

Abstract
Tobacco etch virus NIa proteinase (TEV protease) is an important tool for the removal of fusion tags from recombinant proteins. Production of TEV protease in E. coli has been hampered by insolubility and addressed by many different strategies. However, the best previous results and newer approaches for protein expression have not been combined to test whether further improvements are possible. Here we use a quantitative, high throughput assay for TEV protease activity in cell lysates to evaluate the efficacy of combining several previous modifications with new expression hosts and induction methods. Small-scale screening, purification and mass spectral analysis showed that TEV protease with a C-terminal poly-Arg tag was proteolysed in the cell to remove 4 of the 5 arginine residues. The truncated form was active and soluble but in contrast, the tagged version was also active but considerably less soluble. An engineered TEV protease lacking the C-terminal residues 238-242 was then used for further expression optimization. From this work, expression of TEV protease at high levels and with high solubility was obtained by using auto-induction medium at 37 °C. In combination with the expression work, an automated two-step purification protocol was developed that yielded His-tagged TEV protease with >99% purity, high catalytic activity and purified yields of ~400 mg/L of expression culture (~15 mg pure TEV protease per g of E. coli cell paste). Methods for producing glutathione S-transferase tagged TEV with similar yields (~12 mg pure protease fusion per g of E. coli cell paste) are also reported.
Keywords: TEV protease, protease assays, high throughput assays, MBP, GST, automated protein purification, auto-induction
Introduction
The development of high throughput methods for protein expression and purification is profoundly complicated by the diverse chemical properties of proteins. Since fusion tags can modify the behavior of proteins, they offer the possibility for development of standardized protocols for purification, increased solubility, and detection [1–5]. However, fusion tags can also interfere with protein function and with structural studies [6–8]. Thus it is often advantageous to remove fusion tags prior to use. Proteases such as enterokinase, thrombin, and factor Xa have been used to liberate target proteins from fusion tags. However, these mammalian proteases do not exhibit stringent sequence specificity and often cleave target proteins at advantageous sites [9, 10]. A class of viral proteases that are more specific has emerged as an alternative to these enzymes. These include tobacco etch virus NIa proteinase (TEV protease, [11]), human rhinovirus 14 3C protease (3CP, [12]), and tobacco vein mottling virus protease (TVMV, [13]). Among these, TEV protease has received the most attention because many different small amino acids are tolerated in the P1′ position, allowing target genes to be released from N-terminal fusions with either a native N-terminus or with only a single amino acid substitution [14].
Production of TEV protease in E. coli has been problematic due to three issues, auto-inactivation, codon bias and low solubility. The number of publications describing methods to overcome these problems is an indication of the importance placed on TEV protease as a reagent for proteomics and structural biology. Auto-inactivation has been largely eliminated through substitutions at residue 219 [15, 16]. Codon bias may be addressed through mutations or tRNA supplementation [17]. Solubility has been improved through the use of fusion tags [3], incorporation of mutations [18], co-expression with chaperone proteins [19] or expression at low temperatures [19]. Alternatively, solubility issues can be circumvented by refolding inclusion bodies [15]. These efforts have resulted in improvements in the volumetric productivity of TEV protease production from the first reported values of ~1 mg/L [11] to the best current values of ~50 mg/L [18].
The relative efficacies of the many strategies used to improve TEV protease production have not been systematically compared. Likewise, the best reported results have not been combined to test whether further improvements are possible. Here we report the application of a quantitative, high throughput fluorescence polarization assay to directly measure TEV protease activity in cell lysates. This assay facilitated screening for expression variants and conditions leading to increased activity. By using this assay, we show that multiple factors, including the ability of maltose binding protein (MBP) to promote solubility, removal of deleterious C-terminal residues, modifications of the expression plasmid genotype and use of the auto-induction method may be combined to substantially increase the expression of soluble TEV protease. Furthermore, by coupling the best improvements in bacterial expression with an automated two-step purification protocol to minimize sample handling, TEV protease was obtained in a yield of ~400 mg per L of expression culture with >99% purity. A similar approach was used to optimize the expression of glutathione S-transferase tagged TEV protease (GST-TEV).
Methods
TEV Protease Expression Vectors
Table 1 summarizes the expression plasmids and coding regions used in this work. The expression vector pQE30-S219V containing a TEV protease gene was obtained from Prof. B.F. Volkman and Dr. F.C. Peterson at the Medical College of Wisconsin (Milwaukee, Wisconsin). This pQE30-derived plasmid (Qiagen, Valencia, CA) encoded residues 1-242 of the TEV protease open reading frame, the native residues at the C-terminus and the S219V mutation, which conferred resistance to auto-inactivation [16]. The expression vector pQE30-S219VpR5 was a variant of pQE30-S219V where residues 238-242 were each replaced with arginine residues to create a poly-Arg5 tag (pR5) at the C-terminus. The expression vector pRK793 encoding a self-cleaving MBP-His7-TEV-pR5 protease fusion protein was obtained from Dr. D.S. Waugh at the National Cancer Institute (Frederick, Maryland). pRK793 also encoded the S219V mutation. The MBP-His7-TEV-pR5 fusion can undergo proteolysis in vivo at a TEV protease site in the linker region after MBP to liberate MBP and His7-TEV-pR5.
Table 1.
TEV Protease Coding Sequences Used For Expression Optimization.
| Plasmid or coding sequencea | Anticipated N-terminusb | Anticipated C-terminusc | C-terminal abbreviationd | Solubility enhancing mutationse |
|---|---|---|---|---|
| pQE30-S219V | MRGSHHHHHHGS… | …TQLMNELVYSQ | Full-length | No |
| pQE30-S219V-pR5 | MRGSHHHHHHGS… | …TQLMNRRRRR | pR5 | No |
| pRK793 | GHHHHHHHGE… | …TQLMNRRRRR | pR5 | No |
| MHT | AIAHHHHHHHGE… | …TQ | 233Δ | Yes |
| …TQLMNE | 237Δ | Yes | ||
| …TQLMNELVYSQ | Full-length | Yes | ||
| …TQLMNELVYSQ | Full-length | No | ||
| HT | MGSHHHHHHHHGE… | …TQ | 233Δ | Yes |
| …TQLMNE | 237Δ | Yes | ||
| …TQLMNELVYSQ | Full-length | Yes | ||
| …TQLMNELVYSQ | Full-length | No | ||
| GT | Glutathione-S-transferase-MGILG… | …TQ | 233Δ | Yes |
| …TQLMNE | 237Δ | Yes | ||
| …TQLMNELVYSQ | Full-length | Yes | ||
| …TQLMNELVYSQ | Full-length | No |
Original plasmid or coding sequence for the TEV variant placed into the plasmid shown in Figure 2.
N-terminus anticipated from the sequence-verified expression plasmid including any intentional proteolytic digestion of fusion partners.
C-terminus anticipated from the sequence-verified expression plasmid.
Description used in the text for the C-terminus. 233Δ indicates that all residues after 233 have been deleted. 237Δ indicates all residues after 237 have been deleted.
Presence of solubility enhancing mutations identified in [18].
Figure 1 shows a summary of the PCR primers used to prepare TEV protease variants by overlap extension PCR [20]. All DNA fragments prepared by PCR amplification were sequence verified. The solubility enhancing mutations T17S, N68D, and I77V described previously [18] were incorporated into certain TEV protease variants as indicated below. Separate PCR reactions were used to generate three fragments, one consisting of the N-terminus through T17S, a second between T17S and N68D/I77V, and a third between N68D/I77V and the desired C-terminus.
Figure 1.

Primers used for two-step PCR cloning of TEV protease. The first round of PCR amplification was used to generate DNA fragments at the 5′ end of the gene (A, 5′ fragments), the central portion of the gene (B, central fragments), and the 3′ end of the gene (C, 3′ fragments). The forward primers are shown above the TEV S219V nucleotide sequence and the reverse primers are shown below. Restriction sites are highlighted in blue (5′ SgfI and 3′ PmeI), codons containing solubility enhancing mutations in yellow, and stop codons in green. Overlap extension PCR of the entire coding region was completed by combining the 5′-, central, and 3′-fragments with the appropriate outside primers. The overlapping sequences between the fragments are underlined. The 5′-fragments determined the fusion tag context while 3′-fragments specified the location of the stop codon. For the GT clones, vectors were digested with PacI and PmeI since the PacI and SgfI cleavage sites contain compatible overhanging nucleotides. After ligation, neither restriction site was regenerated.
The PCR primers for the 5′ fragments were designed to produce protein with an N-terminal His7-tag (TEV-For-H7) or protein with no N-terminal tag (TEV-For-NoTag). The 5′ fragment primers also contained the SgfI restriction site (highlighted in blue) for Flexi vector cloning [21]. The PCR primers for the central fragment duplicated the gene from the solubility enhancing mutation T17S (T17S-For) to the other mutations N68D/I77V (N68D-I77V-Rev). The positions of the mutagenic codons are highlighted in yellow. The PCR primers for the 3′ fragments C-terminal fragments were designed to produce protein with different C-terminal extensions. The reverse primers also encoded the PmeI restriction site for use in Flexi vector cloning (highlighted in blue). The primers N68D-I77-For and TEV-Rev-Full were used to generate a full-length 242-residue TEV protease. The TEV protease was also truncated at either residue 238 (protein designated 238Δ, using primers N68D-I77-For and TEV-Rev-L239) or at residue 233 (233Δ, using primers N68D-I77-For and TEV-Rev-L234). The complete coding region was assembled from these fragments by a second round of PCR. Overlapping sequences in the three fragments are underlined in Figure 1.
Figure 2 shows the basic architecture of the expression vectors used. PCR products were incorporated into these expression vectors either directly from the overlap PCR or by transfer from another Flexi vector [21]. The vectors are identical except for the coding region and the promoter used for expression of LacI. The MHT coding region produces an MBP-His7-TEV protease fusion with a TEV protease site (TEVc) in between MBP and the His7 sequence. After cleavage at the TEVc site, the MHT coding region yields AIA-His7-TEV, where the AIA tag originates from the Flexi vector cloning strategy [21]. The HT coding region yields His8-TEV. The GT coding region produces a non-cleavable GST-TEV protease fusion. In some of the vectors, the lacIq promoter was replaced with a wild type lacI promoter in order to increase the level of expression obtained from auto-induction (P.G. Blommel and B.G. Fox, submitted).
Figure 2.

Maps of three expression vectors used in this work. The vectors are identical except for the coding region and the promoter used for expression of LacI. The MHT coding region produces MBP-His7-TEV with a TEV protease site (TEVc) between MBP and the His7 sequence. After cleavage at the TEVc site, the MHT coding region yields Ala-Ile-Ala-His7-TEV. The HT coding region yields His8-TEV. The GT coding region produces a non-cleavable GST-LeuIleAla-TEV protease fusion with no His-tag. Expression levels from auto-induction were increased by replacing the lacIq promoter with a wild type lacI promoter.
Expression Hosts
Escherichia coli BL21 (EMD Biosciences/Novagen, Madison, WI), E. coli BL21 RILP (Stratagene, La Jolla, CA), and E. coli Krx (Promega, Madison, WI) were used as expression hosts. The RILP strain contains a plasmid for codon adaptation that provides constitutive expression of several tRNAs that are in low abundance in E. coli, including argU previously found to be important for TEV expression [17].
TEV Protease Expression
Expression studies were carrier out using either auto-induction [4, 22] or isopropylthiogalactoside (IPTG) induction. Kanamycin (100 μg/mL) was added to all media and chloramphenicol (34 μg/mL) was added to cultures of E. coli BL21 RILP. All starting inocula were grown in chemically defined MDAG medium [22] modified by the addition of 0.375% aspartic acid, 0.8% glucose, and reduction of phosphate to 25 mM. Starting inocula were grown overnight at 25 °C and reached saturation at OD600 of ~10 to 15. The starting inoculum was added at 1/20th the volume of expression medium. Expression medium consisted of terrific broth containing 0.8% glycerol (Sigma, St. Louis, MO) prepared according to the manufacture’s instructions and further supplemented with 2 mM MgSO4 and 0.375% aspartic acid. When used for induction, IPTG was added to a final concentration of 0.5 mM. For auto-induction, the medium also contained 0.5% (w/v) lactose and 0.015% (w/v) glucose.
Small-scale expression screening was conducted in 96-well growth blocks (Qiagen) containing 400 μL of medium. For IPTG induction, the cultures either were grown at 37 °C and treated for 3 h with IPTG or were grown at 25 °C and treated for 5 h with IPTG. The IPTG induction was initiated when culture monitoring showed OD600 ≈ 1.2 – 2.0, which corresponded to early log phase growth. For auto-induction, the expression screening was carried out for either ~12 h at 37 °C or ~24 h at 25 °C. No additional monitoring after inoculation was required. The small-scale cultures were harvested by freezing 100 μL aliquots at −80 °C.
Large-scale expressions were done either in 2-L PET bottles containing 0.5 L of culture medium [4, 23, 24] or in a Bioflow 3000 fermenter (New Brunswick Scientific, Edison, NJ) containing 9.5 L of culture medium. The large-scale cultures were pelleted by centrifuge at 4000 × g for 20 min. The cell pellets were re-suspended in a small volume of 50 mM phosphate, pH 7.5, containing 300 mM NaCl and 20% ethylene glycol and centrifuged again to recover the washed cell paste. The washed cell paste was stored at −80 °C in 50 mL conical tubes.
Preparation of Small-Scale Cell-Free Lysates
The cell cultures frozen in PCR plates were thawed and suspended in lysis buffer to a final volume of 120 μL and a final composition of 20 mM Tris-HCl, pH 7.5, 20 mM NaCl, 0.3 mM (TCEP), 1 mM MgSO4, 3 kU/mL of rLysozyme (EMD Biosciences/Novagen) and 0.7 U/mL of benzonase (EMD Biosciences/Novagen). After 30 min incubation at room temperature, the samples were sonicated on a plate sonicator (Misonix, Farmingdale, NY) for 6 to 10 min. Samples were then centrifuged at 3000 × g for 30 min. The supernatant fraction was retained for protease assay measurements.
TEV Protease Activity Assays
TEV activity was determined using a fluorescence anisotropy based protease assay [9] with the soluble fraction of the cell-free lysate. The assay is based on a reduction in fluorescence anisotropy that occurs when a small fluorescent peptide is liberated from a larger protein [5, 25]. For this work, the substrate reported earlier was modified to minimize the anisotropy upon proteolysis by minimizing the size of the liberated peptide. This fluorescent substrate was produced in E. coli as the fusion protein His8-MBP-3CPc-C4-attB1-TEVc-MBP, where His8 is an N-terminal His-tag, MBP is E. coli maltose binding protein, 3CPc is a human rhinovirus 3C protease cleavage site (LEVLFQ↓GP, where ↓ indicates the 3C protease cleavage site), C4 is the tetraCys motif (CCPGCC), attB1 is the amino acid sequence required for the attB1 site of Gateway cloning (TSLYKKAGS) and TEVc is a TEV protease cleavage site (ENLYFQ↓S).
The fusion protein was expressed and purified as previously reported. After treatment with 3C protease, the substrate protein (27-F) has the N-terminal sequence of GPCCPGCCTSLYKKAGSENLYFQ↓S fused to MBP. FLAsH was synthesized [5] and added to 27-F in an amount sufficient to provide ~5% covalent labeling of the tetraCys motif. The standard proteolysis assay was performed in 20 mM Tris, pH 7.5, containing 100 mM NaCl, 5 mM EDTA, 0.3 mM triscarboxyethylphosphine (TCEP) and 5 μM 27-F with 5% FlAsH labeling at 25 to 28 °C. Proteolysis releases the fluorescently labeled peptide GPCCPGCCTSLYKKAGSENLYFQ. Samples of the fluorescent substrate incubated with TEV protease at conditions known to effect complete cleavage [9] were used to determine the intrinsic anisotropy, mri, of the peptide in the given assay conditions. The time-dependent exponential changes in fluorescence anisotropy were fit by non-linear least squares methods to determine the initial anisotropy, mr0, the final anisotropy, mr∞ and the decay constant (proteolysis rate). The mr0, mr∞ and mri values were used to prepare fractional progress curves [9]. Fitted decay constants were adjusted for the percentage labeling of the substrate. Reported errors for the assay represent two standard deviations of the mean.
Refolded TEV Protease
S219V-TEV protease expressed from IPTG-induced cultures of E. coli BL21 pQE30-S219V was prepared by re-suspension of the inclusion bodies in 6 M guanidinium hydrochloride containing 0.3 mM TCEP to a final protein concentration of 1 mg/mL. This suspension was diluted 20-fold into a refolding buffer containing 50 mM MES, pH 6.5, containing 0.5 M arginine, 0.5 M sucrose, 2 mM MgCl2, and 0.3 mM TCEP. After 1 h, the refolded mixture was subjected to IMAC purification and dialyzed into storage buffer containing 50% glycerol.
Purification of His-TEV Protease
Figure 3 shows a schematic of the instrumentation and buffer compositions used for TEV purification. The Akta Prime system and all other equipment and chromatography resins were from GE Healthcare Life Sciences (Piscataway, NJ). Buffer A was 20 mM phosphate, pH 7.5, containing 500 mM NaCl and 0.3 mM TCEP. Buffer B was 20 mM phosphate, pH 7.5, containing 350 mM NaCl, 500 mM imidazole and 0.3 mM TCEP. Buffer C was 10 mM Tris, pH 7.5, containing 0.3 mM TCEP. Buffer D was 10 mM Tris, pH 7.5, containing 1000 mM NaCl and 0.3 mM TCEP. Control programs were developed to complete consecutive IMAC and cation exchange purifications without user intervention.
Figure 3.

A schematic representation of the equipment used for automated two-step purification of His7-TEV protease. The solid lines in the system injection valves show the flow path during the simultaneous IMAC elution and cation exchange binding phase of the purification. The dotted lines indicate flow paths used during other phases of the purification. Separate control programs were developed for the IMAC and cation exchange steps and were synchronized by starting the programs at the same time. By specifying the timing of steps that require coordinated action of both units, no communication between the purification units was required. Abbreviations: P, pressure sensor; UV, absorbance detector making measurements at 280 nm; C, conductivity detector.
Cell paste (34 g) was re-suspended in 50 mM phosphate, pH 7.5, containing 300 mM NaCl, 20% ethylene glycol and 0.3 mM TCEP at a ratio of 6 mL of buffer per g of wet cell paste. The following protease inhibitors were added to the indicated final concentrations prior to sonication: E-64 (1 μM), EDTA (1 mM) and benzamidine (0.5 mM). The cell suspension was sonicated for 6 min on ice and all subsequent purification steps were conducted at 4 °C. The sonicated cell suspension was centrifuged for 25 min at 95,000 × g and the soluble fraction was retained. The soluble fraction was loaded into either a 50 or 150 mL loading loop and then loaded onto purification system 1 at 3 mL/min. This purifier system had two 5 mL Histrap HP columns arranged in series and equilibrated with buffer A. The columns were washed with eight volumes of a mixture of 85% buffer A and 15% buffer B. During the wash, the flow rate was increased to 5 mL/min.
The bound protease was eluted from purification system 1 by a step-wise change to 100% buffer B. At the start of the elution step, the flow rate of buffer B was decreased to 0.7 mL/min and the flow path was diverted to purification system 2. This purification system had a 2 mL mixing chamber upstream of two 5 mL SP Fast Flow columns arranged in series. The columns were equilibrated with buffer C. The sample from the first purifier was injected into the mixing chamber at 0.7 mL/min, mixed with 100% buffer C at 10 mL/min and loaded onto the columns of purification system 2 at a total flow rate of 10.7 mL/min. The resultant ~15-fold dilution of the sample prior to application to the cation exchange columns ensured that the ionic strength was low enough allow tight binding of the protease to the column.
Upon completion of the IMAC elution, the flow through purifier system 1 was increased to 5 mL/min and directed to waste for column wash and re-equilibration with buffer A prior to the injection of the next aliquot of lysate. The waste sample was collected so that possible losses of TEV protease could be determined. Also upon completion of the IMAC elution, the flow through purifier system 2 was decreased to 5 mL/min and a six column volume gradient from 100% buffer C to a mixture of 40% buffer C and 60% buffer D was started. Fractions containing TEV protease were detected by UV measurement. After elution of the TEV protease, the flow through purification system 2 was directed to waste. The column was then washed with several volumes of 100% buffer D and re-equilibrated with 100% buffer C prior to the start of the next injection from the first purification system. This waste sample was also collected.
Fractions were analyzed by catalytic assays and SDS-PAGE and were pooled based on specific activity and protein purity. The protein concentration of the pooled sample was determined by UV-visible spectroscopy (ε280 = 32770 M−1 cm−1 calculated from the amino acid composition). The pooled TEV protease was diluted with buffer C and storage buffer containing 10 mM Tris, 0.5mM EDTA, 0.3 mM TCEP and 80% (v/v) glycerol to a protein concentration of 1 mg/mL in 50% glycerol. No additional buffer exchange, concentration or dialysis steps were required. The purified TEV protease was stored in this buffer at −20°C.
Purification of GST-TEV Protease
For purification of GST-TEV, the preparation of the cell-free lysate and soluble fraction from 3 g of cell paste were as described above. Ammonium sulfate was added to 55% of saturation in order to precipitate the protease fusion. The pellet from the ammonium sulfate precipitation was re-suspended in 20 mL of 10 mM Tris, pH 7.5, containing 10 mM NaCl and 0.3 mM TCEP. The glutathione Sepharose purification step was completed using an 8 mL gravity flow column at room temperature because the GST-TEV was found to bind slowly to the resin at 4 °C. The column was washed with five column volumes of the re-suspension buffer described above. The protein was then eluted with 50 mM Tris, pH 7.5, containing 2 mM EDTA, 0.3mM TCEP and 10 mM reduced glutathione. The eluted fusion protein was concentrated using an Amicon 10 kDa molecular weight cutoff centrifugal concentrator (Millipore, Billerica, MA) to a concentration of ~18 mg/mL. The concentrated sample was loaded to a Sephacryl S-100 26/10 column equilibrated in 10 mM Tris, pH 7.5, containing 1 mM EDTA and 0.3 mM TCEP at 4°C at a flow rate of 1 mL/min. Fractions were analyzed as described above.
Other Analytical Methods
Protein expression levels were assessed using SDS-PAGE on total cell lysates, and the soluble and insoluble fractions prepared as previously reported [4]. The molecular weight markers shown in gels were from BioRad (Hercules, CA). Mass spectral analyses were determined using a Sciex API 365 triple quadrupole mass spectrometer (Perkin Elmer, Boston, MA) maintained at the University of Wisconsin Biotechnology Center.
Results
Fluorescence Polarization Assay
Figure 4 shows a typical result for an assay of TEV protease with the recombinant protein substrate GPCCPGCCTSLYKKAGSENLYFQ↓S-MBP. In this substrate, the cysteine residues are labeled with the FlAsH fluorophore. No proteolysis was observed from control E. coli cell lysates lacking TEV protease (open circles in Figure 4).
Figure 4.

A representative fluorescence polarization assay of TEV protease activity present in an E. coli cell lysate. Open circles show anisotropy data for an E. coli lysate that did not contain TEV protease. Open triangles show results from expression of MHT238Δ. The gaps in the data occurred when the assay plate was removed from the instrument to add components for additional assays in other wells.
Upon treatment with TEV protease, the N-terminal peptide is released from the remainder of the fusion protein by proteolysis between Q23 and S24. This changes the effective molecular weight of the fluorophore from 43 kDa to 2.5 kDa and also corresponds to a decline in the observed anisotropy from ~230 millianisotropy units (mr) to ~110 mr, depending on buffer conditions. Figure 4 shows that lysates containing recombinant TEV protease give exponential decay in the observed anisotropy (open triangles), corresponding to proteolytic release of the fluorophore-labeled N-terminal peptide from the full substrate. In this work, this assay has been used to investigate the efficacy of various C-terminal modifications, solution conditions and expression methods on the accumulation of active TEV protease in bacterial cell lysates. Moreover, the same assay approach was used to obtain numerical accounting of the results of an automated TEV protease purification described below.
C-Terminal Proteolysis of TEV-pR5 Enhances Solubility
Our initial investigations with TEV protease included the use of three different expression vectors, pQE30-S219V, pQE30-S219VpR5, and MBP-His-TEVS219VpR5 expressed from pRK793. All three vectors produce an N-terminal His-tagged protein, for the third construct this is released upon in vivo proteolysis. The first construct produces a native C-terminus, while the second and third produce a C-terminus where the last five residues are replaced with the pR5 tag. The pR5 tag has been used to enhance purification of TEV protease [16].
TEV protease expressed from pQE30-S219V was primarily present as inclusion bodies despite many attempts to optimize the solubility of the expressed protein. Nevertheless, the fraction of TEV protease that was soluble could be purified using IMAC and high-resolution Mono S cation exchange in a linear salt gradient. Thus the yield of purified TEV protease obtained from the pQE30-S219V was less than 10 mg per liter of expression culture. Figure 5A shows that the elution profile for the TEV protease expressed from pQE30-S219V was a single peak. Moreover, Table 2 shows that the mass spectral analysis (measured mass 28763) was consistent with the presence of the full-length native protein (calculated mass 28751).
Figure 5.

Cation exchange elution absorbance profiles for TEV protease produced from pQE30-S219V (upper trace) and pRK793 (lower trace). The concentration gradient is shown as a dashed line with the concentration indicated by the right axis. Two distinct peaks were evident for protease produced from pRK793, labeled 1 and 2. These peaks contained truncated and full-length protease, respectively, as indicated by ESI mass spectrometry. Protease produced from pQE30-S219V had a mass consistent with full-length protease.
Table 2.
Mass Spectral Analysis Of Expressed and Purified TEV Protease Variants.
| Expression plasmida | Plasmid-encoded N-terminus | Plasmid-encoded C-terminusb | Calculated massc (g/mole) | ESI massd (g/mole) | Deduced C-terminuse | Percent errorf |
|---|---|---|---|---|---|---|
| pQE30-S219V | MRGSHHHHHHGSSL… | …TQLMNELVYSQ | 28751 | 28763 | …TQLMNELVYSQ | 0.04 |
| pQE30-S219VpR5 | MRGSHHHHHHGSSL… | …TQLMNRRRRR | 28187 | 28195 | …TQLMNR | 0.03 |
| pRK793 Peak 1 (Major) | GHHHHHHHGESL… | …TQLMNRRRRR | 27992 | 27988 | …TQLMNR | 0.01 |
| pRK793 Peak 1 (Minor) | GHHHHHHHGESL… | …TQLMNRRRRR | 27477 | 27476 | …TQ | 0.00 |
| pRK793 Peak 2 | GHHHHHHHGESL… | …TQLMNRRRRR | 28617 | 28617 | …TQLMNRRRRR | 0.00 |
| pMHT237Δ | AIAHHHHHHHGESL… | …TQLMNE | 28137 | 28147 | …TQLMNE | 0.04 |
Plasmid used to express the TEV protease sample investigated.
C-terminus anticipated from the sequence-verified expression plasmid.
Mass calculated for the TEV sample with the plasmid-encoded N-terminus and deduced C-terminus.
Mass of the purifed TEV protease sample determined by ESI mass spectrometry. For pRK793, peak 1 and peak 2 refer to the distinct elution peaks shown in Figure 5. Within peak 1, mass peaks for major and minor species were also detected.
Most probable C-terminus deduced from the ESI data.
Percent error between the calculated mass and that determined by ESI mass spectrometry.
For comparison, Figure 5B shows that two distinct peaks were observed from Mono S separation of the TEV protease expressed from pRK793. Activity measurements showed that these two peaks had similar specific activities in the TEV protease assay. Table 2 shows that the majority protein of peak 1 had a mass consistent with proteolysis of four Arg residues from the C-terminus (measured 27988 Da versus calculated 27992 Da), while the protein of peak 2 had a mass consistent with retention of the four Arg residues (measured 28617 Da versus calculated 28617 Da). Moreover, a small fraction of TEV protease present in peak 1 from pRK793 had a mass consistent with truncation after residue 233 (measured 27476 Da versus calculated 27477 Da). Upon purification, the fractions of peak 1 were well behaved and remained in solution over extended periods of time. In contrast, the fractions from peak 2 often contained precipitated protein.
Figure 6 shows SDS-PAGE results for TEV protease expressed from pQE30-S219VpR5. These results provide further corroboration of the lability of the pR5 tag and the insolubility of TEV protease that retains it. Figure 6A shows that expression of His7-TEV-pR5 at 37 °C gave no TEV protease in the soluble fraction. For comparison, expression at 25 °C gave detectable TEV protease activity in the soluble fraction. However, Figure 6B shows that while the soluble fraction contained only a single TEV protease band, the insoluble fraction contained two bands. Mass spectral analysis of the purified soluble fraction was again consistent with proteolysis of four Arg residues from the C-terminus. Thus His7-TEV-R was found in both the soluble and insoluble fractions upon expression at 25 °C. In contrast, His7-TEV-pR5 was exclusively found in the insoluble fraction.
Figure 6.
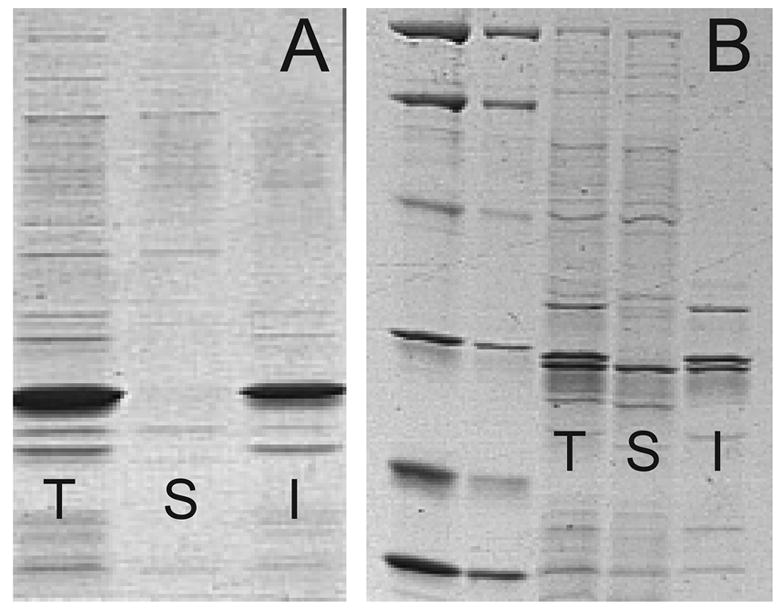
A comparison of the solubility of TEV protease dependent on expression temperature and the nature of the C-terminal tag present. T, total cell lysate; S, soluble fraction of the total cell lysate, and I, the insoluble fraction of the lysate. Gel A shows that His6-TEV-pR5 expressed from pQE30-S219VpR5 at 37 °C is entirely insoluble. Gel B shows that His6-TEV-pR5 expressed from pQE30-S219VpR5 at 25 °C is a doublet that partitioned between the soluble and the insoluble fraction. Other work presented here shows the soluble protein is primarily proteolyzed His6-TEV-R, while the insoluble fraction contains both His6-TEV-R and His6-TEV-pR5.
The results of Figure 5 and Figure 6 indicate that a C-terminal pR5 tag on TEV protease is subject to proteolytic removal. Furthermore, removal of this tag is apparently associated with increased solubility of TEV protease.
Effect of C-Terminal Truncations on TEV Protease Expression
After identifying the importance of the C-terminal region of TEV protease to solubility, we were interested in more fully examining the limits of TEV protease expression. Thus new TEV protease coding sequences shown in Figure 1 were designed to place stop codons after residues 233 and 238. These new coding sequences incorporated three previously discovered solubility enhancing mutations [18]. A full length TEV protease coding sequence was also prepared with and without the solubility enhancing mutations.
Table 1 summarizes the coding sequences, the presence or absence of the solubility enhancing mutations and the three different expression vectors, whose architecture is shown in Figure 2. The MHT vectors incorporate a self-cleaving MBP-His7-TEV coding sequence similar to pRK793. After autocatalytic cleavage, the protease is released with an N-terminal AIA-His7-tag, where AIA comes from the Flexi vector cloning. The HT vectors yield N-terminal His8-TEV with no other fusion tag attachment. The GT vectors yield an N-terminal fusion to GST. This fusion protein has no proteolysis site in the short LIA linker so cannot be separated.
A number of comparative small-scale expression experiments were conducted with these variants. The type of induction was investigated at 25 °C using either auto-induction with lactose or manual induction with IPTG. The role of the expression host was compared using expression strains E. coli BL21 and E. coli Krx. Figure 7 shows the results of the analysis of the cell lysates by catalytic assay and SDS-PAGE. For each coding sequence, the C-terminal 237Δ variants gave the highest catalytic activity, and the best catalytic results (Figure 7A) were obtained from the 237Δ and 233Δ variants expressed from the MHT coding region in E. coli BL21 using auto-induction. At the expression levels obtained in these experiments, SDS-PAGE analysis (Figure 7B) showed no insoluble TEV protease was detected from either the autocatalytic MHT or the HT coding regions, regardless of whether the solubility enhancing mutations were present or not. In contrast, some insoluble protease was observed with the GT coding sequence. However, this fraction was minor compared to the soluble fraction.
Figure 7.

A, activity assays of small-scale expression cultures with fusion tag and C-terminal variants of TEV protease. Solubility enhancing mutations were either present (+SE) or absent (−SE). The expression studies were completed at 25°C in terrific broth using IPTG or auto-induction and E. coli expression strains BL21 or Krx. Error bars represent two standard deviations above and below the mean for measurements conducted in quadruplicate. B, SDS-PAGE gels are shown in Panel B for IPTG induced expression in BL21 for all fusion variants and from expression of MHT variants in E. coli Krx. Total (T), soluble (S), and insoluble fractions (I) were run. The expressed proteins are indicated with arrows.
Optimization of TEV Expression Conditions
Based on the results from Figure 7, the expression conditions were further optimized for the C-terminal 238Δ variant with each coding region. First, each variant was placed into a modified expression vector where the lacIq promoter used to overexpress LacI was replaced with the wild-type lacI promoter. This change helps to optimize protein expression from auto-induction (P.G. Blommel and B.G. Fox, manuscript submitted). Eight different expression conditions were then tested in the lacI context. These were 25 °C versus 37 °C, IPTG versus auto-induction and the presence or absence of the RILP codon adaptation plasmid.
Figure 8 show the results of these comparative studies. The activity results of Figure 8A with all three coding sequences support the value of RILP codon adaptation. Moreover, in most cases the auto-induction method performed better than IPTG induction with respect to cell mass recovered and expression at 37 °C was found to give higher levels of TEV protease activity with both MHT238Δ and HT238Δ. Surprisingly, the level of active TEV protease was not statistically different for either MHT238Δ or HT238Δ (which also include the solubility enhancing mutations and the stabilizing mutation S219V) with auto-induction and codon adaptation. The activity of the GT237Δ variants was uniformly lower than the MHT237Δ or HT237Δ variants. This may reflect steric interactions arising from the fact that GT237Δ is a fusion protein while the other two TEV proteases have only a short His-tag at the N-terminus.
Figure 8.

A, activity assays for expression of TEV237Δ with different fusion tags (MBP, MHT238Δ; His-tag, HT238Δ or GST, GT238Δ) and other different conditions. The enzyme activity in recombinant E. coli BL21 cell lysates was measured in quadruplicate with error bars representing two standard deviations above and below the mean. The inset defines the different expression conditions of the bar graph. B, SDS-PAGE analysis of the protein expression from A. Total (T), soluble (S), and insoluble (I) protein fractions are labeled. The gel lanes used to separate the insoluble protein lanes were constricted by the higher salt concentration present in the total and soluble samples.
Figure 8B shows the SDS-PAGE analysis, and helps to illuminate the tradeoff between solubility and total expression. Total protein expression is higher at 37 °C than 25 °C. For MHT238Δ, all expression conditions at both temperatures yielded soluble protease after in vivo cleavage and the measured activity correlated with the expression level. For HT238Δ, both auto-induction and IPTG induction gave a high level of total expression at 37 °C, but some insoluble protease was also observed. In contrast, expression at 25 °C yielded no insoluble protease. Higher expression (and appearance of insolubility) was also associated with RILP codon adaptation at 37 °C. These gel-deduced differences are corroborated by the assay results.
Figure 8 indicates that the GT237Δ coding sequence expressed comparably with auto-induction at 37 °C or with IPTG induction at 25 °C. However, solubility problems were most apparent for GT238Δ. Insoluble GT238Δ was obtained with IPTG and auto-induction at 37 °C. At 25 °C, the lowest fraction of insoluble GT238Δ was observed with auto-induction. The insolubility of GT237Δ was apparently not remedied by the presence of the solubility enhancing mutations. For GT237Δ, codon adaptation was clearly advantageous as all four comparative conditions containing the RILP plasmid outperformed the corresponding condition without codon adaptation (e.g., condition 8 versus 4 and others). Figure 8 also shows that expression of GT283Δ also corresponded with the accumulation of an unknown protein, possibly derived from GST-TEV. This is particularly evident in condition 8, but also present with conditions 1, 3, 5 and 7). Indeed, only auto-induction at 25 °C seemed to minimize this.
Table 3 summarizes the effects of changes from a starting condition of expression of the full-length TEV protease at 25 °C with auto-induction to inclusion of RILP codon adaptation and a change to the lacI promoter on measured TEV protease activity. For example, change from the full-length TEV protease to TEV237Δ protease gave a 1.7-fold increase in enzyme activity with the MHT coding sequence. The multiplicative fold improvement was most dramatic for HT237Δ, and represented a ~30-fold improvement from the poor activity observed in the starting condition. By contrast, the MHT237Δ and GT237Δ coding sequences gave more modest 5- and 7-fold multiplicative improvements from the starting condition. Table 3 also shows that the highest total units of enzyme activity were obtained from optimized expression with MHT237Δ, arising from the high level of soluble expression and the improvements given by the multiplicative improvements.
Table 3.
Estimation Of The Impact That Individual Factors Have On The Final Activity Improvement Observed with 237Δ-TEV Protease.a
| Coding sequenceb | Protein | Lac repressor promoter | Expression Temperature | Induction Method | RILP codon adaptation | Multiplicative Fold Improvementc | Activity Initial/Finalμmol/hr/Ld |
|---|---|---|---|---|---|---|---|
| Starting Conditione | Full Length | lacIQ | 25 °C | Auto | - | ||
| Alternatef | 237Δ | lacI | 37 °C | IPTG | + | ||
|
| |||||||
| MHT | 1.7 | 1.3 | 1.6 | 0.4 | 1.3 | 5 | 36 / 169 |
| HT | 2.3 | 3.9 | 2.7 | 0.3 | 1.3 | 32 | 4.8 / 154 |
| GT | 2.1 | 1.5 | 0.5 | 1.5 | 1.4 | 7 | 6.5 / 77 |
The improvement was quantified by assay of TEV protease activity in cell lysates.
Coding sequence for the TEV variant placed into the plasmid shown in Figure 2.
The multiplicative fold improvement is the product of the factors of all alternate conditions that led to higher activity.
The initial activity is from the full length TEV protease and the final activity is from the optimized expression conditions.
The starting condition was expression of the full-length TEV protease at 25 °C using auto-induction medium, no RILP codon adaptation and lacIq control of LacI expression.
The initial change to alternate conditions was incorporation of the 237Δ truncation. Other alternate conditions leading to higher activity are shown in bold.
Large-Scale Expression of TEV-protease using Auto-Induction
The combined results of Figure 7, Figure 8 and Table 3 indicated that the highest level TEV protease might be produced from MHT238 at 37 °C using auto-induction, RILP codon adaptation and the lacI promoter for regulation of LacI expression. Figure 9 shows results from performing this expression experiment in a 10-L fermenter. Figure 8A shows the time course of changes in TEV protease activity and cell density. During the auto-induction process, the TEV protease activity was below detection limits until the cell density reached ~6 (3.5 h after inoculation). Thereafter the protease activity increased rapidly with the largest increase occurring between cell densities of 10 and 18 (5 to 7 h after inoculation). Figure 9B shows an SDS-PAGE gel analysis of the expression culture. The SDS-PAGE results are consistent with the assay results, as the protein bands corresponding to both MBP and His-TEV appeared ~4.7 h after induction. The cells were harvested after ~9 h, yielding 23 g of wet cell paste per liter of culture medium (total 220 g of cell paste from 9.5 L of culture medium). The right-most three lanes in Figure 9B show that the TEV protease was almost exclusively soluble, with less than 5% of the protease accumulated in the insoluble fraction based on scanning densitometry (note that the insoluble fraction was loaded at 3× the equivalent volume in the SDS-PAGE to allow better visibility).
Figure 9.

Expression of TEV protease during auto-induction from MHT238Δ in a 10-L fermenter. A, correlation of TEV protease activity and cell density with duration of the fermentation. Error bars for the activity measurements represent two standard deviations above and below the mean. Cell densities are shown as bars and as numbers across the top of the plot. B, SDS-PAGE analysis. Expressed MBP-His7-TEV237Δ fusion protein is cleaved during cell growth to separate MBP and His7-TEV237Δ. Arrows indicate the position of MBP and His7-TEV after in vivo cleavage. The lane marked S contains a sample of the starting inoculum grown in a non-inducing medium. The lanes marked with time correspond to the data points indicated in A. The lanes marked HT, HS, and HI are the total, soluble, and insoluble fractions obtained at harvest, 8.7 h after inoculation. The amount of sample loaded was normalized by cell density for all lanes except the insoluble harvest sample, which was loaded at 3× the normalized amount to allow better visualization.
Purification of His7-TEV Protease
Figure 3 shows a schematic of the instrumentation used for automated purification of His-tagged TEV protease. Two Akta Prime systems were linked together to perform a two-step purification consisting of IMAC followed by cation exchange chromatography. Control software allowed repetitive operation of the linked instruments. The automated procedure allowed analysis of four streams from the purification: starting cell-free lysate, the waste from the IMAC and cation exchange steps and the purified TEV product.
Figure 10A shows elution profiles from the first and second cycles of cation exchange chromatography and the second and third cycles of IMAC chromatography. Table 4 shows results from replicate purification cycles and Table 5 shows the purification table assembled from the pooled results of first four purification cycles.
Figure 10.

Results from the automated two-step purification of MHT238Δ. A, absorbance and gradient profiles for the two-step purification. The lower solid line shows the absorbance profile during the 2nd cycle of sample injection, wash, and elution steps and the 3rd cycle of sample injection for the IMAC purification. The upper solid line shows the absorbance profile for the 1st and 2nd cation exchange steps. The dashed lines indicate the percentage mixture of buffer B during the IMAC step and buffer D during the cation exchange step. The area of elution peaks cannot be directly compared due to the different flow rates during IMAC and cation exchange elution steps. B, characterization of the purification results by SDS-PAGE. Lane 1 is the cell-free lysate and lanes 2 and 3 are the IMAC and cation exchange waste products. The waste products were concentrated using a 10-kDa molecular weight cut off centrifugal concentrator to the same volume as the cell lysate for easier visual comparison. Lanes 4 through 9 are the purified TEV protease obtained from purification cycles 1 through 6. Lane 10 shows a higher loading (50 μg) of the sample from lane 5 (10 μg) and lane 11 contains 312 ng of bovine serum albumin standard run on the same gel as lane 10.
Table 4.
Results of Replicate Automated Purification of His-TEV237Δ.
| Purification cycle | Final Volume mL | Protein Concentration mg/mL | Total Protein mg | Total Activity μmol/hr | Specific Activity μmol/h/mg |
|---|---|---|---|---|---|
| Cycle 1 | 20 | 4.91 ± 0.16 | 98 ± 3 | 33 ± 6 | 0.34 ± 0.06 |
| Cycle 2 | 20 | 5.03 ± 0.26 | 101 ± 5 | 34 ± 8 | 0.34 ± 0.08 |
| Cycle 3 | 20 | 4.97 ± 0.22 | 99 ± 4 | 34 ± 2 | 0.34 ± 0.02 |
| Cycle 4a | 20 | 4.85 ± 0.05 | 97 ± 1 | 32 ± 6 | 0.33 ± 0.06 |
| Cycle 5 | 20 | 5.02 ± 0.21 | 100 ± 4 | 33 ± 3 | 0.33 ± 0.03 |
| Cycle 6b | 10 | 1.05 ± 0.01 | 10 ± 0.1 | 2.4 ± 0.4 | 0.23 ± 0.04 |
In cycle 4, the imidazole concentration in the wash buffer was 100 mM.
In cycle 6, the remaining lysate was used. In addition, the imidazole concentration in the wash buffer was increased to 125 mM.
Table 5.
Purification Of His7-TEV Protease After Auto-Induction Of Expression From Vector pMHT237Δ In A 10-L Fermenter.
| Sample | Volume | Total Protein | Total Activity | Recovery | Specific Activity | Fold-purification |
|---|---|---|---|---|---|---|
| mL | mg | μmol/h | % | μmol/h/mg | ||
| Cell-free lysatea | 168 | 3120 | 156 | 100 | 0.05 | 1 |
| IMAC wasteb | 740 | 2860 | 1.3 | 1 | 0.0005 | 0.01 |
| Cation exchange wastec | 1830 | 15 | 2.4 | 2 | 0.16 | 3.2 |
| Purified His7-TEVd | 80 | 395 | 133 | 85 | 0.34 | 6.7 |
The lysate obtained from 25 g of MHT237Δ cells.
Collected as flow-through from sample injection and column wash prior to elution.
Collected as flow-through from sample injection and column wash prior to elution.
Pooled sample obtained after four cycles of the 2-step automated purification.
For Table 4, six cycles were completed from one batch of cell free lysate. The first five cycles consumed 42 mL of lysate each and produced 20 mL of purified TEV product, while the sixth cycle, using the remaining 15 mL of lysate, was eluted in a 10 mL fraction. The first four purification cycles were conducted starting immediately after the lysate was prepared and showed highly reproducible recovery of total protein, total activity and specific activity. In purification cycle 4, the imidazole concentration in the wash buffer was increased from 75 mM to 100 mM in order to investigate the upper limit of imidazole concentration attainable prior to loss of yield. This change did not decrease the recovery of protease. Purification cycle 5 was undertaken using 75 mM imidazole. Purification cycles 5 and 6 were also begun ~16 h after preparation of the lysate. The yield of TEV protease from purification 5 was similar to the previous four purifications, which indicates that AIA-His7-TEV237Δ protease was stable in the lysate at 4 °C (i. e., no degradation by host proteases, autocatalytic inactivation and no precipitation). In purification 6, the imidazole concentration in the wash buffer was further increased to 125 mM. This led to a partial loss of protease in the wash fraction and decreased recovery.
The purity of the TEV protease obtained from individual purification cycles can be judged from Figure 10B. Lane 1 shows the cell free lysate and over-expressed MBP and TEV protease. Lane 2 shows the flow through from the IMAC column. The TEV protease was completely bound. After the completion of the cation exhange step, it was revealed there was no apparent benefit to changing the IMAC wash buffer from 75 mM (purification cycles 1 to 3 and 5, lanes 4 to 6 and 8) to 100 mM imidazole (purification cycle 4, lane 7), as no contaminants were visible even with the 75 mM imidazole wash. Lane 9 shows the product obtained from purification cycle 6. The yield was diminished because less lysate was used and an exploratory 125 mM imidazole wash of the IMAC column was used resulting in some loss of TEV protease activity to the IMAC wash (Table 4).
The rightmost three lanes of Figure 10B further document the purity of the TEV protease from lane 5. Even with overloading (50 μg, lane 10), no clearly distinguishable contamination products were visible relative to a BSA standard (312 ng, lane 11). Furthermore, scanning densitometry of lane 10 yielded no peaks above background noise outside of the main TEV protease band. Upon the basis of this analysis, the purified TEV protease was judged to be greater than 99% pure. Mass spectrometry confirmed that the purified product had a mass consistent with the MHT238 coding sequence (Table 2).
Table 5 summarizes the combined results of purification cycles 1 to 4. The results obtained from 168 mL of cell lysate correspond to the use of 26 g of cell paste. Overall, ~13% of the total protein present in the cell lysate was recovered as purified AIA-His7-TEV protease. This corresponds well with the 6.7-fold increase in activity during the purification, which suggests that the TEV-protease represented ~15% of the total protein in the original cell lysate. The purified protein sample contained ~85% of the activity detected in the original cell lysate and less than 5% of the TEV protease activity originally detected in the cell-free lysate was accounted for in the waste streams.
Production and Purification of GST-TEV
The analysis of Table 3 suggested that expression of GST-TEV would be preferred using IPTG induction at 25 °C. However, Figure 8 indicates that auto-induction at 25 °C gave nearly equivalent total enzyme activity without the degree of insolubility observed from IPTG and without the appearance of an unknown protein truncation product. For these reasons, GST-TEV was expressed from GT238Δ at 25 °C by auto-induction with RILP codon adaptation and the lacI promoter modification in 2-L PET bottles. The auto-induction yielded 38 g of cell paste per liter of culture medium. Figure 11 shows the SDS-PAGE analysis of a two-step purification of GST-TEV by glutathione Sepharose chromatography and then size exclusion chromatography. The recovery of the partially purified GST-TEV was around 12 mg per g of cell paste. Two minor contaminants, visible in Figure 11 at ~23 kDa and ~32 kDa, were not resolved by the two-step purification.
Figure 11.
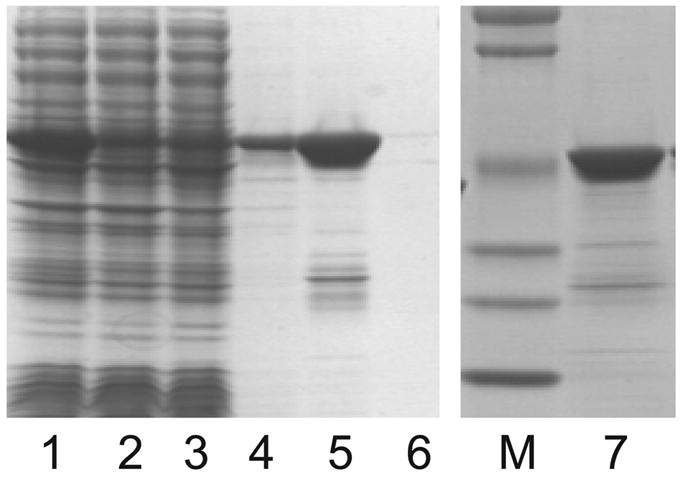
An SDS-PAGE analysis of GST-TEV protease purification. Lane 1, cell lysate. Lane 2, glutathione Sepharose column flow-through. Lane 3, glutathione Sepharose column wash. Lanes 4–6, glutathione Sepharose elution fractions. Lane 7, pooled fractions after size exclusion chromatography.
Comparison of the Activity of Purified His-TEV and GST-TEV
His7-TEV237Δ protease had a kcat/KM value of 0.43 mM−1 s−1, which was similar to the value of 0.27 mM−1 s−1 previously reported for TEV protease acting on a protein substrate [19]. The activities of His7-TEV238Δ protease and refolded His-TEV protease were indistinguishable using the fluorescence polarization assay. In addition, these two TEV preparations had approximately double the kcat/KM of the GST-TEV237Δ protease fusion. The kcat/KM values for the protein substrate were significantly lower than the kcat/KM of 4.6 mM−1 s−1reported for S219V-TEV protease acting on a peptide substrate [16].
Discussion
Fusion tags are important tools to increase solubility, allow standardized purification and improve detection of recombinant proteins [1–5]. However, fusion tags may interfere with either protein function or structural studies. For this reason it is often desirable to remove the fusion tags. TEV protease has received much attention for fusion tag removal because of its high specificity and also because of the tolerance for many amino acids in the P1′ site [14]. Thus considerable efforts have increased the volumetric productivity of TEV protease production from ~1 mg/L [11] to ~50 mg/L [18].
In this work, we asked whether a combination of existing best results with new protein modifications and new expression methods might lead to further improvements in TEV protease production. The cumulative results show that the answer is true and methods documented in this work show how to obtain highly pure, highly active TEV protease in yield of ~400 mg per liter of culture medium (~15 mg per g of cell paste).
C-Terminus
This work revealed that pR5 modification of the C-terminus of TEV protease was removed by proteolysis, and surprisingly, the removal significantly increased the solubility of the truncated protein. The residues adjacent to the major 238Δ truncation product do not appear to be a good substrate for TEV protease based on prior biochemical evidence [26]. However, since the C-terminal residues after 221 are disordered in the TEV protease crystal structure [27], it appears that the C-terminal residues are flexible enough to enter the active site and sufficiently increase the local concentration so that otherwise unfavorable proteolysis reactions can occur [16].
In vivo truncation after residue 238 had no effect on catalytic activity, which contrasts with the 90% reduction in activity after truncation at residue 219 [16, 26]. Therefore, we intentionally truncated the C-terminus to create TEV237Δ. After expression in E. coli BL21, TEV238Δ contained significantly higher enzyme activity in the soluble fraction as compared to the full-length TEV protease regardless of the N-terminal fusion (MBP, GST or His-tag only), or the method used for induction (auto-induction or IPTG). It is also notable that high expression of soluble, active TEV protease could be obtained at 37 °C with both MHT237Δ and HT237Δ (Figure 8). The advantage of the TEV237Δ truncation was enhanced by fusion to MBP and RILP codon adaptation.
Induction Method
Auto-induction cultures attained higher cell density at saturation. In the auto-induction medium, the cultures reached an average of 20 OD600 units. In contrast, IPTG induced cultures typically grew to half of this density and often saturated at densities as low as 5 OD600 units. Sample volumes loaded on the SDS-PAGE gels shown in Figures 7 and 8 were normalized to the volume loaded rather than the cell density. As a result the background of host proteins is more apparent in the auto-induced cultures than IPTG-induced cultures. In many cases, the fraction of protein attributable to TEV protease may be higher for IPTG induced cultures compared to auto-induction but the volumetric productivity is lower due to cell yield. The E. coli expression strains BL21 and Krx were statistically equivalent in their ability to express TEV protease with IPTG induction.
Small-Scale Optimization of Expression Conditions
By combining previous approaches with truncation mutations and screening of expression conditions using a catalytic assay for TEV protease (Table 3), we identified a combination of experimental modifications that gave an ~5-fold increase in TEV protease production over previous reports [18, 19]. The use of the C-terminal deletion 238Δ, a reduction in lac repressor expression, and RILP codon adaptation were beneficial in all cases. The utility of lac repressor reduction during auto-induction will be described elsewhere [P. Blommel and B.G. Fox, submitted]. RILP codon adaptation was in general beneficial to increase expression levels, although not to the extent previously reported [17]. From the experiments conducted, it was not possible to explicitly determine the impact of the solubility-enhancing mutations [18]. Nevertheless, our initial experience with the pQE30 expression vectors (Table 2), are consistent with the utility of the solubility-enhancing mutations for TEV protease obtained from MHT237Δ and HT237Δ.
Large-Scale TEV Protease Production
Since TEV protease is used in many proteomics and structural genomics studies, highly purified and active TEV protease may be required in multi-gram quantities by some researchers. This is true at the University of Wisconsin Center for Eukaryotic Structural Genomics. Previous reported yields of TEV protease include 50 mg/L from solubility enhancement [18], 64 mg/L from chaperone assisted protein production [19], and 100 mg/L from pRK793-derived expression as an MBP-TEV-pR5 fusion [our unpublished results, but here shown to be susceptible to C-terminal proteolysis and precipitation, Figure 5]. To investigate productivity beyond these previous levels, the best condition for TEV protease production identified through small-scale screening (Table 3) was scaled up to a 10-L fermentation. This fermentation yielded 23 g of wet cell paste per liter of culture medium with 15 mg of purified TEV obtained per gram of cell paste. Expression results from Table 3 and Figure 8 were used to test the expression of GST-TEV from GT237Δ in 2-L PET bottles. This work yielded 38 g of wet cell paste per liter of culture medium with ~12 mg of purified GST-TEV obtained per g of cell paste. Thus the defined expression conditions give comparable results from small-scale expression trials in growth blocks, 2-L shaken flask culture and large-volume instrumented fermenters.
Automated Purification
The automated purification was developed to increase the efficiency of TEV protease production. With the four cycles of automated purification described here, 400 mg of TEV protease can be purified in a single day. Following the cation exchange step of the purification, SDS-PAGE and densitometry show that the obtained protease is >99% pure. In addition to the high purity given from the automated protocol, the TEV protease eluted from the cation exchange resin was already in a buffer and concentration suitable for direct dilution with glycerol for long-term storage. Indeed, TEV protease stored in this buffer at −20°C has retained full activity for more than 2 years.
Activity Comparisons
The catalytic activity of AIA-His7-TEV237Δ protease (produced from MHT238Δ to include solubility enhancing mutations [18] and the S219V mutation to minimize autocatalytic inactivation [16]) was identical to that of His-TEV prepared by refolding inclusion bodies obtained from pQE-30 S219V. In contrast, GST-TEV237Δ had only ~50% of the specific activity of AIA-His7-TEV237Δ protease in the assay used here. It is possible that the lower activity may be due to steric hindrance of the active site by GST. To minimize the possibility for proteolysis of the linker, only three residues (Leu-Ile-Ala) were included between GST and TEV. It is possible that extension of the linker may allow greater flexibility between the two domains and increase accessibility of the active site.
Acknowledgments
This work was supported by the NIH Protein Structure Initiative (1 U54 GM 74901, J. L. Markley, Principal Investigator, G. N. Phillips, Jr. and B. G. Fox, Co-Investigators) and by a sponsored research agreement with Promega Corporation (B.G. Fox, Principal Investigator). P.G.B. was a trainee of the NIH Institutional Biotechnology Pre-Doctoral Training Grant T32 GM08349.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Georgiou G, Valax P. Expression of correctly folded proteins in Escherichia coli. Curr Opin Biotechnol. 1996;7:190–197. doi: 10.1016/s0958-1669(96)80012-7. [DOI] [PubMed] [Google Scholar]
- 2.Hammarstrom M, Hellgren N, van Den Berg S, Berglund H, Hard T. Rapid screening for improved solubility of small human proteins produced as fusion proteins in Escherichia coli. Protein Science. 2002;11:313–321. doi: 10.1110/ps.22102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kapust RB, Waugh DS. Escherichia coli maltose-binding protein is uncommonly effective at promoting the solubility of polypeptides to which it is fused. Protein Science. 1999;8:1668–1674. doi: 10.1110/ps.8.8.1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sreenath HK, Bingman CA, Buchan BW, Seder KD, Burns BT, Geetha HV, Jeon WB, Vojtik FC, Aceti DJ, Frederick RO, Phillips GN, Jr, Fox BG. Protocols for production of selenomethionine-labeled proteins in 2-L polyethylene terephthalate bottles using auto-induction medium. Protein Expr Purif. 2005;40:256–267. doi: 10.1016/j.pep.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 5.Adams SR, Campbell RE, Gross LA, Martin BR, Walkup GK, Yao Y, Llopis J, Tsien RY. New biarsenical ligands and tetracysteine motifs for protein labeling in vitro and in vivo: synthesis and biological applications. J Amer Chem Soc. 2002;124:6063–6076. doi: 10.1021/ja017687n. [DOI] [PubMed] [Google Scholar]
- 6.Bucher MH, Evdokimov AG, Waugh DS. Differential effects of short affinity tags on the crystallization of Pyrococcus furiosus maltodextrin-binding protein. Acta Crystallogr D Biol Crystallogr. 2002;58:392–397. doi: 10.1107/s0907444901021187. [DOI] [PubMed] [Google Scholar]
- 7.Chant A, Kraemer-Pecore CM, Watkin R, Kneale GG. Attachment of a histidine tag to the minimal zinc finger protein of the Aspergillus nidulans gene regulatory protein AreA causes a conformational change at the DNA-binding site. Protein Expr Purif. 2005;39:152–159. doi: 10.1016/j.pep.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 8.Smyth DR, Mrozkiewicz MK, McGrath WJ, Listwan P, Kobe B. Crystal structures of fusion proteins with large-affinity tags. Protein Sci. 2003;12:1313–1322. doi: 10.1110/ps.0243403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blommel PG, Fox BG. Fluorescence anisotropy assay for proteolysis of specifically labeled fusion proteins. Anal Biochem. 2005;336:75–86. doi: 10.1016/j.ab.2004.09.023. [DOI] [PubMed] [Google Scholar]
- 10.Jenny RJ, Mann KG, Lundblad RL. A critical review of the methods for cleavage of fusion proteins with thrombin and factor Xa. Protein Expr Purif. 2003;31:1–11. doi: 10.1016/s1046-5928(03)00168-2. [DOI] [PubMed] [Google Scholar]
- 11.Parks TD, Howard ED, Wolpert TJ, Arp DJ, Dougherty WG. Expression and purification of a recombinant tobacco etch virus NIa proteinase: biochemical analyses of the full-length and a naturally occurring truncated proteinase form. Virology. 1995;210:194–201. doi: 10.1006/viro.1995.1331. [DOI] [PubMed] [Google Scholar]
- 12.Cordingley MG, Callahan PL, Sardana VV, Garsky VM, Colonno RJ. Substrate requirements of human rhinovirus 3C protease for peptide cleavage in vitro. J Biol Chem. 1990;265:9062–9065. [PubMed] [Google Scholar]
- 13.Nallamsetty S, Kapust RB, Tozser J, Cherry S, Tropea JE, Copeland TD, Waugh DS. Efficient site-specific processing of fusion proteins by tobacco vein mottling virus protease in vivo and in vitro. Protein Expr Purif. 2004;38:108–115. doi: 10.1016/j.pep.2004.08.016. [DOI] [PubMed] [Google Scholar]
- 14.Kapust RB, Tozser J, Copeland TD, Waugh DS. The P1′ specificity of tobacco etch virus protease. Biochem Biophys Res Commun. 2002;294:949–955. doi: 10.1016/S0006-291X(02)00574-0. [DOI] [PubMed] [Google Scholar]
- 15.Lucast LJ, Batey RT, Doudna JA. Large-scale purification of a stable form of recombinant tobacco etch virus protease. Biotechniques. 2001;30:544–546. 548, 550. doi: 10.2144/01303st06. passim. [DOI] [PubMed] [Google Scholar]
- 16.Kapust RB, Tozser J, Fox JD, Anderson DE, Cherry S, Copeland TD, Waugh DS. Tobacco etch virus protease: mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Engineering. 2001;14:993–1000. doi: 10.1093/protein/14.12.993. [DOI] [PubMed] [Google Scholar]
- 17.Kapust RB, Routzahn KM, Waugh DS. Processive degradation of nascent polypeptides, triggered by tandem AGA codons, limits the accumulation of recombinant tobacco etch virus protease in Escherichia coli BL21(DE3) Protein Expr Purif. 2002;24:61–70. doi: 10.1006/prep.2001.1545. [DOI] [PubMed] [Google Scholar]
- 18.van den Berg S, Lofdahl PA, Hard T, Berglund H. Improved solubility of TEV protease by directed evolution. J Biotechnol. 2006;121:291–298. doi: 10.1016/j.jbiotec.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 19.Fang L, Jia KZ, Tang YL, Ma DY, Yu M, Hua ZC. An improved strategy for high-level production of TEV protease in Escherichia coli and its purification and characterization. Protein Expr Purif. 2006 doi: 10.1016/j.pep.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 20.An Y, Ji J, Wu W, Lv A, Huang R, Wei Y. A rapid and efficient method for multiple-site mutagenesis with a modified overlap extension PCR. Appl Microbiol Biotechnol. 2005;68:774–778. doi: 10.1007/s00253-005-1948-8. [DOI] [PubMed] [Google Scholar]
- 21.Blommel PG, Martin PA, Wrobel RL, Steffen E, Fox BG. High efficiency single step production of expression plasmids from cDNA clones using the Flexi Vector cloning system. Protein Expr Purif. 2006;47:562–570. doi: 10.1016/j.pep.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 22.Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 23.Millard CS, Stols L, Quartey P, Kim Y, Dementieva I, Donnelly MI. A less laborious approach to the high-throughput production of recombinant proteins in Escherichia coli using 2-liter plastic bottles. Protein Expr Purif. 2003;29:311–320. doi: 10.1016/s1046-5928(03)00063-9. [DOI] [PubMed] [Google Scholar]
- 24.Tyler RC, Sreenath HK, Singh S, Aceti DJ, Bingman CA, Markley JL, Fox BG. Auto-induction medium for the production of [U-15N]- and [U-13C, U-15N]-labeled proteins for NMR screening and structure determination. Protein Expr Purif. 2005;40:268–278. doi: 10.1016/j.pep.2004.12.024. [DOI] [PubMed] [Google Scholar]
- 25.Griffin BA, Adams SR, Tsien RY. Specific covalent labeling of recombinant protein molecules inside live cells. Science. 1998;281:269–272. doi: 10.1126/science.281.5374.269. [DOI] [PubMed] [Google Scholar]
- 26.Dougherty WG, Cary SM, Parks TD. Molecular genetic analysis of a plant virus polyprotein cleavage site: a model. Virology. 1989;171:356–364. doi: 10.1016/0042-6822(89)90603-x. [DOI] [PubMed] [Google Scholar]
- 27.Phan J, Zdanov A, Evdokimov AG, Tropea JE, Peters HK, 3rd, Kapust RB, Li M, Wlodawer A, Waugh DS. Structural basis for the substrate specificity of tobacco etch virus protease. J Biol Chem. 2002;277:50564–50572. doi: 10.1074/jbc.M207224200. [DOI] [PubMed] [Google Scholar]