:506–16. doi: 10.1016/j.neuropharm.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 29.Gao ZG, Cui WY, Zhang HT, Liu CG. Effects of nicotine on 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine-induced depression of striatal dopamine content and spontaneous locomotor activity in C57 black mice. Pharmacol Res. 1998;38:101–6. doi: 10.1006/phrs.1998.0337. [DOI] [PubMed] [Google Scholar]
- 30.Janson AM, Fuxe K. Goldstein M, Differential effects of acute and chronic nicotine treatment on MPTP-(1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) induced degeneration of nigrostriatal dopamine neurons in the black mouse. Clin Investig. 1992;70:232–8. doi: 10.1007/BF00184656. [DOI] [PubMed] [Google Scholar]
- 31.Shahi GS, Das NP, Moochhala SM. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity: partial protection against striato-nigral dopamine depletion in C57BL/6J mice by cigarette smoke exposure and by β-naphthoflavone-pretreatment. Neurosci Lett. 1991;127:247–50. doi: 10.1016/0304-3940(91)90804-3. [DOI] [PubMed] [Google Scholar]
- 32.Janson AM, Fuxe K, Agnati L, Sundstrom E, Goldstein M. The effect of chronic nicotine treatment on 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced degneration of nigrostriatal dopamine neurons in the black mouse. Advances in Pharmacological Sciences. 1991;1:323–329. [Google Scholar]
- 33.Behmand RA, Harik SI. Nicotine enhances 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity. J Neurochem. 1992;58:776–9. doi: 10.1111/j.1471-4159.1992.tb09786.x. [DOI] [PubMed] [Google Scholar]
- 34.Hadjiconstantinou M, Hubble JP, Wemlinger TA, Neff NH. Enhanced MPTP neurotoxicity after treatment with isoflurophate or cholinergic agonists. J Pharmacol Exp Ther. 1994;270:639–44. [PubMed] [Google Scholar]
- 35.Ferger B, Spratt C, Earl CD, Teismann P, Oertel WH, Kuschinsky K. Effects of nicotine on hydroxyl free radical formation in vitro and on MPTP-induced neurotoxicity in vivo. Naunyn Schmiedebergs Arch Pharmacol. 1998;358:351–9. doi: 10.1007/pl00005264. [DOI] [PubMed] [Google Scholar]
- 36.Parain K, Hapdey C, Rousselet E, Marchand V, Dumery B, Hirsch EC. Cigarette smoke and nicotine protect dopaminergic neurons against the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine Parkinsonian toxin. Brain Res. 2003;984:224–32. doi: 10.1016/s0006-8993(03)03195-0. [DOI] [PubMed] [Google Scholar]
- 37.Sershen H, Hashim A, Wiener HL, Lajtha A. Effect of chronic oral nicotine on dopaminergic function in the MPTP-treated mouse. Neurosci Lett. 1988;93:270–4. doi: 10.1016/0304-3940(88)90094-8. [DOI] [PubMed] [Google Scholar]
- 38.Carr LA, Rowell PP. Attenuation of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity by tobacco smoke. Neuropharmacology. 1990;29:311–4. doi: 10.1016/0028-3908(90)90019-n. [DOI] [PubMed] [Google Scholar]
- 39.Perry TL, Hansen S, Jones K. Exposure to cigarette smoke does not decrease the neurotoxicity of N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in mice. Neurosci Lett. 1987;74:217–20. doi: 10.1016/0304-3940(87)90152-2. [DOI] [PubMed] [Google Scholar]
- 40.Khwaja M, McCormack A, McIntosh JM, Di Monte DA, Quik M. Nicotine partially protects against paraquat-induced nigrostriatal damage in mice; link to a6b2* nAChRs. Journal of Neurochemistry. 2006;100:180–190. doi: 10.1111/j.1471-4159.2006.04177.x. [DOI] [PubMed] [Google Scholar]
- 41.Quik M, Chen L, Parameswaran N, Xie X, Langston JW, McCallum SE. Chronic oral nicotine normalizes dopaminergic function and synaptic plasticity in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned primates. J Neurosci. 2006;26:4681–9. doi: 10.1523/JNEUROSCI.0215-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bordia T, Parameswaran N, Fan H, Langston JW, McIntosh JM, Quik M. Partial recovery of striatal nicotinic receptors in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-lesioned monkeys with chronic oral nicotine. J Pharmacol Exp Ther. 2006;319:285–92. doi: 10.1124/jpet.106.106997. [DOI] [PubMed] [Google Scholar]
- 43.Quik M, Parameswaran N, McCallum SE, Bordia T, Bao S, McCormack A, Kim A, Tyndale RF, Langston JW, Di Monte DA. Chronic oral nicotine treatment protects against striatal degeneration in MPTP-treated primates. J Neurochem. 2006;98:1866–75. doi: 10.1111/j.1471-4159.2006.04078.x. [DOI] [PubMed] [Google Scholar]
- 44.Balfour DJ, Fagerstrom KO. Pharmacology of nicotine and its therapeutic use in smoking cessation and neurodegenerative disorders. Pharmacol Ther 7. 1996;2:51–81. doi: 10.1016/s0163-7258(96)00099-x. [DOI] [PubMed] [Google Scholar]
- 45.Meshul CK, Kamel D, Moore C, Kay TS, Krentz L. Nicotine alters striatal glutamate function and decreases the apomorphine-induced contralateral rotations in 6-OHDA-lesioned rats. Exp Neurol. 2002;175:257–74. doi: 10.1006/exnr.2002.7900. [DOI] [PubMed] [Google Scholar]
- 46.Cosford ND, Bleicher L, Herbaut A, McCallum JS, Vernier JM, Dawson H, Whitten JP, Adams P, Chavez-Noriega L, Correa LD, Crona JH, Mahaffy LS, Menzaghi F, Rao TS, Reid R, Sacaan AI, Santori E, Stauderman KA, Whelan K, Lloyd GK, McDonald IA. (S)-(-)-5-ethynyl-3-(1-methyl-2-pyrrolidinyl)pyridine maleate (SIB-1508Y): a novel anti-parkinsonian agent with selectivity for neuronal nicotinic acetylcholine receptors. J Med Chem. 1996;39:3235–7. doi: 10.1021/jm960328w. [DOI] [PubMed] [Google Scholar]
- 47.Domino EF, Ni L, Zhang H. Nicotine Alone and in Combination with l-DOPA Methyl Ester or the D(2) Agonist N-0923 in MPTP-Induced Chronic Hemiparkinsonian Monkeys. Exp Neurol. 1999;158:414–421. doi: 10.1006/exnr.1999.7106. [DOI] [PubMed] [Google Scholar]
- 48.Schneider JS, Pope-Coleman A, Van Velson M, Menzaghi F, Lloyd GK. Effects of SIB-1508Y, a novel neuronal nicotinic acetylcholine receptor agonist, on motor behavior in parkinsonian monkeys. Mov Disord. 1998;13:637–42. doi: 10.1002/mds.870130405. [DOI] [PubMed] [Google Scholar]
- 49.Decamp E, Schneider JS. Effects of nicotinic therapies on attention and executive functions in chronic low-dose MPTP-treated monkeys. Eur J Neurosci. 2006;24:2098–104. doi: 10.1111/j.1460-9568.2006.05077.x. [DOI] [PubMed] [Google Scholar]
- 50.Ishikawa A, Miyatake T. Effects of smoking in patients with early-onset Parkinson’s disease. J Neurol Sci. 1993;117:28–32. doi: 10.1016/0022-510x(93)90150-w. [DOI] [PubMed] [Google Scholar]
- 51.Fagerstrom KO, Pomerleau O, Giordani B, Stelson F. Nicotine may relieve symptoms of Parkinson’s disease. Psychopharmacology (Berl) 1994;116:117–9. doi: 10.1007/BF02244882. [DOI] [PubMed] [Google Scholar]
- 52.Kelton MC, Kahn HJ, Conrath CL, Newhouse PA. The effects of nicotine on Parkinson’s disease. Brain Cogn. 2000;43:274–82. [PubMed] [Google Scholar]
- 53.Vieregge A, Sieberer M, Jacobs H, Hagenah JM, Vieregge P. Transdermal nicotine in PD: a randomized, double-blind, placebo-controlled study. Neurology. 2001;57:1032–5. doi: 10.1212/wnl.57.6.1032. [DOI] [PubMed] [Google Scholar]
- 54.Lemay S, Chouinard S, Blanchet P, Masson H, Soland V, Beuter A, Bedard MA. Lack of efficacy of a nicotine transdermal treatment on motor and cognitive deficits in Parkinson’s disease. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28:31–9. doi: 10.1016/S0278-5846(03)00172-6. [DOI] [PubMed] [Google Scholar]
- 55.Ebersbach G, Stock M, Muller J, Wenning G, Wissel J, Poewe W. Worsening of motor performance in patients with Parkinson’s disease following transdermal nicotine administration. Mov Disord. 1999;14:1011–3. doi: 10.1002/1531-8257(199911)14:6<1011::aid-mds1016>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 56.Clemens P, Baron JA, Coffey D, Reeves A. The short-term effect of nicotine chewing gum in patients with Parkinson’s disease. Psychopharmacology (Berl) 1995;117:253–6. doi: 10.1007/BF02245195. [DOI] [PubMed] [Google Scholar]
- 57.Shoulson I. Randomized placebo-controlled study of the nicotinic agonist SIB-1508Y in Parkinson disease. Neurology. 2006;66:408–10. doi: 10.1212/01.wnl.0000196466.99381.5c. [DOI] [PubMed] [Google Scholar]
- 58.Parameswaran N, Cox H, O’Leary K, Langston JW, Di Monte D, Quik M. Nicotine treatment reduces L-dopa-induced dyskinesias in MPTP-treated squirrel monkeys. Soc Neurosci Abstr. 2007 [Google Scholar]
- 59.Fonnum F. Recent developments in biochemical investigations of cholinergic transmission. Brain Res. 1973;62:497–507. doi: 10.1016/0006-8993(73)90714-2. [DOI] [PubMed] [Google Scholar]
- 60.Fuxe K, Hoekfelt T, Nilsson O. Observations on the Cellular Localization of Dopamine in the Caudate Nucleus of the Rat. Z Zellforsch Mikrosk Anat. 1964;63:701–6. doi: 10.1007/BF00339917. [DOI] [PubMed] [Google Scholar]
- 61.Kaiser S, Wonnacott S. α-bungarotoxin-sensitive nicotinic receptors indirectly modulate [3H]dopamine release in rat striatal slices via glutamate release. Mol Pharmacol. 2000;58:312–8. doi: 10.1124/mol.58.2.312. [DOI] [PubMed] [Google Scholar]
- 62.Marchi M, Risso F, Viola C, Cavazzani P, Raiteri M. Direct evidence that release-stimulating α7* nicotinic cholinergic receptors are localized on human and rat brain glutamatergic axon terminals. J Neurochem. 2002;80:1071–8. doi: 10.1046/j.0022-3042.2002.00805.x. [DOI] [PubMed] [Google Scholar]
- 63.Salminen O, Murphy KL, McIntosh JM, Drago J, Marks MJ, Collins AC, Grady SR. Subunit composition and pharmacology of two classes of striatal presynaptic nicotinic acetylcholine receptors mediating dopamine release in mice. Mol Pharmacol. 2004;65:1526–35. doi: 10.1124/mol.65.6.1526. [DOI] [PubMed] [Google Scholar]
- 64.Champtiaux N, Gotti C, Cordero-Erausquin M, David DJ, Przybylski C, Lena C, Clementi F, Moretti M, Rossi FM, Le Novere N, McIntosh JM, Gardier AM, Changeux JP. Subunit composition of functional nicotinic receptors in dopaminergic neurons investigated with knock-out mice. J Neurosci. 2003;23:7820–9. doi: 10.1523/JNEUROSCI.23-21-07820.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wonnacott S. Presynaptic nicotinic ACh receptors. Trends Neurosci. 1997;20:92–8. doi: 10.1016/s0166-2236(96)10073-4. [DOI] [PubMed] [Google Scholar]
- 66.MacDermott AB, Role LW, Siegelbaum SA. Presynaptic ionotropic receptors and the control of transmitter release. Annu Rev Neurosci. 1999;22:443–85. doi: 10.1146/annurev.neuro.22.1.443. [DOI] [PubMed] [Google Scholar]
- 67.Dani JA, Bertrand D. Nicotinic Acetylcholine Receptors and Nicotinic Cholinergic Mechanisms of the Central Nervous System. Annu Rev Pharmacol Toxicol. 2007;47:699–729. doi: 10.1146/annurev.pharmtox.47.120505.105214. [DOI] [PubMed] [Google Scholar]
- 68.Gotti C, Riganti L, Vailati S, Clementi F. Brain neuronal nicotinic receptors as new targets for drug discovery. Curr Pharm Des. 2006;12:407–28. doi: 10.2174/138161206775474486. [DOI] [PubMed] [Google Scholar]
- 69.Gotti C, Zoli M, Clementi F. Brain nicotinic acetylcholine receptors: native subtypes and their relevance. Trends Pharmacol Sci. 2006;27:482–91. doi: 10.1016/j.tips.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 70.Nashmi R, Lester HA. CNS localization of neuronal nicotinic receptors. J Mol Neurosci. 2006;30:181–4. doi: 10.1385/JMN:30:1:181. [DOI] [PubMed] [Google Scholar]
- 71.Quik M, McIntosh JM. Striatal α6* nicotinic acetylcholine receptors: potential targets for Parkinson’s disease therapy. J Pharmacol Exp Ther. 2006;316:481–9. doi: 10.1124/jpet.105.094375. [DOI] [PubMed] [Google Scholar]
- 72.Wada E, Wada K, Boulter J, Deneris E, Heinemann S, Patrick J, Swanson LW. Distribution of α2, α3, α4, and β2 neuronal nicotinic receptor subunit mRNAs in the central nervous system: a hybridization histochemical study in the rat. J Comp Neurol. 1989;284:314–35. doi: 10.1002/cne.902840212. [DOI] [PubMed] [Google Scholar]
- 73.Le Novere N, Zoli M, Changeux JP. Neuronal nicotinic receptor α6 subunit mRNA is selectively concentrated in catecholaminergic nuclei of the rat brain. Eur J Neurosci. 1996;8:2428–39. doi: 10.1111/j.1460-9568.1996.tb01206.x. [DOI] [PubMed] [Google Scholar]
- 74.Klink R, de Kerchove d’Exaerde A, Zoli M, Changeux JP. Molecular and physiological diversity of nicotinic acetylcholine receptors in the midbrain dopaminergic nuclei. J Neurosci. 2001;21:1452–63. doi: 10.1523/JNEUROSCI.21-05-01452.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Charpantier E, Barneoud P, Moser P, Besnard F, Sgard F. Nicotinic acetylcholine subunit mRNA expression in dopaminergic neurons of the rat substantia nigra and ventral tegmental area. Neuroreport. 1998;9:3097–101. doi: 10.1097/00001756-199809140-00033. [DOI] [PubMed] [Google Scholar]
- 76.Elliott KJ, Jones JM, Sacaan AI, Lloyd GK, Corey-Naeve J. 6-hydroxydopamine lesion of rat nigrostriatal dopaminergic neurons differentially affects nicotinic acetylcholine receptor subunit mRNA expression. J Mol Neurosci. 1998;10:251–60. doi: 10.1007/BF02761778. [DOI] [PubMed] [Google Scholar]
- 77.Winzer-Serhan UH, Leslie FM. Codistribution of nicotinic acetylcholine receptor subunit α3 and β4 mRNAs during rat brain development. J Comp Neurol. 1997;386:540–54. doi: 10.1002/(sici)1096-9861(19971006)386:4<540::aid-cne2>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 78.Zoli M, Moretti M, Zanardi A, McIntosh JM, Clementi F, Gotti C. Identification of the nicotinic receptor subtypes expressed on dopaminergic terminals in the rat striatum. J Neurosci. 2002;22:8785–9. doi: 10.1523/JNEUROSCI.22-20-08785.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Luetje CW. Getting past the asterisk: the subunit composition of presynaptic nicotinic receptors that modulate striatal dopamine release. Mol Pharmacol. 2004;65:1333–5. doi: 10.1124/mol.65.6.1333. [DOI] [PubMed] [Google Scholar]
- 80.Whiteaker P, Peterson CG, Xu W, McIntosh JM, Paylor R, Beaudet AL, Collins AC, Marks MJ. Involvement of the α3 subunit in central nicotinic binding populations. J Neurosci. 2002;22:2522–9. doi: 10.1523/JNEUROSCI.22-07-02522.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Clarke PB, Schwartz RD, Paul SM, Pert CB, Pert A. Nicotinic binding in rat brain: autoradiographic comparison of [3H]acetylcholine, [3H]nicotine, and [125I]-α-bungarotoxin. J Neurosci. 1985;5:1307–15. doi: 10.1523/JNEUROSCI.05-05-01307.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Han ZY, Le Novere N, Zoli M, Hill JA, Jr, Champtiaux N, Changeux JP. Localization of nAChR subunit mRNAs in the brain of Macaca mulatta. Eur J Neurosci. 2000;12:3664–74. doi: 10.1046/j.1460-9568.2000.00262.x. [DOI] [PubMed] [Google Scholar]
- 83.Quik M, Polonskaya Y, Gillespie A, Jakowec M, Lloyd GK, Langston JW. Localization of nicotinic receptor subunit mRNAs in monkey brain by in situ hybridization. J Comp Neurol. 2000;425:58–69. doi: 10.1002/1096-9861(20000911)425:1<58::aid-cne6>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 84.Quik M, Polonskaya Y, Kulak JM, McIntosh JM. Vulnerability of 125I-α-conotoxin MII binding sites to nigrostriatal damage in monkey. J Neurosci. 2001;21:5494–500. doi: 10.1523/JNEUROSCI.21-15-05494.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kulak JM, McIntosh JM, Quik M. Loss of nicotinic receptors in monkey striatum after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine treatment is due to a decline in α-conotoxin MII sites. Mol Pharmacol. 2002;61:230–8. doi: 10.1124/mol.61.1.230. [DOI] [PubMed] [Google Scholar]
- 86.Han ZY, Zoli M, Cardona A, Bourgeois JP, Changeux JP, Le Novere N. Localization of [3H]nicotine, [3H]cytisine, [3H]epibatidine, and [125I]α-bungarotoxin binding sites in the brain of Macaca mulatta. J Comp Neurol. 2003;461:49–60. doi: 10.1002/cne.10659. [DOI] [PubMed] [Google Scholar]
- 87.Quik M, Vailati S, Bordia T, Kulak JM, Fan H, McIntosh JM, Clementi F, Gotti C. Subunit composition of nicotinic receptors in monkey striatum: effect of treatments with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine or L-DOPA. Mol Pharmacol. 2005;67:32–41. doi: 10.1124/mol.104.006015. [DOI] [PubMed] [Google Scholar]
- 88.Bordia T, Grady SR, McIntosh JM, Quik M. Nigrostriatal damage preferentially decreases a subpopulation of α6β2* nAChRs in mouse, monkey and Parkinson’s disease striatum. Mol Pharmacol. 2007 doi: 10.1124/mol.107.035998. [DOI] [PubMed] [Google Scholar]
- 89.Court J, Clementi F. Distribution of nicotinic subtypes in human brain. Alzheimer Dis Assoc Disord. 1995;9:6–14. doi: 10.1097/00002093-199501002-00003. [DOI] [PubMed] [Google Scholar]
- 90.Gotti C, Fornasari D, Clementi F. Human neuronal nicotinic receptors. Prog Neurobiol. 1997;53:199–237. doi: 10.1016/s0301-0082(97)00034-8. [DOI] [PubMed] [Google Scholar]
- 91.Hellstrom-Lindahl E, Mousavi M, Zhang X, Ravid R, Nordberg A. Regional distribution of nicotinic receptor subunit mRNAs in human brain: comparison between Alzheimer and normal brain. Brain Res Mol Brain Res. 1999;66:94–103. doi: 10.1016/s0169-328x(99)00030-3. [DOI] [PubMed] [Google Scholar]
- 92.Flora A, Schulz R, Benfante R, Battaglioli E, Terzano S, Clementi F, Fornasari D. Transcriptional regulation of the human α5 nicotinic receptor subunit gene in neuronal and non-neuronal tissues. Eur J Pharmacol. 2000;393:85–95. doi: 10.1016/s0014-2999(00)00040-6. [DOI] [PubMed] [Google Scholar]
- 93.Rubboli F, Court JA, Sala C, Morris C, Perry E, Clementi F. Distribution of neuronal nicotinic receptor subunits in human brain. Neurochem Int. 1994;25:69–71. doi: 10.1016/0197-0186(94)90055-8. [DOI] [PubMed] [Google Scholar]
- 94.Guan ZZ, Nordberg A, Mousavi M, Rinne JO, Hellstrom-Lindahl E. Selective changes in the levels of nicotinic acetylcholine receptor protein and of corresponding mRNA species in the brains of patients with Parkinson’s disease. Brain Res. 2002;956:358–66. doi: 10.1016/s0006-8993(02)03571-0. [DOI] [PubMed] [Google Scholar]
- 95.Whitehouse PJ, Martino AM, Wagster MV, Price DL, Mayeux R, Atack JR, Kellar KJ. Reductions in [3H]nicotinic acetylcholine binding in Alzheimer’s disease and Parkinson’s disease: an autoradiographic study. Neurology. 1988;38:720–3. doi: 10.1212/wnl.38.5.720. [DOI] [PubMed] [Google Scholar]
- 96.Perry DC, Davila-Garcia MI, Stockmeier CA, Kellar KJ. Increased nicotinic receptors in brains from smokers: membrane binding and autoradiography studies. J Pharmacol Exp Ther. 1999;289:1545–52. [PubMed] [Google Scholar]
- 97.Court JA, Martin-Ruiz C, Graham A, Perry E. Nicotinic receptors in human brain: topography and pathology. J Chem Neuroanat. 2000;20:281–98. doi: 10.1016/s0891-0618(00)00110-1. [DOI] [PubMed] [Google Scholar]
- 98.Rinne JO, Myllykyla T, Lonnberg P, Marjamaki P. A postmortem study of brain nicotinic receptors in Parkinson’s and Alzheimer’s disease. Brain Res. 1991;547:167–70. doi: 10.1016/0006-8993(91)90588-m. [DOI] [PubMed] [Google Scholar]
- 99.Lange KW, Wells FR, Jenner P, Marsden CD. Altered muscarinic and nicotinic receptor densities in cortical and subcortical brain regions in Parkinson’s disease. J Neurochem. 1993;60:197–203. doi: 10.1111/j.1471-4159.1993.tb05838.x. [DOI] [PubMed] [Google Scholar]
- 100.Aubert I, Araujo DM, Cecyre D, Robitaille Y, Gauthier S, Quirion R. Comparative alterations of nicotinic and muscarinic binding sites in Alzheimer’s and Parkinson’s diseases. J Neurochem. 1992;58:529–41. doi: 10.1111/j.1471-4159.1992.tb09752.x. [DOI] [PubMed] [Google Scholar]
- 101.Perry EK, Morris CM, Court JA, Cheng A, Fairbairn AF, McKeith IG, Irving D, Brown A, Perry RH. Alteration in nicotine binding sites in Parkinson’s disease, Lewy body dementia and Alzheimer’s disease: possible index of early neuropathology. Neuroscience. 1995;64:385–95. doi: 10.1016/0306-4522(94)00410-7. [DOI] [PubMed] [Google Scholar]
- 102.Sihver W, Gillberg PG, Nordberg A. Laminar distribution of nicotinic receptor subtypes in human cerebral cortex as determined by [3H](-)nicotine, [3H]cytisine and [3H]epibatidine in vitro autoradiography. Neuroscience. 1998;85:1121–33. doi: 10.1016/s0306-4522(97)00652-0. [DOI] [PubMed] [Google Scholar]
- 103.Bohr IJ, Ray MA, McIntosh JM, Chalon S, Guilloteau D, McKeith IG, Perry RH, Clementi F, Perry EK, Court JA, Piggott MA. Cholinergic nicotinic receptor involvement in movement disorders associated with Lewy body diseases. An autoradiography study using [125I]α-conotoxinMII in the striatum and thalamus. Exp Neurol. 2005;191:292–300. doi: 10.1016/j.expneurol.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 104.Quik M, Bordia T, Forno L, McIntosh JM. Loss of α-conotoxinMII- and A85380-sensitive nicotinic receptors in Parkinson’s disease striatum. J Neurochem. 2004;88:668–79. doi: 10.1111/j.1471-4159.2004.02177.x. [DOI] [PubMed] [Google Scholar]
- 105.Gotti C, Moretti M, Bohr I, Ziabreva I, Vailati S, Longhi R, Riganti L, Gaimarri A, McKeith IG, Perry RH, Aarsland D, Larsen JP, Sher E, Beattie R, Clementi F, Court JA. Selective nicotinic acetylcholine receptor subunit deficits identified in Alzheimer’s disease, Parkinson’s disease and dementia with Lewy bodies by immunoprecipitation. Neurobiol Dis. 2006;23:481–9. doi: 10.1016/j.nbd.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 106.Schwartz RD, Lehmann J, Kellar KJ. Presynaptic nicotinic cholinergic receptors labeled by [3H]acetylcholine on catecholamine and serotonin axons in brain. J Neurochem. 1984;42:1495–8. doi: 10.1111/j.1471-4159.1984.tb02818.x. [DOI] [PubMed] [Google Scholar]
- 107.Clarke PB, Pert A. Autoradiographic evidence for nicotine receptors on nigrostriatal and mesolimbic dopaminergic neurons. Brain Res. 1985;348:355–8. doi: 10.1016/0006-8993(85)90456-1. [DOI] [PubMed] [Google Scholar]
- 108.Quik M, Sum JD, Whiteaker P, McCallum SE, Marks MJ, Musachio J, McIntosh JM, Collins AC, Grady SR. Differential declines in striatal nicotinic receptor subtype function after nigrostriatal damage in mice. Mol Pharmacol. 2003;63:1169–79. doi: 10.1124/mol.63.5.1169. [DOI] [PubMed] [Google Scholar]
- 109.Champtiaux N, Han ZY, Bessis A, Rossi FM, Zoli M, Marubio L, McIntosh JM, Changeux JP. Distribution and pharmacology of α6-containing nicotinic acetylcholine receptors analyzed with mutant mice. J Neurosci. 2002;22:1208–17. doi: 10.1523/JNEUROSCI.22-04-01208.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kulak JM, Musachio JL, McIntosh JM, Quik M. Declines in different β2* nicotinic receptor populations in monkey striatum after nigrostriatal damage. J Pharmacol Exp Ther. 2002;303:633–9. doi: 10.1124/jpet.102.039347. [DOI] [PubMed] [Google Scholar]
- 111.Gotti C, Clementi F. Neuronal nicotinic receptors: from structure to pathology. Prog Neurobiol. 2004;74:363–96. doi: 10.1016/j.pneurobio.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 112.Quik M, Bordia T, Okihara M, Fan H, Marks MJ, McIntosh JM, Whiteaker P. L-DOPA Treatment Modulates Nicotinic Receptors in Monkey Striatum. Mol Pharmacol. 2003;64:619–28. doi: 10.1124/mol.64.3.619. [DOI] [PubMed] [Google Scholar]
- 113.McIntosh JM, Azam L, Staheli S, Dowell C, Lindstrom JM, Kuryatov A, Garrett JE, Marks MJ, Whiteaker P. Analogs of α-conotoxin MII are selective for α6-containing nicotinic acetylcholine receptors. Mol Pharmacol. 2004;65:944–52. doi: 10.1124/mol.65.4.944. [DOI] [PubMed] [Google Scholar]
- 114.Gerfen CR, Herkenham M, Thibault J. The neostriatal mosaic: II. Patch- and matrix-directed mesostriatal dopaminergic and non-dopaminergic systems. J Neurosci. 1987;7:3915–34. doi: 10.1523/JNEUROSCI.07-12-03915.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Langer LF, Graybiel AM. Distinct nigrostriatal projection systems innervate striosomes and matrix in the primate striatum. Brain Res. 1989;498:344–50. doi: 10.1016/0006-8993(89)91114-1. [DOI] [PubMed] [Google Scholar]
- 116.German DC, Manaye KF, Sonsalla PK, Brooks BA. Midbrain dopaminergic cell loss in Parkinson’s disease and MPTP-induced parkinsonism: sparing of calbindin-D28k-containing cells. Ann N Y Acad Sci. 1992;648:42–62. doi: 10.1111/j.1749-6632.1992.tb24523.x. [DOI] [PubMed] [Google Scholar]
- 117.Liang CL, Sinton CM, Sonsalla PK, German DC. Midbrain dopaminergic neurons in the mouse that contain calbindin-D28k exhibit reduced vulnerability to MPTP-induced neurodegeneration. Neurodegeneration. 1996;5:313–8. doi: 10.1006/neur.1996.0042. [DOI] [PubMed] [Google Scholar]
- 118.Gerfen CR, Baimbridge KG, Thibault J. The neostriatal mosaic: III. Biochemical and developmental dissociation of patch-matrix mesostriatal systems. J Neurosci. 1987;7:3935–44. doi: 10.1523/JNEUROSCI.07-12-03935.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Herrero MT, Hirsch EC, Kastner A, Ruberg M, Luquin MR, Laguna J, Javoy-Agid F, Obeso JA, Agid Y. Does neuromelanin contribute to the vulnerability of catecholaminergic neurons in monkeys intoxicated with MPTP? Neuroscience. 1993;56:499–511. doi: 10.1016/0306-4522(93)90349-k. [DOI] [PubMed] [Google Scholar]
- 120.Hirsch E, Graybiel AM, Agid YA. Melanized dopaminergic neurons are differentially susceptible to degeneration in Parkinson’s disease. Nature. 1988;334:345–8. doi: 10.1038/334345a0. [DOI] [PubMed] [Google Scholar]
- 121.McCormack AL, Di Monte DA, Delfani K, Irwin I, DeLanney LE, Langston WJ, Janson AM. Aging of the nigrostriatal system in the squirrel monkey. J Comp Neurol. 2004;471:387–95. doi: 10.1002/cne.20036. [DOI] [PubMed] [Google Scholar]
- 122.Zecca L, Zucca FA, Wilms H, Sulzer D. Neuromelanin of the substantia nigra: a neuronal black hole with protective and toxic characteristics. Trends Neurosci. 2003;26:578–80. doi: 10.1016/j.tins.2003.08.009. [DOI] [PubMed] [Google Scholar]