

SUMMARY
The glmS ribozyme is a catalytic RNA that self-cleaves at its 5′-end in the presence of glucosamine-6-phosphate (GlcN6P). We present structures of the glmS ribozyme from Thermoanaerobacter tengcongensis that are bound with the cofactor GlcN6P or the inhibitor glucose-6-phosphate (Glc6P) at 1.7 Å and 2.2 Å resolution, respectively. The two structures are indistinguishable in the conformations of the small molecules and of the RNA. GlcN6P binding becomes apparent crystallographically when the pH is raised to 8.5, where the ribozyme conformation is identical to that observed previously at pH 5.5. A key structural feature of this ribozyme is a short duplex (P2.2) that is formed between sequences just 3′ of the cleavage site and within the core domain, and which introduces a pseudoknot into the active site. Mutagenesis indicates that P2.2 is required for activity in cis- and trans-acting forms of the ribozyme. P2.2 formation in a trans-acting ribozyme was exploited to demonstrate that N1 of the guanine at position 1 contributes to GlcN6P binding by interacting with the phosphate of the cofactor. At neutral pH, RNAs with adenine, 2-aminopurine, dimethyladenine or purine substitutions at position 1 cleave faster with glucosamine than with GlcN6P. This altered cofactor preference provides biochemical support for the orientation of the cofactor within the active site. Our results establish two features of the glmS ribozyme that are important for its activity: a sequence within the core domain that selects and positions the cleavage-site sequence, and a nucleobase at position 1 that helps position GlcN6P.
Keywords: glmS ribozyme, riboswitch, trans-acting ribozyme, glucosamine-6-phosphate, ribozyme cofactor
INTRODUCTION
The glmS ribozyme is present in numerous Gram-positive bacteria as part of the 5′ untranslated region (UTR) of the mRNA that codes for glucosamine-6-phosphate synthase 1,2. This protein enzyme catalyzes formation of glucosamine-6-phosphate (GlcN6P) and glutamate from fructose-6-phosphate and glutamine. The glmS ribozyme domain self-cleaves upon binding GlcN6P 2-3. In a model for genetic regulation proposed by Winkler et al. 2 expression of the glmS gene proceeds when the level of intracellular GlcN6P is low. However, higher concentrations of GlcN6P lead to cleavage of the UTR and a concomitant reduction in GlcN6P synthase production. Thus, the glmS ribozyme was the first example of a naturally occurring ribozyme that also functions as a riboswitch (reviewed in Ref. 4). Structural and biochemical evidence indicates that GlcN6P participates in the catalytic mechanism as a coenzyme rather than functioning as an allosteric activator: the structure of the ribozyme is largely unchanged upon cofactor binding and cleavage 3,5-6 and the 2-amino group of the cofactor is essential for ribozyme activity 7,8.
The secondary structure of the glmS ribozyme as originally proposed 1,2 comprised four helices or paired regions, P1–P4. These were identified and supported by base-pair co-variation between the known ribozyme sequences. However, sequences near the cleavage site proposed to be single-stranded were also invariant in the 18 species where the glmS ribozyme was found 1. Thus, the same comparative phylogenetic approach did not reveal base pairing interactions with sequences proximal to the cleavage site (Figure 1(a)). The cleavage site is located at the extreme 5′-end of the ribozyme domain, and a single nucleotide 5′ of the cleavage site is sufficient for maximum catalytic activity in vitro 2. Therefore, the sequence 3′ to the cleavage site must provide essential interactions that position the nucleotides adjacent to the scissile phosphate. Furthermore, a minimized RNA consisting only of sequences through P2 was catalytically active and GlcN6P-responsive 2. Therefore, if sequences near the cleavage site form a duplex, candidate base pairing sites would be limited to this core domain. Two such sites, both capable of limited base pairing with the sequence immediately 3′ of the cleavage site, are evident in or near the bulged loop of P2 in the original secondary structure (Figure 1(a)). One consists of nucleotides 52-56 (B. anthracis numbering; red, Figure 1(a)), and the alternative site consists of nucleotides 55-60. Based on mutagenesis studies, Soukup 9 previously suggested that nucleotides 55-60 (grey, Figure 1(a)) pair with the sequence just 3′ of the cleavage site.
FIGURE 1.
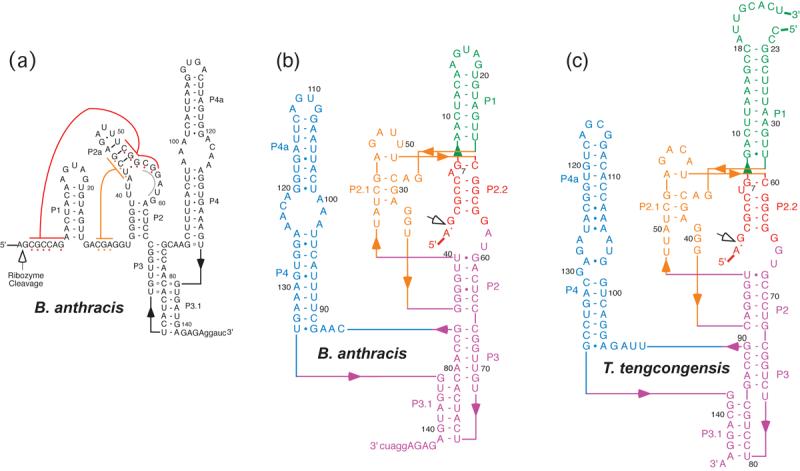
Secondary structures of the Bacillus anthracis and Thermoanaerobacter tengcongensis glmS ribozymes. (a) Proposed secondary structure of the glmS ribozyme from B. anthracis modeled after the B. subtilis structure proposed by Winkler, et al. 2. The site of cleavage is indicated with an open arrow. The position of the P2.2 pairing is shown in red with an alternative-pairing region in the P2 internal loop indicated in gray. Numbering is relative to the cleavage site. The sequence 5' to the cleavage site is not shown; it was 5'-gggaauucAUUGUAAAUUAUAGA-3', where lowercase letters indicate vector-derived sequence. (b) Secondary structure of the B. anthracis glmS ribozyme based on the crystal structure determined by Cochrane et al. 10. (c) Secondary structure of the T. tengcongensis glmS ribozyme construct used for crystal structure determination.
Crystal structures of glmS ribozymes from Thermoanaerobacter tengcongensis 3 and Bacillus anthracis 10 show nucleotides 3′ of the cleavage site forming a duplex with nucleotides 52-56 (red, Figure 1(a), P2.2 in Figure 1(b)). This helix forms part of a solvent-exposed pocket that binds glucose-6-phosphate (Glc6P) 3, an inhibitor of glmS ribozyme cleavage 7, and the catalytic cofactor GlcN6P 10. The crystal structures suggest that selectivity for GlcN6P over non-phosphorylated analogs such as glucosamine (GlcN) could arise from an interaction with the Watson-Crick edge of the guanine at position 1 3,10. This single nucleotide (G1) spans the cleavage site and the 5′-end of P2.2. Despite strong overall similarity between the crystal structures of the glmS ribozymes from the two bacterial species, subtle differences between the conformations of the bound GlcN6P and Glc6P and of some of the nucleotides that form the metabolite-binding pocket were previously noted 10. However, it was unclear whether these structural differences reflected different modes of binding of the two small molecule ligands, or resulted from other factors such as sequence differences between the T. tengcongensis and B. anthracis glmS ribozymes.
Here, we combine crystallographic analysis, structure-guided ribozyme engineering, and kinetic measurements to validate the functional relevance of key interactions observed in the crystal structures. We have refined the structures of the T. tengcongensis glmS ribozyme bound to GlcN6P and Glc6P at 1.7 Å and 2.2 Å resolution, respectively. Through mutagenesis and kinetic measurements, we unambiguously demonstrate that base pairing in P2.2, as seen in the crystal structures, is required for the cis-cleavage activity of the glmS ribozyme. The crystal structures guided the design of a bimolecular version of the B. anthracis glmS ribozyme, in which the 5' side of the P2.2 duplex is provided in trans as a short heptamer (or hexamer) oligoribonucleotide substrate. In this system, sequence specificity for the substrate was predictably altered with mutations in its “binding site” (i.e. the complementary strand of P2.2). This trans-acting ribozyme has allowed us to test a key ribozyme-GlcN6P interaction observed in our high-resolution structure. By employing synthetic oligoribonucleotide substrates with substitutions at G1, we demonstrate that this nucleobase is indeed responsible for the selectivity for phosphorylated amino-sugars displayed by the glmS ribozyme. These results indicate that the orientation of the bound GlcN6P seen in the crystals is required for ribozyme activity, and show that P2.2 is needed for both precise substrate positioning and cofactor binding.
RESULTS
High-resolution structures of activator- and inhibitor-bound glmS ribozymes
Our previous structural analyses of the glmS ribozyme were carried out with crystals grown and stabilized at pH ∼5.5 3. Under these acidic conditions, we were unable to detect electron density corresponding to ribozyme-bound GlcN6P. Instead, we inferred binding from activation of the ribozyme when we soaked our crystals in GlcN6P-containing solutions (at pH 5.5), and deduced the location of GlcN6P binding from a 2.7 Å-resolution structure of the ribozyme bound to the inhibitor Glc6P, which is isosteric to GlcN6P 3.
The catalytic activity of glmS ribozymes has been shown to increase with pH, reaching a maximum above pH 8.0 2,7. We reasoned that tighter binding of GlcN6P might accompany the increased activity observed under basic conditions. We succeeded in transferring our crystals of the T. tengcongensis glmS ribozyme from pH 5.5 to pH 8.5 without loss of crystalline order. Cleavage of the substrate RNA strand in the presence of saturating GlcN6P was blocked by replacing the ribose of residue A(−1) with a 2′-deoxyribose 5. Diffraction data collected from such crystals revealed unambiguous electron density for GlcN6P (Figure 2(a)), allowing us to determine the structure of the glmS ribozyme bound to its catalytic cofactor at 1.7 Å resolution. Subsequent to our initial report 3, we have also improved refinement of the Glc6P-bound T. tengcongensis glmS ribozyme using diffraction data extending to 2.2 Å resolution (Table 1).
FIGURE 2.
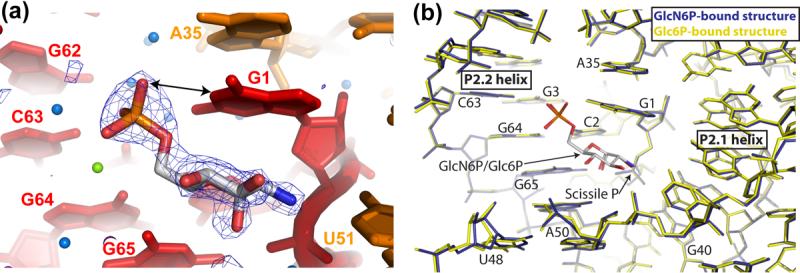
Binding of GlcN6P and Glc6P in high-resolution crystal structures of the T. tengcongensis glmS ribozyme. (a) Simulated annealing omit |Fo|-|Fc| electron density (contoured at 4.0σ) at 1.7 Å resolution for GlcN6P (blue mesh). Ribozyme nucleotides are depicted according to the color scheme in Figure 1(c). Water molecules and metal ions are shown as free floating blue and green spheres, respectively. (b) Superposition of the structure of the T. tengcongensis GlcN6P-bound glmS ribozyme (blue) and Glc6P-bound glmS ribozyme (yellow) at 1.7 Å and 2.2 Å resolution, respectively. GlcN6P and Glc6P are shown in the standard CPK color scheme.
Table 1.
Crystallographic Statistics
| GlcN6P-bound structure |
Glc6P-bound structure |
|
|---|---|---|
| Data collection | ||
| X-ray source | ALS 8.2.2 | ALS 5.0.2 |
| Space group | P21212 | P21212 |
| Cell dimensions | ||
| a, b, c (Å) | 180.9, 39.9, 70.4 | 181.1, 40.1, 70.3 |
| α, β, γ | 90, 90, 90 | 90, 90, 90 |
| Wavelength (Å) | 1.0 | 1.0 |
| Resolution (Å)* | 50-1.70 (1.76-1.70) | 50.0-2.20 (2.28-2.20) |
| Rmerge (%)* | 8.4 (39.0) | 8.4 (42.8) |
| I/σI* | 25.8 (2.5) | 18.8 (1.8) |
| Completeness (%)* | 86.9 (71.8) | 85.1 (68.3) |
| Redundancy* | 3.6 (2.3) | 2.9 (1.9) |
| Refinement | ||
| Resolution (Å) | 30.0-1.70 | 30.0-2.20 |
| No. reflections | 48,439 | 22,256 |
| Rwork/Rfree (%) | 22.6 / 23.9 | 22.2 / 24.5 |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.0044 | 0.0051 |
| Bond angles (°) | 1.21 | 1.25 |
| σ-a coordinate error (Å) | 0.31 | 0.44 |
Highest resolution shell in ().
Our structures of the glmS ribozyme bound to the activator GlcN6P and to the inhibitor Glc6P are identical to within the precision of the atomic coordinates (0.30 Å, and 0.43 Å for the GlcN6P and the Glc6P complexes, respectively, Table 1). The activator and the inhibitor small molecules also superimpose perfectly (Figure 2(b)). Based on our previous 2.7 Å-resolution electron density maps, we had built the atomic model of the ribozyme-bound Glc6P as having a C4-C5-C6-O6 dihedral angle of 180° 3. Examination of high-resolution residual |Fo|-|Fc| Fourier syntheses of the Glc6P and GlcN6P complexes of the T. tengcongensis glmS ribozyme indicates that both Glc6P and GlcN6P are best modeled with a C4-C5-C6-O6 dihedral angle of 60°. Since the positions of the nucleotides that form the cofactor-binding pocket of the ribozyme and of the small molecules are indistinguishable (Figure 2(b)), the inability of Glc6P to activate the ribozyme cannot be explained by conformational differences between its glmS ribozyme complex and that of GlcN6P.
Comparison of T. tengcongensis and B. anthracis glmS ribozymes
Subsequent to our work, a 2.5 Å-resolution crystal structure of the B. anthracis glmS ribozyme bound to GlcN6P was reported 10. Consistent with our observations, the crystals obtained by those authors at pH 6.8 revealed binding of GlcN6P. When the structures of the T. tengcongensis and B. anthracis glmS ribozymes are superimposed using the bound GlcN6P, the only substantive difference in the active site and metabolite-binding pocket is a ∼1 Å shift of the phosphodiester backbone from residues A50 to A54 (A42 to A46, respectively, in the B. anthracis numbering scheme) (Figure 3(a)). A consequence of this shift is a change in the lengths of the hydrogen bonds between the RNA and the 3-OH and 4-OH of GlcN6P. This structural difference appears to result at least partly from deletion of the nucleotide corresponding to T. tengcongensis U48 from the B. anthracis ribozyme 1. In the T. tengcongensis RNA, U48 participates in a cis-Hoogsteen base pair with A50 that caps helix P2. The absence of a nucleotide equivalent to the T. tengcongensis U48 could account for the subtle rearrangements of A42 in the B. anthracis glmS ribozyme, and a more pronounced rotation and displacement of helix P2 between the two glmS ribozymes. This displacement further propagates to the peripheral helices P3, P3.1 and P4 (Figure 3(b)). However, the conserved adenines in P4.1 that stabilize the ribozyme core through A-minor interactions 11 with helix P2.1 occupy similar positions in the T. tengcongensis and B. anthracis glmS ribozymes. Importantly, helix P2.2, which carries the nucleotide bearing the leaving group of the transesterification reaction (G1) on its 5′-terminus, and also lines one side of the metabolite-binding pocket, assumes the same position in the two ribozymes and in both the activator- and inhibitor-bound forms of the T. tengcongensis ribozyme.
FIGURE 3.
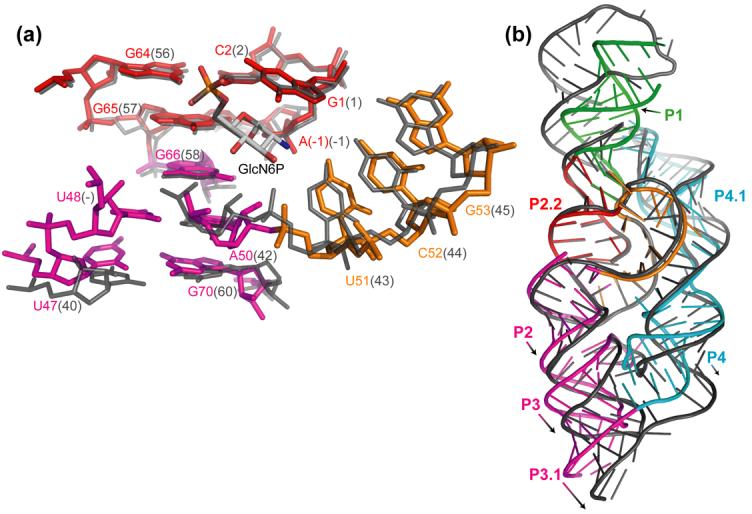
Superposition of the GlcN6P-bound structures of the T. tengcongensis (colored according the scheme in Figure 1(c)) and B. anthracis (dark gray) glmS ribozymes. (a) Detailed view of the nucleotides that comprise the GlcN6P-binding pocket. A ∼1 Å shift of the phosphodiester backbone between the two structures is evident between A50-G53 (A42-G45 in the B. anthracis numbering scheme). Deletion of nucleotide U48 in the B. anthracis ribozyme structure is also apparent. The conformation of the scissile phosphate is very similar between the T. tengcongensis GlcN6P-bound structure (τ = 166°, this work) and the four crystallographically independent structures of the GlcN6P-bound B. anthracis ribozyme (mean τ = 167°, standard deviation 3°, 10). (b) Global view of the overall agreement of the structures of the T. tengcongensis and B. anthracis glmS ribozymes. Large structural variation between the two ribozymes is limited to helical elements distant from the active site core.
P2.2 is required for cis-ribozyme activity
The wild-type cis-cleaving B. anthracis glmS ribozyme used for the biochemical studies, ANX1 (Figure 1(b)), cleaved to 80% (Figure 4(a)) with rate constant of 3.0±0.1 min−1 (Table 2) under standard conditions (2 mM MgCl2, 0.1 mM GlcN6P, pH 7.5, 25 °C). To determine if the base pairing in P2.2 is required for wild-type cleavage rates, two ribozymes, each with two nucleotide changes predicted to disrupt pairing in P2.2, were made and tested for activity. Cleavage activity of ribozymes with mutations in the 5′ side of P2.2 (G3C and C4G; P2.2-5′) or mutations in the 3′ side of P2.2 (G54C and C55G; P2.2-3′) was reduced. The rate constants decreased by a factor of 105 to 106 (1.1±0.1 × 10−5 min−1 and 7.1±0.6 × 10−6 min−1, respectively) relative to the wild-type sequence (Figures 4(a-c), Table 2). A ten-fold increase in the MgCl2 concentration (2 to 20 mM) did not restore cleavage for either P2.2-5′ or P2.2-3′ (Table 2).
FIGURE 4.

Testing the P2.2 pairing. (a) Cleavage of ANX1 and the P2.2 mutant ribozymes. Products were separated on a denaturing polyacrylamide gel and the positions of the precursor and 3' product are indicated. The 5' product is not visible. (b) P2.2 Mutations in the cis-cleaving ribozyme. Mutated regions in P2.2 are boxed and lowercase designates the bases that are non-wild type. (c) Kinetics of the cis-cleaving ribozymes. Cleavage of ribozymes ANX1 (circles) and P2.2-5'/3' (triangles) show first-order kinetics and cleaved rapidly (inset). Cleavage of the P2.2-5' mutant (squares) and P2.2-3' mutant (diamonds) are significantly slower than ANX1 and the compensatory mutant. Ribozyme cleavage reaction conditions were 2 mM MgCl2, 0.1 mM GlcN6P at 25°C, pH 7.5 (HEPES).
Table 2.
Kinetic analysis of the trans-acting B. anthracis glmS ribozyme.
| kobs (min−1) | ||||
|---|---|---|---|---|
| [MgCl2] | ANX1 | P2.2-5′ | P2.2-3′ | P2.2-5′3′ |
| 2 mM | 3.0±0.1 | 1.1±0.1 × 10−5 | 7.1±0.6 × 10−6 | 0.55±0.12 |
| 5 mM | 6.2±0.1 | 1.1±0.2 × 10−5 | 1.5±0.1 × 10−5 | 1.0±0.1 |
| 10 mM | 11±1 | 1.3±0.4 × 10−5 | 3.3±0.2 × 10−5 | 2.2±0.1 |
| 20 mM | 12±1 | 1.2±0.2 × 10−5 | 4.1±0.9 × 10−5 | 2.2±0.1 |
Observed rate constants for self-cleavage of the ANX1 and P2.2 mutant cis-cleaving ribozymes in varying MgCl2 concentrations. Mean values and standard deviations were calculated from at least three independent measurements at 25°C.
If loss of cleavage activity was due to disruption of base pairing in P2.2, then the compensatory mutant designed to restore Watson-Crick base pairing by combining the above changes in P2.2 (P2.2-5′/3′, Figure 4(b)) may restore activity. In this variant, the base identities at the two mutated positions were reversed with respect to the wild-type P2.2. Restoring the potential for base pairing increased the cleavage rate ∼105-fold over that of the mismatch (Figure 4(a,c), Table 2). These rates (0.55 min−1) were within 5-fold of wild-type rates. The decrease in cleavage rate when the sequence of P2.2 was changed was small compared to the effect that disruption of Watson-Crick base pairing had on activity. Importantly, the same base changes that severely reduced activity when introduced in only one strand of P2.2 had little effect on activity when combined such that Watson-Crick pairing was possible. The data from the compensatory change support a model in which pairing proposed for P2.2 was required for ribozyme cleavage. Had activity not been restored in the P2.2-5′/3′ construct, no conclusion about base pairing and sequence requirements in P2.2 could be drawn from the mutagenesis data alone.
A robust trans-acting ribozyme
The P2.2 cleavage-site duplex provided an appealing structural element to be exploited in constructing a trans-acting glmS ribozyme. A P2.2-based trans-acting ribozyme was created by dividing the B. anthracis glmS ribozyme between nucleotides 6 and 7. The “enzyme” portion was made by beginning transcription at G7 (tANX/G7). Winkler et al. 2 showed that ribozyme cleavage requires only one nucleotide 5′ to the cleavage site and we demonstrated that this is also true for the ribozyme in the crystalline state 3, so a heptamer oligoribonucleotide encompassing the wild-type sequence −1 to +6 was used as substrate. Pre-incubation and start conditions were optimized for both reaction rates and the extent of cleavage. Importantly, we observed reaction rates that were too fast to measure by hand (≥15 min−1) when attempting to saturate either MgCl2 or GlcN6P. Therefore, reactions were slowed to measurable rates by using 10 mM MgCl2 and 0.1 mM GlcN6P at 25 °C. Lowering the temperature below 25 °C reduced the extent of cleavage, presumably due to misfolding of a subpopulation of the RNA. Therefore, a full kinetic analysis including measurements of kcat, KM, as well as the apparent KD of GlcN6P for our trans-acting ribozyme will require a rapid quench system to measure reaction rates.
In the presence of the tANX/G7 ribozyme, the 5′ end-labeled substrate released a fast migrating labeled product that separated from the substrate oligoribonucleotide on PEI TLC plates (Figure 5(a)). The product was generated at a rate of 11 min−1 in the presence of 10 mM MgCl2 and 0.1 mM GlcN6P. The ribozyme product co-migrated with a product generated from a U2 nuclease digestion of the substrate (data not shown), verifying that the ribozyme is cleaving between A(−1) and G1. Treatment of the reaction product with acid resulted in a product that, at pH 5, migrated slower than untreated sample (Figure 5(b)). This result was consistent with acid hydrolysis of a 2′,3′-cyclic phosphate group to generate a 2′(3′)-terminal phosphate group. Generation of the 2′,3′-cyclic phosphate group in the ribozyme reaction suggests that the reaction most likely proceeds through a nucleophilic attack of the 2′ oxygen on the phosphorus of the adjacent scissile phosphate. Thus, the trans-cleavage reaction appears to accurately reproduce the cis-cleavage reaction 2.
FIGURE 5.

Cleavage of a short oligonucleotide in trans. (a) Reaction time course for the wild-type sequences. Cleavage products of the 5' end-labeled substrate 7-mer RNA were fractionated on PEI plates in 1M LiCl. The product comigrated with a pAp marker generated by nuclease U2 digestion (not shown). (b) The released 5' nucleotide contains a 2',3'-cyclic phosphate group. Treatment with HCl caused the product of the cleavage reaction to migrate slower at pH 5 (1M LiCl with 0.1M Boric acid pH 5). This result is consistent with a 5' product that contains a cyclic phosphate that is hydrolyzed by acid treatment.
P2.2 is required for trans-cleavage activity
Reconstitution of the cleavage reaction in trans is not, in itself, proof that a particular duplex was responsible for binding of the substrate fragment by the enzyme portion of the ribozyme. To test if the trans-reaction was dependent on formation of P2.2, the enzyme and substrate sequences were altered and tested in the cleavage reaction (Figure 6(a,b)) (sequence changes were equivalent to those made in P2.2 in the cis-cleaving B. anthracis glmS ribozyme). Changes to either the substrate (the 5′ side of P2.2) or the enzyme (3′ side) slowed the cleavage rates to 0.7 × 10−4 min−1 and 1.2 × 10−4 min−1, respectively (Figure 6(b,c)), a decrease of 105. However, when the altered substrate (P2.2 5′-side mutation) and the altered enzyme (P2.2 3′-side mutation) were combined (Figure 6(a)), the cleavage rate increased 2000-fold over that of the mismatched enzyme-substrate combinations. This result suggests that pairing in the alignment of the proposed P2.2 is required for the reaction in trans.
FIGURE 6.

Testing the P2.2 interaction in the trans acting ribozyme. (a) Sequences of the proposed trans interaction with mutated regions boxed. Lowercase designates mutated bases. (b) Reaction time courses. Products were fractionated by TLC (PEI) and positions of the uncleaved substrate and cleavage product are indicated on the left. (c) Kinetics of trans-cleaving ribozymes. Cleavage of wild-type enzyme with wild-type substrate (circles, kobs = 11±1 min−1), wild-type enzyme with altered substrate (squares, 0.7±0.5 × 10−4 min−1), altered enzyme with wild-type substrate (diamonds, 1.2±0.5 × 10−4 min−1), and altered enzyme with altered substrate (triangles, 1.3±0.1 × 10−1 min−1). Values are the average of three independent determinations. Reactions were at 25 °C in 10 mM MgCl2, pH 7.5 (HEPES) and 0.1 mM GlcN6P.
The nucleobase at position 1 is a determinant of cofactor specificity
The P2.2 trans-system was used to probe a specific cofactor-ribozyme interaction inferred from the crystal structures 3,10. A non-bridging phosphate oxygen of GlcN6P is positioned within hydrogen bonding distance (3.0 Å) of ring nitrogen N1 of residue G1 (Figure 2a). This interaction of G1 with the cofactor phosphate group could account for the preference of GlcN6P over the non-phosphorylated amino sugar GlcN in the cleavage reaction 7.
To test if the interaction between G1 and GlcN6P contributes to the cleavage rate, RNA oligonucleotide substrates used in the trans reactions were prepared with purine analogs at G1 (Figure 7). These substrates were tested with the tANX/G7 ribozyme for cleavage activity using four cofactor analogs: GlcN6P, GlcN, Serinol, and TRIS (Figure 7). Increasing enzyme concentrations above 100 nM gave no further increase in the rate of cleavage under the conditions used indicating that the enzyme was in saturating excess over the substrate oligonucleotide. The concentration of both GlcN6P and GlcN in these reactions was 0.1 mM. The concentrations of serinol and TRIS were raised to 10 and 80 mM, respectively. In all cases, the cofactor concentrations were sub-saturating so we report the rates as a second-order rate constant after normalizing for the cofactor concentration (M−1min−1, Figure 7). In these reactions, substrate containing the wild-type G1 was cleaved at a rate of 1.1±0.1 × 105 M−1min−1 in GlcN6P. Under the same conditions, with substrates containing purine analog substitutions for G1, the cleavage rates decreased dramatically. The highest rate in GlcN6P was with the guanosine analog inosine (I) (3.9±0.9 × 103 M−1 min−1) and the slowest rate was with the adenine analog dimethyladenine (DMA) (0.46±0.01 M−1 min−1) (Figure 7). Thus, among the substituted (nonguanosine) substrates, the rates in GlcN6P varied over a range of 104-fold. Notably, with the adenine substitution, the rate constant (1.6±0.1 × 101 M−1 min−1) was down 3 to 4 orders of magnitude relative to the wild-type substrate; this result is consistent with the large loss of activity seen in a cis-cleaving form of the B. subtilis glmS ribozyme with a G1A mutation 2. These data show that nucleobase modifications at position 1 can have a large effect on the rate.
FIGURE 7.
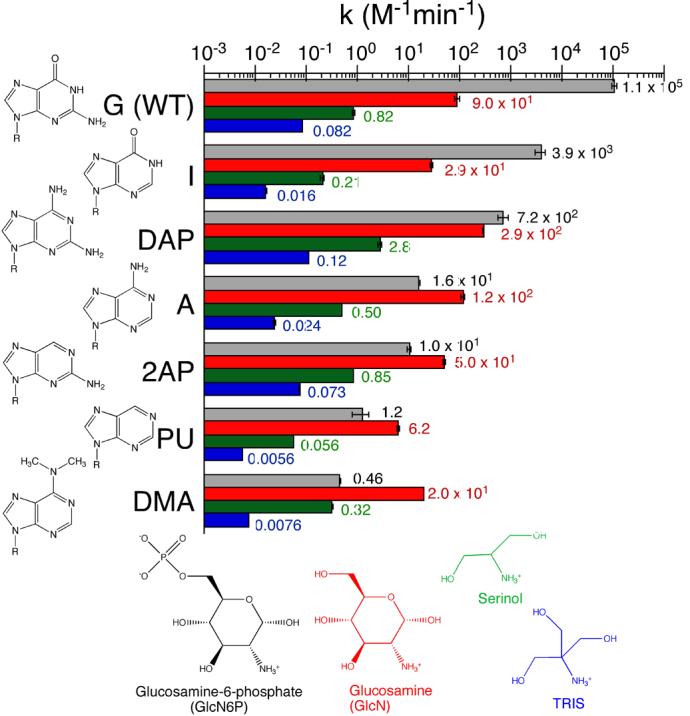
Comparison of cleavage rates of substrates containing nucleobase analogs at G1 with various cofactors. Cleavage reaction conditions were 10 mM MgCl2, 25 °C, pH 7.5 (HEPES) and the cofactor. Cofactor concentrations were 0.1 mM GlcN6P (black), 0.1 mM GlcN (red), 10 mM Serinol (green), and 80 mM TRIS (blue). The observed rate constants are the average of two or more independent determinations and were normalized to cofactor concentration. Structures of each of the nucleobases are shown on the left and the cofactors at the bottom.
To test if the decrease in cleavage activity with position 1 modifications could be attributed to the disruption of the G1-GlcN6P interaction, the effect on the cleavage rates of removing the phosphate group from the cofactor was examined. When GlcN was substituted for GlcN6P, the wild-type sequence cleaved 103-times slower (Figure 7, see also Ref. 7). This preference for GlcN6P over GlcN decreased to a factor of about 100-fold with inosine at position 1, and to only 2-3 fold with diaminopurine (DAP) at that position in the substrate. Moreover, the adenine, 2-aminopurine (2AP), purine (PU), and DMA-containing substrates cleaved faster in GlcN than in GlcN6P. The largest effect was seen with DMA, where the cleavage rate was 40 times faster in GlcN than in GlcN6P. These compensatory effects, seen as the reversal of cofactor preference, were taken as support for an important contribution of the GlcN6P-G1 interaction. Under the conditions used in these reactions, we did not observe saturation with either GlcN6P or GlcN, therefore we have not determined if the changes were due to an effect on binding or catalysis, or both.
The small molecule cofactors serinol and TRIS served as control reactions for possible differences in the reactivity of the substrates. There was moderate variation in cleavage rates among all of the substrates with these two cofactor replacements: ∼20 fold variation in TRIS and ∼50 fold in serinol (Figure 7). A similar variation in rate constants (∼50 fold) was seen in GlcN. We take this as evidence that the effect of nucleobase substitution on intrinsic substrate reactivity was small compared to the large effect (105 fold) that can be attributed to changes in GlcN6P binding or positioning.
DISCUSSION
The initial model for the secondary structure of the glmS ribozyme (Figure 1(a)) was derived in part by comparison of sequences from 18 Gram-positive bacteria 1,2. Sequences near the cleavage site and in and the region flanking P2 were invariant among the species examined 1. As these sequences lacked covariation, pairings involving them could not be determined by phylogenetic analysis. Mutagenesis is an alternative to phylogenetic analysis to test potential duplex elements. This is especially true with ribozymes where the catalytic activity provides a readout for a functional structure. Soukup 9 presented mutagenesis results that he interpreted as evidence for the formation of a duplex between sequences 3′ of the cleavage site and nucleotides in the 3′ side of P2 (Figure 1(a), gray line). Whereas mutations of these sequences that would disrupt the purported duplex did reduce activity, compensatory mutations designed to reestablish pairing did not restore activity 9. Two interpretations are that either there is a strong requirement for duplex of a particular sequence, or the postulated pairing is incorrect and an alternative feature has been disrupted by the mutagenesis. In the latter case, predicted compensatory mutations would fail to rescue activity because base pairing would not be reestablished.
The crystal structures of the glmS ribozyme confirmed the four helices P1, P2, P3, and P4, but in addition revealed that the phylogenetically invariant sequences that had not been assigned pairings participate in two short helices, P2.1 and P2.2 3,10. The presence of these two helices introduces a nested double pseudoknot into the core of the glmS ribozyme, in addition to the peripheral pseudoknot that had been previously discovered 12 near the 3′-end of the ribozyme domain of the UTR. Comparison of the structures of the T. tengcongensis glmS ribozyme in the unliganded pre-cleavage state, in the pre-cleavage state bound to Glc6P, and in the post-cleavage state showed that this catalytic RNA assembles into a remarkably rigid structure 3. Together with previous structural and biochemical studies of other ribozymes, such as the naturally-occurring hepatitis delta virus ribozyme 13,14 and the in vitro evolved Diels-Alderase 15,16, this work suggested that pseudoknotting is a strategy that relatively short catalytic RNAs can adopt in order to achieve a stable fold.
The analysis presented here provides experimental support for the P2.2 duplex seen in the crystal structures of the glmS ribozyme 3; 10 being required for catalytic activity. The key difference between these results and those of Soukup 9 is that mutations designed to restore pairing in P2.2 did restore cleavage activity. Moreover, these results would be inconsistent with the alternative pairing proposed by Soukup 9. For the ribozyme containing P2.2 compensatory mutations, cleavage rates and the extent of cleavage were somewhat lower than for the wild-type ribozyme. The small reduction in cleavage rates suggests that there could be a sequence requirement in P2.2 in addition to the duplex requirement. Cochrane et al. 10 identified two solvated Mg2+ ions that interact with the phosphate group of GlcN6P. While we do not observe clear electron density for these two Mg2+ ions in our native structures, anomalous difference Fourier maps derived from a crystal in which Mg2+ was replaced with Mn2+ confirmed that two divalent cations do coordinate the phosphate of GlcN6P when it is bound to the T. tengcongensis glmS ribozyme (data not shown). As one of these Mg2+ ions interacts with G62 (G54 in B. anthracis numbering) in P2.2, a ribozyme containing a compensatory C mutation in this position may interact more weakly with the cation, thus accounting for the observed reduction in cleavage activity. Nonetheless, and consistent with an absence of an absolute requirement for divalent ions for glmS ribozyme activity 17, the sequence effects observed were small, and relative to ribozymes with a mismatched duplex, the compensatory changes restored nearly full activity.
Unlike other well-characterized naturally occurring ribozymes, the glmS ribozyme requires GlcN6P, a low molecular weight metabolic intermediate, for activity 2. Unlike exogenous guanosine, which is a substrate of the reaction catalyzed by the group I intron 18, GlcN6P appears to function as a coenzyme for the glmS ribozyme 7,3,10. Comparison of the Glc6P-bound T. tengcongensis glmS ribozyme and the GlcN6P-bound B. anthracis glmS ribozyme demonstrated subtle structural differences near the active site 10. These could result from sequence differences between the two RNAs, or could indicate that the inhibitor and activator small molecules bind the ribozyme in slightly different manners. Superposition of the two high-resolution structures of the T. tengcongensis glmS ribozyme presented here shows that, within the precision of the crystallographic coordinates, the two small molecules bind the ribozyme in an identical manner. This further emphasizes the critical catalytic role played by the amine functional group of GlcN6P (the only chemical difference between the two small molecules). Moreover, because GlcN6P- but not Glc6P-binding is pH dependent, it appears that the ribozyme can preferentially bind the neutral amine form of a small molecule. This is consistent with Michaelis-Menten analysis of GlcN6P activation of the B. anthracis glmS ribozyme 10.
Despite some differences in the disposition of peripheral elements, the active site and metabolite-binding pockets of the T. tengcongensis and B. anthracis ribozymes superimpose extremely closely. In both ribozymes, P2.2 forms one of the sides of the metabolite-binding pocket, and is preceded immediately by G1. The purine nucleobase of this residue stacks on top of the pyranose ring of GlcN6P, and the N1 nitrogen of the base is within hydrogen-bonding distance of the phosphate of the activator. The apparent importance of P2.2 in positioning both the cleavage site (which immediately precedes G1) and the cofactor GlcN6P, its high degree of structural conservation in the ribozymes from the two bacterial species, and its proximity to the 5′-end of the ribozyme domain, made this duplex an attractive place to split the RNA in order to develop a trans-cleaving ribozyme. Successful construction of such a ribozyme would further confirm the functional importance of P2.2, but in addition would allow us to test crystallographically observed interactions made by this segment of the RNA using variant small synthetic oligonucleotide substrates.
The crystal structure of the glmS ribozyme guided design of a trans-cleaving ribozyme in which the interaction between substrate and enzyme is predicted to depend exclusively on the P2.2 interaction. A functional ribozyme could be reconstituted, and the reaction rates, under single-turnover conditions, were similar to what was seen in the cis-cleaving forms. In addition, the trans-cleavage reaction appeared to accurately recapitulate the cis-cleavage reaction in that it required GlcN6P and generated a product with a 2′,3′ cyclic phosphate. Several previously reported trans-cleaving forms of the glmS ribozyme have separated the ribozyme from a substrate strand within the hairpin loop at the end of P1 1,3,6. In those ribozymes, the substrate-enzyme interaction would be predicted to be more stable than the P2.2-based ribozyme made here as it involves base pairing in both P2.2 and P1. However, for some studies the version described here could be useful, especially if the use of short substrate RNA is advantageous. In that regard, we note that a hexamer (−1 to 5) was cleaved at the same rate as the heptamer (−1 to 6) used in this study (unpublished data).
We employed our trans-acting ribozyme to examine a possible interaction between G1 and the cofactor phosphate by measuring the effect on reaction rates of replacing G1 with various nucleobases and substituting GlcN6P with GlcN. Previously, it had been reported that a G1A mutation interferes with glmS ribozyme activity 2, and that replacing GlcN6P with GlcN also results in a large decrease in activity 7. Here, we found that replacing G1 with other guanine or adenine analogs also had a large effect on activity, comparable to or greater than what was seen with the GlcN substitution. Importantly, however, the adenine and adenine analog-containing substrates did not cleave appreciably slower in GlcN than they did in GlcN6P. A large preference for GlcN6P over GlcN was only seen with the inosine substitution, a close analog of guanosine. Inosine would be predicted to maintain its N1 hydrogen bond with the phosphate of GlcN6P, similar to what was seen with guanosine. If the decreases in activity with nucleobase substitutions or cofactor substitutions were due to effects that were independent of one another, we might expect an additive effect when they were combined. However, with some of the other nucleobase analogs, notably DMA, higher cleavage rates were seen with GlcN than with GlcN6P. This switch in cofactor preference suggests that there may have been some level of interference that was relieved upon replacing GlcN6P with GlcN. These results are consistent with a model in which the N1 of G1 contributes to the binding or positioning of the GlcN6P in the active site by hydrogen bonding with the phosphate group of the cofactor. Previous nucleotide analog interference mapping (NAIM) and suppression (NAIS) experiments designed to identify interactions between the ribozyme and the phosphate of GlcN6P failed to identify this key interaction involving G1 19. However, in those experiments only guanine analogs that contain an N1 with a pKa similar to the N1 of guanine were tested at position G1.
The results provided here support a model of P2.2, as seen in the crystal structure, being required for cleavage activity of the glmS ribozyme. The trans-reactions in particular suggest that this pairing is largely responsible for positioning the sequence containing the scissile phosphate in the active site. G1, the nucleotide immediately 3′ of the cleavage site, however, is not part of the P2.2 duplex but instead contributes to GlcN6P binding or orientation, or both. The importance of the N1 to phosphate oxygen-hydrogen bonding in the G1-GlcN6P interaction for optimal activity also suggests that it might be possible to design a cofactor that binds tighter than GlcN6P. Replacing the phosphate of GlcN6P with a group more able to take full advantage of the hydrogen bonding positions on the Watson-Crick face of G1 could enhance binding further. Such a molecule might be effective in targeting the ribozyme and causing it to cleave even when intracellular levels of GlcN6P are low. According to the model for how the glmS ribozyme is linked to regulation GlcN6P production in the cell 2, a tight-binding cofactor that enhanced cleavage would lead to decreased synthesis of GlcN6P synthase and thereby slow cell wall synthesis.
MATERIALS AND METHODS
Structure determination
RNA corresponding to the T. tengcongensis glmS ribozyme was prepared, purified, and crystallized as described previously 3. The structure of the glmS ribozyme bound to Glc6P was obtained exactly as described previously using our in-house X-ray source 3. The resolution of this structure was improved to 2.2 Å by collecting diffraction data using synchrotron X-radiation at the Advanced Light Source (ALS). To obtain the structure of the glmS ribozyme bound to GlcN6P, crystals were transferred directly to a dehydrating stabilization solution that contained 25% PEG 4K, 1.5-1.7 M LiCl, 30 mM MgCl2, 15 mM GlcN6P, 100 mM TRIS-HCl, pH 8.5 for 6-8 minutes prior to flash freezing in liquid nitrogen. Occasionally, dehydration caused the crystals to crack in the direction perpendicular to the c-axis. However, such cracks were easily avoided during data collection by aligning the c-axis parallel to the axis of rotation. Diffraction data were collected at 100K at beamline 5.0.2 and 8.2.2 at the ALS and processed with the HKL software package 20. Crystallographic refinement was carried out with CNS 21 using multiple rounds of torsion-angle simulated annealing, energy minimization, and individual B-factor refinement. Statistics for all structures are in Table 1. Structural figures were prepared with PyMOL 22.
Enzymes and Reagents for the Biochemical Studies
T7 RNA polymerase was purified by M. Puttaraju from an over-expressing clone provided by W. Studier 23. Oligodeoxynucleotides for mutagenesis and oligoribonucleotide substrates for the trans reactions were ordered from Integrated DNA Technologies (IDT). Additional substrate oligoribonucleotides with modified bases were ordered from Dharmacon, Inc. Other enzymes and chemicals were purchased from commercial sources.
Plasmid Construction
The plasmid (pANX1) used to prepare the wild-type B. anthracis cis-acting glmS ribozyme (ANX1) has been previously described 12. This sequence was modified by oligonucleotide-directed mutagenesis of a uracil-containing single-stranded form of the pANX1 24-25 to introduce mutations and compensatory changes in the P2.2 duplex region of the cis ribozyme and to generate the “enzyme” construct (ANX/G7) for the trans reactions. The same approach was used for mutagenesis of the P2.2 region in the trans-acting ANX/G7 sequence. Sequences were verified, and plasmid DNA for the mutants was prepared and purified as previously described 12.
In vitro transcription
For the kinetic studies, plasmid DNA was linearized by cutting with BamHI endonuclease and purified by phenol/chloroform extraction and ethanol precipitation. The transcription reactions (50 μl, 15 min, 37°C) contained 5μg of the cut DNA, 40 mM HEPES (pH 7.5), 20 mM MgCl2, 9 mM dithiothreitol, 2 mM spermidine, 1 mM each ATP, UTP, and GTP, 0.5 mM CTP, 10 μCi [α32P] CTP, and ∼300 U T7 RNA polymerase. Reaction products were separated by electrophoresis on a 6% polyacrylamide gel containing 7M urea and precursor RNA was identified by audioradiography, excised, and eluted from a gel slice, desalted by G25 gelfiltration, and recovered by ethanol precipitation. This RNA was stored in 1 mM HEPES (pH 7.5), 0.1 mM EDTA at −20°C. Unlabeled transcripts were prepared in a similar fashion and were located by UV shadowing.
5′-End Labeling
RNA oligonucleotides supplied from Dharmacon were deprotected according to manufacturer instructions. The 5′ ends of the oligonucleotides were labeled with 32P in 10 μl reactions containing 20 pmol RNA oligo, 4 μM γ32P ATP (7000Ci/mmol), 50 mM TRIS-HCl (pH 7.5), 2 mM MgCl2, 50 mM NaCl, and 5U polynucleotide kinase. After 30 min at 30 °C, the reactions were terminated by adding 40μl of formamide containing 100 mM EDTA and were separated by electrophoresis on a 12% polyacrylamide gel with 7M urea. The end-labeled RNA was identified by autoradiography of the gel and eluted from the isolated gel fragment.
Cleavage Assays
For the self-cleavage reactions (cis-cleavage), radiolabeled precursor RNA was first heated to 95°C for 3 min in 1 mM HEPES 7.5, 0.1 mM EDTA and then placed on ice. This RNA was then preincubated in the cleavage cocktail without GlcN6P at 37 °C for 15 min and then shifted to 25 °C for 5 min and a zero time point taken. To start the cleavage reaction, GlcN6P was added. Final concentrations for the cleavage reactions were 50 mM HEPES (pH 7.5), 2 mM MgCl2 (or higher where indicated), and 0.1 mM GlcN6P. Aliquots of the reactions were removed and stopped with EDTA and formamide as described above. The samples were fractionated on 6% polyacrylamide gels containing 7M urea. The gels were dried and the fraction of precursor that cleaved at each time was quantified with a Phosphorimager (Molecular Dynamics).
For the trans reactions, “enzyme” and “substrate” RNAs were first heated separately at 95°C for 3 min in 1 mM HEPES, 0.1 mM EDTA and placed on ice. The RNA was then combined and preincubated at 37 °C for 15 min in the cleavage cocktail minus cofactor and shifted to the reaction temperature of 25 °C for 5 min. For the zero time point, 2 μl of the preincubation was removed and spotted on PEI plate. To start the reaction, cofactor (GlcN6P or other cofactor) was added. Final concentrations for the cleavage reactions were 50 mM HEPES 7.5, 10 mM MgCl2, 100 nM “enzyme” RNA, 0.004-0.02 nM radiolabeled “substrate” RNA (trace amounts), and either 0.1 mM GlcN6P, 0.1 mM GlcN, 10 mM Serinol, or 80 mM TRIS-HCl (pH 7.5) as indicated. Final pH values (measured from mock reactions without RNA) were 7.3, 7.3, 7.6, and 7.5, for GlcN6P, GlcN, Serinol, TRIS-HCl, respectively. Aliquots of the reactions were spotted on PEI plates that had been pre-spotted with 2 μl of 0.5 M EDTA, and fractionated on a half plate (10 cm) using 1M LiCl as the mobile phase. To separate the 2′,3′-cyclic phosphate form of pAp from that with a 2′(3′)-terminal phosphate, 0.1M Boric Acid (pH 5) was included in the mobile phase and a full 20 cm plate was developed. Release of the labeled 5′ nucleotide from the substrate RNA was quantified as above.
All the cleavage reactions followed first-order or pseudo-first-order kinetics and data were fit to the equation ft = F × (1 − e−kt), where ft is the fraction cleaved at time t and F is the extent of reaction or the fraction that cleaved with the observed first-order rate constant k (kobs). For very slow reactions, similar rate constants were estimated from slope of the initial rate or by setting the endpoint (F) to 0.95 and fitting to the above rate equation. Kaleidagraph (Synergy Software) was used for curve fitting and generation of plots.
Acknowledgements
We thank the staff at Advanced Light Source beamlines 5.0.2 and 8.2.2 for assistance data collection. A.R.F. is a Distinguished Young Scholar in Medical Research of the W.M. Keck Foundation. D.J.K. is a Damon Runyon Fellow supported by the Damon Runyon Cancer Research Foundation (DRG-1863-05). This work was also supported by grants from the NIH (GM63576 and GM47233, to A.R.F. and M.D.B., respectively) and the W.M. Keck Foundation. Coordinates and structure factors for the GlcN6P- and Glc6P-bound glmS ribozyme structures have been deposited in the PDB with accession numbers 2Z74 and 2Z75.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Barrick JE, Corbino KA, Winkler WC, Nahvi A, Mandal M, Collins J, Lee M, Roth A, Sudarsan N, Jona I, Wickiser JK, Breaker RR. New RNA motifs suggest an expanded scope for riboswitches in bacterial genetic control. Proc Natl Acad Sci U S A. 2004;101:6421–6. doi: 10.1073/pnas.0308014101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Winkler WC, Nahvi A, Roth A, Collins JA, Breaker RR. Control of gene expression by a natural metabolite-responsive ribozyme. Nature. 2004;428:281–286. doi: 10.1038/nature02362. [DOI] [PubMed] [Google Scholar]
- 3.Klein DJ, Ferré-D'Amaré AR. Structural basis of glmS ribozyme activation by glucosamine-6-phosphate. Science. 2006;313:1752–1756. doi: 10.1126/science.1129666. [DOI] [PubMed] [Google Scholar]
- 4.Edwards TE, Klein DJ, Ferré-D'Amaré AR. Riboswitches: small-molecule recognition by gene regulatory RNAs. Curr. Op. Struct. Biol. 2007;17 doi: 10.1016/j.sbi.2007.05.004. in press. [DOI] [PubMed] [Google Scholar]
- 5.Hampel KJ, Tinsley MM. Evidence for preorganization of the glmS ribozyme ligand binding pocket. Biochemistry. 2006;45:7861–7871. doi: 10.1021/bi060337z. [DOI] [PubMed] [Google Scholar]
- 6.Tinsley RA, Furchak JR, Walter NG. Trans-acting glmS catalytic riboswitch: locked and loaded. RNA. 2007;13:468–477. doi: 10.1261/rna.341807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McCarthy TJ, Plog MA, Floy SA, Jansen JA, Soukup JK, Soukup GA. Ligand requirements for glmS ribozyme self-cleavage. Chem. Biol. 2005;12:1221–1226. doi: 10.1016/j.chembiol.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 8.Lim J, Grove BC, Roth A, Breaker RR. Characteristics of ligand recognition by a glmS self-cleaving ribozyme. Angew Chem Int Ed Engl. 2006;45:6689–93. doi: 10.1002/anie.200602534. [DOI] [PubMed] [Google Scholar]
- 9.Soukup GA. Core requirements for glmS ribozyme self-cleavage reveal a putative pseudoknot structure. Nucleic Acids Res. 2006;34:968–975. doi: 10.1093/nar/gkj497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cochrane JC, Lipchock SV, Strobel SA. Structural investigation of the GlmS ribozyme bound to its catalytic cofactor. Chem. Biol. 2007;14:97–105. doi: 10.1016/j.chembiol.2006.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nissen P, Ippolito JA, Ban N, Moore PB, Steitz TA. RNA tertiary interactions in the large ribosomal subunit: the A-minor motif. Proc. Natl. Acad. Sci. USA. 2001;98:4899–4903. doi: 10.1073/pnas.081082398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilkinson SR, Been MD. A pseudoknot in the 3' non-core region of the glmS ribozyme enhances self-cleavage activity. RNA. 2005;11:1788–1794. doi: 10.1261/rna.2203605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferré-D'Amaré AR, Zhou K, Doudna JA. Crystal structure of a hepatitis delta virus ribozyme. Nature. 1998;395:567–574. doi: 10.1038/26912. [DOI] [PubMed] [Google Scholar]
- 14.Wadkins TS, Perrotta AT, Ferré-D'Amaré AR, Doudna JA, Been MD. A nested double-pseudoknot is required for self-cleavage activity of both the genomic and antigenomic hepatitis delta viurs ribozymes. RNA. 1999;5:720–727. doi: 10.1017/s1355838299990209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Serganov A, Keiper S, Malinina L, Tereshko V, Skripkin E, Hobartner C, Polonskaia A, Tuan Phan A, Wombacher R, Micura R, Dauter Z, Jaschke A, Patel DJ. Structural Basis for Diels-Alder ribozyme catalyzed carbon-carbon bond formation. Nature Struct Mol Biol. 2005;12:218–224. doi: 10.1038/nsmb906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pitt JN, Ferré-D'Amaré AR. How RNA closes a Diel. Nat. Struct. Mol.Biol. 2005;12:206–208. doi: 10.1038/nsmb0305-206. [DOI] [PubMed] [Google Scholar]
- 17.Roth A, Nahvi A, Lee M, Jona I, Breaker RR. Characteristics of the glmS ribozyme suggest only structural roles for divalent metal ions. Rna. 2006;12:607–19. doi: 10.1261/rna.2266506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bass BL, Cech TR. Specific interaction between the self-splicing RNA of Tetrahymena and its guanosine substrate: implications for biological catalysis by RNA. Nature. 1984;308:820–826. doi: 10.1038/308820a0. [DOI] [PubMed] [Google Scholar]
- 19.Jansen JA, McCarthy TJ, Soukup GA, Soukup JK. Backbone and nucleobase contacts to glucosamine-6-phosphate in the glmS ribozyme. Nat Struct Mol Biol. 2006;13:517–23. doi: 10.1038/nsmb1094. [DOI] [PubMed] [Google Scholar]
- 20.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Meth. Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 21.Brünger AT, Adams PD, Clore GM, Gros P, Grosse-Kunstleve RW, Jiang J-S, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography and NMR system: a new software system for macromolecular structure determination. Acta Crystallogr. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 22.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA, USA: 2002. http://www.pymol.org. [Google Scholar]
- 23.Davanloo P, Rosenberg AH, Dunn JJ, Studier FW. Cloning and expression of the gene for bacteriophage T7 RNA polymerase. Proc. Natl. Acad. Sci. USA. 1984;81:2035–2039. doi: 10.1073/pnas.81.7.2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kunkel TA, Roberts JD, Zakour RA. Rapid and efficient site-specific mutagenesis without phenotypic selection. Meth. Enzymol. 1987;154:367–382. doi: 10.1016/0076-6879(87)54085-x. [DOI] [PubMed] [Google Scholar]
- 25.Perrotta AT, Been MD. A pseudoknot-like structure required for efficient self-cleavage of hepatitis delta virus RNA. Nature. 1991;350:434–436. doi: 10.1038/350434a0. [DOI] [PubMed] [Google Scholar]