

Summary
At telomeric heterochromatin in yeast the Sir protein complex spreads from Rap1 sites to silence adjacent genes. This cascade is believed to occur when Sir2, an NAD+-dependent enzyme deacetylates histone H3 and H4 N termini, in particular histone H4 K16, enabling more Sir protein binding. Lysine 56 of histone H3 is located at the entry-exit points of the DNA superhelix surrounding the nucleosome where it may control DNA compaction. We have found that K56 substitutions disrupt silencing severely without decreasing Sir protein binding at the telomere. Our in vitro and in vivo data indicate that Sir2 deacetylates K56 directly in telomeric heterochromatin to compact chromatin and prevent access to RNA polymerase and ectopic bacterial dam methylase. Since the spread of Sir proteins is necessary but not sufficient for silencing we propose that silencing occurs when Sir2 deacetylates H3 K56 to close the nucleosomal entry-exit gates enabling compaction of heterochromatin.
Introduction
Heterochromatin is distinct from euchromatin in the eukaryotic genome in that it appears cytologically condensed throughout the cell cycle and can epigenetically repress genes transposed to its vicinity (Moazed, 2001). Heterochromatin is often very abundant and may encompass large fractions of whole chromosomes in flies and even in humans in the case of the inactive X chromosome (Migeon, 1994). In eukaryotic microorganisms such as Schizosaccharomyces cerevisiae, the fission yeast, heterochromatin is found at centromeres (Allshire et al., 1994) and telomeres (Nimmo et al., 1994). While in Saccharomyces cerevisiae, or budding yeast where centromeres are much smaller in size, heterochromatin-like epigenetically regulated regions include telomeres and the silent mating type loci HML and HMR and may even include the rRNA-encoding DNA (rDNA) (Rusche et al., 2003).
The establishment, maintenance and inheritance of these silent regions in budding yeast require the involvements of cis-acting DNA sequences (silencers), histones H3 and H4 and various trans-acting protein factors (Rusche et al., 2003). Among them, Rap1 and the Silent Information Regulators (SIR proteins) play central roles in the silencing process as the key structural components of silenced chromatin. These proteins are assembled in a stepwise manner. Rap1 is believed to initiate heterochromatin formation by recognizing C1-3A repeat sequences at telomere ends. Rap1 recruits Sir4 and this initial binding between Rap1 and Sir4 is independent of other known silencing factors (Hoppe et al., 2002; Luo et al., 2002; Moretti et al., 1994). Sir2 and Sir3 are recruited to the telomere ends through their interaction with Sir4 (Hoppe et al., 2002; Luo et al., 2002; Moazed et al., 1997; Strahl-Bolsinger et al., 1997). These events are part of a cascade in which Sir2, a conserved NAD+-dependent histone deacetylase that specifically targets H4 lysine 16 (K16) and to a lesser extent K9 and K14 of histone H3 in vitro (Imai et al., 2000) deacetylates the histone tails allowing Sir3, Sir4 and Sir2 to bind successively (Hecht et al., 1995; Liou et al., 2005). One of the metabolites of the Sir2 deacetylation reaction, O-acetyl-ADP-ribose (AAR), promotes the association of extra copies of Sir3 with Sir2/Sir4 and induces a dramatic structural change in the Sir2/3/4 complex in vitro(Liou et al., 2005). Simultaneously, the NAD+-dependent deacetylation reaction also releases free energy from hydrolysis of the glycosidic bond between nicotinamide and ribose, which is comparable to the free energy of hydrolysis of ATP (Rusche et al., 2003). This energy may be required to drive conformational changes in proteins and DNA during the process of heterochromatin formation. Spreading of heterochromatin at telomeres eventually stops due to the acetylation of H4 K16 in adjacent euchromatin at telomeres (Kimura et al., 2002; Suka et al., 2002) or due to the presence of barrier DNA elements at the HM loci (Donze and Kamakaka, 2001).
Although Sir proteins are essential for silencing (Aparicio et al., 1991), the mechanism by which Sir proteins block transcription is still unclear. It has been proposed that the Sir proteins mediate silencing by compacting the chromatin fiber into a more condensed structure, which blocks the association of RNA polymerase II with genes in heterochromatin (Rusche et al., 2003). Supporting evidence for this model comes from the observation that HM loci and telomeres bind less frequently than active regions to various restriction enzymes and foreign DNA methylases (Chen and Widom, 2005; Gottschling, 1992; Loo and Rine, 1994; Singh and Klar, 1992). This may also explain why certain histone post-translational modifications prevalent in euchromatin (e.g. histone H3 K4 and K79 methylation) are absent from heterochromatin (Santos-Rosa et al., 2004; van Leeuwen et al., 2002). However, Sir protein binding may not be sufficient to ensure gene silencing. For instance, Sir2 and Sir4 have been shown to bind continuously from HML to the telomere (Lieb et al., 2001) but reporter genes inserted throughout this region are not silenced (Bi, 2002), In addition, Sir protein spreading is separable from silencing at HMR in G1 arrested cells (Kirchmaier and Rine, 2006). This indicates that the establishment of silencing in heterochromatin requires an unknown but crucial step in addition to Sir protein binding and spreading.
Histone H3 lysine 56 is located near the entry-exit points of the DNA superhelix as it wraps around the histone octamer (Luger et al., 1997). Interestingly, acetylation of this site is uniquely required for histone gene expression (Xu et al., 2005) and DNA damage response (Masumoto et al., 2005). In budding yeast, K56 acetylation is cell cycle regulated, accumulating during S phase and diminishing before mitosis (Xu et al., 2005; Masumoto et al., 2005). This is due at least in part to the global action of the Sir2-related Hst3 and Hst4 histone deacetylases that are expressed outside of S phase and are required for deacetylation of K56Ac in vivo. Whether Hst3 and Hst4 directly deacetylate K56Ac is unknown (Celic et al., 2006; Maas et al., 2006).
In this paper, we ask whether H3 K56 is integral to the mechanism of silencing and the formation of heterochromatin. Using amino acid substitutions we show that H3 K56 is strongly required for the silencing of genes at two distinct telomeres. Our data argue that deacetylation of H3 K56 by Sir2 (as well as through Hst3 and Hst4) is critical for silencing. However, K56 is not required for Sir protein spreading but for the formation of a heterochromatin structure that is inaccessible to RNA polymerase and an ectopic (dam methylase) probe. These data argue for two distinct processes in the formation of silent heterochromatin. The first is distinguished by Sir protein spreading along the deacetylated histone N termini; however this alone is insufficient for silencing. The second process is one controlled by H3 K56 deacetylation that promotes an inaccessible and silent chromatin structure.
Results
Lysine 56 of histone H3 is required for telomeric silencing in Saccharomyces cerevisiae
Given the location of H3 K56 at the entry-exit points of the DNA wrapped around the nucleosome we first asked whether K56 is required for silencing by heterochromatin in Saccharomyces cerevisiae. To do so we generated H3 K56 non-conservative substitutions to glycine or glutamine to simulate the acetylated state or a conservative substitution to arginine to simulate the unacetylated state and examined the expression of a reporter gene URA3, which was transposed to the telomere VII-L or the other silent HM and rDNA loci. In this assay, the expression of URA3 causes the conversion of 5-fluoro-orotic acid (5-FOA) added to the growth medium to toxic 5-fluorouracil. Thus cells defective in silencing URA3 are 5-FOA sensitive. Using this assay, we found that all three K56 mutants cause severe silencing defects at telomere VII-L (Figure 1A and Figure S1), resulting in decreased 5-FOA resistance levels ranging from 4.5×10−5 (K56R) to 1.8×10−6 (K56Q) as compared to wild type cells. The arginine mutant K56R had a reproducibly lower effect on 5-FOA sensitivity and URA3 silencing at the telomere.
Figure 1. Lysine 56 of histone H3 is essential for telomeric silencing in yeast.
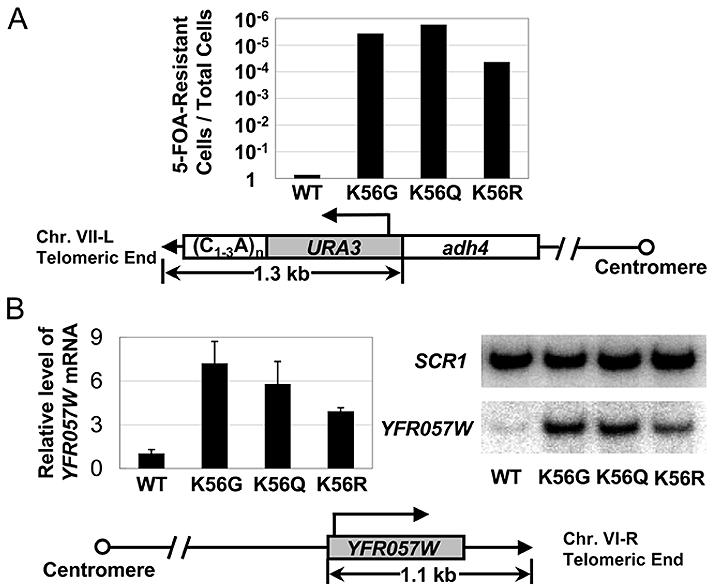
(A) Wild type and K56 substitution strains were assayed for silencing of a telomeric URA3 gene by monitoring cell growth on the 5-FOA plate. Tenfold serial dilutions of WT, K56G, K56Q and K56R cells were spotted onto SD-Trp− plate lacking or containing 0.1% 5-FOA. The fraction of viable cells on the 5-FOA plates was determined as relative to the non-5-FOA plates. Averages of three independent experiments were graphed logarithmically. Position and transcriptional orientation of the telomeric URA3 gene were schematically represented under the graph. (B) Expression of a natural telomeric gene YFR057W was examined by quantitative RT-PCR in wild type and K56 substitution cells. Relative level of YFR057W mRNA in each strain was calculated by first normalizing to the internal control, SCR1, and then to that observed in the WT cells, which has been set to 1. Right panel shows a representative gel. Position and transcriptional orientation of the YFR057W gene were shown under the graph. Values are averages of three independent experiments with error bars shown for standard deviations.
At the silent HM mating loci, where there are redundant silencing mechanisms (Rusche et al., 2003), the effect of K56 substitutions on silencing was less pronounced. At HMR, only K56G substitution had a measurable effect on silencing (∼100 fold increased 5-FOA sensitivity as compared to wild type cells). (Figure S1). We did not detect a silencing defect at HML in any of the K56 substitutions (Figure S1). At the rDNA locus which contains Sir2 but not the other Sir proteins, we observed a mild (∼100 fold) elevated 5-FOA sensitivity in K56G and K56Q mutants as compared to wild type and K56R mutant cells. These data indicate that K56 is critical for telomeric silencing but has a lesser effect at HMRand rDNA and apparently is not required for silencing at HML.
While 5-FOA sensitivity is a very sensitive assay for URA3 expression it is not a linear reflection of the extent of URA3 mRNA synthesis and a small change in expression of URA3 can result in many orders of magnitude change in 5-FOA sensitivity (Thompson et al., 1994). Therefore we also examined the expression of a natural silenced gene YFR057W at the sub-telomeric region of chromosome VI-R in the K56 substitution strains. In this study, we showed by quantitative RTPCR that K56G, K56Q and K56R substitutions result in sizeable increases of YFR057W expression. As compared to the wild type strain, there is a ∼5.9-7.2 fold increased expression of YFR057W in K56G and K56Q mutants (Figure 1B). Again K56R has a lower (∼3.9 fold) effect on expression (Figure 1B). Therefore, H3 K56 is required for silencing at two different telomeres when we examine expression of an artificially transposed reporter gene and a natural sub-telomeric gene. Given the strong effect of K56 mutants on telomeric silencing we focus the remaining experiments on telomeric heterochromatin.
H3 K56 substitutions disrupt telomeric silencing without significantly increasing acetylation of other histone sites or decreasing Sir protein spreading
To rule out the possibility that K56 mutants affect silencing indirectly through their effect on H4 K16 acetylation we examined the K16 acetylation level at telomere VI-R in K56Q and K56R mutants. We found no sizeable change of sub-telomeric K16 acetylation in these mutants as compared to wild type cells (Figure 2A). Similarly, K56G mutants had little effect on K16 acetylation state (Figure S2). Since the H3 N terminus interacts with Sir3 and Sir4 in vitro and is involved in silencing in vivo (Thompson et al., 1994) we also examined H3 N terminal acetylation sites (as well as H2A and H2B sites of acetylation). We found no significant increase in subtelomeric heterochromatin at any of the histone N termini in H3 K56G mutant (Figure S2). Therefore, K56 mutants do not affect silencing indirectly by increasing the acetylation state of H4 K16 or any other N terminal histone acetylation sites.
Figure 2. Lysine 56 of histone H3 is not required for H4 K16 acetylation or Sir2 binding at telomere.
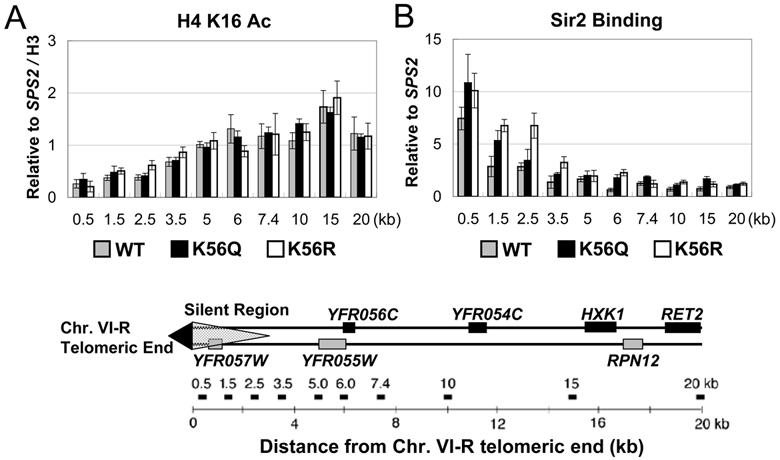
(A) H4 K16 acetylation state and (B) Sir2 binding were assayed by ChIP using antibodies specific for the acetylated H4 K16 and Sir2 protein in WT and K56 substitution strains. Both H4 K16 acetylation and Sir2 binding data were normalized to an internal control (SPS2) and the input DNA. H4 K16 acetylation data were further normalized to histone H3 level, which was examined using an antibody specific to the histone H3 C- terminus. Generally, Sir2 binding at the telomeric silent region and the adjacent euchromatic region doesn't decrease in K56 substitution mutants (K56Q and K56R) as compared to WT cells. H4 K16 acetylation state at this 20-kb region in the mutants is also comparable to the WT strain. Gene map under the graphs shows the positions of fragments amplified in PCR. The results are averages of three independent ChIPs with error bars shown for standard deviations.
Histone H4 K16, K91 and H3 K4, K79 mutants that have been shown to decrease silencing do so by disrupting the binding of Sir proteins (Hecht et al., 1996; Santos-Rosa et al., 2004; Strahl-Bolsinger et al., 1997; van Leeuwen et al., 2002; Ye et al., 2005). To ask if the silencing defects in the K56 substitution strains are the result of Rap1 or Sir protein loss at the telomere, we examined their binding at telomere VI-R using chromatin immunoprecipitation (ChIP). Surprisingly, Sir2 protein binding at the telomere is not decreased and is even slightly increased in K56Q and K56R mutants as compared to the wild type strain (Figure 2B). This is also true for Rap1 (Figure S3A), Sir3 (Figure S3B) and Sir4 binding (Figure S3C). Thus, K56 is not required for Rap1 and Sir protein binding or spreading and Sir protein spreading is not sufficient to ensure gene silencing when K56 is replaced by glutamine or arginine.
K56 acetylation has been shown to be required for histone gene expression (Xu et al., 2005) and expression is accompanied by nucleosome loss (Boeger et al., 2003; Reinke and Horz, 2003). To rule out the possibility that the silencing defect in K56 substitution cells is an indirect effect of decreased histone expression resulting in histone or nucleosome loss at silent loci, we examined the presence of histone H3 at the telomere by ChIP assay using an antibody against the C-terminus of H3 (Gunjan and Verreault, 2003). We observed comparable levels of histone H3 at the telomere VI-R in wild type, K56Q, K56R (Figure S3D) and K56G (data not shown) mutant cells, indicating that histone loss is not involved in the silencing defect in K56 mutant cells.
H3 K56 is hypo-acetylated at silent loci but hyper-acetylated at active genes
Heterochromatin occupies ∼3 kb of subtelomeric chromatin at Chr. VI-R and variable amounts at other telomeres (Hecht et al., 1996; Lieb et al., 2001). We first wished to know the extent of K56 acetylation at the subtelomeric heterochromatin regions of the yeast genome and employed the “ChIP on CHIP” technique using Affimetrix high density (one probe per 5 bp) tiling arrays to address this question. As shown in Figure 3A, when K56 acetylation is calculated in a moving average (window size, 20; step size, 500 bp), K56 acetylation is somewhat lower at the subtelomeric regions as compared to the regions distal to the telomeres. However, heterochromatin contains more densely spaced nucleosomes as is evident when we examine H3 level using an antibody to the C-terminus of histone H3 (Figure 3A). Upon normalizing for histone H3 level we find that telomeric heterochromatin is distinctly hypo-acetylated at K56 (Figure 3A). In contrast, K56 acetylation when normalized to H3 levels is enriched in the coding region of actively transcribed genes (Figure 3B).
Figure 3. H3 K56 is hypo-acetylated at telomeres and hyper-acetylated at active genes.
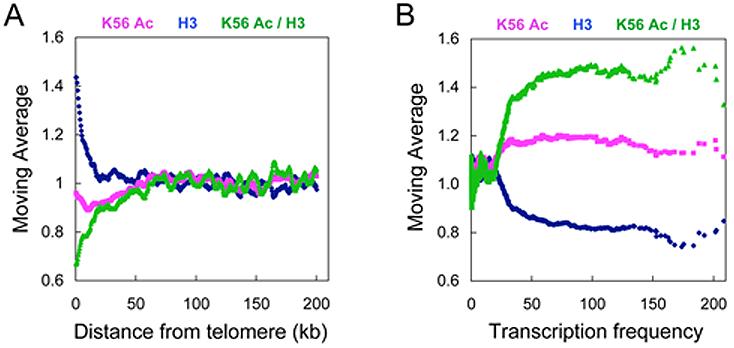
ChIP DNA of K56 acetylation and H3 C-terminus antibodies and input were amplified, fragmented, labeled and hybridized to GeneChip S.cerevisiae Tiling 1.0R Array. Moving averages of K56 acetylation, histone H3 and K56 / H3 data were plotted against (A) the distance from telomere end (window size, 20; step size, 500 bp) or (B) gene transcription rate (window size, 100; step size, 1).
In parallel, we also plotted the tiling array data using a moving average of percentile rank to better illustrate the hypoacetylated nature of K56 at telomeres. In this analysis, we generated a percentile rank for each 500 bp chromosomal region exclusively according to its actual K56 Ac or H3 intensity value and plotted a moving average of this percentile rank (window size, 20; step size, 500 bp) against the distance from telomere. As shown in Figure S4A, regions proximal to telomere have much lower K56 acetylation percentile ranks as compared to chromosomal regions distal to the telomere before and after normalizing to the H3 level. In Figure S4B, we assigned a percentile rank for each gene according to its actual K56 Ac or H3 intensity value and plotted the moving average of this percentile rank against the gene transcription rate (window size, 100; step size, 1). We found that the actively transcribed genes have a higher percentile rank of K56 acetylation both before and after normalization to H3 levels.
Sir2 is required for deacetylating H3 K56 at silent loci in vivo
It is evident that Hst3 and Hst4 affect K56 acetylation globally (Celic et al., 2006; Maas et al., 2006). Given that Sir2 has been shown to bind telomeric heterochromatin (Strahl-Bolsinger et al., 1997) and is related to the NAD+-dependent histone deacetylase Sirtuins, we asked whether Sir2 is required in vivo for H3 K56 deacetylation. Using a sir2 deletion mutant and ChIP, we observed a large increase (∼5.6 fold) of K56 acetylation in Chromosome VI-R subtelomeric heterochromatin (at 0.5 kb from the telomere). Similar increases in K56 acetylation were seen in the sir2 deletion mutant at the HMR and HML silencers E and I (Figure 4). However it should be noted that Sir3 and Sir4 are required for Sir2 binding and spreading at telomeric heterochromatin (Luo et al., 2002) and that we also saw similar increases in K56 acetylation at telomeric heterochromatin in sir3Δ and sir4Δ strains (data not shown). We conclude Sir2 is required for deacetylating H3 K56 in vivo at all three silent loci examined.
Figure 4. Sir2 is required for deacetylating H3 K56 in vivo at telomere and HM loci.
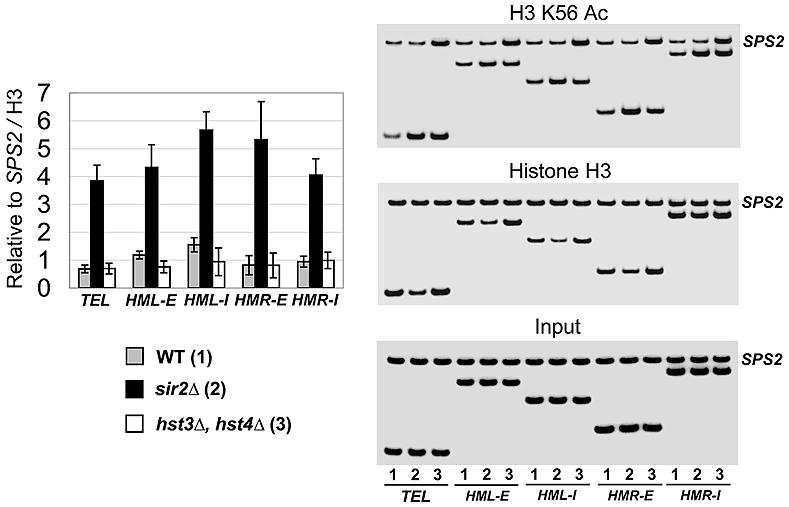
Relative H3 K56 acetylation and histone H3 levels were determined by ChIP assays using K56 acetylation and H3 C-terminus specific antibodies and normalized to an internal control (SPS2) and the input DNA. The relative level of K56 acetylation was further normalized to that of histone H3. K56 acetylation increases dramatically at telomere, E and I silencer of HML and HMR upon SIR2 deletion. In hst3Δ hst4Δ double deletion strain, H3 K56 acetylation increases in parallel at both the heterochromatic and the euchromatic regions, resulting in that the relative levels of K56 acetylation at silent loci to the internal control SPS2 are comparable to the wild type strain. Right panel shows representative gels. Values are averages of three independent experiments with error bars shown for standard deviations.
To compare the effects of Sir2 to those of Hst3 and Hst4 specifically on heterochromatic K56 acetylation we analyzed K56 acetylation using ChIP in the appropriate mutant strains (Figure 4). We found that the hst3Δ hst4Δ deletion did not affect K56 acetylation at the silent loci significantly more than at our internal control, the SPS2 gene. In contrast, sir2 deletion resulted in a 3.6-6.5 fold increase in K56 acetylation at silent loci as compared to SPS2. These data argue that while Hst proteins have a global effect on K56 acetylation, Sir2 is required for heterochromatin specific deacetylation of H3 K56.
To further investigate the roles of Sir2, Hst3 and Hst4 in deacetylating K56, we examined K56 acetylation and H3 levels at the telomere (Chr. VI-R) in the WT, sir2Δ, hst3Δhst4Δ and sir2Δhst3Δhst4Δ strains using real-time PCR and normalized the ChIP data to the corresponding input. As shown in Figure S5, after normalizing to the histone H3 level, K56 acetylation increases significantly in the sir2Δ strain and less so in the hst3Δhst4Δ double mutant as compared to the WT. In the triple mutant, K56 acetylation doesn't increase further than in the sir2Δ strain. This argues that Sir2 is required for deacetylating K56 Ac maximally at the telomere. It also supports a role for Hst3/Hst4 in a different step of the same pathway (for example the histone assembly process).
Sir2 deacetylates H3 K56 in vitro
We next asked whether Sir2 deacetylates K56 directly, in vitro. Sir2 has been shown to be specific for histone H4 K16 and to a lesser extent H3 K9, K14 in vitro (Imai et al., 2000). Using a similar approach we incubated purified recombinant Sir2 in a reaction together with various acetylated peptides in the presence and absence of NAD+ (Experimental Procedures). After the in vitro deacetylation assay, the reaction mixture was subjected to mass spectrometry analysis. As shown in figure 5A(1), no deacetylated K56 peptide was identified in the reaction mix lacking NAD+. In contrast, 95 % of the K56 acetylated peptide (IRRFQKAcSTELLC) was deacetylated in the presence of 1 mM NAD+ (Figure 5A(2)). As a positive control, 100 % of the H4 K16 acetylated peptide was deacetylated in the in vitro assay (Figure 5A(4)), confirming previous observations (Imai et al., 2000). Under the same condition, only 28 % of the H4 K12 acetylated peptide was deacetylated by Sir2 (Figure 5A(6)). In parallel, we used a mock preparation from the expression BL-21 codon (+) strain harboring an empty pET vector in the assay. We failed to detect deacetylated peptide from the reaction (Figure 5A(7)(8)), indicating that the deacetylation activity in the assay is due to the recombinant yeast Sir2 protein functioning in an NAD+ dependent manner. Therefore, using peptide substrates containing a single acetylated site, Sir2 deacetylates H3 K56 and H4 K16 much more effectively than H4 K12 in vitro.
Figure 5. Sir2 deacetylates H3 K56 in vitro.
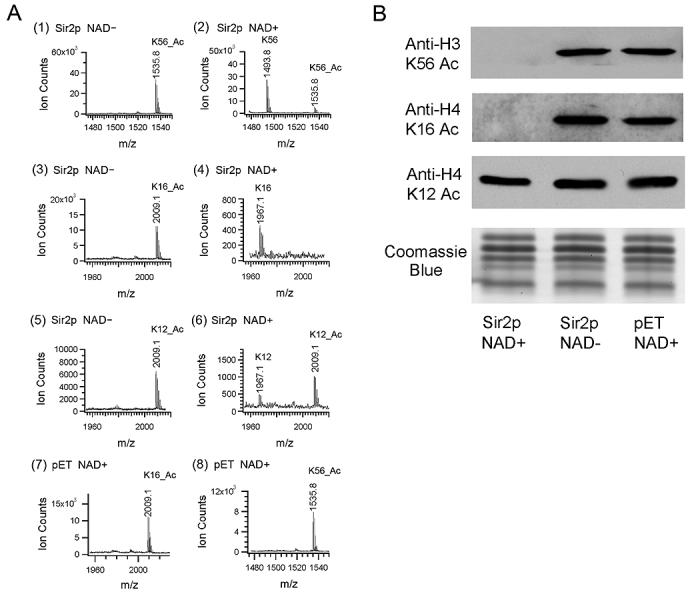
(A) In vitro deacetylation assays were performed by incubating recombinant yeast Sir2 and various acetylated peptides in the presence and absence of NAD+. MALDI-TOF mass spectrometry chromatograms of the reaction mixture of (1) H3 K56 acetylated peptide without NAD+ or (2) with 1 mM NAD+, (3) H4 K16 acetylated peptide without NAD+ or (4) with 1 mM NAD+, (5) H4 K12 acetylated peptide without NAD+ or (6) with 1 mM NAD+, (7) H4 K16 acetylated peptide with vector control (pET) and (8) H3 K56 acetylated peptide with vector control (pET) were shown respectively. (B) Full length wild type core histones were incubated with recombinant yeast Sir2 in the presence and absence of 1 mM NAD+. A vector control experiment (pET) was performed in parallel. Reaction mixtures were resolved by 15% SDS-PAGE and probed with anti-H3 K56 Ac, anti-H4 K16 Ac and anti-H4 K12 Ac antibodies in the western blots. The bottom panel shows Coomassie Blue stained gel as a loading control.
To further investigate the kinetics of the deacetylation reactions carried out by Sir2 on various peptide substrates, we performed a time course experiment by collecting samples at different time points of the in vitro HDAC assay. We then measured the deacetylation level of each sample by mass spectrometry. As shown in Figure S6, the deacetylation of H3 K56 Ac peptide exhibits similar kinetics as that for H4 K16 Ac peptide. The deacetylation of H4 K12 Ac peptide occurred at a much lower rate as compared to that of K56 Ac and K16 Ac peptides, suggesting that Sir2 specifically deacetylates K56 and K16 to a comparable extent.
To determine whether Sir2 also deacetylates full-length histones we purified histones from wild type yeast and incubated the proteins with recombinant Sir2 as described above. The acetylation levels of H3 K56 and the other sites were evaluated using western blots. After incubating recombinant Sir2 with purified core histones, the deacetylated histones were resolved by 15 % SDS-PAGE and probed with anti-K56 Ac, anti-K16 Ac and anti-K12 Ac antibodies (Experimental Procedures). We observed that H3 K56 and H4 K16 in full-length histones were completely deacetylated by Sir2 in an NAD+ dependent manner (Figure 5B). This activity is specific in that H4 K12 acetylation state is hardly changed in the presence of Sir2 (Figure 5B). These assays demonstrate that Sir2 directly deacetylates K56 Ac using both peptide and intact histone protein substrates in vitro.
H3 K56 substitutions alter telomeric heterochromatin structure
The increased transcription of otherwise repressed genes at the telomeres in the K56 mutants indicates that RNA polymerase II has increased access to genes in the mutant telomeric heterochromatin. To determine whether other ectopic factors can access the mutant heterochromatin we utilized the in vivo bacterial dam DNA methylase accessibility assay (Gottschling, 1992) since yeast does not methylate its DNA naturally. Genomic DNA of WT, K56G, K56Q, K56R and sir2Δ cells harboring an integrated dam methylase gene was purified and cleaved with restriction enzymes Dpn I, Mbo I and Sau3 AI that recognize methylated, unmethylated and both methylated and unmethylated GATC, respectively. As shown in Figure 6, the sub-telomeric fragment of chromosome VI-R from the WT strain is largely refractory to Dpn I indicating that the heterochromatin structure is inaccessible to the dam methylase in vivo (Gottschling, 1992). In contrast, we observed ∼ 50-60% Dpn I cleavage or 6-7 fold increase in Dpn I cleavage at telomere VI-R in the K56 mutants as compared to the WT cells. This argues that telomeric heterochromatin structure is more accessible to the dam methylase in the K56 mutants. In parallel experiments, Mbo I cut the WT fragment much more efficiently than the fragments from K56 mutants, which confirms the Dpn I cleavage result (Figure 6). As expected, Sau3 AI cleaves WT and K56 mutant telomeric heterochromatin equally. In sir2Δ cells, ∼95% of the genomic DNA was cleaved by Dpn I, suggesting a nearly complete disruption of the telomeric heterochromatin structure (Figure S7). Meanwhile, ∼5% and ∼98% of the sir2Δ genomic DNA was digested by Mbo I and Sau3 AI, respectively. Together, these data demonstrate that the K56 mutant heterochromatin structure at telomere VI-R is accessible to both RNA polymerase and dam methylase at different locations in telomeric heterochromatin. This argues that intact K56 is required for a generally compacted heterochromatin structure.
Figure 6. Telomeric chromatin is accessible to dam methylase in K56 substitution mutants.
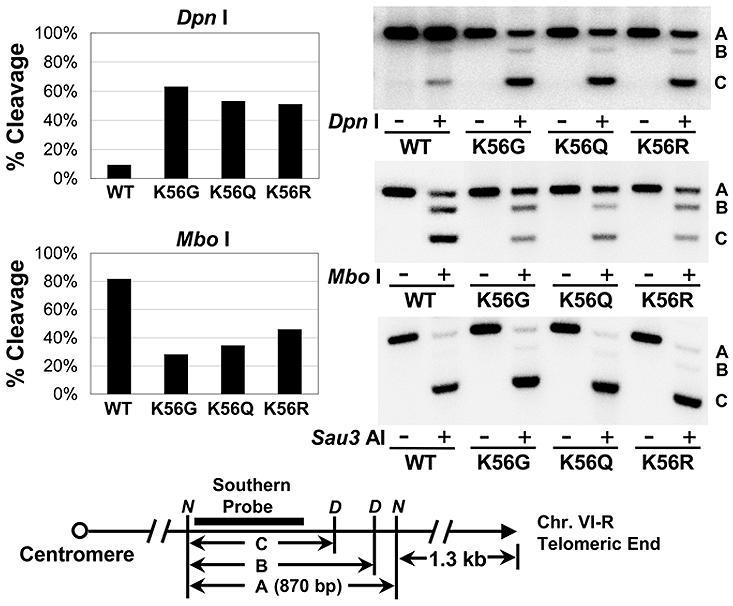
Yeast genomic DNA was isolated from wild type (WT), K56G, K56Q and K56R cells expressing the E.coli dam methyltransferase. DNA samples were first digested with Nde I (lanes −) to yield a 870 bp telomeric fragment (fragment A), then a fraction of Nde I digested DNA was further cleaved with Dpn I or Mbo I or Sau3 AI (lanes +). Enzyme digested DNA samples were subjected to southern blot analysis using a 544 bp telomere VI-R DNA probe (indicated by a solid box in the gene map under the graphs). Percentile of enzyme cleavage in each strain was calculated by dividing the sum of the intensities of B and C bands by the sum of the intensities of all the bands in lane + ( (B+C)/(A+B+C) ). Right panel shows representative southern blots. Position of the telomeric southern blot region was shown under the graph. D, Dpn I, Mbo I and Sau3 AI site; N, Nde I site.
Discussion
We provide evidence that histone H3 K56, whose acetylation is important for the transcription of the histone genes (Xu et al., 2005) and for the DNA damage response (Masumoto et al., 2005), has a unique role in the structure of heterochromatin. K56 is required for silencing telomeric heterochromatin and is deacetylated at the silent loci. However, unlike many other factors that disrupt silencing, K56 mutants do not decrease spreading of Sir proteins and do not result in the increased acetylation of histone N termini. On the basis of in vivo and in vitro data, Sir2 is shown to be the telomere specific NAD+-dependent deacetylase for K56. Thus Sir2 has two major deacetylase functions. It deacetylates H4 K16 providing a binding site for Sir3, enabling Sir protein spreading, but this is insufficient to cause silencing. Silencing also requires the deacetylation of K56. If K56 is mutated, heterochromatin is abnormally accessible and transcription of genes in heterochromatin can occur. Our results suggest that Sir2 promotes telomeric heterochromatin compaction by deacetylating H3 K56 to close the entry-exit gates of DNA wrapped around nucleosomes.
Substitutions of H3 K56 cause severe silencing defects at telomeres and lesser defects at other silent loci
Using quantitative RT-PCR and 5-FOA sensitivity assays, we demonstrated that substitutions of lysine 56 with glycine, glutamine or arginine cause severe silencing defect at telomeres of Chr. VI-R and Chr. VII-L (Figure 1 and S1), while K56G and K56Q mutants showed a lesser effect on silencing at rDNA (Figure S1). At silent mating-type loci, only the K56G mutant has a mild effect on silencing at HMR (Figure S1). Together, K56 mutants showed varying effects on silencing at different silent loci, which reflect in part the differences between the silent loci. For example, at telomeres, silencing requires the coordinate function of Rap1, Sir2, Sir3, Sir4 and the histone tails. In addition to the factors required for telomeric silencing, HM silencing also requires the E and I silencers, SIR1 and ORC genes, resulting in redundant mechanisms of silencing (Rusche et al., 2003). Generally, silencing of telomeres is semistable (Gottschling et al., 1990) and sensitive to genetic and environmental perturbations, while silencing of the HM loci is very stable and more resistant to factors disrupting telomeric silencing (van Leeuwen and Gottschling, 2002). Why rDNA is affected so weakly by K56 mutants is not clear, however the mechanism of silencing there differs most from the other silent loci in that Sir3 and Sir4 are not required for rDNA silencing and many factors at rDNA are absent from the other silent loci (Smith and Boeke, 1997; Straight et al., 1999). Our data shows some disagreement with that of Hyland et al., (2005) whose high throughput analysis also identified K56 as an acetylation site and whose mutagenesis of K56 concluded that rDNA silencing as measured by 5-FOA sensitivity was strongly affected by K56Q while telomeric silencing as measured using an ADE2 cell color reporter assay was only weakly affected. The varied extent of silencing defects in K56 mutants between our results and those of Hyland et al. (2005) may reflect the difference of strain backgrounds and the sensitivity of assays employed. However, it should be stressed that quantitative RT-PCR is the most direct measurement for mRNA levels.
Sir protein spreading is a prerequisite but is not sufficient to establish silencing at telomeres
Sir protein spreading along chromosomes has long been associated with silencing. Loss of any one of the Sir2, Sir3 and Sir4 proteins at telomeres leads to a complete loss of silencing. Moreover, all yeast lesions that prevent Sir protein binding and spreading at silent loci exhibit silencing defects, indicating that Sir protein spreading is a prerequisite for maintaining normal silencing in yeast. On the other hand, although most yeast mutants that have silencing defects also lose Sir proteins at the corresponding silent locus, there are some exceptions. For example, on yeast chromosome III, Sir2 and Sir4 bind continuously from HML to the left telomere (Lieb et al., 2001), but there is no silencing throughout this 7-kb region (Bi, 2002). In addition, a recent study showed that Sir protein binding was partially restored upon histone H3 K9, 14R / H4 K16R mutations in the absence of Sir2's catalytic activity, but silencing is still defective at telomere and HM loci (Yang and Kirchmaier, 2006). Moreover, Kirchmaier et al. (2006) reported that Sir proteins could associate throughout HMR in G1 cells, yet silencing of the HMRa1 gene does not occur, arguing for separable steps in the formation of silent heterochromatin. In this study, we showed by ChIP that Sir protein binding is not decreased at telomere VI-R in the K56 substitution cells as compared to wild type cells (Figure 2 and S3), but telomeric silencing in these mutant strains is disrupted. In summary, our observations, together with previous reports, suggest that Sir protein binding and spreading are insufficient to ensure proper gene silencing in yeast. These data also indicate the existence of one or more additional critical events required for silencing after Sir protein spreading.
Targeted and global deacetylation of H3 K56
Hst3 and Hst4 have been shown to be responsible for the global deacetylation of H3 K56 during the cell cycle (Celic et al., 2006; Maas et al., 2006). However we were unable to detect 13xmyc tagged Hst3 or Hst4 at the telomere by ChIP (data not shown), indicating that they may not act locally at telomeric chromatin. In contrast, Sir2, strongly detectable by ChIP, has been shown to be physically present at the silent loci (Gotta et al., 1997; Strahl-Bolsinger et al., 1997) and the deacetylation of K56 at HML, HMR silencers and telomeric heterochromatin is dependent on SIR2 (Figure 4). Moreover, while the in vitro deacetylation activity of Hst3 and Hst4 on K56 was not reconstituted (Celic et al., 2006; Maas et al., 2006), our data clearly demonstrate that Sir2 deacetylates K56 from both peptide and full length histone substrates (Figure 5). Nevertheless, Sir2 is acting at silent loci preferentially and not throughout the genome since there is no obvious effect of SIR2 deletion on global levels of K56 acetylation (Celic et al., 2006). Based on these observations, we believe that Hst3 and Hst4 function globally while Sir2 is required for the targeted deacetylation of K56 at the silent loci.
Interestingly, yeast Sir2 is required for deacetylating H4 K16 in vivo at silent loci (Suka et al., 2001) and in vitro (Imai et al., 2000), but the bulk level of K16 acetylation in sir2Δ cells is comparable to wild type cells as assayed by western blot (Vaquero et al., 2006). Instead, a large increase of K16 acetylation was observed in the hst2Δ strain, indicating that Hst2 may be the histone deacetylase responsible for deacetylating K16 globally (Vaquero et al., 2006). We conclude that for both H3 K56 acetylation and H4 K16 acetylation there are global mechanisms for their deacetylation. However targeted mechanisms are recruited to ensure heterochromatin specific K56 and K16 deacetylation.
How does deacetylation of H3 K56 modulate silencing in yeast?
Deacetylation of histone N-terminal sites, especially H4 K16, has long been linked to Sir protein binding and spreading at silent loci (Braunstein et al., 1993; Kimura et al., 2002; Suka et al., 2002). Consistent with this function, K16 acetylation increases gradually from the telomere end to adjacent euchromatic region and thus, limits Sir protein binding to silent regions (Kimura et al., 2002; Suka et al., 2002). In contrast, although K56 acetylation is also low at the telomere (Figure 3A and S4A), it doesn't increase gradually after ∼3 kb, like K16 acetylation, at telomere VI-R. The first peak of K56 acetylation appears 7-kb from telomere VI-R, 14-kb from telomere III-L and 12-kb from telomere XI-L (data not shown). Since the substitutions of K56 do not lead to the loss of Sir proteins at the telomere, K56 does not appear to be involved in the recruitment and spreading of Sir proteins or serve as a boundary of heterochromatin in a manner similar to that of H4 K16. Instead, our data suggest that deacetylated K56 has a unique role in the formation of heterochromatin structure. This role is different from that of other histone domains whose absence results in the loss of Sir2 and Sir4 recruitment (Fry et al., 2006).
We observed that the chromatin structure of telomere VI-R is accessible to an ectopic dam methylase in the K56 mutants. RNA polymerase interactions with telomeric heterochromatin are also increased when K56 is mutated. Therefore mutant K56 allows increased productive interactions of two very different protein factors. Our data suggest that the deacetylation of K56 by Sir2 is required to close the entry-exit gates of DNA wrapped around the histone octamer facilitating the compaction of heterochromatin. This is a critical event required for silencing in addition to Sir protein spreading.
To date, histone H3 K56 acetylation has been reported in Saccharomyces cerevisiae (Masumoto et al., 2005; Xu et al., 2005), Schizosaccharomyces pombe (Recht et al., 2006), Tetrahymena thermophila (Garcia et al., 2007), Drosophila (Xu et al., 2005) and humans (Garcia et al., 2007). Further analyses will be helpful to determine if K56 is deacetylated by the corresponding Sir2 homologues and if similar mechanisms of silencing are conserved in these organisms.
Experimental Procedures
Experimental Procedures of Yeast strains and plasmids, Silencing assay and RT-PCR for mRNA quantitation are included in the Supplemental Data
Chromatin immunoprecipitation (ChIP) assay
ChIP assay and the usage of antibodies against individual histone acetylation sites were described earlier (Suka et al., 2001; Xu et al., 2005). Antibodies against Rap1, Sir2, Sir3 and Sir4 were generated in house and used at 5 μl, 2.5 μl, 5 μl and 1 μl per 50 μl lysate, respectively. Histone H3 C-terminus antibody (Gunjan and Verreault, 2003) is a kind gift from Alain Verreault and was used at 3 μl per 50 μl lysate. Sequences of the primers used in ChIP assay were listed in supplemental table 3. For real-time PCR, ChIP and input DNA was analyzed using IQ SYBR Green Supermix (Bio-Rad, Hercules, CA) according to the manufacturer's instruction on an Applied Biosystems 7500 Realtime PCR System. Quantification was performed using the Sequence Detection Software according to the manufacturer's instruction.
Microarray hybridization and data analysis
ChIP DNA of H3 K56 acetylation and histone H3 antibodies and input DNA were amplified in linear range, fragmented, and labeled as described in the GeneChip Mapping 500k Assay Manual (https://www.affymetrix.com/support/downloads/manuals/500k_assay_manual.pdf). Labeled probes were hybridized onto the GeneChip S.cerevisiae Tiling 1.0R Array (Affymetrix, cat # 900645) and washed as described in the GeneChip Mapping 500k Assay Manual. The arrays were scanned with the GeneChip Scanner 3000 7G. Intensities of K56 Ac, H3 and K56 Ac/H3 were calculated by the Two-Sample Analysis method (Affymetrix Tiling Analysis Software v1.1) using the data sets of K56 Ac vs. Input, H3 vs. Input and K56 Ac vs. H3 as the treatment and control group, respectively. The estimator used in this method is the Hodges-Lehmann estimator and is interpreted as the log2 fold change between the treatment and control group signals.
Preparation of recombinant yeast Sir2 protein
The yeast Sir2 expression plasmid pFX21 was transformed into BL21-CodonPlus (DE3)-RIPL E.coli strain (stratagene) and induced with 1 mM IPTG at 37 °C for 1 hour. The induced Sir2-6xHis fusion protein was purified under native conditions by Ni-NTA agarose (Qiagen). The pET mock preparation was performed using the same E.coli strain harboring an empty pET-24a(+) vector.
In vitro deacetylation assay and analysis of deacetylated products
The in vitro assay of Sir2 deacetylase activity was performed essentially as described previously (Imai et al., 2000). Nicotinamide adenine dinucleotide (NAD+, Upstate) was used at the concentration of 1 mM in all reactions. H3 K56 acetylated peptide (IRRFQKAcSTELLC) was synthesized by the Biopolymer Synthesis Center at Caltech (Pasadena, CA). H4 K12 and K16 mono-acetylated peptides were purchased from Upstate. Yeast core histones were purified from wild type asynchronous culture as described (Edmondson et al., 1996).
For peptide deacetylation assay, 30 μg of the purified recombinant Sir2 protein was incubated with 5 μg of various acetylated peptides in the presence and absence of NAD+. After incubation, the reaction mixture was precipitated by 100% trichloroacetic acid (TCA) and reconstituted in 50 μl solvent of CH3CN / H2O / TFA (50%/49%/1%). 0.5 μl of this solution was mixed with 0.5 μl of matrix solution (5 μg of α-cyano-4-hydroxycinamic acid dissolved in the same solvent described above) on the MALDI plate, and then analyzed by MALDI using a voyager DE-STR Biospectrometry Workstation (ABI) with delayed extraction operated in the reflectron mode. In the time course experiment, 10 μg of the Sir2 protein was incubated with 5 μg of various acetylated peptides in the presence of 1 mM NAD+. Samples were taken at 1, 5, 10 and 60 minutes time points and analyzed by mass spectrometry as described above.
For histone deacetylation assay, 10 μg of the purified recombinant Sir2 protein was incubated with 100 μg core histones (∼20 μg of histone H3 or ∼15 μg of histone H4) in the presence and absence of NAD+. The reaction mixtures were precipitated by 100% TCA, reconstituted in 10 mM Tris-HCl buffer (pH 8.0) and resolved by 15% SDS-PAGE. Blots were probed with anti-K56 Ac at 1:2000, anti-H4 K16 Ac (Serotec, cat # AHP1026) at 1:500 and anti-H4 K12 Ac (Upstate, cat # 07-595) at 1:5000.
dam DNA methylase accessibility assay
For dam DNA methylase accessibility assay at telomere VI-R, purified genomic DNA from appropriate yeast strains expressing the E.coli dam methyltransferase were first digested with Nde I to yield a 870 bp telomeric fragment, then a fraction of Nde I digested DNA was further cleaved with Dpn I or Mbo I or Sau3 AI. Enzyme digested DNA samples were subjected to southern blot analysis using a 544 bp telomere VI-R DNA probe. Southern blot hybridization protocol can be found at http://mgwww.mbi.ucla.edu/node/95.
Supplementary Material
Acknowledgements
We are grateful to Siavash Kurdistani and members of the Grunstein laboratory for critical comments and discussion throughout this work. F.X is especially grateful to Li Xin for advice, encouragement and technique help throughout this work. We also thank Zugen Chen in the UCLA Microarray Core Facility, Wei Sun for help with data analysis. We are especially grateful to Alain Verreault for the histone H3 antibody, and to Daniel Gottschling, Jeff Thompson and David Toczyski for plasmids and yeast strains. This work was supported by Public Health Service National Institutes of Health grant GM42421 to MG.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allshire RC, Javerzat JP, Redhead NJ, Cranston G. Position effect variegation at fission yeast centromeres. Cell. 1994;76:157–169. doi: 10.1016/0092-8674(94)90180-5. [DOI] [PubMed] [Google Scholar]
- Aparicio OM, Billington BL, Gottschling DE. Modifiers of position effect are shared between telomeric and silent mating-type loci in S. cerevisiae. Cell. 1991;66:1279–1287. doi: 10.1016/0092-8674(91)90049-5. [DOI] [PubMed] [Google Scholar]
- Bi X. Domains of gene silencing near the left end of chromosome III in Saccharomyces cerevisiae. Genetics. 2002;160:1401–1407. doi: 10.1093/genetics/160.4.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeger H, Griesenbeck J, Strattan JS, Kornberg RD. Nucleosomes unfold completely at a transcriptionally active promoter. Mol Cell. 2003;11:1587–1598. doi: 10.1016/s1097-2765(03)00231-4. [DOI] [PubMed] [Google Scholar]
- Braunstein M, Rose AB, Holmes SG, Allis CD, Broach JR. Transcriptional silencing in yeast is associated with reduced nucleosome acetylation. Genes Dev. 1993;7:592–604. doi: 10.1101/gad.7.4.592. [DOI] [PubMed] [Google Scholar]
- Celic I, Masumoto H, Griffith WP, Meluh P, Cotter RJ, Boeke JD, Verreault A. The sirtuins hst3 and Hst4p preserve genome integrity by controlling histone h3 lysine 56 deacetylation. Curr Biol. 2006;16:1280–1289. doi: 10.1016/j.cub.2006.06.023. [DOI] [PubMed] [Google Scholar]
- Chen L, Widom J. Mechanism of transcriptional silencing in yeast. Cell. 2005;120:37–48. doi: 10.1016/j.cell.2004.11.030. [DOI] [PubMed] [Google Scholar]
- Donze D, Kamakaka RT. RNA polymerase III and RNA polymerase II promoter complexes are heterochromatin barriers in Saccharomyces cerevisiae. Embo J. 2001;20:520–531. doi: 10.1093/emboj/20.3.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmondson DG, Smith MM, Roth SY. Repression domain of the yeast global repressor Tup1 interacts directly with histones H3 and H4. Genes Dev. 1996;10:1247–1259. doi: 10.1101/gad.10.10.1247. [DOI] [PubMed] [Google Scholar]
- Fry CJ, Norris A, Cosgrove M, Boeke JD, Peterson CL. The LRS and SIN domains: two structurally equivalent but functionally distinct nucleosomal surfaces required for transcriptional silencing. Mol Cell Biol. 2006;26:9045–9059. doi: 10.1128/MCB.00248-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia BA, Hake SB, Diaz RL, Kauer M, Morris SA, Recht J, Shabanowitz J, Mishra N, Strahl BD, Allis CD, Hunt DF. Organismal differences in post-translational modifications in histones H3 and H4. J Biol Chem. 2007;282:7641–7655. doi: 10.1074/jbc.M607900200. [DOI] [PubMed] [Google Scholar]
- Gotta M, Strahl-Bolsinger S, Renauld H, Laroche T, Kennedy BK, Grunstein M, Gasser SM. Localization of Sir2p: the nucleolus as a compartment for silent information regulators. Embo J. 1997;16:3243–3255. doi: 10.1093/emboj/16.11.3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottschling DE. Telomere-proximal DNA in Saccharomyces cerevisiae is refractory to methyltransferase activity in vivo. Proc Natl Acad Sci U S A. 1992;89:4062–4065. doi: 10.1073/pnas.89.9.4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottschling DE, Aparicio OM, Billington BL, Zakian VA. Position effect at S. cerevisiae telomeres: reversible repression of Pol II transcription. Cell. 1990;63:751–762. doi: 10.1016/0092-8674(90)90141-z. [DOI] [PubMed] [Google Scholar]
- Gunjan A, Verreault A. A Rad53 kinase-dependent surveillance mechanism that regulates histone protein levels in S. cerevisiae. Cell. 2003;115:537–549. doi: 10.1016/s0092-8674(03)00896-1. [DOI] [PubMed] [Google Scholar]
- Hecht A, Laroche T, Strahl-Bolsinger S, Gasser SM, Grunstein M. Histone H3 and H4 N-termini interact with SIR3 and SIR4 proteins: a molecular model for the formation of heterochromatin in yeast. Cell. 1995;80:583–592. doi: 10.1016/0092-8674(95)90512-x. [DOI] [PubMed] [Google Scholar]
- Hecht A, Strahl-Bolsinger S, Grunstein M. Spreading of transcriptional repressor SIR3 from telomeric heterochromatin. Nature. 1996;383:92–96. doi: 10.1038/383092a0. [DOI] [PubMed] [Google Scholar]
- Hoppe GJ, Tanny JC, Rudner AD, Gerber SA, Danaie S, Gygi SP, Moazed D. Steps in assembly of silent chromatin in yeast: Sir3-independent binding of a Sir2/Sir4 complex to silencers and role for Sir2-dependent deacetylation. Mol Cell Biol. 2002;22:4167–4180. doi: 10.1128/MCB.22.12.4167-4180.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- Kimura A, Umehara T, Horikoshi M. Chromosomal gradient of histone acetylation established by Sas2p and Sir2p functions as a shield against gene silencing. Nat Genet. 2002;32:370–377. doi: 10.1038/ng993. [DOI] [PubMed] [Google Scholar]
- Kirchmaier AL, Rine J. Cell cycle requirements in assembling silent chromatin in Saccharomyces cerevisiae. Mol Cell Biol. 2006;26:852–862. doi: 10.1128/MCB.26.3.852-862.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieb JD, Liu X, Botstein D, Brown PO. Promoter-specific binding of Rap1 revealed by genome-wide maps of protein-DNA association. Nat Genet. 2001;28:327–334. doi: 10.1038/ng569. [DOI] [PubMed] [Google Scholar]
- Liou GG, Tanny JC, Kruger RG, Walz T, Moazed D. Assembly of the SIR complex and its regulation by O-acetyl-ADP-ribose, a product of NAD-dependent histone deacetylation. Cell. 2005;121:515–527. doi: 10.1016/j.cell.2005.03.035. [DOI] [PubMed] [Google Scholar]
- Loo S, Rine J. Silencers and domains of generalized repression. Science. 1994;264:1768–1771. doi: 10.1126/science.8209257. [DOI] [PubMed] [Google Scholar]
- Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- Luo K, Vega-Palas MA, Grunstein M. Rap1-Sir4 binding independent of other Sir, yKu, or histone interactions initiates the assembly of telomeric heterochromatin in yeast. Genes Dev. 2002;16:1528–1539. doi: 10.1101/gad.988802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas NL, Miller KM, DeFazio LG, Toczyski DP. Cell cycle and checkpoint regulation of histone H3 K56 acetylation by Hst3 and Hst4. Mol Cell. 2006;23:109–119. doi: 10.1016/j.molcel.2006.06.006. [DOI] [PubMed] [Google Scholar]
- Masumoto H, Hawke D, Kobayashi R, Verreault A. A role for cell-cycle-regulated histone H3 lysine 56 acetylation in the DNA damage response. Nature. 2005;436:294–298. doi: 10.1038/nature03714. [DOI] [PubMed] [Google Scholar]
- Migeon BR. X-chromosome inactivation: molecular mechanisms and genetic consequences. Trends Genet. 1994;10:230–235. doi: 10.1016/0168-9525(94)90169-4. [DOI] [PubMed] [Google Scholar]
- Moazed D. Common themes in mechanisms of gene silencing. Mol Cell. 2001;8:489–498. doi: 10.1016/s1097-2765(01)00340-9. [DOI] [PubMed] [Google Scholar]
- Moazed D, Kistler A, Axelrod A, Rine J, Johnson AD. Silent information regulator protein complexes in Saccharomyces cerevisiae: a SIR2/SIR4 complex and evidence for a regulatory domain in SIR4 that inhibits its interaction with SIR3. Proc Natl Acad Sci U S A. 1997;94:2186–2191. doi: 10.1073/pnas.94.6.2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti P, Freeman K, Coodly L, Shore D. Evidence that a complex of SIR proteins interacts with the silencer and telomere-binding protein RAP1. Genes Dev. 1994;8:2257–2269. doi: 10.1101/gad.8.19.2257. [DOI] [PubMed] [Google Scholar]
- Nimmo ER, Cranston G, Allshire RC. Telomere-associated chromosome breakage in fission yeast results in variegated expression of adjacent genes. Embo J. 1994;13:3801–3811. doi: 10.1002/j.1460-2075.1994.tb06691.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recht J, Tsubota T, Tanny JC, Diaz RL, Berger JM, Zhang X, Garcia BA, Shabanowitz J, Burlingame AL, Hunt DF, et al. Histone chaperone Asf1 is required for histone H3 lysine 56 acetylation, a modification associated with S phase in mitosis and meiosis. Proc Natl Acad Sci U S A. 2006;103:6988–6993. doi: 10.1073/pnas.0601676103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinke H, Horz W. Histones are first hyperacetylated and then lose contact with the activated PHO5 promoter. Mol Cell. 2003;11:1599–1607. doi: 10.1016/s1097-2765(03)00186-2. [DOI] [PubMed] [Google Scholar]
- Rusche LN, Kirchmaier AL, Rine J. The establishment, inheritance, and function of silenced chromatin in Saccharomyces cerevisiae. Annu Rev Biochem. 2003;72:481–516. doi: 10.1146/annurev.biochem.72.121801.161547. [DOI] [PubMed] [Google Scholar]
- Santos-Rosa H, Bannister AJ, Dehe PM, Geli V, Kouzarides T. Methylation of H3 lysine 4 at euchromatin promotes Sir3p association with heterochromatin. J Biol Chem. 2004;279:47506–47512. doi: 10.1074/jbc.M407949200. [DOI] [PubMed] [Google Scholar]
- Singh J, Klar AJ. Active genes in budding yeast display enhanced in vivo accessibility to foreign DNA methylases: a novel in vivo probe for chromatin structure of yeast. Genes Dev. 1992;6:186–196. doi: 10.1101/gad.6.2.186. [DOI] [PubMed] [Google Scholar]
- Smith JS, Boeke JD. An unusual form of transcriptional silencing in yeast ribosomal DNA. Genes Dev. 1997;11:241–254. doi: 10.1101/gad.11.2.241. [DOI] [PubMed] [Google Scholar]
- Strahl-Bolsinger S, Hecht A, Luo K, Grunstein M. SIR2 and SIR4 interactions differ in core and extended telomeric heterochromatin in yeast. Genes Dev. 1997;11:83–93. doi: 10.1101/gad.11.1.83. [DOI] [PubMed] [Google Scholar]
- Straight AF, Shou W, Dowd GJ, Turck CW, Deshaies RJ, Johnson AD, Moazed D. Net1, a Sir2-associated nucleolar protein required for rDNA silencing and nucleolar integrity. Cell. 1999;97:245–256. doi: 10.1016/s0092-8674(00)80734-5. [DOI] [PubMed] [Google Scholar]
- Suka N, Luo K, Grunstein M. Sir2p and Sas2p opposingly regulate acetylation of yeast histone H4 lysine16 and spreading of heterochromatin. Nat Genet. 2002;32:378–383. doi: 10.1038/ng1017. [DOI] [PubMed] [Google Scholar]
- Suka N, Suka Y, Carmen AA, Wu J, Grunstein M. Highly specific antibodies determine histone acetylation site usage in yeast heterochromatin and euchromatin. Mol Cell. 2001;8:473–479. doi: 10.1016/s1097-2765(01)00301-x. [DOI] [PubMed] [Google Scholar]
- Thompson JS, Ling X, Grunstein M. Histone H3 amino terminus is required for telomeric and silent mating locus repression in yeast. Nature. 1994;369:245–247. doi: 10.1038/369245a0. [DOI] [PubMed] [Google Scholar]
- van Leeuwen F, Gafken PR, Gottschling DE. Dot1p modulates silencing in yeast by methylation of the nucleosome core. Cell. 2002;109:745–756. doi: 10.1016/s0092-8674(02)00759-6. [DOI] [PubMed] [Google Scholar]
- van Leeuwen F, Gottschling DE. Assays for gene silencing in yeast. Methods Enzymol. 2002;350:165–186. doi: 10.1016/s0076-6879(02)50962-9. [DOI] [PubMed] [Google Scholar]
- Vaquero A, Scher MB, Lee DH, Sutton A, Cheng HL, Alt FW, Serrano L, Sternglanz R, Reinberg D. SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes Dev. 2006;20:1256–1261. doi: 10.1101/gad.1412706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu F, Zhang K, Grunstein M. Acetylation in histone H3 globular domain regulates gene expression in yeast. Cell. 2005;121:375–385. doi: 10.1016/j.cell.2005.03.011. [DOI] [PubMed] [Google Scholar]
- Yang B, Kirchmaier AL. Bypassing the catalytic activity of SIR2 for SIR protein spreading in Saccharomyces cerevisiae. Mol Biol Cell. 2006;17:5287–5297. doi: 10.1091/mbc.E06-08-0669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Ai X, Eugeni EE, Zhang L, Carpenter LR, Jelinek MA, Freitas MA, Parthun MR. Histone H4 lysine 91 acetylation a core domain modification associated with chromatin assembly. Mol Cell. 2005;18:123–130. doi: 10.1016/j.molcel.2005.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.