

Abstract
Neuroactive steroids have some of their most potent actions by augmenting the function of GABAA receptors. Endogenous steroid actions on GABAA receptors may underlie important effects on mood and behavior. Exogenous neuroactive steroids have potential as anesthetics, anticonvulsants, and neuroprotectants. We have taken multiple approaches to understand more completely the interaction of neuroactive steroids with GABAA receptors. We have developed many novel steroid analogues in this effort. Recent work has resulted in synthesis of new enantiomer analogue pairs, novel ligands that probe various properties of the steroid pharmacophore, fluorescent neuroactive steroid analogues, and photoaffinity labels. Using these tools, combined with receptor binding and electrophysiological assays, we have begun to untangle the complexity of steroid actions at this important class of ligand-gated ion channel.
Keywords: GABA, neurosteroid, anesthesia, enantiomer, electrophysiology, photoaffinity label
1. Introduction
Strong interest in neuroactive steroids has developed over the last several decades for numerous reasons. Among these are a growing list of neural targets of steroids, increased appreciation that endogenous neuroactive steroids may bind to and modulate these targets during normal and pathological CNS function, and interest in clinical applications of exogenous neuroactive steroids. Our focus has been primarily on anesthetic actions of steroids vis a vis steroid actions at GABAA receptors. Neuroactive steroids exhibit some of their most potent actions at these important ligand-gated ion channels. Arguably, many of the important behavioral effects of neuroactive steroids, including sedative, anxiolytic, and anticonvulsant actions, result from interactions of these steroids with GABAA receptors. This has led us and others to investigate properties of the interactions between steroids and GABAA receptors. These properties include potential binding site(s) on GABAA receptors (or closely related proteins), functional consequences of the interaction, and the importance of aqueous vs. membranous routes of access to the receptor. Our approach has combined molecular biology, biochemistry, pharmacology, single-channel and whole-cell electrophysiology, cellular imaging, and especially medicinal chemistry to elucidate interactions between neuroactive steroids and GABAA receptor-related targets. Here we emphasize recent efforts by our research program in the context of other work in the field. Our recent work emphasizes the likelihood that multiple binding sites for steroids on receptors exist and that there is considerable complexity of actions when GABAergic effects of steroids are examined in detail.
2. The GABAA receptor
Because this review focuses primarily on interactions between neuroactive steroids and GABAA receptors, we first briefly review GABAA receptor properties relevant to the main issues presented in our review. For more extensive discussion of the properties of GABAA receptors, readers are referred to other recent reviews (Akabas, 2004; Ernst et al., 2003; Luscher & Keller, 2004; Mody & Pearce, 2004; Rudolph & Mohler, 2004; Sieghart et al., 1999). GABAA receptors are pentameric heteromers and are members of the cys-loop family of ligand-gated ion channels. This family also includes nicotinic acetylcholine receptors, ionotropic glycine receptors, serotonin 5HT3 receptors and a recently described prokaryotic proton-gated channel (Bocquet et al., 2007). Binding of GABA to the GABAA receptor gates an intrinsic anion-selective channel. Depending on the reversal potential of the permeant ions (chloride and bicarbonate are physiologically most relevant), the postsynaptic GABA response can be excitatory or inhibitory. However, because intracellular chloride in most mature neurons is low, the chloride reversal potential is negative to action potential threshold, so the GABA-gated conductance exerts an inhibitory influence on the cell.
Considerable diversity exists in the subunit structure of GABAA receptors. Functional channels are formed from the assembly of two α subunits (from 6 different gene products, α1-α6) two β subunits (from 3 different gene products, β1-3) plus one additional subunit, often a γ subunit (from γ1-3) (Chang et al., 1996; Tretter et al., 1997) but sometimes a δ, ε, π, or θ subunit. A schematic of a single GABAA receptor subunit is shown in Figure 1A. The pentameric receptor assembly, with several putative sites of action for important modulatory drugs, is shown in Figure 1B.
Figure 1. GABAA receptor schematic and putative binding sites.
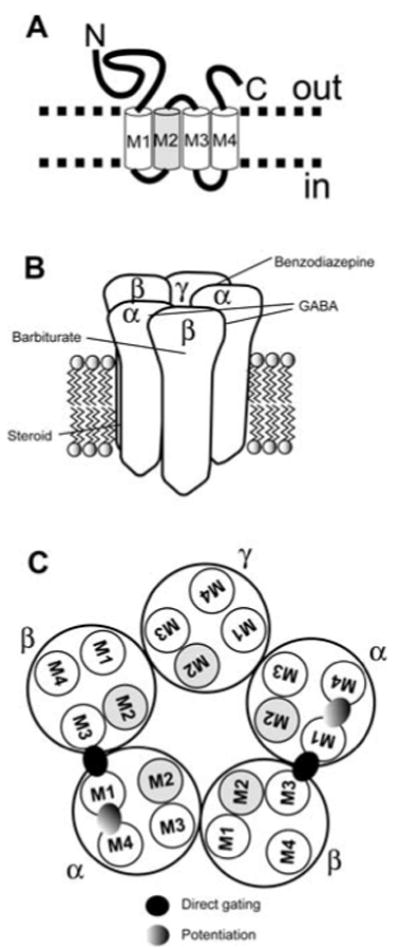
A. A single subunit of the GABAA receptor, highlighting topology. M1-M4 represent transmembrane domains. The M2 transmembrane domain (gray) forms an important part of the chloride channel pore. B. Pentameric structure of a typical GABAA receptor. Several putative sites of GABA and modulatory drugs, including neurosteroids, are shown. Mutations of the β subunit affect barbiturate modulation, but no unequivocal binding site has been identified. The indication that steroids act on the GABAA receptor from within the transmembrane domains is supported by pharmacological studies and by recent site-directed mutagenesis studies (Akk et al., 2005; Hosie et al., 2006; Shu et al., 2004). C. Top-down view of the pentameric receptor showing proposed sites of potentiation and direct gating for neurosteroids, based on site-directed mutagenesis (Hosie et al., 2006).
Multiple splice variants of the subunits also exist, making the combinatorial possibilities for diversity of structure and function quite daunting. Fortunately, nature appears to make use of only a limited number of the subunit combinatorial possibilities, allowing feasible identification and experimental examination of native subunit combinations (Sieghart et al., 1999; Wisden et al., 1992). The α1β2γ2 subunit combination is estimated to be the most widespread combination in the mammalian brain (Fritschy & Mohler, 1995; McKernan & Whiting, 1996; Somogyi et al., 1996). The γ2 subunit contains sequence motifs responsible for synaptic targeting (Essrich et al., 1998), so this subunit appears particularly important for synaptic localization/clustering of GABAA receptors.
GABA gates another ionotropic channel formed from ρ subunits. This channel underlies GABA-C responses in the retina. Although homomeric receptors containing the ρ1 subunit are thought to be most physiologically relevant, there is evidence for heteromeric combinations among ρ subunits 1-3 (Pan et al., 2006), and even among ρ subunits and GABAA subunits (Milligan et al., 2004). Because the GABA-C receptor can function as a homo-oligomer, this in principle could greatly simplify efforts to find steroid binding sites. Use of ρ1 homomers has been exploited by our program and other groups (Goutman & Calvo, 2004; Li et al., 2006b; Morris & Amin, 2004; Morris et al., 1999). Unfortunately, steroid interaction with ρ receptors produces a somewhat different structure-activity relationship (see below) compared with GABAA receptors. Therefore, although some results may generalize to GABAA receptors, this generality needs to be established experimentally.
Although in some important circumstances GABA can mediate excitation (Cherubini et al., 1991; Szabadics et al., 2006), GABAA receptor-mediated signaling constitutes the majority of fast inhibitory transmission in the mature brain. GABA released by local circuit interneurons, or by principal cells in some areas of the nervous system, activates GABAA receptors localized/clustered immediately opposite the synapse. Depending primarily on the kinetics of the receptors underlying the response, the resulting inhibitory postsynaptic current can persist from a few milliseconds to a few hundred milliseconds. The time course of these synaptic currents is generally slower than fast excitation mediated by glutamate acting on AMPA receptors, but slightly faster than excitatory postsynaptic currents mediated by NMDA type glutamate receptors.
In addition to classical phasic inhibition, there is growing appreciation that GABAA receptors also mediate a tonic inhibition, largely generated through high-affinity, extrasynaptic GABAA receptors. Tonic currents have been particularly well studied in cerebellar granule neurons and dentate granule cells of the hippocampus. These cell types share strong expression of δ subunits in extrasynaptic receptor populations (Farrant & Nusser, 2005; Semyanov et al., 2004). Extrasynaptic receptors also contain α4 (dentate granule cells) or α6 (cerebellar granule cells) subunits, which preferentially partner with the δ subunit. The α4/α6 plus δ subunits endow the extrasynaptic GABAA receptors with a high sensitivity to GABA and to neuroactive steroids (Adkins et al., 2001; Brickley et al., 2001; Brown et al., 2002; Mihalek et al., 1999; Semyanov et al., 2004; Stell et al., 2003; Stell & Mody, 2002; Wei et al., 2003; Wohlfarth et al., 2002). Interestingly, the same δ subunit responsible for high steroid sensitivity also has recently been implicated in ethanol sensitivity of GABA receptors (Hanchar et al., 2006; Wallner et al., 2003, 2006a, 2006b), although this relationship has been controversial (Borghese et al., 2006; Yamashita et al., 2006). An additional complexity is added by the recent finding that α1 can partner with the δ subunit in some cell types, notably hippocampal interneurons, to generate ethanol-sensitive tonic current (Glykys et al., 2007).
The tonic GABA current apparently arises from ambient GABA, measured at ≤ 1 μM (Lerma et al., 1986), maintained at steady state by ongoing release balanced by the operation of GABA transporters in the membranes of presynaptic terminals and glia (Farrant & Nusser, 2005; Semyanov et al., 2004). Tonic GABAA receptor mediated currents have also been observed in many other cells types including thalamic neurons (Belelli et al., 2006; Cope et al., 2005), hippocampal pyramidal neurons (Caraiscos et al., 2004a), and interneurons (Semyanov et al., 2003). In the case of hippocampal pyramidal cells, α5-containing subunits appear particularly important in generating the extrasynaptic tonic current (Caraiscos et al., 2004a).
3. Overview of neurosteroid interactions with GABAA receptors
Neurosteroid interactions with GABAA receptors can be grouped qualitatively into potentiating actions and antagonistic actions. The most potent effects, typified by the endogenous steroids (3α,5α)-3-hydroxypregnan-20-one (3α5αP, or allopregnanolone), (3α,5β)-3-hydroxypregnan-20-one (3α5βP, or pregnanolone), (3α,5α)-3,21-dihydroxypregnan-20-one (3α5αTHDOC), and (3α,5β)-3,21-dihydroxypregnan-20-one (3α5βTHDOC), are potentiating. These steroids are synthesized from cholesterol via progesterone of desoxycorticosterone by actions of 5α- or 5β reductases and a 3α-hydroxysteroid dehydrogenase (Mellon & Griffin, 2002; Mellon et al., 2001). Because of the derivation of neurosteroids like 3α5αP (allopregnanolone) from progesterone, fluctuations in progesterone levels may influence the modulatory effects that neurosteroids have over GABAA receptor function. Typical EC50s for GABAA receptor potentiation by neurosteroids are in the high nanomolar range.
The next most potent effects are the non-competitive antagonist actions of steroids sulfated at C3, typified by pregnenolone sulfate and sulfated pregnane steroids, with either α or β stereochemistry at C3 and at C5 (Majewska et al., 1988; Majewska & Schwartz, 1987; Park-Chung et al., 1999; Wang et al., 2002). The IC50 for the action of this class of neuroactive steroids varies with activation state of the receptor (Eisenman et al., 2003; Wang et al., 2002), but typical values are in the high nanomolar to micromolar range. Finally, 3β-OH steroids can also antagonize GABAA receptors in a manner similar to C3 sulfated steroids, but 3β-OH steroid potency, and perhaps efficacy, are weaker than that of sulfated steroids. We recently found that benz[e]indenes, steroid analogues with the A-ring opened and partially removed, also exhibit GABAA receptor antagonist properties (described in detail below). Even certain bile steroids, such as lithocholic acid, with a charge at the opposite end of the molecule, are capable of antagonizing GABA responses (Mennerick et al., 2001). Thus, antagonism tolerates a wide range of structures, albeit with varied potency (Park-Chung et al., 1999). Although important and potentially useful drugs fall into the classes of GABA antagonists, we will not focus on the GABA antagonist steroids in this review, other than in the context of receptor sites for neuroactive steroids. Rather, our primary focus is on actions of the positive modulators of GABAA receptor activity.
Like many GABA-receptor potentiators, including barbiturates, etomidate, and propofol, neuroactive steroids augment the whole-cell response to low concentrations of GABA (Callachan et al., 1987; Cottrell et al., 1987; Majewska et al., 1986; Shu et al., 2004). Depending on subunit composition of the receptor and agonist, another important effect of steroids is to increase the efficacy of agonist actions (Bianchi & Macdonald, 2003; Maksay et al., 2000; Wohlfarth et al., 2002). In addition, in the absence of GABA, higher concentrations of steroids directly gate the GABAA channel, although this action is through a site independent of the GABA binding site (Ueno et al., 1997) and is also likely distinct from the site through which steroid potentiation is mediated.
Very recent results show evidence for two sites of neurosteroid action on GABAA receptors. One site spans the M1 and M4 transmembrane domains of the α subunit and accounts for potentiating actions of some steroids. Another site, between the M1 transmembrane domain of the α subunit and the M3 domain of the β subunit, is responsible for direct gating of the channel by steroids (Hosie et al., 2006). These sites are schematized in Figure 1C.
Direct gating by steroids is inefficient, with maximum responses well below responses generated by saturating GABA (Shu et al., 2004). Nevertheless, even small currents resulting from direct gating can have a significant impact on cellular excitability (Shu et al., 2004). Furthermore, we recently reported that the kinetics of onset and offset of direct gating are particularly slow. This likely has led to an underappreciation of the potency with which neurosteroids directly activate the channel (Shu et al., 2004).
In contrast to the relative structural promiscuity of steroidal GABAA receptor antagonists noted above, positive modulators have several strict rules that apply to the pharmacophore. A hydrogen bond donor at C3 in the alpha configuration (3α-OH) is critical for GABAA receptor activity and anesthesia, as is a hydrogen-bond acceptor attached to C17 on the β face of the steroid (Callachan et al., 1987; Covey et al., 2001; Majewska et al., 1986; Phillipps, 1975). Activity at GABAA receptors is not markedly altered by the stereochemistry at C5; either 5α- or 5β reduction at this position (combined with the other requirements above) results in active steroids. Much of our effort has been aimed at further defining the steroid pharmacophore.
Note that neuroactive steroids are among the most potent and efficacious modulators of GABAA receptors known. Potency is an empirical measure reflecting the concentration of drug required to yield half the maximum response that a saturating concentration of drug is capable of producing. Efficacy is a measure of the maximum effect produced by a saturating drug concentration. Neurosteroid potency rivals that of benzodiazepines (half-maximum effects in the nanomolar concentration range), but benzodiazepines typically generate less than 3-fold maximum potentiation of GABA responses, and therefore are of lower efficacy (Wafford et al., 1993). Neuroactive steroid efficacy, which can approach 10-20-fold maximum potentiation at low GABA concentration (Harrison & Simmonds, 1984; Shu et al., 2004) rivals barbiturate potentiation, but the steroids are much more potent.
Of interest in the pharmacological development of steroids is whether subunit specificity can be achieved, which might allow targeting of particular behaviors with selective neuroactive steroids. Optimism regarding this possibility has been spurred by the finding that the effect of benzodiazepines and other sedatives on specific behaviors can be manipulated by genetic alteration of specific GABAA receptor α subunits (Low et al., 2000; Rudolph et al., 1999). Recent developments in the subunit selectivity of neuroactive steroids have been reviewed (Belelli et al., 2002). In general, all GABAA receptors respond to neuroactive steroids. However, δ-subunit containing receptors are particularly sensitive to the actions of neuroactive steroids. In large part this sensitivity results from the fact that GABA acts as a partial agonist at δ-containing receptors, and a major action of steroids at GABAA receptors appears to be to improve efficacy of the agonist (Bianchi & Macdonald, 2003; Maksay et al., 2000). The net result of these considerations is that δ-containing receptors are not necessarily sensitive to lower concentrations of steroids, but the maximum effect of the steroids is higher at δ-containing receptors compared with other subunit combinations, notably γ-containing receptors (Wohlfarth et al., 2002). As the pharmacopeia of neuroactive steroids grows (our own project has created more than 400 novel analogues), it will be of interest to explicitly screen for subunit selectivity. Another potential approach to achieving selectivity for certain brain regions is to exploit the distribution of enzymes that metabolize neuroactive steroids (Belelli & Herd, 2003; Mellon et al., 2001).
4. Physiological actions of neuroactive steroids
Growth in the interest in neuroactive steroids is driven, in part, by the potential behavioral effects of both endogenous and exogenous neuroactive steroids. Here we review several of these potential actions, with emphasis on those likely relevant to neuroactive steroid effects at GABAA receptors.
4a. Anesthesia
Cellular and molecular mechanisms of anesthesia are far from clear. For instance, some recent research suggests that presynaptic actions may be important for the sedative effects of some anesthetics (Wu et al., 2004). Nevertheless, potentiation of GABAA receptor mediated postsynaptic membrane currents is a primary candidate mechanism shared by such structurally diverse anesthetics as steroid anesthetics, many volatile anesthetics, barbiturates, benzodiazepines, etomidate, and propofol. Traditionally these anesthetics have been thought to act by potentiating the phasic, synaptic actions of GABA, thereby decreasing overall circuit activity. However, it is also possible that tonic GABA currents, described above, may be an important target of at least some anesthetics, including steroids (Belelli & Herd, 2003; Shu et al., 2004; Stell et al., 2003), propofol (Bieda & MacIver, 2004), etomidate (Belelli et al., 2005; Caraiscos et al., 2004a), ethanol (Wei et al., 2004), and volatile anesthetics (Caraiscos et al., 2004b). Neurosteroids potentiate phasic transmission primarily by prolonging the decay time course of the synaptic conductance (Harrison et al., 1987). Neurosteroids may augment tonic currents by both directly activating the GABAA receptor (Lambert et al., 1990; Shu et al., 2004) and also potentiating the effects of ambient GABA on extrasynaptic receptors (Stell et al., 2003).
Use of steroids as anesthetics has arguably the longest history in neuroactive steroid research, and still enjoys interest. Historically, use of steroids as anesthetics may be traced to Hans Selye's observations from the 1940's that progesterone and other steroids can have anesthetic effects. These observations and others led to the development in the 1970's of Althesin, a combination of the steroids alphaxalone and alphadolone, as an intravenous anesthetic. Patient hypersensitivity to the delivery vehicle led to withdrawal of Althesin from clinical use. Nevertheless, Althesin had several favorable properties, including rapid onset and offset and a good therapeutic index. These properties encouraged development of other steroid anesthetics, but the biggest impediment has always been the lack of solubility in aqueous solution. Organon developed two candidates (Org-20599 and Org-21465) with increased water solubility resulting from substitutions at C2, but excitatory actions of the drugs led to discontinuation of clinical trials (Gasior et al., 1999). There is still hope that as we learn more about structure-activity relationships for neuroactive steroids at various neural targets, such adverse effects may be circumvented.
As part of our overall effort to understand neurosteroid actions, particularly as they pertain to anesthetic effects, we have utilized a simple behavioral assay of anesthesia, the Xenopus laevis tadpole loss of righting reflex assay. This assay has enjoyed a long history in anesthesia research, and the advantages of the preparation are well documented (e.g. Downes & Courogen, 1996 and references therein). Chief among these are the equilibration of bath concentrations of anesthetics with the concentration in the aqueous phase of blood in the animals. Therefore, relative potencies among anesthetics can be easily compared.
In our efforts to better understand structure-activity relationships of steroids at GABAA receptors and to relate these with behavioral anesthesia, we have supplemented the tadpole anesthesia assay with two assays of GABAA receptor function that lend themselves to rapid screening of new steroid analogues. First, we use the [35S]TBPS binding assay. TBPS binds to the picrotoxin site of the GABAA receptor/ionophore complex, and neuroactive steroids and other anesthetics are known to allosterically displace TBPS binding (Majewska et al., 1986; Ramanjaneyulu & Ticku, 1984; Ticku & Ramanjaneyulu, 1984). Therefore TBPS displacement can be used as an indirect assay of steroid effects on GABAA receptor function. Second, we use recombinant rat GABAA receptor subunits expressed in Xenopus oocytes for electrophysiological screening of analogue effects on GABA-mediated currents. For screening new steroid analogues, we have used the widespread α1β2γ2L subunit combination (McKernan & Whiting, 1996; Sieghart et al., 1999; Somogyi et al., 1996).
Over the course of more than a decade of investigating the anesthetic properties of neuroactive steroids, we have amassed a large database of steroid analogues examined in all three of these assays. We have published on some of the analogues previously; many we have not yet described. We here include only structures with A,B,C,D steroid ring backbone, although some structures include an additional fifth ring to explore fixed locations for important pharmacophores (Jiang et al., 2003). We do not include sulfated steroids, carboxylated steroids, or steroids with a phenolic A-ring or a phenyl substituent. We evaluated >200 compounds for which we had data in at least two of the screening assays and we examined correlations between TBPS inhibition potency (IC50) and loss of righting reflex (EC50) in Xenopus tadpoles (Fig. 2). The correlation is high (p = 3 × 10-18), with a slope factor of 0.65. The quantitative relationship between the IC50 and EC50 is very close (the geometric mean of the ratio of IC50 to EC50 is 0.7), providing a strong indication that action at the GABAA receptor explains a major part of the anesthetic action of this set of steroids and analogues. The regression of rank potentiation (a three-point concentration response is run in the oocyte assay, insufficient to generate an EC50) on rank loss of righting reflex shows a slope of 0.34 and high significance (p = 7 × 10-16). A similar correlation, with slope closer to unity was reported for propofol analogues (Krasowski et al., 2001). Finally, when rank TBPS potency is correlated with rank electrophysiological potentiation, the slope of the relationship is closer to unity (0.7126), and the correlation is highly significant (p = 2 × 10-35). We interpret these results to mean that effects on GABAA receptors explains most of the behavioral effect of the neuroactive steroid analogues, and that the TBPS binding assay on rat brain membranes correlates very well with a functional readout of recombinant GABAA receptors expressed in oocytes.
Figure 2. Correlation between effects of steroid analogues on TBPS binding in rat brain membranes and loss of righting reflex (LRR) in tadpoles.

This figure shows a scatter plot of data for 212 steroids and analogues. The ordinate gives the IC50 for inhibition of TBPS binding while the abscissa shows the EC50 for producing loss of righting in Xenopus tadpoles. Note that both axes are logarithmic, due to the large range of values observed (about 3.5 orders of magnitude for each parameter). The solid line shows the regression of log(IC50) on log(EC50) (slope 0.65, P = 3×10-18 that the slope does not differ from zero). The dashed line shows the line of equality. Some specific compounds are highlighted. Three pairs of enantiomers are shown, connected by thin solid lines (3α5αP, solid circles; 3α5βP, solid diamonds and ACN, solid triangles). The analogue B285 (X) has been extensively studied in single channel research, while ECN (cross) is an example of a compound with much higher potency at producing loss of righting than at inhibiting TBPS binding.
Further evidence for the GABAA receptor as a target relevant to anesthesia comes from our recent work attempting to develop neurosteroid antagonists. As described further below, the compound (3α,5α)-17-phenylandrost-16-en-3-ol (17PA) behaves as a steroid antagonist and is selective against 5α-reduced steroid potentiation and direct gating. Potentiation of GABA responses and direct gating of receptor currents by 5β-reduced steroids is relatively spared (Mennerick et al., 2004). We found in the tadpole anesthesia assay that 17PA showed the same stereospecificity; the potency of 3α5αP in generating anesthesia was reduced by 17PA, but the potency of 3α5βP was unchanged (Mennerick et al., 2004). Although it is possible that 17PA also blocks the actions of 5α-reduced steroid (but not 5β-reduced steroid) at some other target, this correlative evidence dovetails nicely with elegant work in mammals showing that sedative effects of benzodiazepines are eliminated in animals that have the endogenous α1 GABAA receptor subunit replaced with a functional, but benzodiazepine-insensitive, α1 subunit (Rudolph et al., 1999).
4b. Other neuropsychiatric effects involving GABAA receptors
Although our primary research focus has been on anesthetic actions of neuroactive steroids, there has been interest in the development of neuroactive steroid analogues for other clinical uses. In parallel there has been a growing exploration of the role of endogenous neuroactive steroid levels in normal and abnormal CNS function. We review here some examples of each category, with respect to behavioral effects that may involve interactions of steroids with GABAA receptors.
The epilepsy field serves as a good example of both categories of clinical interest in neuroactive steroids. In the etiology category, evidence has mounted that plummeting levels of progesterone (and progesterone-derived neuroactive steroids) may participate in catamenial epilepsy, a seizure disorder linked to the menstrual cycle (Reddy, 2004; Reddy et al., 2001; Rogawski, 2003; Rosciszewska et al., 1986). Neuroactive steroids have also recently been suggested to participate in dictating the latent period length in status epilepticus models of epileptogenesis (Biagini et al., 2006). Because of the potent actions of neuroactive steroids on GABAA receptors, it is not surprising that there has also been interest in developing neuroactive steroid analogues as anticonvulsants. Ganaxalone, an analogue of 3α5αP with increased metabolic stability, has undergone clinical trials with some success (Monaghan et al., 1999).
It is interesting that endogenous neurosteroid levels may correlate with several mood and anxiety disorders, including major depression (Uzunova et al., 1998), postpartum depression, and panic disorders. Because of the link between progesterone levels and neurosteroid levels, there has been particular interest in the role of neurosteroids in premenstrual syndrome, and post-partum dysphoria/depression (N-Wihlback et al., 2006; Smith, 2001). Antidepressant drugs, notably selective serotonin reuptake inhibitors, may increase neurosteroidogenesis (Griffin & Mellon, 1999; Pinna et al., 2006). Finally, in animal models of anxiety, neuroactive steroid levels increase (Barbaccia et al., 1997), which could underlie an endogenous anxiolytic effect.
4c. Actions of neurosteroids not involving GABAA receptors
Other members of the cys-loop family of ligand-gated ion channels are affected by neuroactive steroid and could help yield insight into steroid actions at GABAA receptors. Some of the same steroids that modulate GABAA receptor function also modulate nicotinic receptor function, but the concentrations needed for the cholinergic effect are much higher than those needed for GABAA receptor effects. Furthermore, structure-activity relationships are different in the case of some steroids; many steroids that potentiate GABAA receptor function inhibit nicotinic function (Paradiso et al., 2000). As for GABAA receptors, several 3β-OH steroids antagonize nicotinic receptor function. Although steroids with nicotinic blocking activity anesthetize tadpoles, the concentrations needed for cholinergic effects are much higher than those needed for anesthesia (Paradiso et al., 2000). Furthermore, estrogen steroids 17β-estradiol and 17α-ethynylestradiol, modulate neuronal nicotinic receptors comprised of α4β2 subunits, but the effect is species specific (Curtis et al., 2002; Paradiso et al., 2001). Human receptors are potentiated, but rat receptor function is inhibited (Paradiso et al., 2001). By using chimeras between rat and human receptors, a sequence of the C-terminus of the α4 subunit was determined to be the binding site for steroids (Paradiso et al., 2001). This result stands as one of few examples in which a steroid binding site has been identified on a ligand-gated receptor. It may be that an homologous site exists on some GABAA receptor subunits (see below).
Ionotropic glycine receptors, which like GABAA receptors are ligand-gated chloride channels, are sensitive to some neuroactive steroids, although again the structure-activity relationship is different, and steroid effects on glycine receptors exhibit more subunit-dependent differences than GABAA receptors (Maksay et al., 2001). Some GABA-potentiating steroids, including endogenous GABA-potentiating steroids, were reported to be largely inactive at glycine receptors (Barker et al., 1987; Belelli et al., 1999b; Harrison et al., 1987; Pistis et al., 1997). On the other hand, some synthetic GABA-potentiating steroids augment glycine responses generated by recombinant glycine receptors (Lambert et al., 2001). Recent papers, however, reported significant antagonistic effects of 3α5βP on native and recombinant glycine receptors (Jiang et al., 2006; Weir et al., 2004). Also, sulfated steroids antagonize glycine receptors (Maksay et al., 2001; Wu et al., 1997), and pregnenolone potentiates glycine receptors in a subunit-specific manner (Maksay et al., 2001). Finally, progesterone inhibits glycine receptors, but by a mechanism apparently different than the sulfated steroids. This complicated pattern of interactions suggests that multiple binding sites for steroids are likely. Another channel type receiving recent attention as a steroid target is the T-type voltage-gated Ca2+ channel (Jevtovic-Todorovic & Todorovic, 2006). Certain steroids and steroid analogues block T-channels with high potency and relatively good selectivity. For instance, 3α5αP inhibits T-channels with somewhat lower potency than its effects on GABAA receptors. However, 3β-OH steroid analogues are also potent T-channel blockers and have little effect on GABAA receptors (or other channels) at IC50 concentrations for T-channels (∼300 nM in some cases). One analogue ((3β,5α,17β)-17-hydroxyestrane-3-carbonitrile, or ECN) with strong T-channel and anesthetic effects, but with little GABAA receptor activity is highlighted in Figure 2. A particular feature of the block of T-channels by steroids is a lack of complete current inhibition at saturating steroid concentrations. 5β-reduced steroid analogues appear to demonstrate improved efficacy of T-channel inhibition compared with 5α-reduced steroids (Todorovic et al., 2004). These pharmacological studies mesh with an increasing appreciation that peripheral T-type Ca2+ channels may be involved in nociception (Jevtovic-Todorovic & Todorovic, 2006). Accordingly, paw injection of steroids active at T-channels results in antinociceptive effects in behavioral studies (Pathirathna et al., 2005; Todorovic et al., 2004). These effects on T-channels are notable because the potency of steroids at these channels apparently approaches the potency for GABAA receptor effects.
5. Steroid interaction with the GABAA receptor
Because of strong steroid behavioral effects, many of which are likely attributable to GABAA receptor modulation, we have become interested in the factors that dictate steroid potency and efficacy at the GABAA receptor. Comprehensive recent reviews of many aspects of steroid/GABAA receptor interactions are available (Belelli et al., 2006; Belelli & Lambert, 2005; Brussaard & Koksma, 2003; Lambert et al., 2001; Lambert et al., 2003; Matsumoto et al., 2005; Reddy, 2004). Here we focus on several aspects of this interaction that have not been the primary focus of recent reviews. Our program's interests have been in elucidating the cellular and molecular events leading to the binding of steroid to a putative site on the receptor and the transduction of this binding into “potentiation” of receptor function.
Our studies encompass several approaches to the question of steroid access, binding, and transduction. First, we review some of our recent work in which we studied whether non-specific properties of steroids, i.e. hydrophobicity, contribute to the potency of analogues. These studies led us to the conclusion that steroids likely access their site through membrane diffusion, which has implications for considering the overall potency of steroid analogues. Second, we have also used conventional molecular biology, combined with pharmacological and biophysical approaches, to try to elucidate candidate steroid sites on the receptor. Finally, we explore several examples of one rather unique facet of our research program: the use of novel medicinal chemistry to study structure-activity relationships. This aspect of our approach has led directly to attempts to develop steroid antagonists to help define the nature of the steroid/receptor interaction, steroid enantiomers to test the specificity of steroid/receptor interactions, and the exploration of benz[e]indenes and benz[f]indenes as steroid analogues.
5a. Access to sites
Although interaction with a site on the receptor is undoubtedly key to defining steroid potency and efficacy (see below, section 5b), we reasoned that lipophilic molecules like steroids may well have a binding site within the transmembrane domains, or other hydrophobic regions of the receptor. This would indicate that lipid solubility is essential to the overall potency of the drug. No matter how well a ligand fits its site, if the ligand can't access the hydrophobic site because of poor lipid solubility, then it will not be an effective drug.
With this rationale in mind, we developed tests of whether steroids access their putative site by typical aqueous access to an extracellular binding site, or whether membrane diffusion might be more important (Akk et al., 2005). A first approach was to examine behavior of individual GABA-gated channels that were either directly exposed to aqueous steroid or sealed off from direct access. As described below, steroids have a defined effect on channel open and closed times within a cluster of channel activity. When supplied directly to receptors from the pipette in a cell-attached recording (along with GABA), steroids are effective modulators of single-channel behavior. However, we also found that steroid quite effectively modulated channels even if it was bath applied but sealed from directly accessing the receptors under the cell-attached recording pipette (Fig. 3). This result suggests that direct aqueous access is not necessary for steroid modulation and that lateral membrane diffusion, or perhaps intracellular access, is important for the steroid to reach its site.
Figure 3. Steroid accesses receptor from membrane diffusion or from intracellular access.
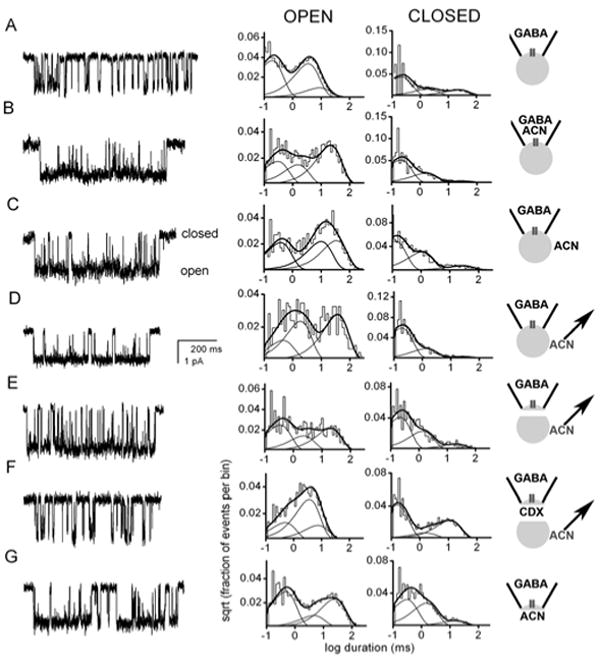
Cell-attached and inside-out recordings from HEK cells transfected with α1β2γ2 subunits were performed under the conditions schematized in the right panels. Downward deflections in the channel records represent channel openings. The graphs are log-binned histograms of open and closed times from the indicated patch. For all recordings, 50 μM GABA was included in the patch pipette to activate the channels. ACN (1 μM) was the steroid analogue used to potentiate channel function. Panels A-D represent cell-attached patch recordings, and panels E-G represent inside-out patch recordings. A. Patch exposed to GABA alone. The open times were 0.16 ms (49 %), 3.1 ms (37 %) and 7.8 ms (14 %). The closed times were 0.19 ms (61 %), 1.7 ms (20 %) and 21.7 ms (18 %). B. Channel activity in a patch exposed to the combination of GABA and ACN in the patch pipette. The open times were 0.27 ms (29 %), 1.4 ms (24 %) and 19.6 ms (47 %). The closed times were 0.19 ms (68 %), 1.5 ms (28 %) and 25.6 ms (5 %). Steroid increases both the duration and the amplitude of the third (longest) component of the open time distribution. Steroid also decreases the relative contribution of the third (longest) component of the closed time distribution (Akk et al., 2004). C. Channel activity in a patch indirectly exposed to ACN. Receptor function was still augmented when ACN was added to the bath solution following seal formation. The open times were 0.34 ms (33 %), 9.2 ms (34 %) and 21.3 ms (33 %). The closed times were 0.11 ms (57 %), 1.0 ms (35 %) and 25.0 ms (8 %). D. Channel activity in a patch recorded from a cell pretreated with ACN. Free bath ACN was removed before the recording was made, but potentiation of channel activity remained. The open times were 0.42 ms (19 %), 1.7 ms (38 %) and 34.5 ms (43 %). The closed times were 0.17 ms (73 %), 1.2 ms (23 %) and 22.7 ms (4 %). E. Channel activity in an excised patch taken from a cell pre-incubated with ACN. The patch was excised into a steroid-free bath, and the record was taken 1 min after excision. Augmented channel function remains despite removal of the channel from intracellular and extracellular sources of ACN. Histograms pertain to data from 0-2 min after excision. The open times were 0.31 ms (42 %), 2.1 ms (23 %) and 18.5 ms (35 %). The closed times were 0.20 ms (58 %), 1.2 ms (32 %) and 18.5 ms (9 %). F. The effect of addition of 5 mM methyl-β-cyclodextrin (CDX) to the bath to remove steroid. Cyclodextrin application immediately removed steroid-mediated potentiation. The open times were 0.46 ms (24 %), 3.4 ms (57 %) and 6.9 ms (19 %). The closed times were 0.15 ms (60 %), 1.5 ms (9 %) and 10.4 ms (31 %). G. Intracellular application of ACN results in potentiation. Channel activity in an inside-out patch exposed to 1 μM ACN applied to the cytoplasmic face of the membrane. The open times were 0.42 ms (48 %), 4.2 ms (15 %) and 23.3 ms (37 %). The closed times were 0.28 ms (48 %), 1.4 ms (42 %) and 14.5 ms (10 %). The figure is taken from data in (Akk et al., 2005). Copyright Society for Neuroscience.
If lateral membrane diffusion (as opposed to intracellular access) is important for steroid access to its site(s), then by excising membrane patches, we might isolate the component resulting from lateral membrane diffusion. We excised membrane patches after loading cells with bath-applied (3α,5α,17β)-3-hydroxyandrostane- 17-carbonitrile (ACN). We found that steroid potentiation persisted when patches were excised into a steroid-free bath (Fig. 3E). This result suggests that plasma-membrane partitioning alone is sufficient to provide steroid access to the receptor. These results are complemented by results in which we found that potentiation of inside-out patches is robust when steroid is applied directly to the inner membrane leaflet (Fig. 3G), and we have synthesized a membrane-impermeant steroid with an ionically charged Alexa group substitution at C17 that only works from the inside of the membrane (Akk et al., 2005).
Conclusions from membrane patch experiments were bolstered by imaging studies of whole cells (Fig. 4 and Akk et al., 2005). We synthesized a fluorescent analogue of 3α5αP that retained activity at GABAA receptors and found that it accumulated in lipophilic regions of the cell, including plasma membrane and lipophilic intracellular compartments (Akk et al., 2005). These imaging results complement the cell-attached patch recording results and suggest the plausibility of membrane and/or intracellular access to steroid receptor sites.
Figure 4. Effect of cyclodextrin on the washout of currents and fluorescence of a tagged steroid analogue.

A1. NBD-3α5αP (300 nM) was applied to neurons in the absence of GABA, and whole-cell currents were monitored. Drug exposure is indicated by the horizontal bar. Despite rapid removal of free drug, the current decayed slowly. A2. Including 500 μM γ-cyclodextrin (CDX) in the wash following drug removal (solid trace) increased the rate of current offset. The dotted trace is a re-plot of A1. B1 and B2. Imaging of a neuron following exposure to fluorescent steroid analogue at the time points denoted by the small letters in A2. The fluorescence intensity is pseudocolored, with warm colors representing high fluorescence intensity. B1 shows loss of fluorescence with normal saline wash. B2 shows intracellular fluorescence loss is speeded by cyclodextrin wash. The figure is taken from (Akk et al., 2005). Copyright Society for Neuroscience.
We noted that steroids have extremely slow deactivation kinetics. Once the cell has been exposed to steroid, washing the effect away is extremely slow (τ ∼ 30 s in the case of direct gating by steroids on cultured hippocampal neurons). We had originally reasoned that such slow deactivation results from an inherently high ligand-receptor affinity, i.e. slow dissociation rate from the receptor. However, the observations above suggest that the inherent steroid dissociation rate may actually be quite rapid. Rather, the long-lived steroid effect may result from the lipophilicity of 3α5αP (estimated logP value of 4.89), coupled with repeated binding and unbinding events occurring from the lipid reservoir. To distinguish whether slow receptor dissociation or lipophilic interactions between steroids and cell membranes cause the slow kinetics of deactivation, we applied γ-cyclodextrin during the deactivation phase of steroid-gated GABAA receptor currents. Cyclodextrins are membrane impermeant and bind steroids by interacting with the plasma membrane and providing a hydrophobic binding pocket for steroids as they laterally diffuse within the membrane. We found that cyclodextrin, at the concentrations we employed, had no effect on normal GABAA receptor function (Shu et al., 2004) but see (Pytel et al., 2006). After forming an inclusion complex with cyclodextrin, the steroids are washed away with aqueous medium. Therefore, cyclodextrin molecules serve as sponges for the non-specific accumulation of steroid. We found that cyclodextrins dramatically speed washout of steroid effects (see Figure 4A), consistent with the idea that membrane interactions rate-limit the steroid effects (Shu et al., 2004).
We returned to the idea that although intracellular steroid is not necessary for GABAA receptor modulation, it may indirectly participate if it equilibrates sufficiently rapidly with the plasma-membrane pool of steroid. We were intrigued by the strong intracellular accumulation observed when cells are exposed to a fluorescent steroid analogue (Akk et al., 2005). After loading with fluorescent steroid, saline wash produces a slow loss of intracellular fluorescence. Interestingly, the slow loss is characterized by a time constant indistinguishable from the slow time constant of current deactivation observed in electrophysiology experiments. Furthermore, cyclodextrin wash speeds the rate of loss of intracellular fluorescence (despite the fact that cyclodextrin is membrane impermeant, see Fig. 4B). This suggests that the two pools of steroid (plasma membrane and intracellular) are in rapid equilibrium. By emptying the plasma membrane pool with cyclodextrin, we observe the net rate of movement of intracellular steroid to the plasma membrane. During cyclodextrin wash, there was significantly faster fade of current than loss of intracellular fluorescence (Akk et al., 2005). Therefore, movement from the intracellular pool is unable to keep up with loss from the plasma membrane when cyclodextrin is used to speed removal from the plasma membrane pool.
Taken together these results suggest that plasma-membrane lateral diffusion is important for steroids to access their site on the GABAA receptor, a conclusion also reached for other modulators of ion channel function (Hille, 1977; Lee & MacKinnon, 2004; Suchyna et al., 2004). Furthermore, intracellular steroid, through rapid equilibration with the plasma-membrane pool, can supply receptors with steroid. There are several implications of these observations. First, although steroids have a high apparent affinity for GABAA receptors, the intrinsic affinity of steroid for the site is likely much lower, since plasma-membrane concentration, the concentration we argue is most directly relevant for the steroid site, is 10,000-100,000 fold greater than the aqueous concentration. As a corollary, we suggest that “non-specific” lipophilicity of steroids will influence overall potency and longevity of steroid actions at GABAA receptors. Given the preference of steroids to affiliate with intracellular and plasma membranes, another implication of these considerations is that autocrine/paracrine actions of endogenous steroids may be particularly important. Recently, it has been suggested that neurosteroid synthetic enzymes are localized within principal, projection neurons in many CNS regions, but not in interneurons (Agis-Balboa et al., 2006). If autocrine actions are particularly important because of lipophilic retention, then we might expect cell-specificity in the actions of endogenous steroids. It also seems possible that beyond simple lipophilicity, different steroids may partition with different orientations within membranes (Alakoskela et al., 2006). Again, such “non-specific” interactions with lipophilic domains near receptor binding sites may influence access to the receptor site and thus potency of steroid effects. Although tools may not always be available to address these issues directly, steroid access should be a serious consideration when interpreting structure-activity relationships and in future drug design.
Our results highlight lipophilicity as an important contributor to the effective lifetime of steroid effects. Other factors may also influence steroid longevity in certain circumstances. For some subunit combinations, inherent steroid affinity may be higher than in subunits we have investigated. Other evidence suggests that regional differences in the presence of degrading enzymes can dictate the lifetime of steroid effects (Belelli & Herd, 2003). Finally, when steroid actions at the systems level are considered, drug pharmacokinetics will influence longevity of action.
5b. Multiple sites for steroids – pharmacological and molecular aspects
Steroid interactions with the GABAA receptor can result in three types of effects: potentiation of currents elicited by GABA or another activator, inhibition of currents elicited by GABA or another activator, or direct activation of the channel. Inhibition manifests itself as a more rapid apparent desensitization rate (Shen et al., 2000). Despite the brief transient of GABA at synapses, desensitized states can be populated during an IPSC (Jones & Westbrook, 1996). Thus, at least part of the action of sulfated steroids may be to enhance receptor accumulation in these non-conducting states and thereby truncate IPSCs. In single-channel recordings, coapplication of an inhibitory steroid, e.g., pregnenolone sulfate with GABA, results in a briefer mean cluster duration without affecting intracluster open or closed time distributions (Akk et al., 2001).
In contrast, coapplication of potentiating steroids, e.g., 3α5αP, along with GABA results in three distinct hallmark effects in the open and closed time distributions (Figs. 3, 5). These three effects presumably combine to augment macroscopic GABA responses described above. The relative frequency and mean duration of the longest-lived open time component (OT3) are enhanced. In addition, exposure to a potentiating steroid leads to a reduction in the relative frequency of the activation-related closed time component, reflecting a reduction in the channel closing rate (Akk et al., 2004; Akk et al., 2005). At higher concentrations, potentiating steroids can elicit activation of the GABAA receptor in the absence of a classic agonist (Callachan et al., 1987; Shu et al., 2004). The channel activity elicited by the application of steroids in the absence of agonists typically consists of brief openings, occurring either as isolated openings or in bursts of a few openings (Akk, unpublished observations).
Figure 5. Coapplication of 3α5αP with GABA affects the open time distributions.

A. Modelled open time histograms for GABA (thin lines) and GABA + 1 μM 3α5αP (thick lines). The longest open time component (OT3) is shown with dashed lines. The presence of steroid results in the increase in the duration and relative frequency of OT3 (shown with arrows). The modelling was conducted using the mean values for control and steroid data from Akk et al. (2005). B. Sample single channel currents recorded in the presence of GABA and GABA + 1 μM 3α5αP. C. Concentration effect curves for the duration and fraction of OT3. The duration of OT3 increases from 5-6 ms to approximately 15 ms, and the relative frequency of OT3 increases from 0.15 to approximately 0.45 in the presence of steroid. In addition to the changes in open times, 3α5αP also affects the relative frequency of the activation-related closed time component (channel closing rate).
The actions of potentiating and inhibitory steroids are likely mediated by steroid interactions with different sites. When both steroids are applied simultaneously, the resulting effects on receptor kinetics mimic the actions of either steroid applied individually without any indication of competition between the two types of steroids. Thus, the presence of 3α5αP does not interfere with the ability of pregnenolone sulfate to reduce cluster duration nor does the presence of pregnenolone sulfate interfere with the ability of 3α5αP to prolong channel open time durations (unpublished observations). This has also been seen for the steroid analogue, ACN, and pregnenolone sulfate (Akk et al., 2001).
Molecular manipulation of receptor structure supports the idea of multiple distinct interaction sites and/or transduction pathways for neuroactive steroids. A mutation to the 2′ residue in the M2 transmembrane domain in the α1 subunit (α1V256S) specifically reduces the ability of inhibitory steroids to modulate receptor function but is ineffective on the actions of potentiating steroids (Akk et al., 2001). Although the 2′ residue is unlikely to constitute a binding site for inhibitory steroids, its preferential action on the effects of inhibitory steroids suggests that the effects of the two types of steroids follow separate transduction pathways. This work is consistent with earlier pharmacological work demonstrating that sulfated steroids likely bind to a site distinct from the potentiating steroids (Park-Chung et al., 1999).
Kinetic analyses of currents recorded in the presence of potentiating steroids with different structures suggest that the potentiating actions of steroids are mediated via steroid interactions with multiple (two or more) distinct sites. While some steroids, e.g., 3α5αP and ACN cause the three hallmark kinetic changes with similar or identical dose-effect relationships, other steroids (e.g., the 5β-reduced, 18-normethyl analogue of ACN, B285) have different EC50s for the increase in the relative frequency of the longest open time component and the decrease in the closing rate on one hand, and, on the other hand, for the prolongation of the longest open time component (Akk et al., 2004). The simplest explanation of the findings is that at least two binding sites, recognizing different features of a steroid, are involved in mediating the effects. We have used the shorthand of naming the site mediating the increase in the frequency of OT3 and the decrease in the channel closing rate as Site A. The site mediating the increase in the duration of OT3 is termed Site B. As will be discussed later, it is quite possible that there may be more subdivisions in these sites.
The individual alterations in channel kinetics that result in current potentiation can be differentially affected by molecular manipulations within the γ subunit. The replacement of the amino acid sequence in the very C′-terminal end of the γ subunit with homologous sequence from the ρ1 subunit results in loss of prolongation of OT3 duration by ACN without affecting the ability of the steroid to modulate the two remaining kinetic components. The effect of the mutation on the prolongation of OT3 duration was specific to steroids and not due to a global change in channel activation because prolongation of OT3 by pentobarbital was not affected. This finding suggests the notion of a separate binding site and/or transduction pathway for OT3 prolongation (Akk et al., 2004).
In addition to different sites mediating different changes in channel kinetics, there is evidence suggesting that the same effects can result from steroid interactions with separate, or possibly overlapping, sites. A recent study showed that mutations to the 407/410 residues (rat amino acid designations) in the M4 domain of the α subunit led to loss of potentiation by steroids such as 3α5αP (Hosie et al., 2006). However, potentiation by B285, a steroid which acts through similar kinetic changes, is not reduced by the mutations (Li et al., 2006a) suggesting that the sites for the two steroids may be distinct.
The concept of multiple steroid binding sites has been touched upon previously. Twyman and Macdonald (Twyman & Macdonald, 1992) demonstrated that at high concentrations, androsterone, in addition to increasing the relative number of long-lived openings, appeared to reduce the mean duration of the long-lived open event class. The latter effect was proposed to result from channel block due to steroid binding to yet another site, distinct from those involved in potentiation or the type of inhibition produced by pregnenolone sulfate. Also, Morrow et al. (Morrow et al., 1990) found that the potentiation curves of muscimol-stimulated chloride uptake into rat brain synaptosomes were biphasic for many steroids suggesting that steroid interactions with multiple binding sites underlie their effects on the receptor, and others have found biphasic effects of 5β-reduced potentiating steroids on TBPS binding (Hawkinson et al., 1994). However, the caveat to these findings is that several subtypes of GABAA receptors, each with their own distinct pharmacological properties, could exist in the synaptoneurosome preparation used.
5c. Steroid antagonists
The elucidation of sites for neuroactive steroids may be aided by the development of pharmacological antagonists selective for one or more steroid effects/sites. We focused our attempts at generating steroid site antagonists on modifications of the two steroid regions that are critical for GABA receptor activity, the 3α-hydrogen bond donor and the 17β-hydrogen bond acceptor. Our first attempts to develop steroid antagonists built upon initial observations suggesting that 3β-OH steroids may antagonize the actions of potentiating 3α-OH steroids (Prince & Simmonds, 1992, 1993). However, when we explored the actions of 3β-OH steroids in detail with electrophysiological techniques, we found that the antagonistic interaction between 3β-OH steroids and 3α-OH steroids is not competitive. Instead, 3β-OH steroids produce direct antagonism of GABAA receptors, with effectiveness of antagonism dependent upon channel activation (Wang et al., 2002). These steroids fail to influence GABA-gated responses at low GABA concentrations. However, when GABA concentration is increased or when potentiating steroid is coapplied with a low GABA concentration, the increased probability of channel opening is associated with more effective antagonism by 3β-OH steroids. Therefore, we conclude that 3β-OH steroids are not useful as ligands for any of the steroid potentiating sites. The effect of 3β-OH steroids, as mentioned above, is qualitatively similar to the effect of steroids sulfated at C3. Although sulfated steroids are more potent antagonists than 3β-OH steroids, they exhibit the same activation dependence as 3β-OH steroids (Eisenman et al., 2003; Wang et al., 2002). Furthermore, the α1V256S mutation that eliminates sulfated steroid antagonism (Akk et al., 2001) also eliminates the antagonistic effect of 3β-OH steroids (Wang et al., 2002).
Although the effect of 3β-OH steroids is indirect with respect to potentiating steroids, these functional antagonists may still be useful compounds. Because antagonism is dependent on receptor activation, these drugs could leave normal GABAergic neuronal communication intact at concentrations that antagonize abnormally strong GABAA receptor activation. It is tempting to speculate that this mechanism participates in the ability of a 3β-OH steroid to reduce ethanol intake in mice (O'Dell et al., 2005).
Because 3β-OH steroids did not represent the steroid-site ligand we ultimately desired, we continued our search for direct antagonists of steroid effects at GABAA receptors by shifting our medicinal chemistry efforts to the C17 position. Using electrophysiological screens and the TBPS binding assay, we identified a series of analogues formed by connecting C21 and C17 via a one carbon linker to form an additional cyclohexyl ring in the steroid. Several of these analogues appeared to have antagonist properties against potentiating steroids. Some of these compounds, for reasons that we do not fully understand, acted as apparent competitive antagonists of steroid potentiation when evaluated on Xenopus oocytes, but in native neurons and in transfected HEK cells, the compounds modestly potentiated GABA responses. These compounds were therefore excluded from further study.
Subsequently, several compounds containing a phenyl group at C17 were identified that had no activity alone or when combined with GABA, but antagonized the effects of potentiating steroids. The most effective of these was (3α,5α)-17-phenylandrost-16-en-3-ol (17PA). We characterized the effects of 17PA on steroid direct gating and potentiation (Mennerick et al., 2004) and found that 17PA antagonized the actions of 3α5αP but only weakly antagonized 3α5βP actions (Fig. 6). GABA receptor potentiation by benzodiazepines and barbiturates was not affected by 17PA. When we evaluated direct gating (in the absence of GABA) by steroids, the selectivity of 17PA effects for 3α5αP over 3α5βP was preserved. In addition 17PA proved to be a stronger antagonist when GABA was absent (against directly gated currents) than when GABA was present (against potentiation). The profile of 17PA effects against 3α5αP potentiation appeared complex. The concentration response relationship for 3α5αP potentiation was shifted to the right by 10 μM 17PA, but the maximum potentiation generated by 3α5αP was also slightly decreased by 17PA. This decrease in the maximum obtainable potentiation could be interpreted to mean that the mechanism of antagonism by 17PA is not purely competitive or that the inhibition at high concentrations of 3α5αP represents an inhibition of direct gating, which as mentioned above, is more sensitive to a given 17PA concentration.
Figure 6. 17PA antagonizes 3α5αP effects on GABA receptors.
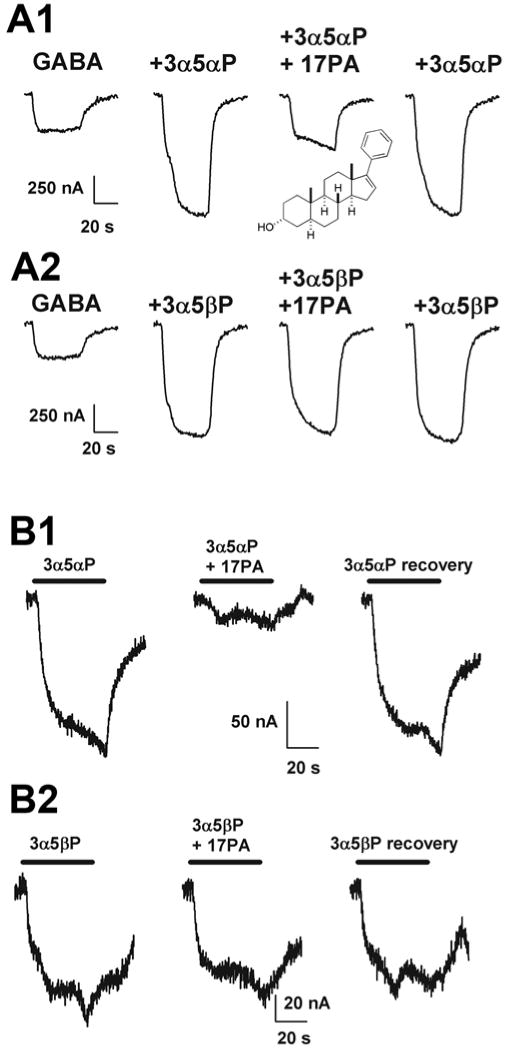
A1 and A2. Comparison of the effect of 10 μM 17PA on potentiation by 0.5 μM 3α5αP (A1) and 3α5βP (A2) on GABA receptors expressed in an oocyte. The inset in panel A1 shows the structure of 17PA. GABA and drugs were co-applied. B1 and B2. Effect of 17PA (10 μM) on steroid (5 μM) activation of receptors in the absence of GABA. Note the nearly complete inhibition of direct gating by 3α5αP despite the high concentration of steroid agonist used. Note that neither potentiation nor direct activation by 3α5βP was depressed by 17PA. The data are taken from (Mennerick et al., 2004). Copyright American Society for Pharmacology and Experimental Therapeutics.
In summary, 17PA represents a direct and selective antagonist of 5α-reduced (but not 5β-reduced) steroid potentiation and direct gating. It is active in the high micromolar concentration range and may bind to a site at least partly overlapping some 5α-reduced potentiating steroids. At face value, the above results demonstrating selective effects of 17PA on 5α vs. 5β diastereomers might suggest different sites for the two broad classes of potentiating steroids. However, it is difficult to reconcile this simple interpretation with other results. For instance, in single-channel recordings, site “A” (increased frequency of long openings and diminished activity-dependent closures) and site “B” (increased duration of long openings) effects are elicited by both 5α-reduced and 5β-reduced steroids, and only under certain conditions can these sites be distinguished (Akk et al., 2004). A key to interpreting the pharmacological effects of 17PA is determining whether the interaction with potentiating steroids is truly competitive.
Although 17PA is an intriguing compound, several limitations reduce its current usefulness. First, it is significantly less potent than the potentiating steroids themselves. It is also poorly water soluble. These features will likely make the compound difficult to use in many applications, including in vivo situations. Second, virtually nothing is known about the selectivity of 17PA for GABAA receptors versus other cellular targets, although our unpublished evidence suggests that gross aspects of synaptic communication are not affected by 17PA. Furthermore, we have shown specificity in 17PA antagonism of steroid effects over two other major classes of GABAA receptor potentiators, benzodiazepines and barbiturates (Mennerick et al., 2004). On the other hand, 17PA effects on other classes of GABAA receptor potentiators have not been exhaustively tested.
5d. Steroid/ethanol interactions
Exposure to ethanol affects multiple cellular processes including the function of the GABAA receptor, and its interactions with neuroactive steroids. The actions of ethanol on receptor function can be broadly divided into two groups. First, there is an indirect effect, where the application of ethanol modifies the level of some endogenous GABAA receptor modulator without affecting the ability of the receptor to respond to the stimulus (Barbaccia et al., 1999; Morrow et al., 2001; O'Dell et al., 2004; Sanna et al., 2004; VanDoren et al., 2000). Second is a direct interaction, where exposure to ethanol modifies the ability of the receptor to respond to a stimulus, which otherwise would have been ineffective at modulating receptor function (Akk et al., 2007; Akk & Steinbach, 2003; Murayama et al., 2006).
VanDoren et al. (2000) showed that administration of ethanol was followed by an increase in the levels of 3α5αP in the rodent cerebral cortex. The elevation in steroid level was time dependent, reaching its peak at approximately 1 hr after injection, and dose dependent, increasing linearly with ethanol dose up to 2-3 g/kg (roughly, 0.2-0.3 % or 35-50 mM). The steroid levels, following ethanol administration, reached concentrations (roughly, 30 nM) that are pharmacologically relevant and capable of modulating GABAA receptor activity. Cortical 3α5αP levels were correlated with ethanol-induced loss of righting reflex, and ethanol administration increased bicuculline-induced seizure threshold with a time course that followed changes in steroid levels. Furthermore, pretreatment of animals with the 5α-reductase inhibitor finasteride (which blocks neurosteroid synthesis) reduced the ability of ethanol to affect steroid levels in the brain, and blocked the anticonvulsant effect of ethanol on bicuculline seizure threshold. Taken together, the data suggest that ethanol-induced changes in the levels of neurosteroids can affect the hypnotic and anticonvulsant effects of ethanol in vivo.
Akk and Steinbach (Akk & Steinbach, 2003) showed another type of interaction between the effects of ethanol and a neuroactive steroid. By examining single-channel recordings obtained in the presence of low concentrations of steroid and/or ethanol, it was demonstrated that co-application of GABA with ethanol (0.05-1 %, 9-170 mM) and 10 nM ACN led to a significant increase in the duration of the longest lived open time component (OT3, see above). Either of the drugs applied separately at these concentrations had no effect on GABA-elicited currents. The other kinetic changes mediating channel potentiation by steroids (relative frequency of OT3, channel closing rate) were not affected by the combination of 10 nM ACN and ethanol suggesting that the binding of ethanol allosterically interacts with the ability of steroid to bind to the site that underlies an increase in the duration of OT3.
A recent study showed a similar interaction between an endogenous neurosteroid, 3α5αP, and ethanol (Akk et al., 2007). Coapplication of the two drugs, at concentrations ineffective individually, resulted in a significant increase in the duration of OT3 without affecting the relative frequency of OT3 or the channel closing rate. Tadpole behavioral assays further demonstrated that a subthreshold dosage of 3α5αP could shift the loss of righting reflex curve for ethanol to lower concentrations. It was concluded that steroid interactions with the classic steroid binding site underlie the phenomenon because the presence of the steroid antagonist 17PA reduced the increase in the OT3 duration and the shift in the tadpole loss of righting reflex.
5e. Insights from enantiomers
There has been a long history of interest in whether anesthetics may act through non-specific membrane perturbation (Cantor, 1998; Kissin & Gelman, 1988) or through specific interactions with proteinaceous binding sites, for instance on ion channels (Belelli et al., 1999a). In the case of neuroactive steroids, some evidence has been interpreted to support the idea that lateral pressure changes within the lipid bilayer are responsible for the actions of steroids on ρ1 (GABA-C) receptors (Morris & Amin, 2004).We have taken the approach of using medicinal chemistry to probe these alternative hypotheses as they pertain to steroid modulation of GABAA receptors.
To determine whether steroids may act at protein sites, Covey and colleagues synthesized and tested a series of steroid enantiomers (Hu et al., 1997; Nilsson et al., 1998; Wittmer et al., 1996; Zorumski et al., 1998). These are important agents because prior studies used diastereomers (or epimers) to probe steroid actions (Im et al., 1990; Woodward et al., 1992). Diastereomers, in which one or a few chiral centers are changed, have different physical properties and perturb lipid membranes differently (Grossley, 1995). Thus, diastereomers cannot be used to distinguish effects in lipid environments from effects at protein sites. Furthermore, different diastereomers have different effects on GABAA receptors. For example, diastereoselectivity is clear at C3 and C17, but there is little diastereoselectivity at C5 (Gee et al., 1988). Unlike steroid diastereomers, steroid enantiomers are mirror image molecules of the natural steroid (Fig. 7), with identical physical properties. The rationale, therefore, is that enantiomers should behave identically in the lipid bilayer (Agarwal et al., 1986a, 1986b; Arnett & Gold, 1982; Ghosh et al., 1971; Guyer & Bloch, 1983; Hermetter & Paltauf, 1982; Mannock et al., 2003; Westover & Covey, 2004). On the other hand, enantioselectivity is likely to be observed in the case of a proteinaceous binding site because proteins are composed entirely of amino acids in the L-configuration. We found that in the 3α5αP series (Fig. 7), potentiation of GABAA receptors, direct gating of chloride channels, effects in the TBPS binding assay, and anesthetic effects in tadpoles and mice show significant enantioselectivity. In some cases enantiomer pairs differ by an order of magnitude or more in potency (Covey et al., 2000; Wittmer et al., 1996; Zorumski et al., 1998). In general, (+)-enantiomers that have the absolute configuration of natural steroids are more potent (and in physiological assays are probably more effective) than (-)-enantiomers. Interestingly, enantiomers tend to show less enantioselectivity in the tadpole swimming assay than in assays of GABA receptor function (Fig. 2).
Figure 7. Structures of enantiomer pairs.

Until the very recent identification of GABAA receptor steroid binding sites by mutagenesis (Hosie et al., 2006), the 3α5α enantiomers (Fig. 7) provided some of the strongest evidence that anesthetic steroids act at chiral sites on the GABAA receptor. Recent biophysical evidence has confirmed that enantiomeric pairs of 3α5αP and 3α5βP do not differ in their interaction with lipid bilayers (Alakoskela et al., 2006). Parameters examined included packing of the lipid interior or head groups, mobility of hydrocarbon chains, hydration of the head group region, phase transitions, surface potential, and association of the steroid with head group or interior region of the leaflet. Potency differences in enantiomer pairs of other lipophilic anesthetic agents (e.g. ketamine, isoflurane, barbiturates) have similarly been used to argue that anesthesia results from specific interactions of drugs with protein receptors (Andrews & Mark, 1982; Franks & Lieb, 1991; Hall et al., 1994). However, the differences in enantiomeric anesthetic potencies observed with these other agents have rarely achieved the order of magnitude separation we found with ACN enantiomers (Wittmer et al., 1996; Zorumski et al., 1998).
In the case of GABA-C receptors, the issue of lateral membrane pressure effects of steroids (Morris & Amin, 2004) was recently re-investigated using the enantiomer approach. Li et al. (2006b) found enantioselectivity of 3α5αP and 3α5βP in both potentiating and inhibiting effects of steroids on ρ1 receptors (Li et al., 2006b). Coupled with evidence that interactions of these steroids lack enantioselectivity in their interaction with lipid bilayers (Alakoskela et al., 2006), these studies suggest that membrane effects alone (Morris & Amin, 2004) are unlikely to account for steroid effects on GABA-C receptors.
There are, however, GABA-potentiating steroids that show much less enantioselectivity at the GABAA receptor. For instance pregnanolone (3α5βP) exhibits much less enantioselectivity compared with 3α5αP (Covey et al., 2000). We are in the process of developing enantiomers of other steroids and steroid analogues. Surprisingly, some of these compounds behave differently than either 3α5αP or 3α5βP. A particularly striking example occurs for enantiomeric pairs of steroids in the androgen class of steroids. Naturally-occurring androsterone (3α5αA; Fig. 7) and etiocholanolone (3α5βA) are very weak potentiators of GABAA receptor activity, whereas their unnatural enantiomers are more effective potentiators. Therefore, these pairs exhibit reverse enantioselectivity (unnatural > natural).
In summary, exploration of enantiomers has yielded evidence for strong enantioselectivity (nat > ent), weak enantioselectivity (nat ≈ ent), and reverse enantioselectivity (ent > nat). How are these results to be understood in terms of a comprehensive hypothesis of steroid actions on GABAA receptors? Of course, a real possibility is that there may be multiple binding sites on GABAA receptors that accommodate steroids of different structures. Some sites may exhibit little enantioselectivity (e.g. the 3α5βP site on the GABAA receptor), and others may exhibit strong enantioselectivity (the 3α5αP site), or there may be an “enantiomer” site that accommodates ent-androgens especially well. Recent identification of putative steroid binding sites on GABAA receptors (Hosie et al., 2006) will undoubtedly lead to future studies with mutant receptors to test the hypothesis of heterogeneous binding sites.
6. Probing the pharmacophore with novel steroid analogues
6a. Insights from benz[e]indenes
Our program has synthesized tricyclic steroid analogues with the intent of mapping binding sites for steroids on GABAA receptors (Covey et al., 1993; Han et al., 1995; Hu et al., 1993; Zorumski et al., 1996). Benz[e]indenes are steroid-like molecules in which the A-ring has been opened and partially removed (Fig. 8). This creates tricyclic agents with considerable molecular flexibility at the positions occupied by the critical 3α hydrogen-bond donor. We found that certain benz[e]indenes are potent and effective modulators of GABAA receptor responses with effects akin to steroid anesthetics (Rodgers-Neame et al., 1992). As with 3α5αP, the benz[e]indene analogues exhibit significant enantioselectivity in their actions on GABAA receptors (Zorumski et al., 1996). In the next several paragraphs, using an example of BI-2, we will describe the functional effects that benz[e]indenes have on the α1β2γ2 GABAA receptor function (Li et al., 2006a). BI-2 is an analogue with a carbonitrile substitution at the position analogous to the C17 position in steroids, and thus is a benz[e]indene mimic of ACN.
Figure 8. Structures of benz[e]indene and benz[f]indenes.

BI-2 has dual effects on GABAA receptor function. At low micromolar concentrations, BI-2 acts by enhancing the peak macroscopic current. The apparent EC50 for the macroscopic dose-response curve is ∼0.4 μM. However, the true half-maximal concentration may be masked by an antagonist effect of the compound. This antagonism becomes apparent at concentrations above 10 μM, and at even higher concentrations, antagonism dominates the drug's net effect. The nature of inhibition is similar to that of 3β-hydroxy (epipregnanolone, 3β5βP) or sulfated steroids (pregnenolone sulfate), and manifests itself as an enhancement in the rate of desensitization.
The kinetic details of potentiation and inhibition have been examined from single-channel recordings. Potentiation is a result of BI-2 affecting the channel open times: the mean duration and relative contribution of the longest-lived open time component are increased in the presence of BI-2. Qualitatively these effects are similar to those observed in the presence of potentiating neurosteroids, e.g., 3α5αP. However, in contrast to potentiating steroids, BI-2 does not affect the intracluster closed time distributions. Specifically, BI-2 does not reduce the relative frequency of the activation-related closed time component that is a hallmark effect of all potentiating neurosteroids studied by us using single-channel kinetic analysis. Furthermore, BI-2 is unable to prevent the ability of B285 (a 5β carbonitrile steroid) to act on the closed times, suggesting that BI-2, and possibly other benz[e]indenes do not interact with the site through which the closed time effect is mediated. The inhibitory effect of BI-2 manifests itself as shortened cluster durations in single-channel recordings, similar to that observed in the presence of pregnenolone sulfate.
There is evidence suggesting that the potentiating and inhibitory effects of BI-2 are mediated by actions through classic steroid binding sites and mechanisms. First, mutations to the sites shown to form the potentiating steroid site (α1Q241 and α1N407/Y410; (Hosie et al., 2006) abolish potentiation by BI-2 (Li et al., 2006a). Second, 17PA, a steroid capable of blocking the actions of many 5α-reduced potentiating steroids, also reduces the ability of BI-2 to potentiate receptor function. The involvement of the classic pathway for the inhibitory effect of BI-2 is demonstrated by the observation that the α1V256S mutation, a mutation that blocks inhibition by pregnenolone sulfate and 3β-hydroxy steroids, also blocks the inhibitory actions of BI-2.
As a result of the structure with a partial A ring, benz[e]indenes are more flexible than steroids, and, unlike steroids, can mimic steroids having the 3-hydroxyl group in either the 3α or the 3β configuration. It is likely that such flexibility is the reason for the presence of both potentiating and inhibitory actions of this compound: BI-2 when mimicking the 3α configuration interacts with the potentiating site, and BI-2 when mimicking the 3β configuration binds to the inhibitory site.
Studies with benz[e]indenes have made several important contributions to characterization of steroid sites on the GABAA receptor. First, the results tell us that the steroid binding pocket does not require interactions with a full steroid backbone. Instead it allows molecules with only partial steroid structures to potently and efficaciously cause changes in receptor function. The second, and rather consequential finding is that BI-2 lacks one of the kinetic hallmarks of steroid effects (reduction in channel closing rate); BI-2 is also unable to block this effect when elicited by B285. Previous studies with steroids had attributed the reduction in channel closing rate along with an increase in the relative frequency of long openings to being a result of steroid interactions with a single site. The experiments with BI-2, however, suggest that these two kinetic effects are mediated by steroid interactions with two distinct sites.
6b. Benz[f]indenes
Recently we also analyzed the actions of benz[f]indenes (Scaglione et al., 2006). These are tricyclic steroid analogues in which the rings are aligned in a linear fused carbocyclic structure (Fig. 8). Thus, these molecules are unique and differ from the benz[e]indenes that we previously characterized. We found that certain trans-trans benz[f]indenes are GABA potentiators, but are less active than their steroid and benz[e]indene counterparts. In contrast, non-planar cis-trans compounds have little activity at GABAA receptors. Interestingly, the effects of the trans-trans compounds were not blocked by 17PA, suggesting that their effects may more closely resemble effects of 5β-steroids (Scaglione et al., 2006).
Two important conclusions may be drawn from structure-activity relationships for benz[f]indenes. First, the geometrical arrangement of the steroid rings is clearly an important part of the pharmacophore for steroid action at GABAA receptors. For benz[f]indenes in which all three rings are in the same plane (trans-trans ring fusions), these compounds are less potent than steroids or benz[e]indenes as positive modulators of GABAA receptors. For benz[f]indenes in which the first six-membered ring is orthogonal to the other two rings (cis-trans ring fusions), the compounds have little if any activity at GABAA receptors. The cis-trans arrangement of rings in benz[f]indenes is likely not tolerated for steric reasons. Several factors may contribute to the diminished actions of the trans-trans benz[f]indenes at GABAA receptors. These include: 1) the flexibility of the hydroxyethyl chain; 2) the location of the first six-membered ring in a forbidden region of space (i.e., space already occupied by the receptor amino acids); and 3) the loss of a favorable hydrophobic contact normally present for the steroid B-ring in its interactions with GABAA receptors. The last possible explanation was suggested by other studies that explored the effect of methyl group substitution at C6 and C7 of the steroid B-ring (Zeng et al., 2005).
A second conclusion from the benz[f]indene structure-activity study involved antagonism results obtained with 17PA. The previously discussed benz[e]indenes were effectively antagonized by 17PA whereas the trans-trans benz[f]indenes were not. One explanation is that the benz[e]indenes bind to sites for 5α-reduced steroids which are effectively antagonized by 17PA and the trans-trans benz[f]indenes bind to sites for 5β-reduced steroids, sites which are not effectively antagonized by 17PA (see above). Alternatively, the trans-trans benz[f]indenes may bind to a unique site because of their altered shape. More experimentation will be needed to distinguish between these possibilities.
6c. Insights from cyclohexylsteroid analogues
The synthesis of a series of 5α- and 5β-reduced 17,18-cyclohexylsteroids (Fig. 9) was conducted with the rationale of determining, using conformationally constrained analogues, possible positions for the C17 hydrogen-bond acceptor, a known critical component of the steroid pharmacophore. These analogues contain carbons C17 and C18 (according to the IUPAC rules for steroid nomenclature C18 has to be renumbered as C24 in these cyclosteroids) in an additional six-membered ring. A hydrogen-bond-accepting group was systematically placed at each of the four carbon atoms on the new ring. The synthesis and pharmacological evaluation of these analogues with a ketone functional group at each of the four positions on the newly formed bridge has been published (Jiang et al., 2003).
Figure 9. Structures of indicated analogues.

These analogues provided an opportunity to search, in a spatially restricted fashion, for alternate locations of the D-ring hydrogen-bond-accepting group normally located at steroid position C17. When a carbonyl group is present at each position, we found that the most active compounds in both the 5α- and 5β-reduced series of analogues had the carbonyl group at C24. In this position, the carbonyl group is oriented over the steroid C-ring pointing in a direction nearly opposite to the direction of the carbonyl group found in 3α5αP and 3α5βP. This finding was completely unexpected. Moreover, we found that when a double bond was placed in the ring in conjugation with the C24 carbonyl group, these new enone analogues had activities comparable to those of 3α5αP and 3α5βP. We hypothesize that these results may indicate that there are multiple hydrogen-bond-donor groups in the receptor arranged in a band over the steroid D-ring that have the potential to interact with hydrogen-bond-acceptor groups present in different locations on the steroid. Alternatively, it may be that the same receptor hydrogen-bond donor interacts through an intervening water molecule with steroid hydrogen-bond-acceptor groups positioned in different locations. Additional studies with mutated GABAA receptors that are insensitive to 3α5αP and 3α5βP potentiation should provide a better understanding of the molecular interactions of the 17,18-cyclohexylsteroids with the receptor.
An interesting auxiliary result from studies of cyclosteroids is that all of the above conclusions apply equally to 5α- and 5β-reduced steroid analogues. In other words, as constrained placement of the hydrogen-bond accepting group is systematically altered, the same rank order of activity at GABAA receptors is obtained in both the 5α-reduced and the 5β reduced series of analogues (Jiang et al., 2003). These results suggest that binding sites for 5α- and 5β- reduced steroids are quite similar, a conclusion somewhat at odds with the differences in enantiomer behavior described above.
6d. Insights from C6 and C7 substitutions
These results again raise the puzzle of similarity of steroid structure-activity relationships in both planar 5α-reduced potentiating steroids and nonplanar 5β-reduced steroids. Systematic positioning of the hydrogen-bond acceptor using the cyclosteroid strategy affected rank-order of GABAergic effects similarly in the 5α-reduced and 5β-reduced series of analogues. However, structural changes at the D ring are relatively distant from where the stereochemistry of the A/B ring fusion is determined at C5. To test more directly the hypothesis that different steroid binding sites accommodate 5α- vs. 5β-reduced steroids, we examined a series of C6 and C7 methyl substituted analogues of 3α5αP and 3α5βP (Fig. 9). We chose C6 and C7 because these positions are relatively close to the A/B ring fusion, the distinguishing part of 5α- and 5β-reduced steroids. Several previous reports had indicated that this area is sensitive to modification, with both active and inactive compounds obtained (Han et al., 1996; Nicoletti et al., 2000; Veleiro et al., 2003). For instance, 6-oxa analogues of 3α5αP have diminished activity at GABAA receptors (Nicoletti et al., 2000). Our hypothesis was that if a common binding site accommodates 5α- and 5β-reduced steroids, then methyl substitutions in both 5α- and 5β-reduced steroids should produce similar changes in the structure-activity relationships. If different binding sites accommodate the two classes, then methyl substitutions in the 5α-reduced steroids may produce a different functional effect than the same substitutions in the 5β-reduced steroid.
Overall, when assessed with the TBPS binding assay or with electrophysiological recordings in oocytes, 6β-methyl groups resulted in high activity, and those containing a 6α-, 7β-, or 7α-methyl group had diminished activity. As with the cyclosteroids, the structure-activity relationship for analogues in each series of C5 epimers was very similar. The results, then, fail to support the existence of separate binding sites for 5α-reduced and 5β-reduced steroids. In summary, despite considerable synthetic effort including efforts with cyclosteroid analogues and hydrophobic substitutions at C6 and C7, structural attributes that might contribute in a decisive way to differentiating sites for 5α- and 5β-reduced potentiating steroids have not been identified. Clearly, portions of the receptor in contact with the steroid B, C, and D rings are similar for the sites recognizing 5α- and 5β epimers. Structural modifications of the steroid A ring might provide new information regarding whether or not the steroid A ring can be accommodated in two different locations in the same binding pocket.
6e. Insights from photoaffinity labels
Photoaffinity labeling has successfully been used to locate the benzodiazepine (Davies et al., 1996; Duncalfe et al., 1996; Sawyer et al., 2002; Wieland et al., 1992) and muscimol (Smith & Olsen, 1994) binding sites on the GABAA receptor, and sites for etomidate (Husain et al., 2006; Ziebell et al., 2004) and octanol (Pratt et al., 2000) on the Torpedo nicotinic acetylcholine receptor. In principle photoaffinity labeling techniques should provide a comprehensive search of all possible protein targets and should not be confounded by either multiple regions of protein contributing to a binding site or by the existence of multiple binding sites. Photoaffinity labeling reagents also provide an indirect test to distinguish whether mutations that affect a ligand's action in functional assays do so by preventing ligand binding or by interfering with signal transduction; i.e. if a mutation causes loss of photo-labeling, this supports an altered binding site. The success of the approach is critically dependent on designing photolabeling reagents in which the photolabeling moiety is appropriately positioned on the drug analogue (i.e. facing a protein wall of the binding site) and can efficiently label any nearby amino acid.
We have synthesized (3α,5β)-6-azi-3-hydroxypregnan-20-one (6-AziP; Fig. 9) to try to identify neuroactive steroid binding sites in rat brain membranes (Darbandi-Tonkabon et al., 2003). 6-AziP retains biological activity in TBPS binding assays and electrophysiological screens for GABA potentiation. It also produces time and concentration-dependent photolabeling of protein bands at 35 kDa and 60 kDa. We found the 35 kDa band to represent Voltage-Dependent Anion Channel-1 (VDAC-1). VDAC-1 was previously shown to co-precipitate with GABAA receptors (Bureau et al., 1992; Darbandi-Tonkabon et al., 2003), but the protein is currently thought to represent a mitochondrial membrane protein (Colombini et al., 1996). To test relevance of VDAC-1 to steroid modulation of GABA receptors and to anesthesia, we analyzed VDAC-deficient animals and cells. These studies show that the binding of neuroactive steroid to VDAC protein is not related to GABA modulation or to anesthetic effects of steroids (Darbandi-Tonkabon et al., 2004), but the studies raise the general point that steroid modulation need not occur through direct interaction with the GABAA receptor. In principle steroid actions could instead result from binding to an accessory protein.
Based on the data showing unaltered steroid modulation of GABAA receptors in VDAC-deficient cells, we reasoned that the most likely binding site for neuroactive steroids is the GABAA receptor itself. We considered reasons that 6-AziP may not have labeled one or more subunits of the GABAA receptor. Irradiation of a diazirine can, via elimination of molecular nitrogen, generate a carbene, which then participates in either an internal rearrangement reaction (intramolecular) or a bimolecular insertion reaction. If an intramolecular reaction is energetically favored, the carbene that is generated will be unavailable for bimolecular insertion. Carbenes can insert into any C-H bond and are thus not selective for specific amino acids. Irradiation can also convert a diazirine to a diazo intermediate that can then form a carbonium ion; the carbonium ion acts as an electrophile and can selectively label charged amino acid side chains (Brunner et al., 1980). Recent work using azi-etomidate (Ziebell et al., 2004) and azi-octanol (Pratt et al., 2000) as photolabeling reagents indicates that the labeling mechanism for these aliphatic diazirines is through the diazo intermediate. The inability of 6-AziP to label the neuroactive steroid binding sites responsible for GABAA receptor modulation could be explained if the carbene generated by photolysis preferentially participates in intramolecular reactions.
To test this possibility we examined the ability of 6-AziP to form a unique adduct with cyclohexane (a solvent in which the only bond available for attack is a C-H) or with ethanol (a solvent which can react as a nucleophile). The photolysis products of [3H]6-AziP generated in ethanol are indistinguishable from those generated in cyclohexane, indicating that the products result from intramolecular rearrangement rather than bimolecular solvent reaction (unpublished results). This conclusion was also confirmed by photolysis studies of 20-AziP in which the major photolysis products were identified by 13C- and 1H-NMR spectroscopy as a mixture of the cis and trans olefins resulting from carbene insertion at C17 of 20-AziP. Based on these findings we conclude that: (1) The major photolysis products of 6-AziP and 20-AziP result from intramolecular rearrangement; (2) Photolysis of 6-AziP probably also generates a diazo intermediate (as a minor product) that is responsible for the photolabeling of VDAC and tubulin; (3) 6-AziP reversibly modulates GABAA receptor function but does not photolabel the protein either because the neuroactive steroid binding site(s) do not contain reactive amino acids or because the inefficiency of the diazo pathway only allows detectable labeling of abundant proteins. Our research program is the process of developing new photolabeling reagents that do not suffer from the limitations of 6-AziP.
In the process of developing new tools for labeling and imaging neurosteroid analogues, we recently discovered that several of our novel fluorescent neurosteroid analogues can be photoactivated on live cells. These compounds are largely inert at GABAA receptors until cells exposed to the analogues are illuminated with fluorescence excitation wavelengths (Eisenman et al., 2007). This photo-potentiation of GABA-gated currents is similar in many respects to natural steroid potentiation. However, photopotentiaion is very long-lived and resistant to cyclodextrin removal. This suggests the possibility of a covalent modification of the receptor, although the photochemistry and precise relationship of this phenomenon to steroid potentiation are still unclear. Further characterization may reveal that these compounds are novel photoaffinity reagents. At the very least, they represent interesting photolabile tools for manipulating GABAA receptor function.
7. Summary and future prospects
Our efforts have centered on understanding anesthetic steroid actions at GABAA receptors using a multifaceted approach anchored by medicinal chemistry. This approach has yielded data consistent with the likelihood of multiple binding sites on GABAA receptors. Evidence for this comes from the finding that some steroids exhibit different concentration dependences for distinguishable steroid effects on GABAA channel function. More evidence comes from the differences in enantioselectivity of various steroid analogues. Enantiomers remain the cleanest means of distinguishing contributions of membrane effects from “site” effects. Because recent evidence favors membrane diffusion as a route to the putative receptor site, interpreting affinity differences among other analogue pairs is complicated. Selectivity of the steroid antagonist 17PA is also consistent with multiple steroid sites and/or actions. On the other hand, some data are still hard to reconcile with multiple sites for steroid modulation of GABAA receptors. Structure-activity relationships, for instance, that run strikingly parallel for 5α-reduced and 5β-reduced steroids suggest that despite heterogeneity, binding pockets may share similar recognition sites for steroid B, C, and D rings. There is optimism that the recent identification of steroid binding sites through mutagenesis and future photoaffinity labeling will help clarify the observed complexity. Finally, although complexity of steroid actions poses challenges experimentally, there is hope that this same complexity, once it is better understood, can be exploited for developing specific analogues directed toward specific GABAA receptor subunits and selective behavioral effects.
Acknowledgments
This work was supported by National Institutes of Health Grants GM47969 (J.H.S., C.F.Z., D.F.C.), AA14707 (G.A.), AA12951 (C.F.Z.), AA12952, NS54174, and MH78823 (S.M.) and by grants from the American Health Assistance Foundation (S.M.) and the Bantly Foundation (C.F.Z.). J.H.S. is the Russell and Mary Shelden Professor of Anesthesiology. We thank the colleagues from our Program who have participated in the studies cited herein.
Abbreviations
- ACN
(3α,5α,17β)-3-hydroxyandrostane-17-carbonitrile
- B285
(3α,5β,17β)-3-hydroxy-18-norandrostane-17-carbonitrile
- ECN
(3β,5α,17β)-17-hydroxyestrane-3-carbonitrile
- GABA
γ-aminobutyric acid
- 3α5αP
(3α,5α)-3-hydroxypregnan-20-one
- 3α5βP
(3α,5β)-3-hydroxypregnan-20-one
- 17PA
(3α5α)-17-phenylandrost-16-en-3-ol
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adkins CE, Pillai GV, Kerby J, Bonnert TP, Haldon C, McKernan RM, Gonzalez JE, Oades K, Whiting PJ, Simpson PB. α4β3δ GABAA receptors characterized by fluorescence resonance energy transfer-derived measurements of membrane potential. J Biol Chem. 2001;276:38934–38939. doi: 10.1074/jbc.M104318200. [DOI] [PubMed] [Google Scholar]
- Agarwal K, Bali A, Gupta CM. Effect of phospholipid structure on stability and survival times of liposomes in circulation. Biochim Biophys Acta. 1986a;883:468–475. doi: 10.1016/0304-4165(86)90286-2. [DOI] [PubMed] [Google Scholar]
- Agarwal K, Bali A, Gupta CM. Influence of the phospholipid structure on the stability of liposomes in serum. Biochim Biophys Acta. 1986b;856:36–40. doi: 10.1016/0005-2736(86)90006-4. [DOI] [PubMed] [Google Scholar]
- Agis-Balboa RC, Pinna G, Zhubi A, Maloku E, Veldic M, Costa E, Guidotti A. Characterization of brain neurons that express enzymes mediating neurosteroid biosynthesis. Proc Natl Acad Sci U S A. 2006;103:14602–14607. doi: 10.1073/pnas.0606544103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akabas MH. GABAA receptor structure-function studies: a reexamination in light of new acetylcholine receptor structures. Int Rev Neurobiol. 2004;62:1–43. doi: 10.1016/S0074-7742(04)62001-0. [DOI] [PubMed] [Google Scholar]
- Akk G, Bracamontes J, Steinbach JH. Pregnenolone sulfate block of GABAA receptors: mechanism and involvement of a residue in the M2 region of the α subunit. J Physiol. 2001;532:673–684. doi: 10.1111/j.1469-7793.2001.0673e.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akk G, Bracamontes JR, Covey DF, Evers A, Dao T, Steinbach JH. Neuroactive steroids have multiple actions to potentiate GABAA receptors. J Physiol. 2004;558:59–74. doi: 10.1113/jphysiol.2004.066571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akk G, Li P, Manion BD, Evers AS, Steinbach JH. Ethanol modulates the interaction of the endogenous neurosteroid allopregnanolone with the α1β2γ2L GABAA receptor. Mol Pharmacol. 2007;71:461–472. doi: 10.1124/mol.106.029942. [DOI] [PubMed] [Google Scholar]
- Akk G, Shu HJ, Wang C, Steinbach JH, Zorumski CF, Covey DF, Mennerick S. Neurosteroid access to the GABAA receptor. J Neurosci. 2005;25:11605–11613. doi: 10.1523/JNEUROSCI.4173-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akk G, Steinbach JH. Low doses of ethanol and a neuroactive steroid positively interact to modulate rat GABAA receptor function. J Physiol. 2003;546:641–646. doi: 10.1113/jphysiol.2002.032300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alakoskela JM, Covey DF, Kinnunen PK. Lack of enantiomeric specificity in the effects of anesthetic steroids on lipid bilayers. Biochim Biophys Acta. 2006 doi: 10.1016/j.bbamem.2006.07.011. [DOI] [PubMed] [Google Scholar]
- Andrews PR, Mark LC. Structural specificity of barbiturates and related drugs. Anesthesiology. 1982;57:314–320. doi: 10.1097/00000542-198210000-00014. [DOI] [PubMed] [Google Scholar]
- Arnett EM, Gold JM. Chiral aggregation phenomena. 4. A search for stereospecific interactions between highly purified enantiomeric and racemic dipalmitoyl phosphatidylcholines and other chiral surfactants in monolayers, vesicles, and gels. J Am Chem Soc. 1982;104:636–639. [Google Scholar]
- Barbaccia ML, Affricano D, Trabucchi M, Purdy RH, Colombo G, Agabio R, Gessa GL. Ethanol markedly increases “GABAergic” neurosteroids in alcohol-preferring rats. Eur J Pharmacol. 1999;384:R1–2. doi: 10.1016/s0014-2999(99)00678-0. [DOI] [PubMed] [Google Scholar]
- Barbaccia ML, Roscetti G, Trabucchi M, Purdy RH, Mostallino MC, Concas A, Biggio G. The effects of inhibitors of GABAergic transmission and stress on brain and plasma allopregnanolone concentrations. Br J Pharmacol. 1997;120:1582–1588. doi: 10.1038/sj.bjp.0701046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker JL, Harrison NL, Lange GD, Owen DG. Potentiation of γ-aminobutyric-acid-activated chloride conductance by a steroid anaesthetic in cultured rat spinal neurones. J Physiol. 1987;386:485–501. doi: 10.1113/jphysiol.1987.sp016547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belelli D, Casula A, Ling A, Lambert JJ. The influence of subunit composition on the interaction of neurosteroids with GABAA receptors. Neuropharmacology. 2002;43:651–661. doi: 10.1016/s0028-3908(02)00172-7. [DOI] [PubMed] [Google Scholar]
- Belelli D, Herd MB. The contraceptive agent Provera enhances GABAA receptor-mediated inhibitory neurotransmission in the rat hippocampus: evidence for endogenous neurosteroids? J Neurosci. 2003;23:10013–10020. doi: 10.1523/JNEUROSCI.23-31-10013.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belelli D, Herd MB, Mitchell EA, Peden DR, Vardy AW, Gentet L, Lambert JJ. Neuroactive steroids and inhibitory neurotransmission: mechanisms of action and physiological relevance. Neuroscience. 2006;138:821–829. doi: 10.1016/j.neuroscience.2005.07.021. [DOI] [PubMed] [Google Scholar]
- Belelli D, Lambert JJ. Neurosteroids: endogenous regulators of the GABAA receptor. Nat Rev Neurosci. 2005;6:565–575. doi: 10.1038/nrn1703. [DOI] [PubMed] [Google Scholar]
- Belelli D, Peden DR, Rosahl TW, Wafford KA, Lambert JJ. Extrasynaptic GABAA receptors of thalamocortical neurons: a molecular target for hypnotics. J Neurosci. 2005;25:11513–11520. doi: 10.1523/JNEUROSCI.2679-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belelli D, Pistis M, Peters JA, Lambert JJ. General anaesthetic action at transmitter-gated inhibitory amino acid receptors. Trends Pharmacol Sci. 1999a;20:496–502. doi: 10.1016/s0165-6147(99)01405-4. [DOI] [PubMed] [Google Scholar]
- Belelli D, Pistis M, Peters JA, Lambert JJ. The interaction of general anaesthetics and neurosteroids with GABAA and glycine receptors. Neurochem Int. 1999b;34:447–452. doi: 10.1016/s0197-0186(99)00037-6. [DOI] [PubMed] [Google Scholar]
- Biagini G, Baldelli E, Longo D, Pradelli L, Zini I, Rogawski MA, Avoli M. Endogenous neurosteroids modulate epileptogenesis in a model of temporal lobe epilepsy. Exp Neurol. 2006;201:519–524. doi: 10.1016/j.expneurol.2006.04.029. [DOI] [PubMed] [Google Scholar]
- Bianchi MT, Macdonald RL. Neurosteroids shift partial agonist activation of GABAA receptor channels from low- to high-efficacy gating patterns. J Neurosci. 2003;23:10934–10943. doi: 10.1523/JNEUROSCI.23-34-10934.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieda MC, MacIver MB. Major role for tonic GABAA conductances in anesthetic suppression of intrinsic neuronal excitability. J Neurophysiol. 2004;92:1658–1667. doi: 10.1152/jn.00223.2004. [DOI] [PubMed] [Google Scholar]
- Bocquet N, Prado de Carvalho L, Cartaud J, Neyton J, Le Poupon C, Taly A, Grutter T, Changeux JP, Corringer PJ. A prokaryotic proton-gated ion channel from the nicotinic acetylcholine receptor family. Nature. 2007;445:116–119. doi: 10.1038/nature05371. [DOI] [PubMed] [Google Scholar]
- Borghese CM, Storustovu S, Ebert B, Herd MB, Belelli D, Lambert JJ, Marshall G, Wafford KA, Harris RA. The δ subunit of γ-aminobutyric acid type A receptors does not confer sensitivity to low concentrations of ethanol. J Pharmacol Exp Ther. 2006;316:1360–1368. doi: 10.1124/jpet.105.092452. [DOI] [PubMed] [Google Scholar]
- Brickley SG, Revilla V, Cull-Candy SG, Wisden W, Farrant M. Adaptive regulation of neuronal excitability by a voltage-independent potassium conductance. Nature. 2001;409:88–92. doi: 10.1038/35051086. [DOI] [PubMed] [Google Scholar]
- Brown N, Kerby J, Bonnert TP, Whiting PJ, Wafford KA. Pharmacological characterization of a novel cell line expressing human α4β3δ GABAA receptors. Br J Pharmacol. 2002;136:965–974. doi: 10.1038/sj.bjp.0704795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner J, Senn H, Richards FM. 3-Trifluoromethyl-3-phenyldiazirine. A new carbene generating group for photolabeling reagents. J Biol Chem. 1980;255:3313–3318. [PubMed] [Google Scholar]
- Brussaard AB, Koksma JJ. Conditional regulation of neurosteroid sensitivity of GABAA receptors. Ann N Y Acad Sci. 2003;1007:29–36. doi: 10.1196/annals.1286.003. [DOI] [PubMed] [Google Scholar]
- Bureau MH, Khrestchatisky M, Heeren MA, Zambrowicz EB, Kim H, Grisar TM, Colombini M, Tobin AJ, Olsen RW. Isolation and cloning of a voltage-dependent anion channel-like Mr 36,000 polypeptide from mammalian brain. J Biol Chem. 1992;267:8679–8684. [PubMed] [Google Scholar]
- Callachan H, Cottrell GA, Hather NY, Lambert JJ, Nooney JM, Peters JA. Modulation of the GABAA receptor by progesterone metabolites. Proc R Soc Lond B Biol Sci. 1987;231:359–369. doi: 10.1098/rspb.1987.0049. [DOI] [PubMed] [Google Scholar]
- Cantor RS. The lateral pressure profile in membranes: a physical mechanism of general anesthesia. Toxicol Lett. 1998;100-101:451–458. doi: 10.1016/s0378-4274(98)00220-3. [DOI] [PubMed] [Google Scholar]
- Caraiscos VB, Elliott EM, You-Ten KE, Cheng VY, Belelli D, Newell JG, Jackson MF, Lambert JJ, Rosahl TW, Wafford KA, MacDonald JF, Orser BA. Tonic inhibition in mouse hippocampal CA1 pyramidal neurons is mediated by α5 subunit-containing γ-aminobutyric acid type A receptors. Proc Natl Acad Sci U S A. 2004a;101:3662–3667. doi: 10.1073/pnas.0307231101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caraiscos VB, Newell JG, You-Ten KE, Elliott EM, Rosahl TW, Wafford KA, MacDonald JF, Orser BA. Selective enhancement of tonic GABAergic iInhibition in murine hippocampal neurons by low concentrations of the volatile anesthetic isoflurane. J Neurosci. 2004b;24:8454–8458. doi: 10.1523/JNEUROSCI.2063-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y, Wang R, Barot S, Weiss DS. Stoichiometry of a recombinant GABAA receptor. J Neurosci. 1996;16:5415–5424. doi: 10.1523/JNEUROSCI.16-17-05415.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherubini E, Gaiarsa JL, Ben-Ari Y. GABA: an excitatory transmitter in early postnatal life. Trends Neurosci. 1991;14:515–519. doi: 10.1016/0166-2236(91)90003-d. [DOI] [PubMed] [Google Scholar]
- Colombini M, Blachly-Dyson E, Forte M. VDAC, a channel in the outer mitochondrial membrane. Ion Channels. 1996;4:169–202. doi: 10.1007/978-1-4899-1775-1_5. [DOI] [PubMed] [Google Scholar]
- Cope DW, Hughes SW, Crunelli V. GABAA receptor-mediated tonic inhibition in thalamic neurons. J Neurosci. 2005;25:11553–11563. doi: 10.1523/JNEUROSCI.3362-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottrell GA, Lambert JJ, Peters JA. Modulation of GABAA receptor activity by alphaxalone. Br J Pharmacol. 1987;90:491–500. doi: 10.1111/j.1476-5381.1987.tb11198.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covey DF, Evers AS, Mennerick S, Zorumski CF, Purdy RH. Recent developments in structure-activity relationships for steroid modulators of GABAA receptors. Brain Res Brain Res Rev. 2001;37:91–97. doi: 10.1016/s0165-0173(01)00126-6. [DOI] [PubMed] [Google Scholar]
- Covey DF, Hu Y, Bouley MG, Holland KD, Rodgers-Neame NT, Isenberg KE, Zorumski CF. Modulation of GABAA receptor function by benz[e]indenes and phenanthrenes. J Med Chem. 1993;36:627–630. doi: 10.1021/jm00057a012. [DOI] [PubMed] [Google Scholar]
- Covey DF, Nathan D, Kalkbrenner M, Nilsson KR, Hu Y, Zorumski CF, Evers AS. Enantioselectivity of pregnanolone-induced γ-aminobutyric acidA receptor modulation and anesthesia. J Pharmacol Exp Ther. 2000;293:1009–1016. [PubMed] [Google Scholar]
- Curtis L, Buisson B, Bertrand S, Bertrand D. Potentiation of human α4β2 neuronal nicotinic acetylcholine receptor by estradiol. Mol Pharmacol. 2002;61:127–135. doi: 10.1124/mol.61.1.127. [DOI] [PubMed] [Google Scholar]
- Darbandi-Tonkabon R, Hastings WR, Zeng CM, Akk G, Manion BD, Bracamontes JR, Steinbach JH, Mennerick SJ, Covey DF, Evers AS. Photoaffinity labeling with a neuroactive steroid analogue. 6-azipregnanolone labels voltage-dependent anion channel-1 in rat brain. J Biol Chem. 2003;278:13196–13206. doi: 10.1074/jbc.M213168200. [DOI] [PubMed] [Google Scholar]
- Darbandi-Tonkabon R, Manion BD, Hastings WR, Craigen WJ, Akk G, Bracamontes JR, He Y, Sheiko TV, Steinbach JH, Mennerick SJ, Covey DF, Evers AS. Neuroactive steroid interactions with voltage-dependent anion channels: lack of relationship to GABAA receptor modulation and anesthesia. J Pharmacol Exp Ther. 2004;308:502–511. doi: 10.1124/jpet.103.058123. [DOI] [PubMed] [Google Scholar]
- Davies M, Martin IL, Bateson AN, Hadingham KL, Whiting PJ, Dunn SM. Identification of domains in human recombinant GABAA receptors that are photoaffinity labelled by [3H]flunitrazepam and [3H]Ro15-4513. Neuropharmacology. 1996;35:1199–1208. doi: 10.1016/s0028-3908(96)00085-8. [DOI] [PubMed] [Google Scholar]
- Downes H, Courogen PM. Contrasting effects of anesthetics in tadpole bioassays. J Pharmacol Exp Ther. 1996;278:284–296. [PubMed] [Google Scholar]
- Duncalfe LL, Carpenter MR, Smillie LB, Martin IL, Dunn SM. The major site of photoaffinity labeling of the γ-aminobutyric acid type A receptor by [3H]flunitrazepam is histidine 102 of the α subunit. J Biol Chem. 1996;271:9209–9214. doi: 10.1074/jbc.271.16.9209. [DOI] [PubMed] [Google Scholar]
- Eisenman LN, He Y, Fields C, Zorumski CF, Mennerick S. Activation-dependent properties of pregnenolone sulfate inhibition of GABAA receptor-mediated current. J Physiol. 2003;550:679–691. doi: 10.1113/jphysiol.2003.043810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenman LN, Shu HJ, Akk G, Wang C, Manion B, Kress G, Evers AS, Steinbach JH, Covey DF, Zorumski CF, Mennerick S. Anticonvulsant and anesthetic effects of a fluorescent neurosteroid analogue activated by visible light. Nature Neurosci. 2007 doi: 10.1038/nn1862. In Press. [DOI] [PubMed] [Google Scholar]
- Ernst M, Brauchart D, Boresch S, Sieghart W. Comparative modeling of GABAA receptors: limits, insights, future developments. Neuroscience. 2003;119:933–943. doi: 10.1016/s0306-4522(03)00288-4. [DOI] [PubMed] [Google Scholar]
- Essrich C, Lorez M, Benson JA, Fritschy JM, Luscher B. Postsynaptic clustering of major GABAA receptor subtypes requires the γ2 subunit and gephyrin. Nat Neurosci. 1998;1:563–571. doi: 10.1038/2798. [DOI] [PubMed] [Google Scholar]
- Farrant M, Nusser Z. Variations on an inhibitory theme: phasic and tonic activation of GABAA receptors. Nat Rev Neurosci. 2005;6:215–229. doi: 10.1038/nrn1625. [DOI] [PubMed] [Google Scholar]
- Franks NP, Lieb WR. Stereospecific effects of inhalational general anesthetic optical isomers on nerve ion channels. Science. 1991;254:427–430. doi: 10.1126/science.1925602. [DOI] [PubMed] [Google Scholar]
- Fritschy JM, Mohler H. GABAA-receptor heterogeneity in the adult rat brain: differential regional and cellular distribution of seven major subunits. J Comp Neurol. 1995;359:154–194. doi: 10.1002/cne.903590111. [DOI] [PubMed] [Google Scholar]
- Gasior M, Carter RB, Witkin JM. Neuroactive steroids: potential therapeutic use in neurological and psychiatric disorders. Trends Pharmacol Sci. 1999;20:107–112. doi: 10.1016/s0165-6147(99)01318-8. [DOI] [PubMed] [Google Scholar]
- Gee KW, Bolger MB, Brinton RE, Coirini H, McEwen BS. Steroid modulation of the chloride ionophore in rat brain: structure-activity requirements, regional dependence and mechanism of action. J Pharmacol Exp Ther. 1988;246:803–812. [PubMed] [Google Scholar]
- Ghosh D, Lyman RL, Tinoco J. Behavior of specific natural lecithins and cholesterol at the air water-interface. Chem Phys Lipids. 1971;7:173–184. doi: 10.1016/0009-3084(71)90030-2. [DOI] [PubMed] [Google Scholar]
- Glykys J, Peng Z, Chandra D, Homanics GE, Houser CR, Mody I. A new naturally occurring GABA(A) receptor subunit partnership with high sensitivity to ethanol. Nat Neurosci. 2007;10:40–48. doi: 10.1038/nn1813. [DOI] [PubMed] [Google Scholar]
- Goutman JD, Calvo DJ. Studies on the mechanisms of action of picrotoxin, quercetin and pregnanolone at the GABA rho 1 receptor. Br J Pharmacol. 2004;141:717–727. doi: 10.1038/sj.bjp.0705657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin LD, Mellon SH. Selective serotonin reuptake inhibitors directly alter activity of neurosteroidogenic enzymes. Proc Natl Acad Sci U S A. 1999;96:13512–13517. doi: 10.1073/pnas.96.23.13512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossley R. Chirality and the Biological Activity of Drugs. New York: CRC Press; 1995. [Google Scholar]
- Guyer W, Bloch K. Phosphatidylcholine and cholesterol interactions in model membranes. Chem Phys Lipids. 1983;33:313–322. doi: 10.1016/0009-3084(83)90025-7. [DOI] [PubMed] [Google Scholar]
- Hall AC, Lieb WR, Franks NP. Stereoselective and non-stereoselective actions of isoflurane on the GABAA receptor. Br J Pharmacol. 1994;112:906–910. doi: 10.1111/j.1476-5381.1994.tb13166.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han M, Hu Y, Zorumski CF, Covey DF. Neurosteroid analogues. 3. The synthesis and electrophysiological evaluation of benz[e]indene congeners of neuroactive steroids having the 5β-configuration. J Med Chem. 1995;38:4548–4556. doi: 10.1021/jm00022a021. [DOI] [PubMed] [Google Scholar]
- Han M, Zorumski CF, Covey DF. Neurosteroid analogues. 4. The effect of methyl substitution at the C-5 and C-10 positions of neurosteroids on electrophysiological activity at GABAA receptors. J Med Chem. 1996;39:4218–4232. doi: 10.1021/jm960304p. [DOI] [PubMed] [Google Scholar]
- Hanchar HJ, Chutsrinopkun P, Meera P, Supavilai P, Sieghart W, Wallner M, Olsen RW. Ethanol potently and competitively inhibits binding of the alcohol antagonist Ro15-4513 to α4/6β3δ GABAA receptors. Proc Natl Acad Sci U S A. 2006;103:8546–8551. doi: 10.1073/pnas.0509903103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison NL, Simmonds MA. Modulation of the GABA receptor complex by a steroid anaesthetic. Brain Res. 1984;323:287–292. doi: 10.1016/0006-8993(84)90299-3. [DOI] [PubMed] [Google Scholar]
- Harrison NL, Vicini S, Barker JL. A steroid anesthetic prolongs inhibitory postsynaptic currents in cultured rat hippocampal neurons. J Neurosci. 1987;7:604–609. doi: 10.1523/JNEUROSCI.07-02-00604.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkinson JE, Kimbrough CL, McCauley LD, Bolger MB, Lan NC, Gee KW. The neuroactive steroid 3α-hydroxy-5β-pregnan-20-one is a two-component modulator of ligand binding to the GABAA receptor. Eur J Pharmacol. 1994;269:157–163. doi: 10.1016/0922-4106(94)90082-5. [DOI] [PubMed] [Google Scholar]
- Hermetter A, Paltauf F. Interaction of enantiomeric alkylacyl and diacylglycero-phosphocholines with cholesterol in bilayer membranes. Chem Phys Lipids. 1982;31:283–289. doi: 10.1016/0009-3084(82)90063-9. [DOI] [PubMed] [Google Scholar]
- Hille B. Local anesthetics: hydrophilic and hydrophobic pathways for the drugreceptor reaction. J Gen Physiol. 1977;69:497–515. doi: 10.1085/jgp.69.4.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosie AM, Wilkins ME, da Silva HMA, Smart TG. Endogenous neurosteroids regulate GABAA receptors via two discrete transmembrane sites. Nature. 2006;444:486–489. doi: 10.1038/nature05324. [DOI] [PubMed] [Google Scholar]
- Hu Y, Wittmer LL, Kalkbrenner M, Evers AS, Zorumski CF, Covey DF. Neurosteroid analogues. Part 5. Enantiomers of neuroactive steroids and benz[e]indenes: total synthesis, electrophysiological effects on GABAA receptor function and anesthetic actions in tadpoles. J Chem Soc, Perkin Trans. 1997;1:3665–3671. [Google Scholar]
- Hu Y, Zorumski CF, Covey DF. Neurosteroid analogues: structure-activity studies of benz[e]indene modulators of GABAA receptor function. 1. The effect of 6-methyl substitution on the electrophysiological activity of 7-substituted benz[e]indene-3-carbonitriles. J Med Chem. 1993;36:3956–3967. doi: 10.1021/jm00076a025. [DOI] [PubMed] [Google Scholar]
- Husain SS, Nirthanan S, Ruesch D, Solt K, Cheng Q, Li GD, Arevalo E, Olsen RW, Raines DE, Forman SA, Cohen JB, Miller KW. Synthesis of trifluoromethylaryl diazirine and benzophenone derivatives of etomidate that are potent general anesthetics and effective photolabels for probing sites on ligand-gated ion channels. J Med Chem. 2006;49:4818–4825. doi: 10.1021/jm051207b. [DOI] [PubMed] [Google Scholar]
- Im WB, Blakeman DP, Davis JP, Ayer DE. Studies on the mechanism of interactions between anesthetic steroids and γ-aminobutyric acidA receptors. Mol Pharmacol. 1990;37:429–434. [PubMed] [Google Scholar]
- Jevtovic-Todorovic V, Todorovic SM. The role of peripheral T-type calcium channels in pain transmission. Cell Calcium. 2006;40:197–203. doi: 10.1016/j.ceca.2006.04.024. [DOI] [PubMed] [Google Scholar]
- Jiang P, Yang CX, Wang YT, Xu TL. Mechanisms of modulation of pregnanolone on glycinergic response in cultured spinal dorsal horn neurons of rat. Neuroscience. 2006;141:2041–2050. doi: 10.1016/j.neuroscience.2006.05.009. [DOI] [PubMed] [Google Scholar]
- Jiang X, Manion BD, Benz A, Rath NP, Evers AS, Zorumski CF, Mennerick S, Covey DF. Neurosteroid analogues. 9. Conformationally constrained pregnanes: structure-activity studies of 13,24-cyclo-18,21-dinorcholane analogues of the GABA modulatory and anesthetic steroids (3α,5α)- and (3α,5β)-3-hydroxypregnan-20-one. J Med Chem. 2003;46:5334–5348. doi: 10.1021/jm030302m. [DOI] [PubMed] [Google Scholar]
- Jones MV, Westbrook GL. The impact of receptor desensitization on fast synaptic transmission. Trends Neurosci. 1996;19:96–101. doi: 10.1016/s0166-2236(96)80037-3. [DOI] [PubMed] [Google Scholar]
- Kissin I, Gelman S. Components of anaesthesia. Br J Anaesth. 1988;61:237–238. doi: 10.1093/bja/61.2.237. [DOI] [PubMed] [Google Scholar]
- Krasowski MD, Jenkins A, Flood P, Kung AY, Hopfinger AJ, Harrison NL. General anesthetic potencies of a series of propofol analogs correlate with potency for potentiation of γ-aminobutyric acid (GABA) current at the GABAA receptor but not with lipid solubility. J Pharmacol Exp Ther. 2001;297:338–351. [PubMed] [Google Scholar]
- Lambert JJ, Belelli D, Harney SC, Peters JA, Frenguelli BG. Modulation of native and recombinant GABAA receptors by endogenous and synthetic neuroactive steroids. Brain Res Brain Res Rev. 2001;37:68–80. doi: 10.1016/s0165-0173(01)00124-2. [DOI] [PubMed] [Google Scholar]
- Lambert JJ, Belelli D, Peden DR, Vardy AW, Peters JA. Neurosteroid modulation of GABAA receptors. Prog Neurobiol. 2003;71:67–80. doi: 10.1016/j.pneurobio.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Lambert JJ, Peters JA, Sturgess NC, Hales TG. Steroid modulation of the GABAA receptor complex: electrophysiological studies. Ciba Found Symp. 1990;153:56–71. doi: 10.1002/9780470513989.ch4. discussion 71-82. [DOI] [PubMed] [Google Scholar]
- Lee SY, MacKinnon R. A membrane-access mechanism of ion channel inhibition by voltage sensor toxins from spider venom. Nature. 2004;430:232–235. doi: 10.1038/nature02632. [DOI] [PubMed] [Google Scholar]
- Lerma J, Herranz AS, Herreras O, Abraira V, Martin del Rio R. In vivo determination of extracellular concentration of amino acids in the rat hippocampus. A method based on brain dialysis and computerized analysis. Brain Res. 1986;384:145–155. doi: 10.1016/0006-8993(86)91230-8. [DOI] [PubMed] [Google Scholar]
- Li P, Covey DF, Steinbach JH, Akk G. Dual potentiating and inhibitory actions of a benz[e]indene neurosteroid analog on recombinant α1β2γ2 GABAA receptors. Mol Pharmacol. 2006a;69:2015–2026. doi: 10.1124/mol.106.022590. [DOI] [PubMed] [Google Scholar]
- Li W, Covey DF, Alakoskela JM, Kinnunen PK, Steinbach JH. Enantiomers of neuroactive steroids support a specific interaction with the GABAC receptor as the mechanism of steroid action. Mol Pharmacol. 2006b;69:1779–1782. doi: 10.1124/mol.106.022863. [DOI] [PubMed] [Google Scholar]
- Low K, Crestani F, Keist R, Benke D, Brunig I, Benson JA, Fritschy JM, Rulicke T, Bluethmann H, Mohler H, Rudolph U. Molecular and neuronal substrate for the selective attenuation of anxiety. Science. 2000;290:131–134. doi: 10.1126/science.290.5489.131. [DOI] [PubMed] [Google Scholar]
- Luscher B, Keller CA. Regulation of GABAA receptor trafficking, channel activity, and functional plasticity of inhibitory synapses. Pharmacol Ther. 2004;102:195–221. doi: 10.1016/j.pharmthera.2004.04.003. [DOI] [PubMed] [Google Scholar]
- Majewska MD, Harrison NL, Schwartz RD, Barker JL, Paul SM. Steroid hormone metabolites are barbiturate-like modulators of the GABA receptor. Science. 1986;232:1004–1007. doi: 10.1126/science.2422758. [DOI] [PubMed] [Google Scholar]
- Majewska MD, Mienville JM, Vicini S. Neurosteroid pregnenolone sulfate antagonizes electrophysiological responses to GABA in neurons. Neurosci Lett. 1988;90:279–284. doi: 10.1016/0304-3940(88)90202-9. [DOI] [PubMed] [Google Scholar]
- Majewska MD, Schwartz RD. Pregnenolone-sulfate: an endogenous antagonist of the γ-aminobutyric acid receptor complex in brain? Brain Res. 1987;404:355–360. doi: 10.1016/0006-8993(87)91394-1. [DOI] [PubMed] [Google Scholar]
- Maksay G, Laube B, Betz H. Subunit-specific modulation of glycine receptors by neurosteroids. Neuropharmacology. 2001;41:369–376. doi: 10.1016/s0028-3908(01)00071-5. [DOI] [PubMed] [Google Scholar]
- Maksay G, Thompson SA, Wafford KA. Allosteric modulators affect the efficacy of partial agonists for recombinant GABAA receptors. Br J Pharmacol. 2000;129:1794–1800. doi: 10.1038/sj.bjp.0703259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannock DA, McIntosh TJ, Jiang X, Covey DF, McElhaney RN. Effects of natural and enantiomeric cholesterol on the thermotropic phase behavior and structure of egg sphingomyelin bilayer membranes. Biophys J. 2003;84:1038–1046. doi: 10.1016/S0006-3495(03)74920-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto K, Pinna G, Puia G, Guidotti A, Costa E. Social isolation stress-induced aggression in mice: a model to study the pharmacology of neurosteroidogenesis. Stress. 2005;8:85–93. doi: 10.1080/10253890500159022. [DOI] [PubMed] [Google Scholar]
- McKernan RM, Whiting PJ. Which GABAA-receptor subtypes really occur in the brain? Trends Neurosci. 1996;19:139–143. doi: 10.1016/s0166-2236(96)80023-3. [DOI] [PubMed] [Google Scholar]
- Mellon SH, Griffin LD. Neurosteroids: biochemistry and clinical significance. Trends Endocrinol Metab. 2002;13:35–43. doi: 10.1016/s1043-2760(01)00503-3. [DOI] [PubMed] [Google Scholar]
- Mellon SH, Griffin LD, Compagnone NA. Biosynthesis and action of neurosteroids. Brain Res Brain Res Rev. 2001;37:3–12. doi: 10.1016/s0165-0173(01)00109-6. [DOI] [PubMed] [Google Scholar]
- Mennerick S, He Y, Jiang X, Manion B, Wang M, Shute AA, Benz A, Evers AS, Covey DF, Zorumski C. Selective antagonism of 5α-reduced neurosteroid potentiation of GABAA receptors. Mol Pharmacol. 2004;65:1191–1197. doi: 10.1124/mol.65.5.1191. [DOI] [PubMed] [Google Scholar]
- Mennerick S, Zeng CM, Benz A, Shen W, Izumi Y, Evers AS, Covey DF, Zorumski CF. Effects on γ-aminobutyric acid (GABA)A receptors of a neuroactive steroid that negatively modulates glutamate neurotransmission and augments GABA neurotransmission. Mol Pharmacol. 2001;60:732–741. [PubMed] [Google Scholar]
- Mihalek RM, Banerjee PK, Korpi ER, Quinlan JJ, Firestone LL, Mi ZP, Lagenaur C, Tretter V, Sieghart W, Anagnostaras SG, Sage JR, Fanselow MS, Guidotti A, Spigelman I, Li Z, DeLorey TM, Olsen RW, Homanics GE. Attenuated sensitivity to neuroactive steroids in γ-aminobutyrate type A receptor δ subunit knockout mice. Proc Natl Acad Sci U S A. 1999;96:12905–12910. doi: 10.1073/pnas.96.22.12905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan CJ, Buckley NJ, Garret M, Deuchars J, Deuchars SA. Evidence for inhibition mediated by coassembly of GABAA and GABAC receptor subunits in native central neurons. J Neurosci. 2004;24:7241–7250. doi: 10.1523/JNEUROSCI.1979-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mody I, Pearce RA. Diversity of inhibitory neurotransmission through GABAA receptors. Trends Neurosci. 2004;27:569–575. doi: 10.1016/j.tins.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Monaghan EP, McAuley JW, Data JL. Ganaxolone: a novel positive allosteric modulator of the GABAA receptor complex for the treatment of epilepsy. Expert Opin Investig Drugs. 1999;8:1663–1671. doi: 10.1517/13543784.8.10.1663. [DOI] [PubMed] [Google Scholar]
- Morris KD, Amin J. Insight into the mechanism of action of neuroactive steroids. Mol Pharmacol. 2004;66:56–69. doi: 10.1124/mol.66.1.56. [DOI] [PubMed] [Google Scholar]
- Morris KD, Moorefield CN, Amin J. Differential modulation of the γ-aminobutyric acid type C receptor by neuroactive steroids. Molecular Pharmacology. 1999;56:752–759. [PubMed] [Google Scholar]
- Morrow AL, Montpied P, Lingford-Hughes A, Paul SM. Chronic ethanol and pentobarbital administration in the rat: effects on GABAA receptor function and expression in brain. Alcohol. 1990;7:237–244. doi: 10.1016/0741-8329(90)90012-2. [DOI] [PubMed] [Google Scholar]
- Morrow AL, VanDoren MJ, Penland SN, Matthews DB. The role of GABAergic neuroactive steroids in ethanol action, tolerance and dependence. Brain Res Brain Res Rev. 2001;37:98–109. doi: 10.1016/s0165-0173(01)00127-8. [DOI] [PubMed] [Google Scholar]
- Murayama K, Zorumski CF, Izumi Y. Effects of neurosteroid 3α-hydroxy-5α-pregnan-20-one on ethanol-mediated paired-pulse depression of population spikes in the CA1 region of rat hippocampal slices. Neurosci Lett. 2006;394:28–32. doi: 10.1016/j.neulet.2005.09.062. [DOI] [PubMed] [Google Scholar]
- N-Wihlback A, Sundstrom-Poromaa I, Backstrom T. Action by and sensitivity to neuroactive steroids in menstrual cycle related CNS disorders. Psychopharmacology (Berl) 2006;186:388–401. doi: 10.1007/s00213-005-0185-2. [DOI] [PubMed] [Google Scholar]
- Nicoletti D, Ghini AA, Furtmuller R, Sieghart W, Dodd RH, Burton G. Synthesis and GABAA receptor activity of 6-oxa-analogs of neurosteroids. Steroids. 2000;65:349–356. doi: 10.1016/s0039-128x(00)00088-x. [DOI] [PubMed] [Google Scholar]
- Nilsson KR, Zorumski CF, Covey DF. Neurosteroid analogues. 6. The synthesis and GABAA receptor pharmacology of enantiomers of dehydroepiandrosterone sulfate, pregnenolone sulfate, and (3α,5β)-3-hydroxypregnan-20-one sulfate. J Med Chem. 1998;41:2604–2613. doi: 10.1021/jm980148h. [DOI] [PubMed] [Google Scholar]
- O'Dell LE, Alomary AA, Vallee M, Koob GF, Fitzgerald RL, Purdy RH. Ethanol-induced increases in neuroactive steroids in the rat brain and plasma are absent in adrenalectomized and gonadectomized rats. Eur J Pharmacol. 2004;484:241–247. doi: 10.1016/j.ejphar.2003.11.031. [DOI] [PubMed] [Google Scholar]
- O'Dell LE, Purdy RH, Covey DF, Richardson HN, Roberto M, Koob GF. Epipregnanolone and a novel synthetic neuroactive steroid reduce alcohol self-administration in rats. Pharmacol Biochem Behav. 2005;81:543–550. doi: 10.1016/j.pbb.2005.03.020. [DOI] [PubMed] [Google Scholar]
- Pan Y, Ripps H, Qian H. Random assembly of GABA ρ1 and ρ2 subunits in the formation of heteromeric GABAC receptors. Cell Mol Neurobiol. 2006;26:289–305. doi: 10.1007/s10571-006-9001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradiso K, Sabey K, Evers AS, Zorumski CF, Covey DF, Steinbach JH. Steroid inhibition of rat neuronal nicotinic α4β2 receptors expressed in HEK 293 cells. Mol Pharmacol. 2000;58:341–351. doi: 10.1124/mol.58.2.341. [DOI] [PubMed] [Google Scholar]
- Paradiso K, Zhang J, Steinbach JH. The C terminus of the human nicotinic α4β2 receptor forms a binding site required for potentiation by an estrogenic steroid. J Neurosci. 2001;21:6561–6568. doi: 10.1523/JNEUROSCI.21-17-06561.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park-Chung M, Malayev A, Purdy RH, Gibbs TT, Farb DH. Sulfated and unsulfated steroids modulate γ-aminobutyric acidA receptor function through distinct sites. Brain Res. 1999;830:72–87. doi: 10.1016/s0006-8993(99)01381-5. [DOI] [PubMed] [Google Scholar]
- Pathirathna S, Brimelow BC, Jagodic MM, Krishnan K, Jiang X, Zorumski CF, Mennerick S, Covey DF, Todorovic SM, Jevtovic-Todorovic V. New evidence that both T-type calcium channels and GABAA channels are responsible for the potent peripheral analgesic effects of 5α-reduced neuroactive steroids. Pain. 2005;114:429–443. doi: 10.1016/j.pain.2005.01.009. [DOI] [PubMed] [Google Scholar]
- Phillipps GH. Structure-activity relationships in steroidal anaesthetics. J Steroid Biochem. 1975;6:607–613. doi: 10.1016/0022-4731(75)90041-2. [DOI] [PubMed] [Google Scholar]
- Pinna G, Costa E, Guidotti A. Fluoxetine and norfluoxetine stereospecifically and selectively increase brain neurosteroid content at doses that are inactive on 5-HT reuptake. Psychopharmacology (Berl) 2006;186:362–372. doi: 10.1007/s00213-005-0213-2. [DOI] [PubMed] [Google Scholar]
- Pistis M, Belelli D, Peters JA, Lambert JJ. The interaction of general anaesthetics with recombinant GABAA and glycine receptors expressed in Xenopus laevis oocytes: a comparative study. Br J Pharmacol. 1997;122:1707–1719. doi: 10.1038/sj.bjp.0701563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt MB, Husain SS, Miller KW, Cohen JB. Identification of sites of incorporation in the nicotinic acetylcholine receptor of a photoactivatible general anesthetic. J Biol Chem. 2000;275:29441–29451. doi: 10.1074/jbc.M004710200. [DOI] [PubMed] [Google Scholar]
- Prince RJ, Simmonds MA. 5β-pregnan-3β-ol-20-one, a specific antagonist at the neurosteroid site of the GABAA receptor-complex. Neurosci Lett. 1992;135:273–275. doi: 10.1016/0304-3940(92)90454-f. [DOI] [PubMed] [Google Scholar]
- Prince RJ, Simmonds MA. Differential antagonism by epipregnanolone of alphaxalone and pregnanolone potentiation of [3H]flunitrazepam binding suggests more than one class of binding site for steroids at GABAA receptors. Neuropharmacology. 1993;32:59–63. doi: 10.1016/0028-3908(93)90130-u. [DOI] [PubMed] [Google Scholar]
- Pytel M, Mercik K, Mozrzymas JW. Interaction between cyclodextrin and neuronal membrane results in modulation of GABAA receptor conformational transitions. Br J Pharmacol. 2006;148:413–422. doi: 10.1038/sj.bjp.0706747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanjaneyulu R, Ticku MK. Binding characteristics and interactions of depressant drugs with [35S]t-butylbicyclophosphorothionate, a ligand that binds to the picrotoxinin site. J Neurochem. 1984;42:221–229. doi: 10.1111/j.1471-4159.1984.tb09721.x. [DOI] [PubMed] [Google Scholar]
- Reddy DS. Role of neurosteroids in catamenial epilepsy. Epilepsy Res. 2004;62:99–118. doi: 10.1016/j.eplepsyres.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Reddy DS, Kim HY, Rogawski MA. Neurosteroid withdrawal model of perimenstrual catamenial epilepsy. Epilepsia. 2001;42:328–336. doi: 10.1046/j.1528-1157.2001.10100.x. [DOI] [PubMed] [Google Scholar]
- Rodgers-Neame NT, Covey DF, Hu Y, Isenberg KE, Zorumski CF. Effects of a benz[e]indene on γ-aminobutyric acid-gated chloride currents in cultured postnatal rat hippocampal neurons. Mol Pharmacol. 1992;42:952–957. [PubMed] [Google Scholar]
- Rogawski MA. Progesterone, neurosteroids, and the hormonal basis of catamenial epilepsy. Ann Neurol. 2003;53:288–291. doi: 10.1002/ana.10534. [DOI] [PubMed] [Google Scholar]
- Rosciszewska D, Buntner B, Guz I, Zawisza L. Ovarian hormones, anticonvulsant drugs, and seizures during the menstrual cycle in women with epilepsy. J Neurol Neurosurg Psychiatry. 1986;49:47–51. doi: 10.1136/jnnp.49.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph U, Crestani F, Benke D, Brunig I, Benson JA, Fritschy JM, Martin JR, Bluethmann H, Mohler H. Benzodiazepine actions mediated by specific γ-aminobutyric acidA receptor subtypes. Nature. 1999;401:796–800. doi: 10.1038/44579. [DOI] [PubMed] [Google Scholar]
- Rudolph U, Mohler H. Analysis of GABAA receptor function and dissection of the pharmacology of benzodiazepines and general anesthetics through mouse genetics. Annu Rev Pharmacol Toxicol. 2004;44:475–498. doi: 10.1146/annurev.pharmtox.44.101802.121429. [DOI] [PubMed] [Google Scholar]
- Sanna E, Talani G, Busonero F, Pisu MG, Purdy RH, Serra M, Biggio G. Brain steroidogenesis mediates ethanol modulation of GABAA receptor activity in rat hippocampus. J Neurosci. 2004;24:6521–6530. doi: 10.1523/JNEUROSCI.0075-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer GW, Chiara DC, Olsen RW, Cohen JB. Identification of the bovine γ-aminobutyric acid type A receptor α subunit residues photolabeled by the imidazobenzodiazepine [3H]Ro15-4513. J Biol Chem. 2002;277:50036–50045. doi: 10.1074/jbc.M209281200. [DOI] [PubMed] [Google Scholar]
- Scaglione JB, Manion BD, Benz A, Taylor A, DeKoster GT, Rath NP, Evers AS, Zorumski CF, Mennerick S, Covey DF. Neurosteroid analogues. 11. Alternative ring system scaffolds: γ-aminobutyric acid receptor modulation and anesthetic actions of benz[f]indenes. J Med Chem. 2006;49:4595–4605. doi: 10.1021/jm0602920. [DOI] [PubMed] [Google Scholar]
- Semyanov A, Walker MC, Kullmann DM. GABA uptake regulates cortical excitability via cell type-specific tonic inhibition. Nat Neurosci. 2003;6:484–490. doi: 10.1038/nn1043. [DOI] [PubMed] [Google Scholar]
- Semyanov A, Walker MC, Kullmann DM, Silver RA. Tonically active GABAA receptors: modulating gain and maintaining the tone. Trends Neurosci. 2004;27:262–269. doi: 10.1016/j.tins.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Shu HJ, Eisenman LN, Jinadasa D, Covey DF, Zorumski CF, Mennerick S. Slow actions of neuroactive steroids at GABAA receptors. J Neurosci. 2004;24:6667–6675. doi: 10.1523/JNEUROSCI.1399-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieghart W, Fuchs K, Tretter V, Ebert V, Jechlinger M, Hoger H, Adamiker D. Structure and subunit composition of GABAA receptors. Neurochem Int. 1999;34:379–385. doi: 10.1016/s0197-0186(99)00045-5. [DOI] [PubMed] [Google Scholar]
- Smith GB, Olsen RW. Identification of a [3H]muscimol photoaffinity substrate in the bovine γ-aminobutyric acidA receptor α subunit. J Biol Chem. 1994;269:20380–20387. [PubMed] [Google Scholar]
- Smith SS. Pre-menstrual steroids. Cell Mol Life Sci. 2001;58:1263–1275. doi: 10.1007/PL00000938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somogyi P, Fritschy JM, Benke D, Roberts JD, Sieghart W. The γ2 subunit of the GABAA receptor is concentrated in synaptic junctions containing the α1 and β2/3 subunits in hippocampus, cerebellum and globus pallidus. Neuropharmacology. 1996;35:1425–1444. doi: 10.1016/s0028-3908(96)00086-x. [DOI] [PubMed] [Google Scholar]
- Stell BM, Brickley SG, Tang CY, Farrant M, Mody I. Neuroactive steroids reduce neuronal excitability by selectively enhancing tonic inhibition mediated by δ subunit-containing GABAA receptors. Proc Natl Acad Sci U S A. 2003;100:14439–14444. doi: 10.1073/pnas.2435457100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stell BM, Mody I. Receptors with different affinities mediate phasic and tonic GABAA conductances in hippocampal neurons. J Neurosci. 2002;22:RC223. doi: 10.1523/JNEUROSCI.22-10-j0003.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchyna TM, Tape SE, Koeppe RE, 2nd, Andersen OS, Sachs F, Gottlieb PA. Bilayer-dependent inhibition of mechanosensitive channels by neuroactive peptide enantiomers. Nature. 2004;430:235–240. doi: 10.1038/nature02743. [DOI] [PubMed] [Google Scholar]
- Szabadics J, Varga C, Molnar G, Olah S, Barzo P, Tamas G. Excitatory effect of GABAergic axo-axonic cells in cortical microcircuits. Science. 2006;311:233–235. doi: 10.1126/science.1121325. [DOI] [PubMed] [Google Scholar]
- Ticku MK, Ramanjaneyulu R. Differential interactions of GABA agonists, depressant and convulsant drugs with [35S]-t-butylbicyclophosphorothionate binding sites in cortex and cerebellum. Pharmacol Biochem Behav. 1984;21:151–158. doi: 10.1016/0091-3057(84)90145-x. [DOI] [PubMed] [Google Scholar]
- Todorovic SM, Pathirathna S, Brimelow BC, Jagodic MM, Ko SH, Jiang X, Nilsson KR, Zorumski CF, Covey DF, Jevtovic-Todorovic V. 5β-reduced neuroactive steroids are novel voltage-dependent blockers of T-type Ca2+ channels in rat sensory neurons in vitro and potent peripheral analgesics in vivo. Mol Pharmacol. 2004;66:1223–1235. doi: 10.1124/mol.104.002402. [DOI] [PubMed] [Google Scholar]
- Tretter V, Ehya N, Fuchs K, Sieghart W. Stoichiometry and assembly of a recombinant GABAA receptor subtype. J Neurosci. 1997;17:2728–2737. doi: 10.1523/JNEUROSCI.17-08-02728.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twyman RE, Macdonald RL. Neurosteroid regulation of GABAA receptor single-channel kinetic properties of mouse spinal cord neurons in culture. J Physiol. 1992;456:215–245. doi: 10.1113/jphysiol.1992.sp019334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno S, Bracamontes J, Zorumski C, Weiss DS, Steinbach JH. Bicuculline and gabazine are allosteric inhibitors of channel opening of the GABAA receptor. J Neurosci. 1997;17:625–634. doi: 10.1523/JNEUROSCI.17-02-00625.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uzunova V, Sheline Y, Davis JM, Rasmusson A, Uzunov DP, Costa E, Guidotti A. Increase in the cerebrospinal fluid content of neurosteroids in patients with unipolar major depression who are receiving fluoxetine or fluvoxamine. Proc Natl Acad Sci U S A. 1998;95:3239–3244. doi: 10.1073/pnas.95.6.3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanDoren MJ, Matthews DB, Janis GC, Grobin AC, Devaud LL, Morrow AL. Neuroactive steroid 3α-hydroxy-5α-pregnan-20-one modulates electrophysiological and behavioral actions of ethanol. J Neurosci. 2000;20:1982–1989. doi: 10.1523/JNEUROSCI.20-05-01982.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veleiro AS, Rosenstein RE, Jaliffa CO, Grilli ML, Speroni F, Burton G. Synthesis and GABAA receptor activity of a 6,19-oxido analogue of pregnanolone. Bioorg Med Chem Lett. 2003;13:343–346. doi: 10.1016/s0960-894x(02)01019-3. [DOI] [PubMed] [Google Scholar]
- Wafford KA, Whiting PJ, Kemp JA. Differences in affinity and efficacy of benzodiazepine receptor ligands at recombinant γ-aminobutyric acidA receptor subtypes. Mol Pharmacol. 1993;43:240–244. [PubMed] [Google Scholar]
- Wallner M, Hanchar HJ, Olsen RW. Ethanol enhances α4 β3 δ and α6β3δ γ-aminobutyric acid type A receptors at low concentrations known to affect humans. Proc Natl Acad Sci U S A. 2003;100:15218–15223. doi: 10.1073/pnas.2435171100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallner M, Hanchar HJ, Olsen RW. Low-dose alcohol actions on α4β3δ GABAA receptors are reversed by the behavioral alcohol antagonist Ro15-4513. Proc Natl Acad Sci U S A. 2006a;103:8540–8545. doi: 10.1073/pnas.0600194103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallner M, Hanchar HJ, Olsen RW. Low dose acute alcohol effects on GABAA receptor subtypes. Pharmacol Ther. 2006b doi: 10.1016/j.pharmthera.2006.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, He Y, Eisenman LN, Fields C, Zeng CM, Mathews J, Benz A, Fu T, Zorumski E, Steinbach JH, Covey DF, Zorumski CF, Mennerick S. 3β -hydroxypregnane steroids are pregnenolone sulfate-like GABAA receptor antagonists. J Neurosci. 2002;22:3366–3375. doi: 10.1523/JNEUROSCI.22-09-03366.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Faria LC, Mody I. Low ethanol concentrations selectively augment the tonic inhibition mediated by δ subunit-containing GABAA receptors in hippocampal neurons. J Neurosci. 2004;24:8379–8382. doi: 10.1523/JNEUROSCI.2040-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Zhang N, Peng Z, Houser CR, Mody I. Perisynaptic localization of δ subunit-containing GABAA receptors and their activation by GABA spillover in the mouse dentate gyrus. J Neurosci. 2003;23:10650–10661. doi: 10.1523/JNEUROSCI.23-33-10650.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir CJ, Ling AT, Belelli D, Wildsmith JA, Peters JA, Lambert JJ. The interaction of anaesthetic steroids with recombinant glycine and GABAA receptors. Br J Anaesth. 2004;92:704–711. doi: 10.1093/bja/aeh125. [DOI] [PubMed] [Google Scholar]
- Westover EJ, Covey DF. The enantiomer of cholesterol. J Membr Biol. 2004;202:61–72. doi: 10.1007/s00232-004-0714-7. [DOI] [PubMed] [Google Scholar]
- Wieland HA, Luddens H, Seeburg PH. A single histidine in GABAA receptors is essential for benzodiazepine agonist binding. J Biol Chem. 1992;267:1426–1429. [PubMed] [Google Scholar]
- Wisden W, Laurie DJ, Monyer H, Seeburg PH. The distribution of 13 GABAA receptor subunit mRNAs in the rat brain. I. Telencephalon, diencephalon, mesencephalon. J Neurosci. 1992;12:1040–1062. doi: 10.1523/JNEUROSCI.12-03-01040.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittmer LL, Hu Y, Kalkbrenner M, Evers AS, Zorumski CF, Covey DF. Enantioselectivity of steroid-induced γ-aminobutyric acidA receptor modulation and anesthesia. Mol Pharmacol. 1996;50:1581–1586. [PubMed] [Google Scholar]
- Wohlfarth KM, Bianchi MT, Macdonald RL. Enhanced neurosteroid potentiation of ternary GABAA receptors containing the δ subunit. J Neurosci. 2002;22:1541–1549. doi: 10.1523/JNEUROSCI.22-05-01541.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward RM, Polenzani L, Miledi R. Effects of steroids on γ-aminobutyric acid receptors expressed in Xenopus oocytes by poly(A)+ RNA from mammalian brain and retina. Mol Pharmacol. 1992;41:89–103. [PubMed] [Google Scholar]
- Wu FS, Chen SC, Tsai JJ. Competitive inhibition of the glycine-induced current by pregnenolone sulfate in cultured chick spinal cord neurons. Brain Research. 1997;750:318–320. doi: 10.1016/s0006-8993(97)00053-x. [DOI] [PubMed] [Google Scholar]
- Wu XS, Sun JY, Evers AS, Crowder M, Wu LG. Isoflurane inhibits transmitter release and the presynaptic action potential. Anesthesiology. 2004;100:663–670. doi: 10.1097/00000542-200403000-00029. [DOI] [PubMed] [Google Scholar]
- Yamashita M, Marszalec W, Yeh JZ, Narahashi T. Effects of Ethanol on Tonic GABA Currents in Cerebellar Granule Cells and Mammalian Cells Recombinantly Expressing GABAA Receptors. J Pharmacol Exp Ther. 2006;319:431–438. doi: 10.1124/jpet.106.106260. [DOI] [PubMed] [Google Scholar]
- Zeng CM, Manion BD, Benz A, Evers AS, Zorumski CF, Mennerick S, Covey DF. Neurosteroid analogues. 10. The effect of methyl group substitution at the C-6 and C-7 positions on the GABA modulatory and anesthetic actions of (3α,5α)- and (3α,5β)-3-hydroxypregnan-20-one. J Med Chem. 2005;48:3051–3059. doi: 10.1021/jm049027+. [DOI] [PubMed] [Google Scholar]
- Ziebell MR, Nirthanan S, Husain SS, Miller KW, Cohen JB. Identification of binding sites in the nicotinic acetylcholine receptor for [3H]azietomidate, a photoactivatable general anesthetic. J Biol Chem. 2004;279:17640–17649. doi: 10.1074/jbc.M313886200. [DOI] [PubMed] [Google Scholar]
- Zorumski CF, Mennerick SJ, Covey DF. Enantioselective modulation of GABAergic synaptic transmission by steroids and benz[e]indenes in hippocampal microcultures. Synapse. 1998;29:162–171. doi: 10.1002/(SICI)1098-2396(199806)29:2<162::AID-SYN7>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Zorumski CF, Wittmer LL, Isenberg KE, Hu YF, Covey DF. Effects of neurosteroid and benz[e]indene enantiomers on GABAA receptors in cultured hippocampal neurons and transfected HEK-293 cells. Neuropharmacology. 1996;35:1161–1168. doi: 10.1016/s0028-3908(96)00035-4. [DOI] [PubMed] [Google Scholar]