

Abstract
Estrogen is an important hormone signal that regulates multiple tissues and functions in the body. This review focuses on the neurotrophic and neuroprotective actions of estrogen in the brain, with particular emphasis on estrogen actions in the hippocampus, cerebral cortex and striatum. Sex differences in the risk, onset and severity of neurodegenerative disease such as Alzheimer’s disease, Parkinson’s disease and stroke are well known, and the potential role of estrogen as a neuroprotective factor is discussed in this context. The review assimilates a complex literature that spans research in humans, non-human primates and rodent animal models and attempts to contrast and compare the findings across species where possible. Current controversies regarding the WHI (Women’s Health Initiative) study, its ramifications, concerns and the new studies needed to address these concerns are also addressed. Signaling mechanisms underlying estrogen-induced neuroprotection and synaptic plasticity are reviewed, including the important concepts of genomic versus nongenomic mechanisms, types of estrogen receptor involved and their subcellular targeting, and implicated downstream signaling pathways and mediators. Finally, a multicellular mode of estrogen action in the regulation of neuronal survival and neurotrophism is discussed, as are potential future directions for the field.
Keywords: Estrogen, steroid, spine density, synaptic plasticity, cognition, neuroprotection, brain, stroke, Alzheimer’s disease, Parkinson disease.
Introduction
Estrogen has been one of the most studied hormones in the scientific arena. A search of PubMed using the keyword “estrogen” reveals over 113,000 citations/publications since 1934, with more than 40% of the published papers appearing in the last 10 years. Thus, the area of estrogen research has seen a relative explosion in the last decade. Much of the interest has been sparked by a desire to understand the actions and mechanisms of estrogen throughout the body, and to formulate better diagnoses and treatment for breast cancer, osteoporosis, cardiovascular and neurological disease and a multitude of postmenopausal symptoms. Classically, estrogen is considered a “reproductive” hormone, due to its well-known role in feedback signaling in the hypothalamic-pituitary-ovarian axis. The reproductive actions and roles of estrogen have been reviewed extensively previously, and the reader is referred to several excellent reviews on this subject [1-5]. This review will focus on the “non-reproductive” effects of estrogen in the brain, specifically on the neuroprotective and neurotrophic/synaptic plasticity actions of estrogen. Much of the work in these areas has been conducted in rodent animal models; however, there is a growing body of work in humans and non-human primates, which will also be presented and discussed in context with the findings in rodents. There is also a small but growing literature on estrogen analogues, such as the clinically relevant selective estrogen receptor modulators (SERMs), and their effects on brain plasticity and neuroprotection, which will also be discussed. A major goal of the review is to discuss potential sites and mechanisms of action for estrogen in the modulation of plasticity, cognition and neuroprotection, and project possible future directions for these important fields of research. An additional goal is to discuss current controversies in the field, and propose potential future directions for research.
ESTROGEN AND NEUROPROTECTION
Cerebral Ischemia
Sex Differences and Neuroprotection
Indirect evidence that estrogen may be neuroprotective first arose from studies in intact animals on sex differences in brain injury. Studies on cerebral ischemia-induced brain damage in gerbils revealed that female gerbils had a lower incidence and less severe brain damage following carotid artery occlusion than male gerbils [6]. Subsequent studies in rats and mice found a similar sex difference, with young adult female rats and mice having smaller infarct volume as compared to young adult males following middle cerebral artery occlusion (MCAO) [7,8]. Female rats have also been reported to have greater survival rates as compared to males following diffuse traumatic brain injury [9]. Studies on sex differences in stroke in humans are few, focusing primarily on incidence, age of first stroke and outcome. A number of studies have documented that women are “protected” against stroke relative to men – at least until the years of menopause, when estrogen levels decline due to follicular depletion and stroke incidence increases in women [10-13]. Intriguingly, stroke outcome in postmenopausal women has been shown in several studies to be worse as compared to males, with postmenopausal women having a significantly higher disability and fatality rate as compared to men [11-14]. While there may be many reasons for the worse stroke outcome in women, it is interesting that the onset and diminished outcome of stroke in women parallels the time period of falling estrogen levels that occurs after menopause. This issue will be discussed more in the subsequent section on evidence for estrogen as a neuroprotective factor.
Estrogen as a Neuroprotective Factor in Cerebral Ischemia
The above-cited studies provided support for a potential “neuroprotective” factor in female animal studies and possibly in humans. That the protective factor is estrogen was supported by studies showing that removal of the ovaries in rats and mice eliminates the protective effect observed in females following cerebral ischemia [7,8]. Furthermore, many groups have shown that exogenous administration of estrogen dramatically reduces infarct volume following focal or global cerebral ischemia in ovariectomized female mice, rats and gerbils [8,15-24], in male rats and gerbils [25,26], and in aged, reproductively senescent female rats [27,28]. In fact Gibson et. al. [29] performed a systematic review of over 161 publications on the effect of estrogen treatment before and after cerebral ischemia in animals, which included 1304 experimental subjects. The systematic review revealed that estrogen dose-dependently reduced lesion volume after either transient or permanent ischemia. A number of studies also examined estrogen effect upon cerebral blood flow and showed it had little or no significant effect, implying that the neuroprotective effect of estrogen occurs directly at the level of the brain. The neuroprotective effect of estrogen in global cerebral ischemia, which affects primarily the CA1 region of the hippocampus, was shown to be correlated with significant improvements in recognition, working memory and spatial memory [30-32]. Likewise, the sensorimotor deficits observed in MCAO have been reported to be significantly reduced by estradiol treatment, an effect that correlated with estrogen reduction of infarct size [33]. As a whole, the above studies provide strong evidence that estrogen is a major neuroprotective factor that contributes to sex differences in cerebral ischemia damage in intact male and female animals.
Additional studies provided more support for “endogenous” estrogen functioning as a neuroprotective factor in females, as it was demonstrated that serum estrogen levels are inversely correlated with ischemic stroke damage in intact animals, and treatment of intact female mice with an anti-estrogen receptor compound, ICI182,780, significantly enhanced stroke infarct size [34,35]. Furthermore, elegant studies in an estrogen-deplete animal model (e.g. mice in which the estrogen biosynthetic gene, aromatase has been knocked out) revealed that intact and ovariectomized aromatase knockout mice have increased MCAO-induced infarct size [36]. Additional studies showed that aromatase inhibitors caused a similar increase in cortical and striatal damage following cerebral ischemia [36], and that hippocampal sensitivity to kainic acid-induced neuronal death was markedly increased in gonadectomized rats that received an aromatase inhibitor as compared to vehicle-treated controls [37]. Intriguingly, brain aromatase levels were shown to increase in astrocytes in the peri-infarct area following MCAO, which may facilitate survival of neurons following the insult [38]. As a whole, these findings emphasize the importance of gonadal-derived circulating estrogen in protecting the brain against ischemic insult. They also support a potential role as well for brain-derived estrogen synthesis in neuroprotection, as evidenced by the finding that ischemic brain injury was smaller in ovariectomized wild type animals than in aromatase knockout mice [36], and that aromatase expression increases in the peri-infarct area following cerebral ischemia [38].
SERMs and Neuroprotection in Cerebral Ischemia
It is well known that estrogen can have undesired stimulatory effects on the breast and uterus, which raises concern for a potential increased risk of developing breast and uterine cancers and increased risk for stroke. These potential limitations have kindled interest in the development and therapeutic use of nonsteroidal SERMs. A number of studies have now appeared in the literature demonstrating neuroprotective actions of nonsteroidal SERMs. For instance, pretreatment with the raloxifene analogue LY353381.HCl, a raloxifene analogue, protected the caudate-putamen region of the brain of OVX female rats in an ischemia-reperfusion model of ischemic stroke [39]. LY353381.HCl did not affect cerebral blood flow, suggesting a potential direct neuroprotective effect of this SERM in the brain. Additionally, several studies have shown that the SERM, tamoxifen also significantly reduces infarct size in both transient and permanent occlusion/reperfusion models of cerebral ischemia [40-43]. Like the raloxifene analogue, the protective effect of tamoxifen was independent of cerebral blood flow changes, indicating a potential direct neuroprotective effect of this SERM in the brain. Interestingly, intravenous injection of tamoxifen was protective even up to three hours after cerebral ischemia [43]. These studies suggest that development of nonsteroidal SERMs that specifically target the brain for neuroprotection may be possible [44], and should be a focus for future studies in the area.
Potential Mechanisms of Estrogen Neuroprotection in Cerebral Ischemia
Several studies have shown that a 24h pretreatment and enhanced gene expression is needed to observe the protective effects of estrogen in cerebral ischemia [24,32,45], although there are dissenting reports in the literature [31,46,47]. Several groups have thus explored the potential role of estrogen receptors in mediation of the neuroprotective effects of estrogen. Administration of the potent ER antagonist, ICI182,780, dramatically increased infarct size in intact female rats following MCAO [35]. ICI182,780 also blocked neuroprotection by estrogen in global ischemia and in cortical explant studies [25,48]. Two ER isoforms have been identified to date, ERα and ERβ, both of which are expressed in the adult brain and thus could mediate the neuroprotection by estrogen. Evidence supporting a neuroprotective role for ER-α has come from studies demonstrating that estrogen-mediated neuroprotection was lost in OVX estrogen receptor-α knockout (ERKO) mice [49,50]. ER-α mRNA was also up-regulated in the penumbra region following MCAO in rats, further suggesting a possible role for ER-α in neuroprotection [51]. In contrast, estrogen protected the brain of OVX estrogen receptor-β knockout (BERKO) mice in a manner similar to that observed in OVX wild-type mice. Together, these findings suggest estrogen is neuroprotective via mediation by ER-α. However, these findings are in contrast to those of Sampei et al. [52], who failed to demonstrate a critical role for ERα in neuroprotection following ischemic stroke. Intact wild-type and ERKO mice sustained a similar infarct volume following ischemic stroke, leading to the conclusion that ERα is dispensable for protection by endogenous estrogen. Interestingly, other studies using ER-subtype selective agonists have implicated ER-β [53] or ER-α and ER-β [25] in estrogen mediated protection against global ischemia. ER-α and ER-β selective agonists have also been shown to be protective in hippocampal neurons against glutamate-induced cell death [54]. A third potential estrogen receptor is the recently discovered GPR30. GPR30 is a g-protein-coupled receptor which has been reported to bind estrogen with high affinity, and has been shown to be expressed in breast cancer cells and various tissues in the body, including the brain [55-57]. The role, if any, of GPR30 in estrogen actions in the brain is unknown. However, preliminary work by our laboratory and others has shown that GPR30 is expressed in various regions of the brain including the hippocampus, cortex and striatum, and may thus have a role in mediating estrogen actions [58-60]. Further work is needed to address this issue. As a whole, the current data suggests that ERα and ERβ may exert neuroprotection in the brain, with the role of GPR30 unexplored.
With respect to genes regulated by estrogen that may facilitate its neuroprotection, estrogen has been shown to increase the expression of the anti-apoptotic gene, bcl-2, in the ischemic penumbra following MCAO and global ischemia [51]. Furthermore, ovariectomized transgenic bcl-2 over-expressing mice have a significant decreased infarct volume following MCAO as compared to wild type ovariectomized mice [61]. Estrogen also increases bcl-2 in vitro in rat hippocampal neurons [54,62] and human NT2 neurons [63], while it inhibits expression of proapoptotic BAD (bcl-2-antagonist of cell death) [51,54,61]. Additionally, estrogen has also been demonstrated to reduce cytochrome c translocation [64,65], as well as caspase 3 activation and DNA fragmentation [21,65,66], further implicating an anti-apoptotic action of estrogen in cerebral ischemia.
In addition to a genomic effect, nongenomic effects of estrogen may also play a role in mediating its neuroprotective effects in the brain. For instance, estrogen can rapidly activate the extracellular signal-regulated kinases (ERK) and phosphoinositol-3-kinase (PI3K)-Akt pathways in cortical and hippocampal cells in vitro, effects implicated in estrogen neuroprotection action [67-69]. Estrogen has also been shown to enhance Akt activation in the cerebral cortex and CA1 of the hippocampus following focal or global cerebral ischemia [65,70]. Akt can phosphorylate two downstream death kinases to inactivate them, BAD and GSK-3β (glycogen synthase kinase-3β), which could be another important mechanism of estrogen neuroprotection [71-73]. Additionally, recent work by Wang et. al. [70] revealed that estradiol benzoate treatment-induced phosphorylation of Akt in the CA1 following cerebral ischemia is associated with inhibition of the prosapoptotic MLK3-MKK4/7-JNK1/2 (mixed lineage kinase-3/MAP kinase kinase-4-7/c-jun-N-terminal kinase) pathway, an effect blocked by pretreatment with the ER antagonist ICI182,780. These findings suggest that kinase activation/deactivation is an important component in estrogen-induced neuroprotection, and that both nongenomic and genomic mechanisms contribute to estrogen protective actions in the brain.
The mechanism whereby, estrogen interacts with ERK and Akt is unclear, but work in breast cancer cells has shown that ER-α can interact with the cytoskeleton protein, p130Cas in a complex containing Src and PI3K (which are upstream regulators of ERK and Akt activation, respectively), and siRNAs to p130Cas attenuated the ability of estrogen to increase ERK activation [74]. Additionally, ER-α has also been shown to interact with the calmodulin-binding protein, striatin, in vascular cells, which facilitates its targeting to the cell membrane and is critical for estrogen activation of Akt and eNOS (endothelial nitric oxide synthase) [75]. Striatin has been shown to be highly expressed in the striatum, with moderate expression in the cortex and hippocampus [76], while p130Cas localization in the brain has not been described in detail to our knowledge. The colocalization of striatin or p130Cas with estrogen receptors in the brain is unknown, as is their precise role in estrogen signaling and effects in the brain. Studies on these issues are needed. Finally, our laboratory recently cloned a protein in the rat and monkey called MNAR (modulator of nongenomic activity of estrogen receptor), which has been shown to serve as a scaffold protein in human breast cancer cell studies to link ER-α and ER-β to various kinases, including Src and PI3K, and is important in their activation by estrogen [77-79]. Further work by our laboratory showed that MNAR is expressed in both neurons and astrocytes in the brain and is highly colocalized with ER-α (77,78). Thus, MNAR may be a key factor linking ER to its kinase targets in the brain and may facilitate nongenomic signaling by estrogen. Work is underway in our laboratory to further explore the role of MNAR in estrogen actions in the brain, including its neuroprotective and plasticity effects.
Astrocytes, Microglia and Estrogen Neuroprotection
Astrocytes
In addition to a potential direct protective action on neurons, there is evidence that estrogen may also have indirect effects on other cell types such as astrocytes and microglia that may facilitate its neuroprotective actions in cerebral ischemia and other neurodegenerative disorders. With respect to astrocytes, physiological concentrations of estrogen are clearly protective in organotypic cortical explants and neuronal-astrocyte cocultures, and there is evidence of astrocyte mediation of estrogen effects [80-85]. For instance, work by our laboratory and others has provided evidence of a role for astrocyte-derived transforming growth factor-β (TGF-β) in the protection of cortical and hippocampal neurons from serum deprivation- or β-amyloid-induced cell death [82,83]. Furthermore, TGF-β has also been shown to be neuroprotective in cerebral ischemia, while conversely antagonists of TGF-β increase neuronal cell death and infarct size [86,87; 88, for review]. Work from our laboratory recently provided evidence of an estrogen-astrocyte-TGF-β1 neuroprotective signaling pathway. Along these lines, both estrogen receptor-α and −β were shown to be expressed in cortical astrocytes in vitro [85]. Furthermore, incubation of cultured rat cortical astrocytes with estradiol (10 nM) or TMX (1 μM) induced the release of both TGF-β1 and TGF-β2 from 6h-36h after treatment [85].
The effect of estradiol on TGF-β1 release from cultured cortical astrocytes following 18 h treatment and the role of nongenomic cell signaling is illustrated in Figure 1. As shown in Figure 1, estradiol (E2) or the cell impermeable estradiol-BSA (E2-BSA) induced a significant increase in TGF-β1 release from the cortical astrocytes and treatment of astrocytes with LY294002 or wortmannin, specific PI3K inhibitors, or Akt inhibitor, which directly prevents Akt activation, completely blocked the induction of TGF-β1 release by E2 or E2-BSA (Fig. 1A). Assays of cell death did not reveal a loss of cell viability due to LY294002 or Akt inhibitor I pretreatment, suggesting that this effect is not due to passive growth factor release after cell death induced membrane disruption. In contrast, the MAPK (MEK) inhibitors, PD98059 and U0126, were ineffective at inhibiting E2-or E2-BSA-induced TGF-β1 release (Fig. 1B). To implicate Akt in the induction of TGF-β1 release by E2, astrocytes were transfected with a dnAkt, which contains a Ser3Ala mutation at amino acid 473, rendering Akt incapable of activation and thereby preventing its actions downstream. Over-expression of dnAkt completely inhibited the ability of E2 or E2-BSA to induce TGF-β1 release and resulted in TGF-β1 levels equivalent to those in vehicle-treated controls (Fig.1C). Conversely, over-expression of myrAkt increased TGF-β1 to levels significantly higher than those in E2- or E2-BSA-treated cultures. Empty vector transfection had no effect on TGF-β1 release after E2 or E2-BSA treatment, confirming the specificity of the dnAkt and myrAkt effects (Fig.1C) and implicating the PI3K pathway in TGF-β1 release. Further work suggested that the enhanced release of TGF-β1 was important for the neuroprotective effect of estrogen in mixed astrocyte-neuronal cell cultures (Figure 2) [84,85]. Using a neuronal-glial coculture model, pretreatment with physiological levels of E2 [85], therapeutic levels of TMX, or E2-BSA rescued cultures from camptothecin, a neuronal-selective apoptosis inducer (Fig. 2A). This effect required E2/TMX pretreatment, because co-treatment at the time of camptothecin exposure failed to prevent cell death (Fig. 2B). These effects did not occur directly at the level of the neuron, as neither E2, TMX, nor E2-BSA reversed camptothecin-induced cell death in purified cortical neuronal cultures(which contain ~97% neurons and ~2% astrocytes), suggesting that glial cells may be involved in the neuroprotective effect (Fig. 2C). The neuroprotective effects also were dependent, at least in part, on TGF-β1 release, because immunoneutralization of TGF-β using a pan-specific TGF-β antibody or a TGF-β1-specific immunoneutralization antibody, reversed the observed protection by E2/TMX/E2BSA (Fig. 2D). In contrast, basic fibroblast growth factor immunoneutralization had no effect on E2- or TMX-induced neuroprotection, demonstrating the specificity of the TGF-β effect (data not shown). The protective effect of E2/TMX was also ER and PI3K mediated, because both ICI182,780 and LY294002 inhibited neuroprotection, implicating the E2-induced activation of Akt and subsequent release of TGF-β in neuroprotection (Fig. 2E).
FIG. 1. Effect of PI3-K/Akt on E2-induced TGF-β release in rat cortical astrocytes.
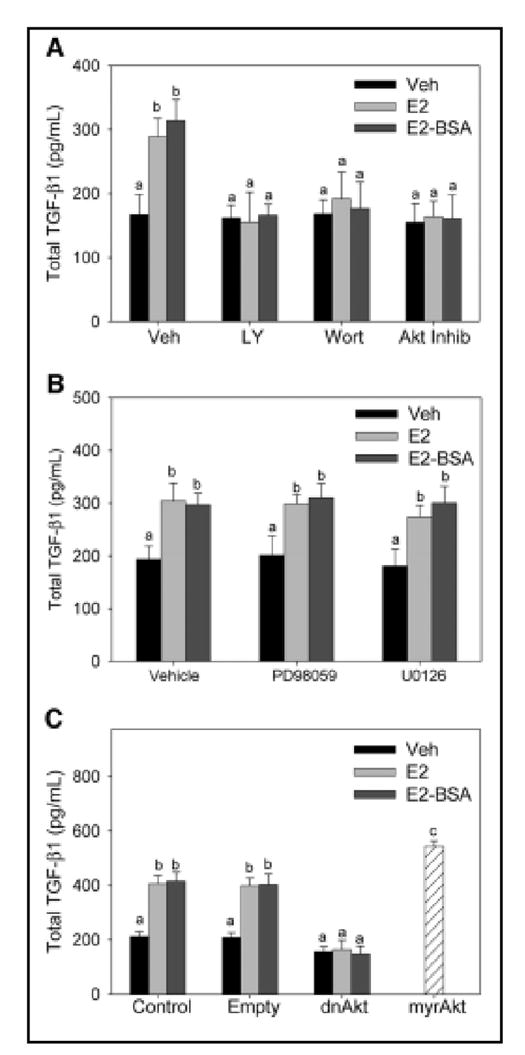
A, Rat cortical astrocytes were cotreated with E2 in the presence or absence or the PI3K inhibitors, LY294002 (20 μM) and wortmannin (200 nM), or Akt inhibitor prevented the induction of TGF-β1 release by E2 after an 18-h treatment. B, Cotreatment of rat cortical astrocytes with E2 in the presence of MAPK kinase inhibitors, PD98059 (30 μM) and U0126 (10 μM), was without effect on E2-induced TGF-β1 release. C, Overexpression of a dnAkt construct prevented E2-induced release of TGF-β1 in rat cortical astrocytes. Conversely, myrAkt significantly increased TGF-β1 release compared with that by empty vector-transfected astrocytes. For all studies, there were six wells per group, and experiments were performed in three independent cultures. Different superscripts denote significant differences between groups (P < 0.05, by one-way ANOVA, Student-Newman-Keuls post hoc test). Reproduced with permission from: Dhandapani, K. M. et al. Endocrinology 2005;146:2749-2759.
FIG. 2. Effect of astrocyte-derived TGF-β on E2- and TMX-mediated neuroprotection from camptothecin (CPT) in mixed cortical cultures.
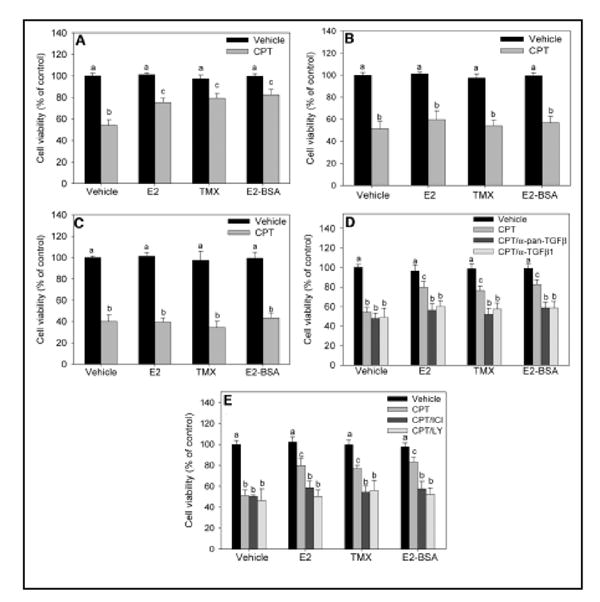
A, Effects of E2, TMX, and E2-BSA on cell death induced by CPT in mixed glial-neuronal cultures. Mixed cultures were pretreated for 24 h with 10 nM E2, 1 μM TMX, or 100 nM E2-BSA before 10 μM CPT treatment for 24 h before determination of cell viability. B, Treatment of glial-neuronal mixed cultures with E2, TMX, or E2-BSA at the time of CPT treatment does not affect cell death 24 h later. C, Pretreatment of purified cortical neurons with E2, TMX, or E2-BSA does not reverse the cell death induced by 24 h 10 μM CPT. D, Mixed cultures were pretreated with E2, TMX, or E2-BSA in the presence or absence of a pan-specific TGF-β isoform-neutralizing antibody (α-pan-TGFβ) or a TGF-β1-specific neutralizing antibody (α-TGF-β1), followed by a 24-h CPT exposure. Cell viability was determined 24 h after treatment with CPT. E, Effects of the ER antagonist, ICI182,780, and the PI3-K inhibitor, LY294002, on E2- and TMX-mediated rescue. Mixed cultures were pretreated with E2, TMX, or E2-BSA in the presence or absence of 1 μM ICI182,780 or 20 μM LY294002 for 24 h. Cultures were then exposed to CPT for another 24 h, followed by determination of cell viability. For all studies, cellular viability was determined using the MTT assay. Vehicle-treated cultures were considered to be 100% viable, and all treatment groups were compared with these control cultures. Viability was also confirmed using lactate dehydrogenase release assays (data not shown). In all panels, data are expressed as the mean ± SEM, and groups with different superscripts are significantly different from each other (P < 0.05, by one-way ANOVA, Student-Newman-Keuls post hoc test). For all studies, there were six wells per treatment group and experiments were performed in three independent sets of cultures for verification of results. Reproduced with permission from: Dhandapani, K. M. et al. Endocrinology 2005;146:2749-2759.
Estrogen has also been implicated in regulating other astrocyte-specific factors that could facilitate its neuroprotective effects. For instance, estrogen has been shown to enhance glutamine synthetase, an astrocyte-specific enzyme which is critical for producing glutamine [89]. Astrocyte-derived glutamine is taken up by neurons, and is critical for their survival as it is used to produce the key neurotransmitter glutamate [89]. Estradiol also enhances the expression of the astrocyte glutamate transporters, GLAST (glutamate/aspartate transporter) and GLT-1 (glutamate transporter-1), which could serve as a key neuroprotective effect in the brain by reducing damaging extracellular glutamate levels [90]. As mentioned previously, brain-derived estrogen may play an important role in protecting the brain, as evidenced by the aromatase mutant mouse studies described previously [28]. Astrocytes may participate in the production of brain-derived estrogen as they have been shown to express aromatase and this expression is enhanced following cerebral ischemia and following brain injury [31,91]. Thus, local production of estrogen by astrocytes could be another mechanism of astrocyte-mediated protection of neurons. As a whole, these studies suggest that astrocytes may play an important role in mediating the protective effects of estrogen. Since much of this work was performed in vitro, novel approaches to study the role of astrocytes in estrogen neuroprotective effects in vivo are clearly needed. While this has been a difficult problem to address in the past, it is hoped that mutant animal approaches that allow specific ablation of astrocyte factors and pathways may further facilitate our understanding of the role of astrocytes in estrogen neuroprotection.
Microglia
After neuronal injury and in many neurodegenerative disorders, activated microglia are known to secrete proinflammatory factors that can contribute to the progressive neural damage. A number of studies have suggested that estrogen may act upon microglia to suppress their activation, an effect that could help mediate estrogen neuroprotection [92-96]. Along these lines, Wen et. al. [92] recently demonstrated that estradiol reduces inflammatory responses during transient cerebral ischemia, including reductions in IkappaB phosphorylation, NF-kappaB activation and iNOS (inducible nitric oxide synthase) over-expression. The inhibition of iNOS over-expression is intriguing, as microglia are an important site for iNOS production, and microglia induction of neuronal cell death has been suggested to be due to a “proximity-dependent” mechanism involving nitric oxide [97]. Other work has shown that estrogen pretreatment in vitro attenuates immunostimulated microglial superoxide anion release, phagocytic activity and inflammatory markers in primary rat microglial and N9 microglia cell lines [93,94].
Recent work has further implicated reactive oxygen species (ROS), particularly superoxide, as playing a key role in neuronal cell death following cerebral ischemia [98-103]. The superoxide anion radical (O2) is the product of a one electron reduction of oxygen and it is the precursor of most ROS and a mediator in oxidative chain reactions [104, for review]. O2- combines spontaneously or is dismutated with H+ to produce H2O2, which readily breaks down in the presence of metals Fe++ and Cu+ or O2to form hydroxyl (OH) radicals. OH radicals are one of the strongest oxidants in nature and are highly injurious to adjacent structures, including lipid membranes, DNA, and proteins [99]. In both permanent and transient cerebral ischemia, ROS have been shown to increase significantly following onset of cerebral ischemia [100,102,103]. Along these lines, online in vivo ROS chemiluminescence measurement shows a marked steady elevation of ROS in the penumbra (infarct border) of the parietal cortex during a 3h measurement period post ischemia in permanent cerebral ischemia [100]. Likewise, studies using a marker of O2- Z production, hydroethidine (HEt), have yielded a similar pattern of increased superoxide production in male mice within 2 hrs of permanent cerebral ischemia [102,103]. HEt is freely diffusible into the CNS parenchyma after an intravenous injection and is selectively oxidized to ethidium (Et) by O2-, but not by other ROS such as hydrogen peroxide, hydroxyl radical, or peroxynitrite [105,106]. The oxidized HEt is detected by fluorescent microscopy, allowing in situ detection of O2- production following cerebral ischemia. We therefore sought to assess whether physiological (proestrus) levels of estradiol (80-100 pg/ml) achieved by subcutaneously estradiol pellet implants 1 week prior to pMCAO can decrease O2- production in the ipsilateral cortex of ovariectomized rats. HEt (1mg/ml) was administered intravenously 15 minutes prior to pMCAO in placebo and estradiol-treated ovariectomized rats, and the animals were sacrificed at 1h following cerebral ischemia for fluorescent microscopic analysis of oxidized HEt signal. As shown in Figure 3, there was little oxidized HEt in the placebo contralateral (non-injured) cortex 1h following cerebral ischemia, demonstrating that O2- production was low in the non-injured cortex. In contrast, the ipsilateral (injured) cortex showed a robust O2- production 1h following cerebral ischemia as indicated by high expression of oxidized HEt. Of significant interest, estradiol-treated animals had a significant reduction in O2- production in the ipsilateral cortex at 1h following cerebral ischemia, as indicated by a significant reduction in oxidized HEt fluorescent signal and the number of oxidized Het-positive cells (Figure 3). The ability of estradiol to reduce damaging O2- production in the cortex penumbra following cerebral ischemia could play a role in its neuroprotective effects following cerebral ischemia. Likewise, Kii et. al. [107] reported that estradiol significantly attenuated hydrogen peroxide production in the striatum following cerebral ischemia, which was correlated with the neuroprotective effect of estrogen. Prokai et. al. [108] also recently reported that estrogen (estradiol and estrone) has intrinsic free radical scavenging capability through its capture of hydroxyl radicals, which produces a nonphenolic quinol. The quinol is then converted back to estrogen by using NAD(P)H as a coenzyme, without production of reactive oxygen species. Estradiol quinol and estrone quinol were shown to be neuroprotective in hippocampal neuronal cells in vitro and in cerebral ischemia, respectively [108]. That the estrogen receptor may not be essential for all estrogen neuroprotection was also suggested by the work of Rothman and coworkers [109], who showed that an estrogen analog (ZYC-5), that lacks activity at estrogen receptors, was neuroprotective via a free radical scavenging mechanism. Other studies have confirmed estrogen neuroprotection against oxidative stress in various cell and animal models of neurodegenerative disorders, most prominently Parkinson’s disease, which will be discussed in the next section [110-113].
FIG. 3. Effect of 17β-Estradiol (E2) on superoxide anion O2- production in the ipsilateral cortex in ovariectomized animals 1h following permanent middle cerebral artery occlusion (pMCAO).
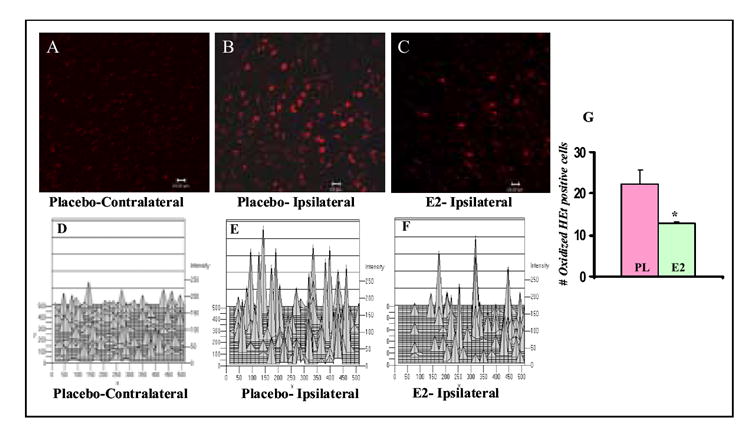
Panels A-C: Representative photomicrographs showing O2- production 1h after pMCAO in contralateral (non-injured) (Panel A) and ipsilateral (injured) cortex (Panel B) of placebo-treated ovariectomized adult rat, and in ipsilateral cortex of E2-treated ovariectomized adult rat (Panel C). O2- production was shown by oxidized HEt signals (bright red). Panels D-F: Corresponding 2-dimensional histograms of the fluorescence intensity for the images shown in panels A-C. Panel G: Graph showing difference in number of oxidized HEt-positive cells in the ipsilateral cortex of Placebo- vs. E2-treated animals at 1h following pMCAO. The results show that E2 markedly decreases O2- production as measured by oxidized HEt intensity and number of oxidized HEt positive cells in the ipsilateral cortex at 1h after pMCAO. N = 3 animals per group, 5-6 sections counted per group, and the experiment was repeated twice with similar results. P < 0.05 vs. Placebo group.
Figure 4 shows a summary diagram for the proposed multi-cell, multi-mechanism protection afforded by estrogen in cerebral ischemia. In addition to a direct neuroprotective effect upon neurons, the model proposes that estrogen acts on astrocytes to enhance release of protective growth factors and to regulate other astrocyte genes and proteins such as glutamine synthetase and glutamate transporters that facilitate protection of neurons by ensuring adequate precursor pools for glutamate transmitter production and helps remove excess damaging glutamate from synaptic clefts. Astrocytes have also been suggested to be sites for local brain production of estrogen, which may provide a mechanism for local protection during insults to the brain. The model also proposes an anti-inflammatory effect of estrogen which inhibits microglia (and possibly reactive astrocyte) production of inflammatory cytokines and free radical production that cause inflammatory damage to neurons. Estrogen has also been demonstrated to have important protective effects on cerebral endothelial cells by increasing mitochondrial efficiency, decreasing free radical production, promoting cell survival, and stimulating angiogenesis [114, and 115 for review]. Finally, the hypothetical model further proposes that genomic regulation of anti-apoptotic genes such as those in the Bcl family helps mediate estrogen neuroprotection, as does a nongenomic rapid regulation of activation of kinases and associated downstream cell signaling. A free radical scavenging effect of estrogen has also been demonstrated in the literature and may additionally contribute to overall estrogen neuroprotection. This hypothetical model of estrogen neuroprotection has applicability to not only protection from cerebral ischemia damage, but also other neurodegenerative disorders such as Parkinson’s disease and Alzheimer’s disease, which will be discussed in the following sections.
FIG. 4. Proposed multicellular mechanism of estrogen neuroprotection.
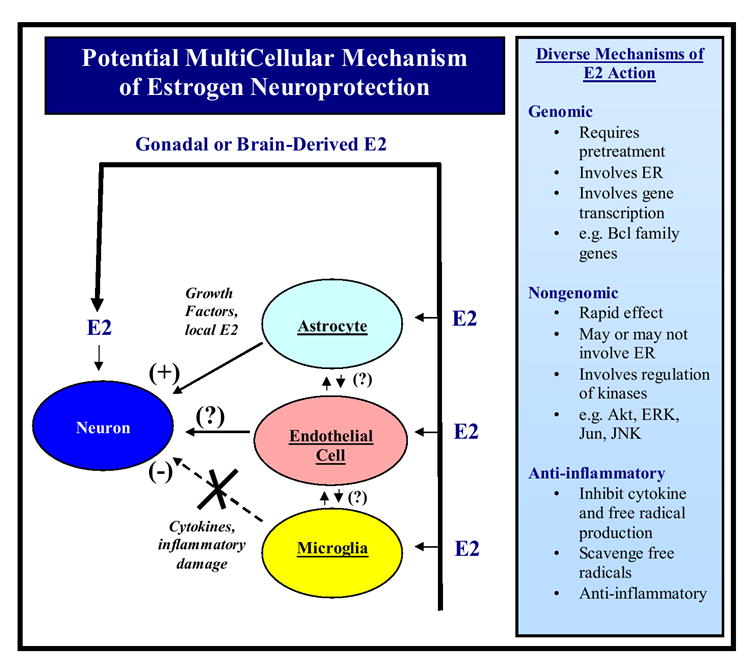
Estrogen neuroprotection is proposed to be mediated by genomic, nongenomic and anti-inflammatory mechanisms. Both direct effects on neurons and indirect effects mediated via astrocytes, endothelial cells and microglia are suggested to contribute to the overall protective actions of estrogen in the brain. E2 = 17β-Estradiol, ER = estrogen receptor, ERK = extracellular signal regulated kinase, JNK = Jun NH(2)-terminal kinase, (?) = unknown mediators. See text for further detailed description.
Parkinson’s Disease
Parkinson’s Disease
Parkinson’s disease (PD) is a chronic neurodegenerative disorder of unknown etiology. It is characterized by destruction of dopaminergic neurons in the substantia nigra, which leads to the hallmark symptoms of tremor, slowness of movement, rigidity and difficulty with balance. Recent studies have suggested there may be sex differences in PD risk, symptom severity, and treatment outcome. For instance, a number of studies have reported a higher incidence of PD in men as compared to women [116-118], with a recent meta-analysis reporting the relative risk for PD being 1.5 times greater in men than women [119]. A number of studies have also reported sex differences in symptom severity among male and female PD patients [120-123]. For instance, Rajput et. al. [120] examined 794 PD patients (58% males, 42% females) and found that women had significantly less severe rigidity as compared to males. Likewise, Lyons et. al. [121] examined 630 PD patients and also found that men exhibited more severe parkinsonian motor features but that women exhibited more levodopa-induced dyskinesia. Other studies have similarity reported worse rigidity in male PD patients [122], and that males also had more frequent symptoms such as writing difficulties, fumblingness, speech problems, and gait problems than females [123].
Sex differences in response to antiparkinsonism medications have also been reported. Along these lines, analysis of data from one of the largest trials conducted on PD, the deprenyl and tocopherol therapy of parkinsonism (DATATOP) study, found that women treated with levodopa had more significant improvement of motor function than men treated with levodopa, but women were found to be more likely to develop drug-related dyskinesias than men [124]. Another study found that levodopa area under the curve was higher in women than men and that women had significantly higher incidence of peak-dose dyskinesia [125]. Arabia et. al. [126] found in a study on 164 PD patients that levodopa area under the curve and body weight were significantly and inversely correlated. They suggested that lighter PD patients, especially women may receive a greater cumulative dosage of levodopa per kilogram of body weight, and that this may explain the higher incidence of levodopa-induced peak dose dyskinesias in women. Martinelli et. al. [127] in a study of 116 PD patients also found that women had a higher bioavalibity of levodopa as compared to men. Other studies have also found a sex difference in bioavailability of the dopaminergic agonist, pramipexole, which is often used in conjunction with levodopa to potentiate the dopaminergic effect. In these studies, females were demonstrated to have a 24-27% lower oral clearance for pramipexole as compared to males [128]. This further suggests there is a sex difference in the clearance of dopaminergic agents that may play a role in sex-specific dosing of PD medications. Finally, in addition to these reports on sex differences in PD patients on prevalence, symptom severity and drug effectiveness/bioavailability, there are reports of sex differences in treatment outcomes. Hariz et. al. [129] examined the clinical status, disability and health-related quality of life of 38 PD (24 male and 14 female) patients before and 11 months after they had undergone either pallidotomy, thalamotomy or deep brain stimulation. Using a variety of standard assessment tools such as the Unified Parkinson’s Disease Rating Scale, the ADL Taxonomy, the Nottingham Health Profile, the Life Satisfaction Questionnaire, and a Visual Analogue Scale, the results of the study revealed that both men and women showed improvement following surgery, but women showed greater benefit than men in emotional health, social functioning and activities of daily living.
While the reason for the above-described sex differences in PD remains to be elucidated, there is growing evidence that estrogen may have a neuroprotective role in PD. Along these lines, a number of studies have demonstrated that estrogen is neuroprotective in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced nigrostriatal lesions, an animal model of idiopathic PD [130-140]. Estrogen prevented MPTP-induced depletion of striatal dopamine, dopamine transporter binding and expression, decreased the MPTP-induced tyrosine hydroxylase-immunoreactive neuron loss and attenuated the glial activation induced by MPTP [130-140]. The estrogenic steroid principally used in these studies was 17β-estradiol (E2), and its effect on dopamine loss was shown by several studies to be stereospecific, as an isomer with weak estrogenic activity, 17α-estradiol, was ineffective at preventing MPTP-induced dopamine loss [131-133]. Work by Shughrue [136] has shown that ER-α and ER-β are sparsely localized in the striatum and subtantia nigra of the mouse, and treatment with MPTP or estrogen did not change the localization pattern or density of the estrogen receptors. Thus, estrogen protects in the striatum against MPTP-induced loss of dopaminergic neurons despite having only a sparse localization of estrogen receptors in this region of the brain. However, the sparse localization of ER-α seems sufficient to mediate estrogen neuroprotection in the striatum, as evidenced by the study of D’Astous et. al. [138] who demonstrated that an ER-α agonist (PPT) exerts protection against MPTP-induced dopamine depletion in the striatum, while ER-β agonists (DPN, Delta-3-diol) were ineffective. This preliminary evidence suggests that estrogen protection in the striatum may involve ER-α, but additional studies such as examining estrogen ability to protect the striatum against MPTP-induced damage in ER-knockout mice would help add important corroborating evidence for the role of ER in estrogen action in the striatum. Furthermore, alternative pathways for estrogen neuroprotection must be considered as well, as work by Quesada and Micevych [141] has provided evidence that estrogen interacts with the IGF-1 system to protect nigrostriatal dopaminergic neurons and maintain motor function after 6-hydroxydopamine lesion animals. Finally, it should be added that estrogen has also been shown to reduce the dyskinetic effects produced by MPTP in monkeys, suggesting that estrogen’s beneficial effects on the dopaminergic system applies to higher species [142].
In human studies, Ragonese et. al. [143] found that PD was associated with factors that reduced estrogen stimulation during life (e.g. PD was significantly associated with a fertile life length shorter than 36 years and a cumulative length of pregnancies longer than 30 months). Currie et. al. [144] also reported that women who had taken postmenopausal estrogen replacement therapy were less likely to develop PD than those who had not. Furthermore, a single nucleotide polymorphism allele of the estrogen receptor-β gene was recently shown to be more frequent in patients that have an early age of onset of PD [145]. Of significant interest and paralleling results in animal studies, a pilot study in women revealed that short term estrogen treatment in postmenopausal women increased dopamine transporter availability in the caudate putamen [146]. Thus, both animal and human studies suggest a beneficial regulatory effect of estrogen on the nigrostriatal dopaminergic system, which may underlie putative protective effects of estrogen in PD. A corollary and exciting finding was the recent discovery that estrogen treatment induces differentiation of human neural stem cells (NSCs), giving rise to tyrosine hydroxylase (dopaminergic) neurons, an effect blocked by use of an estrogen receptor antagonist [147]. Estrogen also increased the number of dopaminergic neurons derived from human NSCs in vivo when the cells were grafted into mouse brains. This finding reveals another layer to the beneficial regulation by estrogen of the dopaminergic neuronal system, and raises the possibility of a potential role for estrogen in transplantation of NSCs for PD. Finally, oxidative stress is suggested to play an important role in neuronal degeneration in PD [148]. It is thus intriguing that estrogen has been shown to suppress free radical production and protect striatal neurons against oxidative stress [111-113], potentially providing an additional mechanism for estrogen neuroprotection in PD.
SERMs
The beneficial effect of estrogen in animal models of PD has recently been extended to clinically relevant SERMs, such as tamoxifen and raloxifene. For instance, several studies have shown that tamoxifen can protect the striatum against methamphetamine-induced toxicity and prevent striatal dopamine depletion in male and female animals [149-152]. Tamoxifen also had a significant beneficial effect upon locomotor activities in MPTP-treated animals, increasing horizontal activities, number of movements and total distance traveled [151]. The effect of tamoxifen may be mediated by its active metabolite, 4-OH-tamoxifen, which exerts similar protective effects in mouse models of PD [152]. Interestingly, 4-OH-tamoxifen was shown to have potent superoxide anion scavenging ability in vitro, which may explain its protective actions in vivo in the PD animal models [152]. Furthermore, a recent study by Obata [153] using in vivo microdialysis in anesthetized rats, found that tamoxifen infusion (100 μM) in the striatum blocked the ability of a dopaminergic neurotoxin, 1-methyl-4-phenylpyridinium to enhance hydroxyl radical production. Of significant interest, the neuroprotective effect of tamoxifen has been implicated to extend to the vascularture and astrocytes in the striatum, as tamoxifen treatment was shown to significantly reduce 3-nitropropionic acid-induced lesions of the lateral striatal artery and astrocyte cell death [154]. Finally, additional work has shown that the SERM raloxifene can prevent MPTP-induced decreases in striatal dopamine concentrations and dopamine transporter binding in mice [151,152]. This effect was dose dependent, with a beneficial effect of raloxifene observed at a dose of 5 mg/kg/day but not at a dose of 1 mg/kg/day. Furthermore, raloxifene did not affect NMDA or AMPA receptor sensitive binding in the striatum in MPTP-treated mice, suggesting that raloxifene does not regulate the concentration of glutamate receptors in MPTP-treated mice [151]. However, work in a clonal substania nigra cell line, SN4741 showed that raloxifene, like E2 can significantly reduce neuronal cell death induced by oxidative stress, suggesting that an antioxidant capability may be involved in at least part of its neuroprotective effects of raloxifene [110].
As a whole, these studies suggest that estrogen and SERMs may have an important protective effect in PD. However, further clinical epidemiological studies are needed to study relationships between sex or estrogen/SERM and PD incidence. It should also be pointed out that the majority of the studies in the literature on estrogen and PD used exogenous hormone replacement paradigms, which are poorly suited to assess the role of endogenous estrogen as a neuroprotective factor in PD animal models. Studies using transgenic animal models such as the aromatase and/or FSH-β receptor knockout mice, which exhibit estrogen depletion, would help address this important issue. Use of estrogen receptor knockout animals may also be beneficial in elucidating whether the effects of estrogen in the striatum involve an ER-dependent or ER-independent mechanism. In the next section, the potential protective role of estrogen in Alzheimer’s disease will be discussed.
Alzheimer’s Disease
Alzheimer’s disease (AD) is the leading cause of dementia in the elderly and is characterized by the presence of extensive plaque deposition and neurofibrillary pathology. A number of clinical studies have suggested that estrogen therapy may delay the onset or contribute to the prevention and/or attenuation of Alzheimer’s disease (AD) [155-163, and 164 for review]. As reviewed by Henderson (165), 12 case-control and cohort studies were conducted in the 1990s on estrogen use and AD. All but two studies found a significantly reduced risk of the incidence of AD in women who were estrogen users versus non-users. In fact, meta-analysis studies suggest overall Alzheimer risk reductions of about 29–44% [161,162]. However, several studies showed that estrogen treatment after the disease process has started was generally unsuccessful in ameliorating the decline in cognitive function in AD that inevitably occurs over time (166-168), although there are dissenting studies in the literature [169-171].
WHI and the Healthy Cell Bias Hypothesis
Chief among the studies reporting negative effects of estrogen on dementia (and stroke) is the Women’s Health Initiative (WHI) study, which reported that chronic treatment with conjugated equine estrogens (Premarin) plus MPA (Provera) increased the relative risk (hazard ratio) of probable dementia in postmenopausal women age 65 years or older [172]. Premarin alone also had an elevated relative risk of dementia, which however was not statistically significant [173]. There was also an increased stroke risk in women 60-80 who received the Premarin alone or in combination with Provera (e.g. from an incidence of 21 in 10,000 women (control) to 29 in 10,000 women for Premarin and Provera combined therapy) [174,175].
As reviewed recently by Henderson, Clark and others working in the area [164,176-182], several caveats/potential limitations to the study should be noted. First, as reviewed recently by Clark [176], one of the major problems with the interpretation of the study was that the authors emphasized the use of relative risks rather than absolute risks. For instance, the final hazard ratio (relative risk) for stroke for the Premarin and Provera combined therapy group was determined by the WHI authors to be 1.41 with an adjusted 95% confidence interval (CI) of 0.86-2.31. As pointed out by Clark [176], such broad confidence intervals which include 1.0 are not significant. The absolute risk was low (0.08%), making it highly unlikely that the data demonstrate any clinically significant risk. Similarly, for the estrogen alone study the final hazard ratio (relative risk) was 1.39 based on a non-adjusted 95% CI. When the adjusted 95%CI is used (0.97-1.99), the hazard ratio becomes statistically insignificant. The Consolidated Standards of Reporting Trials (CONSORT) indicated that authors should state their results as absolute, not just relative numbers [183]. The WHI studies have also been criticized for being 10-fold underpowered to show cardioprotection of women starting hormone treatment during the menopausal transition [177]. Other concerns raised included the fact that progestin should have been administered in cyclic fashion rather than chronic and that estrogen rather than Premarin should have been tested, with estrogen given transdermally rather than orally – as oral estrogen is known to increase blood clot risk – which could have explained the increased stroke risk in the WHI study and the increased risk of dementia, which could have been secondary to blood clots in the brain [177,178,182]. A major concern shared by many investigators working in the area was that the average age of women in the study was 63.3 yrs, with two thirds between the ages of 60-70, which is far past the menopause. Several groups have proposed that early initiation of estrogen replacement therapy at the inception of menopause is necessary to provide neural and cardiovascular benefit [164,178,179,181,182].
In fact, Brinton and others have proposed a “healthy cell bias of estrogen benefit”, in which it is suggested that as neurological health progresses from healthy to unhealthy, so too do the benefits of estrogen or hormone therapy [178,182,184]. Stated more concisely, if neurons are healthy at the time of estrogen exposure, their response to estrogen or hormone therapy is beneficial for both neurological function and survival. In contrast, if neurological health is compromised, estrogen exposure over time exacerbates neurological demise. In support of this concept, Brinton and coworkers created a “prevention versus treatment” situation using rat hippocampal neuronal cultures, and found that estrogen was protective against Abeta1-42-induced neurodegeneration when administered before or during Abeta1-42 insult, but treatment after Abeta1-42 insult was not beneficial and actually led to enhanced cell death - findings consistent with a healthy cell bias of estrogen benefit [185]. Also consistent with the healthy cell bias hypothesis of estrogen benefit is the recent MIRAGE study conducted by Henderson and coworkers [186]. The MIRAGE study examined the relation between estrogen containing hormone therapy used for more than six months in different aged women and AD risk, with the goal of ascertaining whether hormone therapy at the time of menopause would be beneficial in reducing AD risk. The study revealed that a significant protective effect was observed only in the youngest age group studied (e.g. the 50-63 years old age group). The authors interpreted the study results to mean that hormone therapy may protect younger women from AD or reduce the risk of early onset AD or that hormone therapy during the early menopause may reduce AD risk. An additional new clinical study underway to study effect of estrogen treatment at the time of onset of menopause is the Kronos Early Estrogen Prevention Study (KEEPS) [187]. The KEEPS study is a 5 year study initiated in 2005 to reexamine estrogen beneficial effects when initiated at the beginning of menopause, and which will use both conjugated equine estrogens (CEE) and estrogen and will test cyclic progestin use, overcoming many of the previous proposed limitations of the WHI study. This study and hopefully more like it in the future should help clarify many of the unknowns and clear up remaining controversies concerning beneficial effects of estrogen therapy.
Gene Mutations, Aromatase and Potential Mechanisms of Estrogen Neuroprotection in AD
Recent studies in both animals and humans have provided additional evidence supporting a potential beneficial protective role for estrogen in AD. Along these lines, a novel study in transgenic mice was conducted in which mice were produced by crossing aromatase knock out mice with APP23 transgenic mice, a mouse model of AD [188]. The aromatase knockout-APP23 transgenic crossed mice allowed the investigators to study the effect of estrogen depletion on A-beta plaque formation in a well-characterized model of AD. The results revealed that compared to the APP-23 transgenic control mice, the estrogen-deficient APP23 transgenic mice exhibited greatly reduced brain estrogen levels and early-onset and increased beta amyloid peptide deposition. In contrast, ovariectomized APP23 mice exhibited plaque pathology similar to the APP23 transgenic control mice. This suggests that estrogen depletion in the brain may be a significant risk factor for developing AD neuropathology. Intriguingly, human genetic studies have also revealed evidence of a relationship between mutations of the aromatase gene and AD risk. Ivivonen et. al. [189] reported an approximate 60% increased risk for AD in subjects who had single nucleotide polymorphisms (SNPs) in the CYP19 gene which encodes aromatase. Likewise, Huang and Poduslo [190] found a two-fold increased risk for AD in APOE 4 carriers who had SNPs in the CYP19 gene. Of significant interest, Ishunina et. al. [191] found that ER-α and aromatase expression increased with age in the hippocampus of women, and women with AD had down-regulated expression of these genes, suggesting a potential deficit in local brain production of estrogen and altered estrogen signaling in AD.
Brain imaging studies have revealed that one of the first regions to show cerebral metabolism decline in AD is the posterior cingulate cortex [192]. Rasgon et. al. [193] thus examined cerebral glucose metabolic changes in the posterior cingulate cortex in postmenopausal estrogen users and nonusers at baseline and two years later. The results revealed that nonusers had a significant decline in cerebral metabolism in the posterior cingulate cortex at the two year follow up, while estrogen users had no such decline. The authors suggested that estrogen may preserve regional cerebral metabolism and prevent metabolic decline in postmenopausal women, especially in the posterior cingulate cortex, a region found to decline in the early stages of AD. Other studies in postmenopausal women revealed similar effects in the cortex, with women receiving estrogen replacement therapy having higher cerebral metabolism in the inferior frontal cortex and temporal cortex with respect to non-estrogen users [194]. Interestingly, postmenopausal women with breast cancer taking tamoxifen had widespread areas of hypometabolism in the inferior and dorsal lateral frontal lobes [194]. This is intriguing as it may support a role for brain-derived estrogen in cerebral metabolism/activity, since postmenopausal women generally have low serum estrogen levels due to ovarian follicle depletion.
The mechanisms of how estrogen may exert protection in AD are not clear. However, a number of studies have shown that estrogen can protect neurons against β-amyloid toxicity [73,185,195-199], oxidative stress [110-113,195,200] and excitotoxicity [67,195,201,202], events suggested to participate in the pathology of AD. Additionally, estrogen has been shown to induce dephosphorylation of the tau protein and prevent its hyperphosphorylation in neurons [203]. At the molecular level, estrogen has been shown to enhance activation of the survival factor, Akt, while inducing phosphorylation and deactivation of GSK-3β and BAD, known death signals in neurons [67-73,204,205]. Collectively, these effects could explain the reported protective actions of estrogen in AD. It should be mention that the SERM, raloxifene has also been shown to protect against β-amyloid-induced cell death [199], and it has been reported to decrease risk of AD and cognitive impairment in postmenopausal women [206]. As a whole, the above studies support a potential protective role for estrogen in AD, which deserves further study. The concept that brain derived estrogen may be neuroprotective is exciting and adds a new realm of study to the field. In the remaining sections of the review, we will turn our attention to the regulatory actions of estrogen on synaptic plasticity and cognition, which could be especially relevant to AD, but also to the other neurodegenerative disorders as well.
ESTROGEN, PLASTICITY AND COGNITION
Estrogen and Memory
A growing body of evidence has accumulated over several decades that support a regulatory role for estrogen on cognition/memory. With respect to humans, this body of evidence springs from several sources, including sex differences in cognitive function, human menstrual cycle fluctuations in cognitive performance, randomized controlled trials and observational studies on effects of estrogen depletion and replacement in postmenopausal women, as well as imaging studies on estrogen and cognition. While qualitative differences in cognitive skills between the sexes do not exist, quantitatively, women tend to excel on tasks of verbal skills and memory, on perceptual speed and accuracy, and on fine motor skills [207-210]. Additionally, improved verbal working memory has been reported to be associated with periods of high estrogen levels in the menstrual cycle [211]. In fact, the majority of recent studies that precisely identified cycle stage, excluded anovulatory women, and used gender-sensitive cognitive tests found that women perform best on sexually dimorphic tests during the midluteal stage, further suggesting that estrogen facilitates verbal memory and fine motor skills [207,211-214]. Randomized control trials, while clearly not entirely consistent, nevertheless have shown that performance on 47% of memory measures was better in postmenopausal women who received estrogen replacement therapy [215]. Likewise, most observational and longitudinal studies also show that estrogen users perform better on cognitive tests [207]. In fact, when considered in totality, 71% of the studies that examined the effect of estrogen on cognitive functioning in humans found significant beneficial effects on one or more neuropsychological tests of cognition [207]. There is also accumulating evidence that there may be a critical period during the immediate postmenopausal years for protective effect of estrogen on cognition [216]. Treatment too far past the menopausal transition may have less beneficial effect. The beneficial effect of estrogen upon cognition is not restricted to humans, as it is also observed in lower species. For instance, cyclic estrogen replacement has been shown to significantly improve cognitive function in ovariectomized female monkeys [217]. Furthermore, estrogen replacement has been shown to improve the performance of ovariectomized rats in memory tasks such as radial mazes and water mazes [218-225], as well as in the operant alternation task [226,227] and the active avoidance task [228]. A number of studies have also provided evidence that estrogen can enhance working memory in ovariectomized animals [229-232]. The potential site(s) of action and underlying mechanisms for estrogen beneficial effects on cognition will be reviewed in the subsequent sections.
The Hippocampus and Prefrontal Cortex as Important Sites of Estrogen Action on Cognition
Hippocampus
The hippocampus has long been presumed the primary site of action of estrogen on cognition; and explicit memory is considered the cognitive function most vulnerable to menopausal loss of estrogen [222,233-235]. Supportive evidence includes increased hippocampal neuronal excitability after estradiol administration to ovariectomized rats [233], and a positive correlation of hippocampal CA1 dendritic spine and synapse density with estrogen levels, in which exogenous estrogen was administered in ovariectomized rats and monkeys [236-245], or endogenous estrogen levels fluctuated across the rat estrous cycle [235]. The beneficial effect of estrogen upon hippocampal synaptic plasticity was confirmed via a variety of approaches in these studies, including Golgi analysis, light microscopic or electron microscopic analysis of synapses or synaptic proteins, dye-filling techniques, and radioimmunochemistry [235-245]. Estrogen has also been shown to increase spine density and neurite outgrowth in vitro in hippocampal neuronal cultures [239,246]. Additionally, estrogen enhances long term potentiation (LTP) in the hippocampus, which is a postulated model for the synaptic changes that may underlie learning and memory [237-250]. Interestingly, induction of LTP in the CA1 region of the hippocampus has been shown to be enhanced on proestrus, which correlates with peak serum estrogen levels and spine density enhancement observed on proestrus [251,252]. It should be mentioned that despite more than a century of fascination with dendritic spines, little is known about their assumed role in memory storage and learning. However, there is growing evidence that dendritic spines are indeed involved in learning and memory [see 253-257, for review]. Several lines of evidence support this contention. First, experimental manipulations that affect dendritic spine number in the hippocampus have been shown to affect various types of learning processes [253,254, for review]. Secondly, learning itself affects the presence of dendritic spines in the hippocampus [253,258]. Furthermore, with respect to estrogen, recent work has shown that ovariectomized (estrogen deficient) rats have significant reductions in spine densities in the pyramidal neurons of the prefrontal cortex and CA1 region of the hippocampus that is correlated with a significant decline in performance in memory tests [259]. Additional behavior studies have shown that estrogen treatment in ovariectomized animals enhances learning and memory of rats when assessed using hippocampal dependent memory tasks [222,253,260], which further suggests a functional relevance for the spine density enhancements reported in the CA1 region of the hippocampus following estrogen treatment [235-245]. Taken as a whole, available evidence suggests that dendritic spines provide the anatomical structure necessary for the processing of novel information used in memory formation, e.g. they allow the maintenance of the hippocampal or cortical system in a “learning mode” in which memories can be acquired rapidly and efficiently [see 253,255, for review]. This postulated function of dendritic spines still requires further elaboration and confirmation, but studies using transgenic animals and modern time-lapsing imaging methods in vivo and in vitro are starting to allow a more detailed examination of this question, which will hopefully lead to further clarification to the precise roles of dendritic spine changes in learning and memory [254,256,257, for review].
Prefrontal Cortex
While there is a large amount of evidence in rodents and primates supporting estrogen modulation of hippocampal plasticity and activity, few neuroimaging studies in humans show a significant effect of estrogen on hippocampal integrity/activity; rather the preponderance of studies show significant effects of estrogen on frontal, parietal and temporal lobes of the cerebral cortex, areas responsible for executive function (prefrontal cortex [PFC]) and storage of memories (temporal/parietal cortex [T/P cortex]) [261-265]. This has led to the proposal that menopausal cognitive decline in humans may be secondary to executive dysfunction [266]. Learning and memory are enhanced by frontally mediated processes that collectively are referred to as “executive functioning”, which includes such cognitive processes as working memory, directed attention, response inhibition, dual task coordination, cognitive set switching, and behavioral monitoring [266-267]. The integrity of the prefrontal cortex is critical to intact executive functions, as is its complex neural circuitry that consolidates input from various modalities via cortical, subcortical, and limbic connections.
Keenan et. al. [266] recently provided evidence for executive dysfunction in untreated menopausal women, as women with hormone replacement therapy (HRT) outperformed women without HRT on tests requiring directed attention, inhibition of inappropriate responses, and cognitive set switching. A number of other studies have also provided evidence that estrogen enhances prefrontal cognitive processes such as working memory and attention [267-274], although there are dissenting reports [275]. For instance, a recent study by Joffe et. al. [263] found that estrogen replacement in postmenopausal women enhanced executive function, an effect that was strongly correlated with functional magnetic resonance imaging (fMRI)-documented increases in frontal lobe activity. Interestingly, six week treatment with soy phytoestrogens has also been shown to enhance prefrontal cognitive functions, including planning ability and mental flexibility [276]. Several other functional neuroimaging studies have provided additional support for estrogen dependent alterations in frontal lobe functioning. For instance, menopausal women treated for 21 days with conjugated equine estrogens were shown to have increased activation in the superior frontal lobe during a verbal working memory task as assessed with fMRI [262]. Similarly, Berman et al. [261] demonstrated that suppression of estrogen levels with Lupron, a GnRH agonist, led to a loss of the previously observed regional cerebral blood flow (rCBF) increase in prefrontal cortex in young women during the Wisconsin Card Sorting Test. Normalization of the activation pattern in the prefrontal cortex was noted when estrogen was added to Lupron regimen. Augmentation of temporal and parietal cortex was also observed, which fits with the current hypothesis that memory consolidation can occur throughout the cortex [277]. In a recent study, Erickson et. al. [278] used high resolution magnetic resonance imaging and an optimized voxel-based morphometric technique to examine the effects of HRT on brain volume in postmenopausal women. The study revealed that HRT was associated with sparing of grey matter in prefrontal, temporal and parietal cortical regions, and that longer duration of therapy was associated with greater sparing. A sparing effect of HRT was also observed in the anterior hippocampus. The results of the study suggest that estrogen has a protective sparing role in cortical regions during aging that may provide cognitive benefit, as well as protection from neurodegenerative disorders. Finally, estrogen has also been shown to enhance dendritic spine density in the prefrontal cortex of young and aged monkeys and in young rats [279-281], which may explain the prefrontal cognitive enhancing effects of estrogen. As a whole, these findings provide support for the prefrontal cortex being considered, along with the hippocampus, as a potential key target for estrogen actions on cognition. The mechanisms that underlie estrogen regulation of synaptic plasticity will be discussed in the following section.
Mechanisms of Estrogen Regulation of Synaptic Plasticity
Localization and Role of Estrogen Receptors
In situ hybridization and immunohistochemical studies have revealed that both ER-α and ER-β are expressed in various regions of the hippocampus and prefrontal cortex in a variety of species [282-286]. In the female rat, mRNA for both estrogen receptors has been reported to be expressed in the hippocampus and cortex, with ER-β mRNA reportedly predominating over ER-α [282]. Immunohistochemistry studies revealed that ER-α and ER-β positive neurons are present in all subfields of the hippocampus in the female rat, with maximum levels observed in the stratum pyramidale of CA3 [283]. Some ER-α and ER-β positive neurons in the CA3 exhibited pyramidal morphology. ER-α expression has also been reported in inhibitory interneurons in the hippocampus of the rat [287]. Interestingly, the number of ER-α and ER-β positive neurons in the hippocampus has been reported to decrease significantly in aged animals [281]. Furthermore, Adams et. al. [288] has also reported that the aged rat hippocampus has 50% fewer spines containing ER-α than the young rat hippocampus.
In addition to being expressed in hippocampal neurons, both ER-α and ER-β have also been reported to be expressed in glia in the CA1-CA3 [284,289], which is of interest due to the growing appreciation of astrocytes as a component of the tripartite synapse. In higher primates such as the monkey, ER-β mRNA has been reported to be dense in the dentate gyrus, CA1-CA4 and the prosubiculum/subiculum areas of the hippocampus [290]. There are only a few studies in the literature on the distribution of ER-α in the monkey hippocampus. One study reported a failure to detect ER-α in monkey hippocampus using in situ hybridization or immunohistochemistry [290]. However, another study did detect ER-α mRNA in the female monkey hippocampus using RT-PCR, but there was much higher ER-β expression as compared to ER-β [291]. With respect to the cerebral cortex, Wang et. al. [285] found that ER-α mRNA is widely expressed in neurons in the monkey prefrontal cortex with the largest concentration present in layers IV-VI. Studies in humans have shown that both ER-α and ER-β mRNA are expressed in the hippocampus and cortex, with the hippocampus displaying higher expression for ER-β than ER-α [286]. Thus, there seems to be good agreement throughout the species of higher ER-β expression in the hippocampus as compared to ER-α. Finally, as discussed previously, the putative membrane estrogen receptor, GPR30 is also reported to be expressed in both the hippocampus and cortex in rodents, and could have a role in mediating estrogen actions in these regions of the brain [58-60].
An important point to consider is the subcellular localization of estrogen receptors, especially as estrogen action in the regulation of synaptic plasticity has been implicated to involve both genomic as well as nongenomic mechanisms [289, for review]. In addition to nuclear localization, there are now a number of reports that ER-α and ER-β are localized at extranuclear sites in neurons in the hippocampus and cortex [281,287,289,292-294]. For instance, ER-α and ER-β have been demonstrated by ultrastructural studies to be localized at plasma membrane, dendritic shafts, and dendritic spines in neurons in the hippocampus and cortex [289,292,294]. Preliminary reports also show localization of GPR30 at plasma membrane, endoplasmic reticulum, dendritic shafts and dendritic spines in neurons in the rat hippocampus and cortex [58-60]. The localization of estrogen receptors at extranuclear locations in neurons suggests that estrogen may have local effects in dendrites and spines to regulate synaptic plasticity. It also provides a morphological basis for nongenomic actions of estrogen that have been reported in these tissues and which have been implicated in estrogen-induced synaptic plasticity [204,240,281,289,294]. Surprisingly, there are only a few studies that have attempted to explore the role of ER-α and ER-β in estrogen-induced synaptic plasticity and memory. Murakami et. al. [295] found that estrogen rapidly increased spine density in CA1 pyramidal neurons in hippocampal slices and this effect was shared by the ER-α selective agonist PPT, and conversely blocked by the estrogen receptor antagonist, ICI182,780. This study showed a very rapid effect on spine density by estrogen, e.g. induction of spines was observed as early as two hours after estrogen treatment, which suggests a potential nongenomic effect of estrogen. In vivo studies by McEwen and coworkers [296] also showed that an estrogen receptor antagonist that is permeable to the brain, CI-628 blocked the ability of estrogen to increase spine density in CA1 pyramidal neurons. Estrogen receptor knockout mice have also been examined functionally using memory testing. Fugger et. al. [297] demonstrated that ER-α but not ER-β knockout mice have impaired performance upon inhibitory avoidance, a hippocampal-dependent task. Day et. al. [298] recently demonstrated that hippocampal slices from ER-β knockout mice have significant attenuations of LTP, and that ER-β knockout mice display significant impairment in hippocampal-mediated fear conditioning. Unfortunately, none of these studies examined spine density or other measures of synaptic plasticity in the ER knockout mice. Such studies are needed, as are more studies using ER selective agonists and antagonists to help delineate the role of ER-α and ER-β in estrogen-induced synaptic plasticity. Parallel studies are also needed to determine the role, if any of the putative membrane ER, GPR30.
NMDA, Calcium and Kinase Mediated Signaling
A growing body of evidence suggests that NMDA neurotransmission mediates the spine density and synapse changes induced by estrogen in the hippocampus. Along these lines, estrogen has been demonstrated to increase NMDA receptor binding [234,299-301], increase NMDAR1 and NMDAR2b gene expression in CA1 region of the hippocampus of rats [302,303], and enhance NMDA-dependent calcium signals in CA1 spines and dendrites [304]. Furthermore, NMDA receptor antagonists have been shown to block the estrogen-induced increase in spine density in the CA1 of the hippocampus [239,295,296]. A role for NMDAR2b receptors was also recently implicated in estrogen induction of LTP, as NMDAR2b specific antagonists blocked the ability of estrogen to enhance the magnitude of LTP induction in the hippocampus [250]. A single report claims a role for AMPA receptors in estrogen-induced spine density changes in the CA1 stratum oriens [295]. In addition to increasing sensitivity of hippocampal neurons to excitatory input, estrogen has also been shown to transiently suppress GABA-A-mediated inhibition of CA1 pyramidal cells [306,307]. This suggests that disinhibition of CA1 pyramidal cells by estrogen is also a component of estrogen action on synaptic plasticity in the hippocampus.
Calcium is an important regulator of cell signaling and neurite outgrowth via its ability to activate various kinases and transcription factors, including calcium-calmodulin kinase (CaMKII and IV) and cyclic AMP response element-binding protein (CREB) [246,308,309]. Estrogen has been shown to rapidly enhance calcium influx in hippocampal neurons in not only the cytoplasm, but also in the nucleus and dendrites [246], and this calcium influx was found critical for estrogen activation of CREB, a key transcription factor implicated in synaptic plasticity [246,310,311]. Further work showed that activation of CREB is important for estrogen-induced synaptic plasticity in hippocampal neurons, as administration of H89, a specific protein kinase A antagonist or CREB antisense oligonucleotides blocked estrogen-induced spine formation [312]. Furthermore, the activation of CREB was shown to be downstream of NMDA receptor activation and the resultant subsequent calcium influx [312]. Calcium also activates CaM kinases, and CaMKII has been shown to be rapidly activated by estrogen in hippocampal neurons [310,313]. The activation of CaMKII was shown to be critical for estrogen-induced activation of CREB and estrogen-enhanced expression of the spine specific protein, spinophilin, as these effects of estrogen were blocked by treatment with a CaMKII inhibitor [310].
Estrogen has also been shown to rapidly activate the serine/threonine kinase, Akt in hippocampal and cortical neurons [65,69,70,80,204,205,240,314]. The activation of Akt was recently shown to occur in the same cortical neurons as activation of extracellular signal-regulated kinases (ERKs) [314], which is intriguing as both Akt and ERK have been implicated to play an important role in regulation of translation in neurons [315 and 316, for review]. Akama and McEwen [240] have shown that estrogen rapidly phosphorylates Akt in hippocampal neurons, which is correlated with phosphorylation of the downstream translation repressor, eukaryotic initiation factor 4E-binding protein (4E-BP1). Further work showed that estrogen stimulated new protein synthesis of post-synaptic density-95 (PSD95), a dendritic spine scaffold protein, and this translational effect of estrogen was shown to be Akt-dependent [240]. The role of ERKs in estrogen-induced plasticity in the hippocampus and cortex has also been suggested based on the findings that ERK1/2 are rapidly activated following estrogen treatment in neurons [67-69,310] and administration of MEK inhibitors significantly attenuated the neurotrophic actions of estrogen in hippocampal and cortical neurons [281,295]. The findings suggest estrogen exerts a rapid kinase regulatory effect that leads to enhanced protein synthesis for dendritic function. It is currently unclear how estrogen activates these multiple kinase signaling pathways. Studies to examine whether scaffold proteins such as MNAR, striatin or p130Cas play a role in linking membrane estrogen receptors to kinase targets in the brain and thereby facilitate kinase activation are clearly needed. Also unclear is what role does rapid kinase signaling have in the genomic effects of estrogen, and are genomic effects critical for estrogen induction of synaptic plasticity? Finally, more work is needed on the precise identity and targeting of the membrane receptor involved in estrogen-induced synaptic plasticity – e.g. is it ER-α, ER-β, GPR30 or an as yet unidentified new estrogen receptor, and how are membrane estrogen receptors targeted to the membrane? Further work on these important issues should help enhance our understanding of basic mechanisms of estrogen signaling in the brain and may provide insights into potential therapies.
Finally, endogenous production of estrogen by brain cells has recently been suggested to have a role in local regulation of synaptic plasticity in the brain [317-319]. Hojo et. al. [317] recently showed that there is significant localization of cytochromes P450 aromatase and P45017alpha in pyramidal neurons in the CA1-CA3 and granule cells in the dentate gyrus. P450 aromatase localization has also been reported in astrocytes and oligodendrocytes [37,38,317]. Furthermore, stimulation of hippocampal neurons with NMDA was shown to induce a significant increase in net production of estradiol from cholesterol, an effect abolished by P450 inhibitors [317]. Further work by Kretz et. al. [318] showed that treatment of hippocampal slice cultures with an aromatase inhibitor caused a dose-dependent decrease in estradiol production, which was accompanied by a significant decrease in the density of synaptic spines and presynaptic boutons. A correlated down-regulation of the spine protein, spinophilin, and the presynaptic vesicular protein, synaptophysin was observed with aromatase inhibitor treatment [318]. The studies suggest that endogenous estrogen produced in brain cells may exert a local regulatory effect on synaptic plasticity [318,319]. However, it should be pointed out that work in aromatase knockout mice have failed to show any significant change in one type of memory - spatial memory, which one might expect to be altered if brain-derived estrogen had a significant role in regulating synaptic plasticity [320]. However, before definitive conclusions can be drawn on the role of endogenous brain-derived estrogen in plasticity and cognition, further work is needed to examine synaptic plasticity in aromatase knockout mice and to conduct more extensive learning and memory testing. Clearly, the possibility of synaptic regulation by locally produced estrogen in the brain is an exciting concept worthy of further study. If demonstrated to have an important biological role, additional studies on what factors and situations modulate local estrogen production in the brain would be extremely interesting and worthy of pursuing.
Conclusions
This review demonstrates the remarkable body of work that has been conducted on the neuroprotective and neurotrophic actions of estrogen in the brain. Clearly much progress in our understanding of estrogen actions in the brain has been derived from this body of work that spans decades. Just as clear is the need for continued focused research in the area to resolve lingering controversies in the field and to address complex mechanisms that underlie estrogen neuroprotective and neurotrophic actions. More clinical studies to address the concerns raised over the WHI study are needed, as is a renewed focus on development of brain selective SERMs that maintain the beneficial effects of estrogen without its negative side effects. From a signaling standpoint, there is a significant need for targeted research to better elucidate the mechanisms of estrogen activation of nongenomic kinase signaling pathways in neurons and non-neuronal cells, e.g. studies to identify the involved membrane estrogen receptors, mechanisms of targeting of these receptors to the plasma membrane, and further identification of scaffold proteins in the brain that may link the membrane estrogen receptor to its kinase targets and facilitate their activation. Further studies to elucidate whether the nongenomic and genomic signaling pathways induced by estrogen in the brain display crosstalk or are separate regulatory pathways are also needed, as well as a careful determination of the contribution of each pathways to the synaptic plasticity and cognitive effects of estrogen. In closing, while much has been learned in the past several decades concerning estrogen neuroprotective and neurotrophic actions in the brain, significant work lies ahead to fully unravel the complexities of estrogen action in the brain and to fully understand its mechanisms and implications. Nevertheless, the continued use of multiple species, multiple models/approaches, and the strategy of employing technological advances as they become available should continue to speed progress in this exciting area of research and propel the field toward providing answers to many of the key questions that remain.
Acknowledgments
This research was supported by Research Grants (HD28965 and NS050730) from the National Institute of Child Health and Human Development, and National Institutes of Neurological Disorders and Stroke, National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Petersen SL, Ottem EN, Carpenter CD. Direct and indirect regulation of gonadotropin-releasing hormone neurons by estrogen. Biol Reprod. 2003;69:17771–1778. doi: 10.1095/biolreprod.103.019745. [DOI] [PubMed] [Google Scholar]
- 2.Kelly MJ, Qiu J, Ronnekleiv OK. Estrogen signaling in the hypothalamus. Vitam Horm. 2005;71:123–145. doi: 10.1016/S0083-6729(05)71005-0. [DOI] [PubMed] [Google Scholar]
- 3.Brann DW, Mills TM, Mahesh VB. Female reproduction the ovulatory cycle . In: Witorsch RJ, editor. Reproductive Toxicology. 2. Raven Press; 1995. pp. 23–44. [Google Scholar]
- 4.Hewitt SC, Korach KS. Oestrogen receptor knockout mice: roles for oestrogen receptors alpha and beta in reproductive tissues. Reproduction. 2003;125:143–149. doi: 10.1530/rep.0.1250143. [DOI] [PubMed] [Google Scholar]
- 5.Gore AC. Gonadotropin-releasing hormone neurons, NMDA receptors, and their regulation by steroid hormones across the reproductive life cycle. Brain Res Brain Res Rev. 2001;37:235–248. doi: 10.1016/s0165-0173(01)00121-7. [DOI] [PubMed] [Google Scholar]
- 6.Hall ED, Pazara KE, Linseman KL. Sex differences in postischemic neuronal necrosis in gerbils. J Cereb Blood Flow Metab. 1991;11:292–298. doi: 10.1038/jcbfm.1991.61. [DOI] [PubMed] [Google Scholar]
- 7.Alkayed NJ, Harukuni I, Kimes AS, London ED, Traystman RJ, Hurn PD. Gender-linked brain injury in experimental stroke. Stroke. 1998;29:159–166. doi: 10.1161/01.str.29.1.159. [DOI] [PubMed] [Google Scholar]
- 8.Park EM, Cho S, Frys KA, Glickstein SB, Zhou P, Anrather J, Ross ME, Iadecola C. Inducible nitric oxide synthase contributes to gender differences in ischemic brain injury. J Cereb Blood Flow Metab. 2006;26:392–401. doi: 10.1038/sj.jcbfm.9600194. [DOI] [PubMed] [Google Scholar]
- 9.Roof RL, Hall ED. Estrogen-related gender difference in survival rate and cortical blood flow after inpact-acceleration head injury in rats. J Neurotrauma. 2000;17:1159–1169. doi: 10.1089/neu.2000.17.1155. [DOI] [PubMed] [Google Scholar]
- 10.Murphy SJ, McCullough LD, Smith JM. Stroke in the female: role of biological sex and estrogen. ILAR J. 2004;45:147–159. doi: 10.1093/ilar.45.2.147. [DOI] [PubMed] [Google Scholar]
- 11.Roquer J, Campello AR, Gomis M. Sex differences in first-ever acute stroke. Stroke. 2003;34:1581–1585. doi: 10.1161/01.STR.0000078562.82918.F6. [DOI] [PubMed] [Google Scholar]
- 12.Niewada M, Kobayashi A, Sandercock PA, Kaminski B, Czlonkowska A. Influence of gender on baseline features and clinical outcomes among 17,370 patients with confirmed ischemic stroke in the international stroke trial. Neuroepidemiology. 2005;24:123–128. doi: 10.1159/000082999. [DOI] [PubMed] [Google Scholar]
- 13.Di Carlo A, Lamassa M, Baldereschi M, Pracucci G, Basile AM, Wolfe CD, Giroud M, Rudd A, Ghetti A, Inzitari D. Sex differences in the clinical presentations, resource use, and 3-month outcome of acute stroke in Europe: data from a multi-center multi-national hospital-based registry. Stroke. 2003;34:1114–1119. doi: 10.1161/01.STR.0000068410.07397.D7. [DOI] [PubMed] [Google Scholar]
- 14.Hochner-Celnikier D, Manor O, Garbi B, Chajek-Shaul T. Gender gap in cerebrovascular accidents: comparison of the extent, severity, and risk factors in men and women aged 45-65. Int J Fertile Womens Med. 2005;50:122–128. [PubMed] [Google Scholar]
- 15.Simpkins JW, Rajakumar G, Zhang YQ, Simpkins CE, Greenwald D, Yu CJ, Bodor N, Day AL. Estrogens may reduce mortality and ischemic damage caused by middle cerebral artery occlusion in the female rat. J Neurosurg. 1997;87:724–730. doi: 10.3171/jns.1997.87.5.0724. [DOI] [PubMed] [Google Scholar]
- 16.Dubal DB, Kashon ML, Pettigrew LC, Ren JM, Finklestein SP, Rau SW, Wise PM. Estrogen protects against ischemic injury. J Cereb Blood Flow Metab. 1998;18:1253–1258. doi: 10.1097/00004647-199811000-00012. [DOI] [PubMed] [Google Scholar]
- 17.Shi J, Panickar KS, Yang SH, Rabbani O, Day AL, Simpkins JW. Estrogen attenuates over-expression of β-amyloid precursor protein messenger RNA in an animal model of focal ischemia. Brain Res. 1998;810:87–92. doi: 10.1016/s0006-8993(98)00888-9. [DOI] [PubMed] [Google Scholar]
- 18.Zhang YQ, Shi J, Rajakumar G, Day AL, Simpkins JW. Effects of gender and estrogen treatment on focal brain ischemia. Brain Res. 1998;784:321–324. doi: 10.1016/s0006-8993(97)00502-7. [DOI] [PubMed] [Google Scholar]
- 19.Rusa R, Alkayed NJ, Crain BJ, Traystman RJ, Kimes AS, London ED, Klaus JA, Hurn PD. 17β-Estrogen reduces stroke injury in estrogen-deficient animals. Stroke. 1999;30:1665–1670. doi: 10.1161/01.str.30.8.1665. [DOI] [PubMed] [Google Scholar]
- 20.McCullough LD, Zeng Z, Blizzard KK Debchoudhury I, Hurn PD. Ischemic nitric oxide and poly (ADP-ribose) polymerase-1 in cerebral ischemia: male toxicity, female protection. J Cereb Blood Flow Metab. 2005;25:502–512. doi: 10.1038/sj.jcbfm.9600059. [DOI] [PubMed] [Google Scholar]
- 21.Shughrue PJ, Merchenthaler I. Estrogen prevents the loss of CA1 hippocampal neurons in gerbils after ischemic injury. Neuroscience. 2003;116:851–861. doi: 10.1016/s0306-4522(02)00790-x. [DOI] [PubMed] [Google Scholar]
- 22.Shi J, et al. Estrogens decrease reperfusion-associated cortical ischemic damage: an MRI analysis in a transient focal ischemia model. Stroke. 2001;32:987–992. doi: 10.1161/01.str.32.4.987. [DOI] [PubMed] [Google Scholar]
- 23.Plahta WC, Clark DL, Colbourne F. 17Beta-Estrogen pretreatment reduces CA1 sector cell death and the spontaneous hyperthermia that follows forebrain ischemia in the gerbil. Neuroscience. 2004;129:187–193. doi: 10.1016/j.neuroscience.2004.07.037. [DOI] [PubMed] [Google Scholar]
- 24.Jover T, Tanaka H, Calderone A, Oguro K, Bennett MV, Etgen AM, Zukin RS. Estrogen protects against global ischemia-induced neuronal death and prevents activation of apoptotic signaling cascades in the hippocampal CA1. J Neurosci. 2002;22:2115–2124. doi: 10.1523/JNEUROSCI.22-06-02115.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miller NR, Jover T, Cohen HW, Zukin RS, Etgen AM. Estrogen can act via estrogen receptor alpha and beta to protect hippocampal neurons against global ischemia-induced cell death. Endocrinology. 2005;146:3070–3079. doi: 10.1210/en.2004-1515. [DOI] [PubMed] [Google Scholar]
- 26.Toung TJK, Traystman RJ, Hurn PD. Estrogen-mediated neuroprotection after experimental stroke in males. Stroke. 1998;29:1666–1670. doi: 10.1161/01.str.29.8.1666. [DOI] [PubMed] [Google Scholar]
- 27.Alkayed NJ, Crain BJ, Traystman RJ, Hurn PD. Estrogen-mediated neuroprotection in reproductively senescent rats. Stroke. 1999;30:274. [Google Scholar]
- 28.Dubal DB, Wise PM. Neuroprotective effects of estrogen in middle aged female rats. Endocrinology. 2001;142:43–48. doi: 10.1210/endo.142.1.7911. [DOI] [PubMed] [Google Scholar]
- 29.Gibson CL, Gray LJ, Murphy SP, Bath PM. Estrogens and experimental ischemic stroke: a systematic review. J Cereb Blood Flow Metab. 2006;26:1103–1113. doi: 10.1038/sj.jcbfm.9600270. [DOI] [PubMed] [Google Scholar]
- 30.Kondo Y, Suzuki K, Sakuma Y. Estrogen alleviates cognitive dysfunction following transient brain ischemia in ovariectomized gerbils. Neurosci Lett. 1997;238:45–48. doi: 10.1016/s0304-3940(97)00847-1. [DOI] [PubMed] [Google Scholar]
- 31.Gulinello M, Lebesgue D, Jover-Mengual T, Zukin RS, Etgen AM. Acute and chronic estrogen treatments reduce memory deficits induced by transient global ischemia in female rats. Horm Behav. 2006;49:246–260. doi: 10.1016/j.yhbeh.2005.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Plamondon H, Morin A, Charron C. Chronic 17beta-estrogen pretreatment and ischemia-induced hippocampal degeneration and memory impairments: a 6-month survival study. Horm Behav. 2006;50:361–369. doi: 10.1016/j.yhbeh.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 33.Li X, Blizzard KK, Zeng Z, DeVries AC, Hurn PD, McCullough LD. Chronic behavioral testing after focal ischemia in the mouse: functional recovery and the effects of gender. Exp Neurol. 2004;187:94–104. doi: 10.1016/j.expneurol.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 34.Liao SL, Chen WY, Kuo JS, Chen CJ. Association of serum estrogen level and ischemic neuroprotection in female rats. Neurosci Lett. 2001;297:159–162. doi: 10.1016/s0304-3940(00)01704-3. [DOI] [PubMed] [Google Scholar]
- 35.Sawada M, Alkayed NJ, Goto S, Crain BJ, Traystman RJ, Shaivitz A, Nelson RJ, Hurn PD. Estrogen receptor antagonist ICI182,780 exacerbates ischemic injury in female mouse. Stroke. 2000;20:112–118. doi: 10.1097/00004647-200001000-00015. [DOI] [PubMed] [Google Scholar]
- 36.McCullough LD, Blizzard K, Simpson ER, Oz OK, Hurn PD. Aromatase cytochrome P450 and extragonadal estrogen play a role in ischemic neuroprotection. J Neurosci. 2003;23:8701–8705. doi: 10.1523/JNEUROSCI.23-25-08701.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Veiga S, Azcoiitia I, Garcia-Segura LM. Extragonadal synthesis of estrogen is protective against kainic acid excitotoxic damage to the hippocampus. Neuroreport. 2005;16:1599–1603. doi: 10.1097/01.wnr.0000179081.39659.7d. [DOI] [PubMed] [Google Scholar]
- 38.Carswell HV, Dominiczak AF, Garcia-Segura LM, Harada N, Hutchison JB, Macrae IM. Brain aromatase expression after experimental stroke: topography and time course. J Steroid Biochem Mol Biol. 2005;96:89–91. doi: 10.1016/j.jsbmb.2005.02.016. [DOI] [PubMed] [Google Scholar]
- 39.Rossberg M, Murphy S, Traystman R, Hurn PD. LY353381.Hcl, a selective estrogen receptor modulator, and experimental stroke. Stroke. 2000;31:3041–3046. doi: 10.1161/01.str.31.12.3041. [DOI] [PubMed] [Google Scholar]
- 40.Kimelberg H, Feurstel P, Jin Y, Paquette J, Boulos A, Keller RW, Trammer BI. Acute treatment with tamoxifen reduces ischemic damage following middle cerebral artery occlusion. Neuroreport. 2000;11:2675–2679. doi: 10.1097/00001756-200008210-00014. [DOI] [PubMed] [Google Scholar]
- 41.Osuka K, Feustel PJ, Mongin AA, Tranmer BI, Kimelberg HL. Tamoxifen inhibits nitroytrosine formation after reversible middle cerebral artery occlusion in the rat. J Neurochem. 2001;76:1842–1850. doi: 10.1046/j.1471-4159.2001.00198.x. [DOI] [PubMed] [Google Scholar]
- 42.Mehta SH, Dhandapani KM, De Sevilla LM, Webb RC, Mahesh VB, Brann DW. Tamoxifen, a selective estrogen receptor modulator, reduces ischemic damage caused by middle cerebral artery occlusion in the ovariectomized female rat. Neuroendocrinology. 2003;77:44–50. doi: 10.1159/000068332. [DOI] [PubMed] [Google Scholar]
- 43.Zhang Y, Jin Y, Behr MJ, Feustel PJ, Morrison JP, Kimelberg HK. Behavioral and histological neuroprotection by tamoxifen after reversible focal cerebral ischemia. Exp Neurol. 2005;196:41–46. doi: 10.1016/j.expneurol.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 44.Zhao L, O’Neill K, Brinton RD. Selective estrogen receptor modulators (SERMs) for the brain: Current status and remaining challenges for developing NeuroSERMs. Brain Res Rev. 2005;49:472–493. doi: 10.1016/j.brainresrev.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 45.Dubal DB, Kashon ML, Pettigrew LC, Ren JM, Finklestein SP, Rau SW, Wise PM. Estrogen protects against ischemic injury. J Cereb Blood Flow Metab. 1998;18:1253–1258. doi: 10.1097/00004647-199811000-00012. [DOI] [PubMed] [Google Scholar]
- 46.Simpkins JW, Rajakumar G, Zhang YQ, Simpkins CE, Greenwald D, Yu CJ, Bodor N, Day AL. Estrogens may reduce mortality and ischemic damage caused by middle cerebral artery occlusion in the female rat. J Neurosurg. 1997;87:724–730. doi: 10.3171/jns.1997.87.5.0724. [DOI] [PubMed] [Google Scholar]
- 47.Yang SH, Shi J, Day AL, Simpkins JW. Estrogen exerts neuroprotective effects when administered after ischemic stroke. Stroke. 2000;31:745–749. doi: 10.1161/01.str.31.3.745. [DOI] [PubMed] [Google Scholar]
- 48.Wilson ME, Dubal DB, Wise PM. Estrogen protects against injury induced cell death in cortical explant cultures: a role for estrogen receptors. Brain Res. 2000;873:235–242. doi: 10.1016/s0006-8993(00)02479-3. [DOI] [PubMed] [Google Scholar]
- 49.Dubal DB, Zhu H, Yu J, Rau SW, Shughrue PJ, Merchenthaler I, Kindy MS, Wise PM. Estrogen receptor alpha, not beta, is a critical link in estrogen-mediated protection against brain injury. Proc Natl Acad Sci U S A. 2001;98:1952–1957. doi: 10.1073/pnas.041483198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Merchenthaler I, Dellovade TL, Shughrue PJ. Neuroprotection by estrogen in animal models of global and focal ischemia. Ann NY Acad Sci. 2003;1007:89–100. doi: 10.1196/annals.1286.009. [DOI] [PubMed] [Google Scholar]
- 51.Dubal DB, Shughrue PJ, Wilson ME, Merchenthaler I, Wise PM. Estrogen modulates bcl-2 in cerebral ischemia: a potential role for estrogen receptors. J Neurosci. 1999;19:6385–6393. doi: 10.1523/JNEUROSCI.19-15-06385.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sampei K, Goto S, Alkayed NJ, Crain BJ, Korach KS, Traystman RJ, Demas GE, Nelson RJ, Hurn PD. Stroke in estrogen receptor-alpha-deficient mice. Stroke. 2000;31:738–743. doi: 10.1161/01.str.31.3.738. [DOI] [PubMed] [Google Scholar]
- 53.Carswell HV, Macrae IM, Gallagher L, Harrop E, Horsburgh KJ. Neuroprotection by a selective estrogen receptor beta agonist in a mouse model of global ischemia. Am J Physiol Heart Circ Physiol. 2004;287:H1501–H1504. doi: 10.1152/ajpheart.00227.2004. [DOI] [PubMed] [Google Scholar]
- 54.Zhao L, Wu TW, Brinton RD. Estrogen receptor subtypes alpha and beta contribute to neuroprotection and increased Bcl-2 expression in primary hippocampal neurons. Brain Res. 2004;1010:22–34. doi: 10.1016/j.brainres.2004.02.066. [DOI] [PubMed] [Google Scholar]
- 55.Carmeci C, Thompson DA, Ring HZ, Francke U, Weigel RJ. Identification of a gene (GPR30) with homology to the G-protein-coupled receptor superfamily associated with estrogen receptor expression in breast cancer. Genomics. 1997;45:607–617. doi: 10.1006/geno.1997.4972. [DOI] [PubMed] [Google Scholar]
- 56.Filardo EJ, Thomas P. GPR30: a seven-transmembrane-spanning estrogen receptor that triggers EGF release. Trends Endocrinol Metab. 2005;16:362–367. doi: 10.1016/j.tem.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 57.O’Dowd BF, Nguyen T, Marchese A, Cheng R, Lynch KR, Heng HH, Kolakowski L, Jr, George SR. Discovery of three novel G-protein-coupled receptor genes. Genomics. 1988;47:310–313. doi: 10.1006/geno.1998.5095. [DOI] [PubMed] [Google Scholar]
- 58.Funakoshi T, Yanai A, Shinoda K, Kawano MM, Mizukami Y. G-protein-coupled receptor 30 is an estrogen receptor in the plasma membrane. Biochem Biophys Res Commun. 2006;346:904–910. doi: 10.1016/j.bbrc.2006.05.191. [DOI] [PubMed] [Google Scholar]
- 59.Khan MM, De Sevilla LM, Hadman M, Brann DW. Cellular and subcellular organization of a novel G-protein coupled estrogen receptor (GPR30) in brain: implication for synaptic functions. 2006; Neuroscience Meeting Planner; Atlanta, GA. 2006. Program No. 659.19. Online. [Google Scholar]
- 60.Perry AN, Cushing BS. Membrane estrogen receptor (GPR30): bridging the gap between rapid estrogen signaling and dopamine neurotransmission. 2006; Neuroscience Meeting Planner; Atlanta, GA. 2006. Program No. 153.20. Online. [Google Scholar]
- 61.Alkayed NJ, Goto S, Sugo N, Joh HD, Klaus J, Crain BJ, Bernard O, Traystman RJ, Hurn PD. Estrogen and Bcl-2: gene induction and effect of transgene in experimental stroke. J Neurosci. 2001;21:7543–7550. doi: 10.1523/JNEUROSCI.21-19-07543.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wu TW, Wang JM, Chen S, Brinton RD. 17 Beta-estrogen induced Ca2+ influx via L-type calcium channels activates the Src/ERK/cyclic-AMP response element binding protein signal pathway and BCL-2 expression in rat hippocampal neurons: a potential initiation mechanism for estrogen-induced neuroprotection. Neuroscience . 2005;135:59–72. doi: 10.1016/j.neuroscience.2004.12.027. [DOI] [PubMed] [Google Scholar]
- 63.Singer CA, Rogers KL, Dorsa DM. Modulation of Bcl-2 expression: a potential component of estrogen protection in NT2 neurons. Neuroreport. 1998;9:2565–2568. doi: 10.1097/00001756-199808030-00025. [DOI] [PubMed] [Google Scholar]
- 64.Bagetta G, Chiappetta O, Amantea D, Iannone M, Rotiroti D, Costa A, Nappi G, Corasaniti MT. Estrogen reduces cytochrome c translocation and minimizes hippocampal damage caused by transient global ischemia in rat. Neurosci Lett. 2004;368:87–91. doi: 10.1016/j.neulet.2004.06.062. [DOI] [PubMed] [Google Scholar]
- 65.Choi YC, Lee JH, Hong KW, Lee KS. 17Beta-estrogen prevents focal cerebral ischemic damages via activation of Akt and CREB in association with reduced PTEN phosphorylation in rats. Fundam Clin Pharmacol. 2004;18:547–557. doi: 10.1111/j.1472-8206.2004.00284.x. [DOI] [PubMed] [Google Scholar]
- 66.Rau SW, Dubal DB, Bottner M, Gerhold LM, Wise PM. Estrogen attenuates programmed cell death after stroke-like injury. J Neurosci. 2003;23:11420–11426. doi: 10.1523/JNEUROSCI.23-36-11420.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Singer CA, Figueroa-Masot XA, Batchelor RH, Dorsa DM. The mitogen-activated protein kinase pathway mediates estrogen neuroprotection after glutamate toxicity in primary cortical neurons. J Neurosci. 1999;19:2455–2463. doi: 10.1523/JNEUROSCI.19-07-02455.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Singh M, Setalo G, Jr, Guan X, Warren M, Toran-Allerand CD. Estrogen-induced activation of mitogen-activated protein kinase in cerebral cortical explants: convergence of estrogen and neurotrophin signaling pathways. J Neurosci. 1999;19:1179–1186. doi: 10.1523/JNEUROSCI.19-04-01179.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Singh M. Ovarian hormones elicit phosphorylation of Akt and extracellular signal regulated kinase in explants of the cerebral cortex. Endocrine. 2001;14:407–415. doi: 10.1385/ENDO:14:3:407. [DOI] [PubMed] [Google Scholar]
- 70.Wang R, Zhang QZ, Han D, Lu Q, Zhang GY. Inhibition of MLK3-MKK4/7-JNK1/2 pathway by Akt1 in exogenous estrogen-induced neuroprotection against transient global cerebral ischemia by a non-genomic mechanism in male rats. J Neurochem. 2006 doi: 10.1111/j.1471-4159.2006.04201.x. epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 71.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 72.Cross Da, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 73.Goodenough S, Schleusner D, Pietrzik C, Skutella T, Behl C. Glycogen synthase kinase 3beta links neuroprotection by 17beta-estrogen to key Alzheimer processes. Neuroscience. 2005;132:581–589. doi: 10.1016/j.neuroscience.2004.12.029. [DOI] [PubMed] [Google Scholar]
- 74.Cabodi S, et al. p130Cas interacts with estrogen receptor alpha and modulates non-genomic estrogen signaling in breast cancer cells. J Cell Sci. 2004;117:1603–1611. doi: 10.1242/jcs.01025. [DOI] [PubMed] [Google Scholar]
- 75.Lu Q, Pallas DC, Surks HK, Baur WE, Mendelsohn ME, Karas RH. Striatin assembles a membrane signaling complex necessary for rapid, nongenomic activation of endothelial NO synthase by estrogen receptor alpha. Proc Natl Acad Sci USA. 2004;101:17126–17131. doi: 10.1073/pnas.0407492101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Castets F, et al. A novel calmodulin-binding protein, belonging to the WD-repeat family, is localized in dendrites of a subset of CNS neurons. J Cell Biolo. 1996;134:1051–1062. doi: 10.1083/jcb.134.4.1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Khan MM, Hadman M, Wakade C, De Sevilla LM, Dhandapani KM, Mahesh VB, Vadlamudi RK, Brann DW. Cloning, expression, and localization of MNAR/PELP1 in rodent brain: colocalization in estrogen receptor-alpha - but not in gonadotropin-releasing hormone-positive neurons. Endocrinology. 2005;146:215–5227. doi: 10.1210/en.2005-0276. [DOI] [PubMed] [Google Scholar]
- 78.Khan MM, Hadman M, De Sevilla LM, Mahesh VB, Buccafusco J, Hill WD, Brann DW. Cloning, distribution and colocalization of MNAR/PELP1 with glucocorticoid receptors in primate and non-primate brain. Neuroendocrinology. 2007 doi: 10.1159/000097746. in press. [DOI] [PubMed] [Google Scholar]
- 79.Vadlamudi RK, Wang RA, Mazumdar A, Kim Y, Shin J, Sahin A, Kumar R. Molecular cloning and characterization of PELP1, a novel human coregulator of estrogen receptor alpha. J Biol Chem. 2001;276:38272–38279. doi: 10.1074/jbc.M103783200. [DOI] [PubMed] [Google Scholar]
- 80.Wilson ME, Liu Y, Wise PM. Estrogen enhances Akt activation in cortical explant cultures following neuronal injury. Brain Res Mol Brain Res. 2002;102:48–54. doi: 10.1016/s0169-328x(02)00181-x. [DOI] [PubMed] [Google Scholar]
- 81.Wilson ME, Dubal DB, Wise PM. Estrogen protects against injury induced cell death in cortical explant cultures: a role for estrogen receptors. Brain Res. 2000;873:235–242. doi: 10.1016/s0006-8993(00)02479-3. [DOI] [PubMed] [Google Scholar]
- 82.Dhandapani KM, Hadman M, De Sevilla L, Wade MF, Mahesh VB, Brann DW. Astrocyte protection of neurons: role of transforming growth factor-beta signaling via a c-Jun-AP-1 protective pathway. J Biol Chem. 2003;278:43329–43339. doi: 10.1074/jbc.M305835200. [DOI] [PubMed] [Google Scholar]
- 83.Sortino MA, Chisari M, Merlo S, Vancheri C, Caruso M, Nicoletti F, Canonico PL, Copani A. Glia mediates the neuroprotective action of estrogen on beta-amyloid-induced neuronal death. Endocrinology. 2004;145:5080–5086. doi: 10.1210/en.2004-0973. [DOI] [PubMed] [Google Scholar]
- 84.Dhandapani K, Brann DW. Neuroprotective effects of estrogen and tamoxifen in vitro: a facilitative role for glia? Endocrine. 2003;21:59–66. doi: 10.1385/endo:21:1:59. [DOI] [PubMed] [Google Scholar]
- 85.Dhandapani KM, Wade FM, Mahesh VB, Brann DW. Astrocyte-derived transforming growth factor-{beta} mediates the neuroprotective effects of 17{beta}-estrogen: involvement of nonclassical genomic signaling pathways. Endocrinology. 2005;146:2749–2759. doi: 10.1210/en.2005-0014. [DOI] [PubMed] [Google Scholar]
- 86.Zhu Y, Yang GY, Ahlemeyer B, Pang L, Che XM, Culmsee C, KlumppS, Krieglstein J. Transforming growth factor-beta 1 increases Bad phosphorylation and protects neurons against damage. J Neurosci. 2002;22:3898–3909. doi: 10.1523/JNEUROSCI.22-10-03898.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ruocco A, et al. A transforming growth factor-beta antagonist unmasks the neuroprotective role of this endogenous cytokine in excitotoxic and ischemic brain injury. J Cereb Blood Flow Metab. 1999;19:1345–1353. doi: 10.1097/00004647-199912000-00008. [DOI] [PubMed] [Google Scholar]
- 88.Dhandapani KM, Brann DW. Transforming growth factor-β. A neuroprotective factor in cerebral ischemia. Cell Biochem Biophys. 2003;39:13–22. doi: 10.1385/CBB:39:1:13. [DOI] [PubMed] [Google Scholar]
- 89.Mong JA, Blutstein T. Estrogen modulation of astrocytic form and function: implications for hormonal control of synaptic communication. Neuroscience. 2006;138:967–975. doi: 10.1016/j.neuroscience.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 90.Pawlak J, Brito V, Kuppers E, Beyer C. Regulation of glutamate transporter GLAST and GLT-1 expression in astrocytes by estrogen. Brain Res Mol Brain Res. 2005;138:1–7. doi: 10.1016/j.molbrainres.2004.10.043. [DOI] [PubMed] [Google Scholar]
- 91.Saldanha CJ, Rohmann KN, Coomaralingam L, Wynne RD. Esrogen provision by reactive glia decreases apoptosis in the zebra finch (Taeniopygia guttata) J Neurobiol. 2005;64:192–201. doi: 10.1002/neu.20147. [DOI] [PubMed] [Google Scholar]
- 92.Wen Y, Yang S, Liu R, Perez E, Yi KD, Koulen P, Simpkins JW. Estrogen attenuates nuclear factor-kappa B activation induced by transient cerebral ischemia. Brain Res. 2004;1008:147–154. doi: 10.1016/j.brainres.2004.02.019. [DOI] [PubMed] [Google Scholar]
- 93.Bruce-Keller AJ, Keeling JL, Keller JN, Huang FF, Camondola S, Mattson MP. Antiinflammatory effects of estrogen on microglia activation. Endocrinology. 2000;141:3646–3456. doi: 10.1210/endo.141.10.7693. [DOI] [PubMed] [Google Scholar]
- 94.Vegeto E, et al. Estrogen prevents the lipopolysaccharide-induced inflammatory response in microglia. J Neurosci. 2001;21:1809–1818. doi: 10.1523/JNEUROSCI.21-06-01809.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tripanichkul W, Sripanichkulchai K, Finkelstein DI. Estrogen down-regulates glial activation in male mice following 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine intoxication. Brain Res. 2006;1084:28–37. doi: 10.1016/j.brainres.2006.02.029. [DOI] [PubMed] [Google Scholar]
- 96.Morale MC, et al. Estrogen, neuroinflammation and neuroprotection in Parkinson’s disease: glia dictates resistance versus vulnerability to neurodegneration. Neuroscience. 2006;138:869–878. doi: 10.1016/j.neuroscience.2005.07.060. [DOI] [PubMed] [Google Scholar]
- 97.Gibbons HM, Dragunow M. Microglia induce neural cell death via a proximity-dependent mechanism involving nitric oxide. Brain Res. 2006;1084:1–15. doi: 10.1016/j.brainres.2006.02.032. [DOI] [PubMed] [Google Scholar]
- 98.Sugawara T, Noshita N, Lewen A, Gasche Y, Ferrand-Drake M, Fujimura M, Morita-Fujimura Y, Chan PH. Overexpression of copper/zinc superoxide dismutase in transgenic rats protects vulnerable neurons against ischemic damage by blocking the mitochondrial pathway of caspase activation. J Neurosci. 2002;22:209–217. doi: 10.1523/JNEUROSCI.22-01-00209.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chan PH. Role of oxidants in ischemic brain damage. Stroke. 1996;27:1124–1129. doi: 10.1161/01.str.27.6.1124. [DOI] [PubMed] [Google Scholar]
- 100.Peters O, Back T, Lindauer U, Busch C, Megow D, Dreier J, Dirnagl U. Increased formation of reactive oxygen species after permanent and reversible middle cerebral artery occlusion in the rat. J Cereb Blood Flow Metab. 1998;18:196–205. doi: 10.1097/00004647-199802000-00011. [DOI] [PubMed] [Google Scholar]
- 101.Nagayama T, Lan J, Henshall DC, Chen D, O’Horo C, Simon RP, Chen J. Induction of oxidative DNA damage in the peri-infarct region after permanent focal cerebral ischemia. J Neurochem. 2000;75:1716–1728. doi: 10.1046/j.1471-4159.2000.0751716.x. [DOI] [PubMed] [Google Scholar]
- 102.Fujimura M, Morita-Fujimura Y, Kawase M, Copin J, Calagui B, Epstein CJ, Chan PH. Maganese superoxide dismutase mediates the early release of mitochrondrial cytochrome c and subsequent DNA fragmentation after persistent focal cerebral ischemia in mice. J Neurosci. 1999;19:3414–3422. doi: 10.1523/JNEUROSCI.19-09-03414.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mehta SH, Webb RC, Ergul A, Tawfik A, Dorrance AM. Neuroprotection by tempol in a model of iron-induced oxidative stress in acute ischemic stroke. Am J Physiol Regul Integr Comp Physiol. 2004;286:R283–R288. doi: 10.1152/ajpregu.00446.2002. [DOI] [PubMed] [Google Scholar]
- 104.Matttson MP, Culmsee C, Yu ZF. Apoptotic and antiapoptotic mechanisms in stroke. Cell Tissue Res. 2000;301:173–187. doi: 10.1007/s004419900154. [DOI] [PubMed] [Google Scholar]
- 105.Bindokas VP, Jordan J, Lee CC, Miller RJ. Superoxide production in rat hippocampal neurons: selective imaging with hydroethidine. J Neurosci. 1996;16:1324 –1336. doi: 10.1523/JNEUROSCI.16-04-01324.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Murakami K, Kondo T, Kawase M, Li Y, Sato S, Chen SF, Chan PH. Mitochondrial susceptibility to oxidative stress exacerbates cerebral infarction that follows permanent focal cerebral ischemia in mutant mice with manganese superoxide dismutase deficiency. J Neurosci. 1998;18:205–213. doi: 10.1523/JNEUROSCI.18-01-00205.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kii N, Adachi N, Liu K, Arai T. Acute effects of 17beta-estrogen on oxidative stress in ischemic rat striatum. J Neurosurg Anesthesiol. 2005;17:27–32. [PubMed] [Google Scholar]
- 108.Prokai L, Prokai-Tatrai K, Perjesi P, Zharikova AD, Perez EJ, Liu R, Simpkins JW. Quinol-based cyclic antioxidant mechanism in estrogen neuroprotection. Proc Natl Acad Sci USA. 2003;100:11741–11746. doi: 10.1073/pnas.2032621100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Xia S, Cai ZY, Thio LL, Kim-Han JS, Dugan LL, Covey DF, Rothman SM. The estrogen receptor is not essential for all estrogen neuroprotection: new evidence from a new analog. Neurobiol Dis. 2002;9:282–293. doi: 10.1006/nbdi.2002.0478. [DOI] [PubMed] [Google Scholar]
- 110.Biewenga E, Cabell L, Audesirk T. Estrogen and raloxifene protect cultured SN4741 neurons against oxidative stress. Neurosci Lett. 2005;373:179–183. doi: 10.1016/j.neulet.2004.09.067. [DOI] [PubMed] [Google Scholar]
- 111.Angoa-Perez M, Jiang H, Rodriguez AI, Lemini C, Levine RA, Rivas-Arancibia S. Estrogen counteracts ozone-induced oxidative stress and nigral neuronal death. Neuroreport. 2006;17:629–633. doi: 10.1097/00001756-200604240-00014. [DOI] [PubMed] [Google Scholar]
- 112.Wallace DR, Dodson S, Nath A, Booze RM. Estrogen attenuates gp120- and tat1-72-induced oxidative stress and prevents loss of dopamine transporter function. Synapse. 2006;59:51–60. doi: 10.1002/syn.20214. [DOI] [PubMed] [Google Scholar]
- 113.Sawada H, Ibi M, Kihara T, Urushitani M, Akaike A, Shimohama S. Estrogen protects mesencephalic dopaminergic neurons from oxidative stress-induced neuronal death. J Neurosci Res. 1998;54:709–719. doi: 10.1002/(SICI)1097-4547(19981201)54:5<707::AID-JNR16>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 114.Shi J, Zhang YQ, Simpkins JW. Effects of 17beta-estradiol on glucose transporter 1 expression and endothelial cell survival following focal ischemia in the rats. Exp Brain Res. 1997;117:200–206. doi: 10.1007/s002210050216. [DOI] [PubMed] [Google Scholar]
- 115.Krause DN, Duckles SP, Pellingrino DA. Influence of sex steroid hormones on cerebrovascular function. J Appl Physiol. 2006;101:1252–1261. doi: 10.1152/japplphysiol.01095.2005. [DOI] [PubMed] [Google Scholar]
- 116.Shulman LM, Bhat V. Gender disparities in Parkinson’s disease. Expert Rev Neurother. 2006;6:407–416. doi: 10.1586/14737175.6.3.407. [DOI] [PubMed] [Google Scholar]
- 117.Bower JH, Maraganore DM, McDonnell SK, Rocca WA. Incidence and distribution of parkinsonism in Olmstead County, Minnesota, 1976-1990. Neurology. 1999;52:1214–1220. doi: 10.1212/wnl.52.6.1214. [DOI] [PubMed] [Google Scholar]
- 118.Baldereschi M, Di Carlo A, Rocca WA, Vanni P, Maggi S, Perissinotto E, Grigoletto F, Amaducci L, Inzitari D. Parkinson’s disease and parkinsonism in a longitudinal study: two-fold higher incidence in men. ILSA Working Group. Italian Longitudinal Study on Aging. Neurology. 2000;55:1358–1363. doi: 10.1212/wnl.55.9.1358. [DOI] [PubMed] [Google Scholar]
- 119.Wooten GF, Currie LJ, Bovbjerg VE, Lee JK, Patrie J. Are men at greater risk for Parkinson’s disease than women? J Neurol Neurosurg Psychiatry. 2004;75:637–639. doi: 10.1136/jnnp.2003.020982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Rajput MI, Rajput A, Rajput AH. Gender differences in Parkinson’s disease presentation in a movement disorder clinic. Mov Disorder. 2004;19:1118–1134. [Google Scholar]
- 121.Lyons KE, Hubble JP, Troster AI, Pahwa R, Koller WC. Gender differences in Parkinson’s disease. Clin Neuropharmacol. 1998;21:118–121. [PubMed] [Google Scholar]
- 122.Baba Y, Putzke JD, Whaley NR, Wszolek ZK, Uitti RJ. Gender and the Parkinson’s disease phenotype. J Neurol. 2005;252:1201–1205. doi: 10.1007/s00415-005-0835-7. [DOI] [PubMed] [Google Scholar]
- 123.Scott B, Borgman A, Engler H, Johnels B, Aquilonius SM. Gender differences in Parkinson’s disease symptom profile. Acta Neurol Scand. 2000;102:37–43. doi: 10.1034/j.1600-0404.2000.102001037.x. [DOI] [PubMed] [Google Scholar]
- 124.Parkinson Study Group. Impact of deprenyl and tocopherol treatment on Parkinson’s disease in DATATOP patients requiring levodopa. Ann Neurol. 1996;39:37–45. doi: 10.1002/ana.410390107. [DOI] [PubMed] [Google Scholar]
- 125.Zappia M, Crescibene L, Arabia G, et al. Body weight influences pharmacodynamics of levodopa in Parkinson’s disease. Clin Neuropharmacol. 2002;25:79–82. doi: 10.1097/00002826-200203000-00004. [DOI] [PubMed] [Google Scholar]
- 126.Arabia G, Zappia M, Bosco D, Crescibene L, Bagala A, Bastone L, Caracciolo M, Scornalenghi M, Quattrone A. Body weight, levodopa pharmokinetics and dysknesia in Parkinson’s disease. Neurol Sci. 2002 ;2:S53–S54. doi: 10.1007/s100720200066. [DOI] [PubMed] [Google Scholar]
- 127.Martinelli P, Contin M, Scaglione RC, Riva R, Albani F, Baruzzi A. Levodopa pharmacokinetics and dyskinesias: are there sex-related differences? Neurol Sci. 2003;24:192–193. doi: 10.1007/s10072-003-0125-z. [DOI] [PubMed] [Google Scholar]
- 128.Kompoliti K. Gender and pramipexole effects on levodopa pharmacokinetics and pharmocodynamics. Neurology. 2002;58:1412–1422. doi: 10.1212/wnl.58.9.1418. [DOI] [PubMed] [Google Scholar]
- 129.Hariz GM, Lindberg M, Hariz MI, Bergenheim AT. Gender differences in disability and health-related quality of life in patients with parkinson’s disease treated with stereotaxic surgery. Acta Neurol Scand. 2003;108:28–37. doi: 10.1034/j.1600-0404.2003.00092.x. [DOI] [PubMed] [Google Scholar]
- 130.Dluzen DE, McDermott JL, Liu B. Estrogen as a neuroprotectant against MPTP-induced neurotoxicity in C57/B1 mice. Neurotoxicol Teratol. 1996;18:603–606. doi: 10.1016/0892-0362(96)00086-4. [DOI] [PubMed] [Google Scholar]
- 131.Callier S, Morissette M, Grandbois M, Pelaprat D, Di Paolo T. Neuroprotective properties of 17beta-estrogen, progesterone, and raloxifene in MPTP C57Bl/6 mice. Synapse. 2001;41:131–138. doi: 10.1002/syn.1067. [DOI] [PubMed] [Google Scholar]
- 132.Grandbois M, Morissette M, Callier S, Di Paolo T. Ovarian steroids and raloxifene prevent MPTP-induced dopamine depletion in mice. Neuroreport. 2000;11:343–346. doi: 10.1097/00001756-200002070-00024. [DOI] [PubMed] [Google Scholar]
- 133.Callier S, Morissette M, Grandbois M, Di Paolo T. Stereospecific prevention by 17beta-estradiol of MPTP-induced dopamine depletion in mice. Synapse. 2000;37:245–251. doi: 10.1002/1098-2396(20000915)37:4<245::AID-SYN1>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 134.D’Astous M, Morissette M, Tanguay B, Callier S, Di Paolo T. Dehydroepiandrosterone (DHEA) such as 17beta-estradiol prevents MPTP-induced dopamine depletion in mice. Synapse. 2003;47:10–14. doi: 10.1002/syn.10145. [DOI] [PubMed] [Google Scholar]
- 135.Ramirez AD, Liu X, Menniti FS. Repeated estradiol treatment prevents MPTP-induced dopamine depletion in male mice. Neuroendocrinology. 2003;77:223–231. doi: 10.1159/000070277. [DOI] [PubMed] [Google Scholar]
- 136.Shughrue PJ. Estrogen attenuates the MPTP-induced loss of dopamine neurons from the mouse SNc despite a lack of estrogen receptors (ERalpha and ERbeta) Exp Neurol. 2004;190:468–477. doi: 10.1016/j.expneurol.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 137.Dluzen D, Horstink M. Estrogen as neuroprotectant of nigrostriatal dopaminergic system: laboratory and clinical studies. Endocrine. 2003;21:67–75. doi: 10.1385/endo:21:1:67. [DOI] [PubMed] [Google Scholar]
- 138.D’Astous M, Morissete M, Di Paolo T. Effect of estrogen receptor agonists treatment in MPTP mice: evidence of neuroprotection by an ER alpha agonist. Neuropharmacology. 2004;47:1180–1188. doi: 10.1016/j.neuropharm.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 139.D’Astous M, Mendez P, Morissette M, Garcia-Segura LM, Di Paolo T. Implication of the phosphatidylinositol-3 kinase/protein kinase B signaling pathway in the neuroprotective effect of estrogen in the striatum of 1 methyl-4phenyl-1,2,3,6-tetrahydropyridine mice. Mol Pharmacol. 2006;69:1492–1498. doi: 10.1124/mol.105.018671. [DOI] [PubMed] [Google Scholar]
- 140.Tripanichkul W, Sripanichkulchai K, Finkelstein DI. Estrogen down-regulates glial activation in male mice following 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine intoxication. Brain Res. 2006;1084:28–37. doi: 10.1016/j.brainres.2006.02.029. [DOI] [PubMed] [Google Scholar]
- 141.Quesada A, Micevych PE. estrogen interacts with the IGF-1 system to protect nigrostriatal dopamine and maintain motoric behavior after 6-hydroxydopamine lesions. J Neurosci Res. 2004;75:107–116. doi: 10.1002/jnr.10833. [DOI] [PubMed] [Google Scholar]
- 142.Gomez-Mancilla B, Bedard PJ. Effect of estrogen and progesterone on L-dopa induced dyskinesia in MPTP-treated monkeys. Neurosci Lett. 1992;135:129–32. doi: 10.1016/0304-3940(92)90152-w. [DOI] [PubMed] [Google Scholar]
- 143.Ragonese P, D’Armelio M, Aridon P, Gammino M, Epifanio A, Morgante L, Savettieri G. Risk of Parkinson disease in women: effect of reproductive characteristics. Neurology. 2004;62:2010–2014. doi: 10.1212/wnl.62.11.2010. [DOI] [PubMed] [Google Scholar]
- 144.Currie LJ, Harrison MB, Trugman JM, Bennet JP, Wooten GF. Postmenopausal estrogen use affects risk for Parkinson disease. Arch Neurol. 2004;61:886–888. doi: 10.1001/archneur.61.6.886. [DOI] [PubMed] [Google Scholar]
- 145.Westberg L, et al. Association between the estrogen receptor beta gene and age of onset of Parkinson’s disease. Psychoneuroendocrinology. 2004;29:993–998. doi: 10.1016/j.psyneuen.2003.08.010. [DOI] [PubMed] [Google Scholar]
- 146.Gardiner SA, Morrison MF, Mozley PD, Mozley LH, Brensinger C, Bilker W, Newberg A, Battistini M. Pilot study on the effect of estrogen replacement therapy on brain dopamine transporter availability in healthy, postmenopausal women. Am J Geriatr Psychiatry. 2004;12:621–630. doi: 10.1176/appi.ajgp.12.6.621. [DOI] [PubMed] [Google Scholar]
- 147.Kishi Y, et al. Estrogen promotes differentiation and survival of dopaminergic neurons derived from human neural stem cells. J Neurosci Res. 2005;79:279–286. doi: 10.1002/jnr.20362. [DOI] [PubMed] [Google Scholar]
- 148.Przedborski S. Parthogenesis of nigral cell death in Parkinson’s disease. Parkinsonism Relat Disord. 2005;11(Suppl 1):S3–S7. doi: 10.1016/j.parkreldis.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 149.Dluzen DE, Mcdermott JL, Andersen LI. Tamoxifen diminishes methamphetamine-induced striatal dopamine depletion in intact female and male mice. J Neuroendocrinol. 2001;13:618–624. doi: 10.1046/j.1365-2826.2001.00675.x. [DOI] [PubMed] [Google Scholar]
- 150.D’Astous M, Mickley KR, Dluzen DE, Di Paolo T. Differential protective properties of estrogen and tamoxifen against methamphetamine-induced nigrostriatal dopaminergic toxicity in mice. Neuroendocrinology. 2005;82:111–120. doi: 10.1159/000091206. [DOI] [PubMed] [Google Scholar]
- 151.Dluzen E, Mickley KR. Gender differences in modulatory effects of tamoxifen upon the nigrostriatal dopamnergic system. Pharmacol Biochem Behav. 2005;80:27–33. doi: 10.1016/j.pbb.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 152.Kuo YM, Chen HH, Shieh CC, Chuang Cherng CG, Yu L. 4-Hydroxytamoxifen attenuates methamphetamine-induced nigrostriatal dopaminergic toxicity in intact and gonadectomized mice. J Neurochem. 2003;87:436–443. doi: 10.1046/j.1471-4159.2003.02089.x. [DOI] [PubMed] [Google Scholar]
- 153.Obata T. Tamoxifen protect against hydroxyl radical generation induced by phenelzine in rat striatum. Toxicology. 2006;222:46–52. doi: 10.1016/j.tox.2006.01.023. [DOI] [PubMed] [Google Scholar]
- 154.Nishino H, Nakajima K, Kumazaki M, Fukuda A, Muramatsu K, Deshpande SB, Inubushi T, Morikawa S, Borlongan CV, Sanberg PR. Estrogen protects against while testosterone exacerbates vulnerability of the lateral striatal artery to chemical hypoxia by 3-nitropropionic acid. Neurosci Res. 1998;30:303–312. doi: 10.1016/s0168-0102(98)00010-8. [DOI] [PubMed] [Google Scholar]
- 155.Fillit H, Weinreb H, Cholst I, Luine V, McEwen B, Amador R, Zabriskie J. Observations in a preliminary open trial of estrogen therapy for senile dementia-Alzheimer’s type. Psychoneuroendocrinology. 1986;11:337–345. doi: 10.1016/0306-4530(86)90019-3. [DOI] [PubMed] [Google Scholar]
- 156.Ohkura T, Isse K, Akazawa K, Hamamoto M, Yaoi Y, Hagino N. Evaluation of estrogen treatment in female patients with dementia of the Alzheimer type. Endocr J. 1994;41:361–371. doi: 10.1507/endocrj.41.361. [DOI] [PubMed] [Google Scholar]
- 157.Kawas C, Resnick S, Morrison A, Brookmeyer R, Corrada M, Zonderman A, Bacal C, Lingle DD, Metter E. A prospective study of estrogen replacement therapy and the risk of developing Alzheimer’s disease: the Baltimore Longitudinal Study of Aging. Neurology. 1997;48:1517–1521. doi: 10.1212/wnl.48.6.1517. published erratum appears in Neurology 51:654, 1998. [DOI] [PubMed] [Google Scholar]
- 158.Paganini-Hill A, Henderson V. Estrogen deficiency and risk of Alzheimer’s disease in women. Am J Epidemiol. 1994;140:256–261. doi: 10.1093/oxfordjournals.aje.a117244. [DOI] [PubMed] [Google Scholar]
- 159.Paganini-Hill A, Henderson VW. Estrogen replacement therapy and risk of Alzheimer disease. Arch Intern Med. 1996;156:2213–2217. [PubMed] [Google Scholar]
- 160.Tang MX, Jacobs D, Stern Y, Marder K, Schofield P, Gurland B, Andrews H, Mayeux R. Effect of oestrogen during menopause on risk and age at onset of Alzheimer’s disease. Lancet. 1996;348:429–432. doi: 10.1016/S0140-6736(96)03356-9. [DOI] [PubMed] [Google Scholar]
- 161.Yaffe K, Sawaya G, Lieberburg I, Grady D. Estrogen therapy in postmenopausal women. JAMA. 1998;279:688–695. doi: 10.1001/jama.279.9.688. [DOI] [PubMed] [Google Scholar]
- 162.Hogervorst E, Williams J, Budge M, Riedel W, Jolles J. The nature of the effect of female gonadal hormone replacement therapy on cognitive function in post-menopausal women: a metaanalysis. Neuroscience. 2000;101:485–512. doi: 10.1016/s0306-4522(00)00410-3. [DOI] [PubMed] [Google Scholar]
- 163.Sundermann E, Gilbert PE, Murphy C. Estrogen and performance in recognition memory for olfactory and visual stimuli in females diagnosed with Alzheimer’s disease. J Int Neuropsychol Soc. 2006;12:400–404. doi: 10.1017/s1355617706060474. [DOI] [PubMed] [Google Scholar]
- 164.Henderson VW. Estrogen-containing hormone therapy and Alzheimer’s disease risk: understanding discrepant inferences from observational and experimental research. Neuroscience. 2006;138:1031–1039. doi: 10.1016/j.neuroscience.2005.06.017. [DOI] [PubMed] [Google Scholar]
- 165.Henderson VW. The role of sex steroids in Alzheimer’s disease: prevention and treatment. In: Lobo RA, editor. Treatment of the Postmenopausal Woman: Basic and Clinical Aspects. 2. Lippincott: Williams and Wilkins; 1999. pp. 269–276. [Google Scholar]
- 166.Mulnard RA, et al. Estrogen replacement therapy for treatment of mild to moderate Alzheimer’s disease. JAMA. 2000;283:1007–1015. doi: 10.1001/jama.283.8.1007. [DOI] [PubMed] [Google Scholar]
- 167.Henderson VW, et al. Estrogen for Alzheimer’s disease in women randomized, double-blind, placebo-controlled trial. Neurology. 2000;54:295–301. doi: 10.1212/wnl.54.2.295. [DOI] [PubMed] [Google Scholar]
- 168.Brenner DE, et al. Postmenopausal estrogen replacement therapy and the risk of Alzheimer’s disease: a population-based case-control study. Am J Epidimiol. 1994;140:262–267. doi: 10.1093/oxfordjournals.aje.a117245. [DOI] [PubMed] [Google Scholar]
- 169.Asthana S, Craft S, Baker LD, Raskind MA, Birnbaum RS, Lofgreen CP, Veith RC, Plymate SR. Cognitive and neuroendocrine response to transdermal estrogen in postmenopausal women with Alzheimer’s disease: results of a placebo-controlled, double-blind, pilot study. Psychoneuro-endocrinology. 1999;24:657–677. doi: 10.1016/s0306-4530(99)00020-7. [DOI] [PubMed] [Google Scholar]
- 170.Honjo H, Tanaka K, Kashiwagi T, Urabe H, Hayashi M, Hayashi K. (1995) Senile dementia – Alzheimer’s type and estrogen. Horm Metabol Res. 1995;27:204–207. doi: 10.1055/s-2007-979941. [DOI] [PubMed] [Google Scholar]
- 171.Fillit H, Weinreb H, Cholst I, Luine V, McEwen B, Amador R, Zabriskie J. Observations in a preliminary open trial of estrogen therapy for senile dementia-Alzhemier’s type. Psychoneuro-endocrinology. 1986;11:337345. doi: 10.1016/0306-4530(86)90019-3. [DOI] [PubMed] [Google Scholar]
- 172.Shumaker, et al. Estrogen plus progestin and the incidence of dementia and mild cognitive impairment in postmenopausal women: the Women’s Health Initiative memory study: a randomized controlled trial. JAMA. 2003;289:2651–2662. doi: 10.1001/jama.289.20.2651. [DOI] [PubMed] [Google Scholar]
- 173.Espeland JE, et al. Conjugated equine estrogens and global cognitive function in postmenopausal women: Women’s Health Initiative Memory Study. JAMA. 2004;291:2959–2968. doi: 10.1001/jama.291.24.2959. [DOI] [PubMed] [Google Scholar]
- 174.Wassertheil-Smoller S, et al. Effect of estrogen plus progestin on stroke in postmenopausal women: the Women’s Health Initiative: a randomized trial. JAMA. 2003;289:2673–2684. doi: 10.1001/jama.289.20.2673. [DOI] [PubMed] [Google Scholar]
- 175.Anderson GL, et al. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women’s Health Initiative randomized controlled trial. JAMA. 2004;291:1701–1712. doi: 10.1001/jama.291.14.1701. [DOI] [PubMed] [Google Scholar]
- 176.Clark JH. A critique of Women’s Health initiative studies (2002-2006) Nuclear Receptor Signaling. 2006;4:e023. doi: 10.1621/nrs.04023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Naftolin F, et al. The Women’s Health Initiative could not have detected cardioprotective effects of starting hormone therapy during the menopausal transition. Fertil Steril. 2004;81:1498–1502. doi: 10.1016/j.fertnstert.2004.02.095. [DOI] [PubMed] [Google Scholar]
- 178.Harman SM, Brinton EA, Clarkson T, Heward CB, Hecht HS, Karas RH, Judelson DR, Naftolin F. Is the WHI relevant to HRT started in the perimenopause? Endocrine. 2004;24:195–202. doi: 10.1385/ENDO:24:3:195. [DOI] [PubMed] [Google Scholar]
- 179.Gambrell RD., Jr The Women’s Health Initiative reports in perspective: facts or fallacies? Climateric. 2004;7:225–228. doi: 10.1080/13697130400001349. [DOI] [PubMed] [Google Scholar]
- 180.Wise PM, Dubal DB, Rau SW, Brown CM, Suzuki S. Are estrogens protective or risk factors in brain injury and neurodegeneration? Reevaluation after the Women’s Health Initiative. Endocrine Rev. 2005;26:308–312. doi: 10.1210/er.2004-0014. [DOI] [PubMed] [Google Scholar]
- 181.Harman SM, Naftolin F, Brinton EA, Judelson DR. Is the estrogen controversy over? Deconstructing the Women’s Health Initiative study: a critical evaluation of the evidence. Ann NY Acad Sci. 2005;1052:43–56. doi: 10.1196/annals.1347.004. [DOI] [PubMed] [Google Scholar]
- 182.Klaiber EL, Vogel W, Rako S. A critique of the Women’s Health Initiative hormone therapy study. Fertil Steril. 2005;84:1589–1601. doi: 10.1016/j.fertnstert.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 183.Moher D, Schulz K, Altman D. The CONSORT statement: revised recommendations for improving the quality of reports of parallel-group randomized trials. JAMA. 2001;285:1987–1991. doi: 10.1001/jama.285.15.1987. [DOI] [PubMed] [Google Scholar]
- 184.Brinton RD. Investigative models for determining hormone therapy-induced outcome in brain: evidence in support of a healthy cell bias of estrogen action. Ann NY Acad Sci. 2005;1052:57–74. doi: 10.1196/annals.1347.005. [DOI] [PubMed] [Google Scholar]
- 185.Chen S, Nilsen J, Brindon RD. Dose and temporal pattern of estrogen exposure determines neuroprotective outcome in hippocampal neurons: therapeutic implications. Endocrinology. 2006;147:5303–5313. doi: 10.1210/en.2006-0495. [DOI] [PubMed] [Google Scholar]
- 186.Henderson VW, Benke KS, Green RC, Cupples LA, Farrer LA. Postmenopausal hormone therapy and Alzheimer’s disease risk: interaction with age. J Neurol Neurosurg Psychiatry. 2005;76:103–105. doi: 10.1136/jnnp.2003.024927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Harman SM, Brinton EA, Cedars M, Lobo R, Manson JE, Merriam GR, Miller VM, Nafolin F, Santoro N. The Kronos Early Estrogen Prevention Study. Climateric. 2005;8:3–12. doi: 10.1080/13697130500042417. [DOI] [PubMed] [Google Scholar]
- 188.Yue X, Lu M, Lancaster T, Cao P, Hond S, Staufenbiel M, Harada N, Zhong Z, Shen Y, Li R. Brain estrogen deficiency accelerates Abeta plaque formation in an Alzheimer’s disease animal model. Proc Natl Acad Sci USA. 2005;102:19198–19203. doi: 10.1073/pnas.0505203102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Ivivonen S Corder E, Lehtovirta M, Helisalmi S, Mannermaa A, Vespalainen S, Hanninen T, Soininen H, Hiltunen M. 2004 Polymorphisms in the CYP19 gene confer increased risk for Alzheimer’s disease. Neurology. 2004;62:1170–1176. doi: 10.1212/01.wnl.0000118208.16939.60. [DOI] [PubMed] [Google Scholar]
- 190.Huang R, Poduslo SE. CYP19 haplotype increase risk for Alzheimer’s disease. J Med Genet. 2006;43:e42. doi: 10.1136/jmg.2005.039461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Ishunina TA, Fischer DF, Swaab DF. Estrogen receptor alpha and its splice variants in the hippocampus in aging and Alzheimer’s disease. Neurobiol Aging. 2006 doi: 10.1016/j.neurobiolaging.2006.07.024. epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 192.Small, et al. Cerebral metabolic and cognitive decline in persons at risk for Alzheimer’s disease. Proc Natl Acad Sci USA. 2000;97:6037–6042. doi: 10.1073/pnas.090106797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Ragson, et al. Estrogen use and brain metabolic change in postmenopausal women. Neurobiol Aging. 2005;26:229–235. doi: 10.1016/j.neurobiolaging.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 194.Eberling JL, Wu C, Tong-Turnbeaugh R, Jagust WJ. Estrogen- and tamoxifen-associated effects on brain structure and function. Neuroimage. 2004;21:364–371. doi: 10.1016/j.neuroimage.2003.08.037. [DOI] [PubMed] [Google Scholar]
- 195.Goodman Y, Bruce AJ, Cheng B, Mattson MP. Estrogens attenuate and corticosterone exacerbates excitotoxicity, oxidative injury, and amyloid-β-peptide toxicity in hippocampal neurons. J Neurochem. 1996;66:1836–1844. doi: 10.1046/j.1471-4159.1996.66051836.x. [DOI] [PubMed] [Google Scholar]
- 196.Green SG, Gridley KE, Simpkins JW. Estradiol protects against β-amyloid (25-35)-induced toxicity in SK-N-SH human neuroblastoma cells. Neurosci Lett. 1996;218:165–168. doi: 10.1016/s0304-3940(96)13148-7. [DOI] [PubMed] [Google Scholar]
- 197.Pike CJ. Estrogen modulates neuronal Bcl-XL expression and β-amyloid-induced apoptosis: relevance to Alzheimer’s disease. J Neurochem. 1999;72:1552–1563. doi: 10.1046/j.1471-4159.1999.721552.x. [DOI] [PubMed] [Google Scholar]
- 198.Brinton RD, Chen S, Montoya M, Hsieh D, Minaya J. The estrogen replacement therapy of the Women’s health Initiative promotes the cellular mechanisms of memory and neuronal survival in neurons vulnerable to Alzheimer’s disease. Maturitas. 2000;34(Suppl 2):S35–S52. doi: 10.1016/s0378-5122(00)00107-9. [DOI] [PubMed] [Google Scholar]
- 199.Du, et al. Both estrogen and raloxifene protect against beta-amyloid-induced neurotoxicity in estrogen receptor alpha-transfected PC12 cells by activation of telomerase activity via Akt cascade. J Endocrinol. 2004;183:605–615. doi: 10.1677/joe.1.05775. [DOI] [PubMed] [Google Scholar]
- 200.Behl C, Widmann M, Trapp T, Holsboer F. 17β-estradiol protects neurons from oxidative stress-induced cell death in vitro. Biochem Biophys Res Commun. 1998;216:473–482. doi: 10.1006/bbrc.1995.2647. [DOI] [PubMed] [Google Scholar]
- 201.Singer CA, Rogers KL, Strickland TM, Dorsa DM. Estrogen protects primary cortical neurons from glutamate toxicity. Neurosci Lett. 1996;212:13–16. doi: 10.1016/0304-3940(96)12760-9. [DOI] [PubMed] [Google Scholar]
- 202.Weaver CE, Jr, Park-Chung M, Gibbs TT, Farb DH. 17β-Estradiol protects against NMDA-induced excitotoxicity by direct inhibition of NMDA receptors. Brain Res. 1997;761:338–341. doi: 10.1016/s0006-8993(97)00449-6. [DOI] [PubMed] [Google Scholar]
- 203.Alvarez-de-la-Rosa M, Silva I, Nilsen J, Perez MM, Garcia-Segura LM, Avila J, Naftolin F. Estradiol prevents neural tau hyperphosphorylation characteristic of Alzheimer’s disease. Ann NY Acad Sci. 2005;1052:210–224. doi: 10.1196/annals.1347.016. [DOI] [PubMed] [Google Scholar]
- 204.Znamensky V, Akama KT, McEwen BS, Milner TA. Estrogen levels regulate the subcellular distribution of phosphorylated Akt in hippocampal CA1 dendrites. J Neurosci. 2003;23:2340–2347. doi: 10.1523/JNEUROSCI.23-06-02340.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Zhang L, Rubinow DR, Xaing G, Li BS, Chang YH, Maric D, Barker JL, Ma W. Estrogen protects against beta-amyloid-induced neurotoxicity in rat hippocampal neurons by activation of Akt. Neuroreport. 2001;12:1919–1923. doi: 10.1097/00001756-200107030-00030. [DOI] [PubMed] [Google Scholar]
- 206.Yaffe K, Krueger K, Cummings SR, Blackwell T, Henderson VW, Sarkar S, Ensrud K, Grady D. Effect of raloxifene on prevention of dementia and cognitive impairment in older women: the multiple outcomes of raloxifene evaluation (MORE) randomized trial. Am J Psychiatry. 2005;162:683–690. doi: 10.1176/appi.ajp.162.4.683. [DOI] [PubMed] [Google Scholar]
- 207.Sherwin BB. Estrogen and cognitive function in women. Endocr Rev. 2003;24:133–151. doi: 10.1210/er.2001-0016. [DOI] [PubMed] [Google Scholar]
- 208.Halpern DF. Sex differences in cognitive abilities. 2. Hillsdale NJ: Lawrence Erlbaum Associates; 1992. [Google Scholar]
- 209.Kimura D, Clarke PG. Women’s advantage on verbal memory is not restricted to concrete words. Psychol Rep. 2002;91:1137–1142. doi: 10.2466/pr0.2002.91.3f.1137. [DOI] [PubMed] [Google Scholar]
- 210.Speck O, Ernst T, Braun J, Koch C, Miller E, Chang I. Gender differences in the functional organization of the brain for working memory. Neuroreport. 2000;11:2581–2585. doi: 10.1097/00001756-200008030-00046. [DOI] [PubMed] [Google Scholar]
- 211.Rosenberg L, Park S. Verbal and spatial functions across the menstrual cycle in healthy young women. Psychoneuroendocrinology. 2002;27:835–841. doi: 10.1016/s0306-4530(01)00083-x. [DOI] [PubMed] [Google Scholar]
- 212.Broverman DM, Vogel W, Klaiber EL, Majcher D, Shea D, Paul V. Changes in cognitive task performance across the menstrual cycle. J Comp Physiol Psychol. 1981;95:646–654. doi: 10.1037/h0077796. [DOI] [PubMed] [Google Scholar]
- 213.Hampson E. Variations in sex-related cognitive abilities across the menstrual cycle. Brain Cogn. 1990;14:26–43. doi: 10.1016/0278-2626(90)90058-v. [DOI] [PubMed] [Google Scholar]
- 214.Hampson E, Kimura D. Reciprocal effects of hormonal fluctuations on human motor and perceptual-spatial skills. Behav Neurosci. 1988;102:456–459. doi: 10.1037//0735-7044.102.3.456. [DOI] [PubMed] [Google Scholar]
- 215.Zec RF, Trivendi MA. The effects of estrogen replacement therapy on neuropsychological function in postmenopausal women with and without dementia: a critical and theoretical review. Neuropsychol Rev. 2002;12:65–109. doi: 10.1023/a:1016880127635. [DOI] [PubMed] [Google Scholar]
- 216.Matthews K, Cauley J, Yaffe K, Zmuda JM. Estrogen replacement therapy and cognitive decline in older community women. J Am Geriatr Soc. 1999;47:518–523. doi: 10.1111/j.1532-5415.1999.tb02563.x. [DOI] [PubMed] [Google Scholar]
- 217.Rapp PR, Morrison JH, Roberts JA. Cyclic estrogen replacement improves cognitive function in aged ovariectomized rhesus monkeys. J Neurosci. 2003;23:5708–5714. doi: 10.1523/JNEUROSCI.23-13-05708.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Daniel JM, Fader AJ, Spencer AL, Dohanich GP. Estrogen enhances performance of female rats during acquisition of a radial arm maze. Horm Behav. 1997;32:217–225. doi: 10.1006/hbeh.1997.1433. [DOI] [PubMed] [Google Scholar]
- 219.Fader AJ, Johnson PE, Dohanich GP. Estrogen improves working but not reference memory and prevents amnestic effects of scopolamine of a radial-arm maze. Pharmacol Biochem Behav. 1999;62:711–717. doi: 10.1016/s0091-3057(98)00219-6. [DOI] [PubMed] [Google Scholar]
- 220.Luine VN, Richards ST, Wu VY, Beck KD. Estradiol enhances learning and memory in a spatial memory task and effects levels of monoaminergic transmitters. Horm Behav. 1998;34:149–162. doi: 10.1006/hbeh.1998.1473. [DOI] [PubMed] [Google Scholar]
- 221.O’Neal MF, Means LW, Poole MC, Hamm RJ. Estrogen affects performance of ovariectomized rats in a two-choice water escape working memory task. Psychoneuroendocrinology. 1996;21:51–65. doi: 10.1016/0306-4530(95)00032-1. [DOI] [PubMed] [Google Scholar]
- 222.Sandstrom NJ, Williams CL. Memory retention is modulated by acute estrogen and progesterone replacement. Behav Neurosci. 2001;115:384–393. [PubMed] [Google Scholar]
- 223.Bimonte-Nelson HA, Francis KR, Umphlet CD, Granholm AC. Progesterone reverses spatial memory enhancements initiated by tonic and cyclic oestrogen therapy in middle-aged ovariectomized female rats. Eur J Neurosci. 2006;24:229–242. doi: 10.1111/j.1460-9568.2006.04867.x. [DOI] [PubMed] [Google Scholar]
- 224.Xu X, Zhang Z. Effects of estradiol benzoate on learning-memory behavior and synaptic structure in ovariectomized mice. Life Sci. 2006;79:1553–1560. doi: 10.1016/j.lfs.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 225.Rhodes ME, Frye CA. ERbeta-selective SERMs produce mnemonic-enhancing effects in the inhibitory avoidance and water maze tasks. Neurobiol Learn Mem. 2006;85:183–190. doi: 10.1016/j.nlm.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 226.Dohanich GP, Fader AJ, Javorsky DJ. Estrogen and estrogen-progesterone treatments counteract the effect of scopolamine on reinforced T-maze alternation in female rats. Behav Neurosci. 1994;108:988–992. doi: 10.1037//0735-7044.108.5.988. [DOI] [PubMed] [Google Scholar]
- 227.Fader AJ, Hendriecson AW, Dohanich GP. Estrogen improves performance of reinforced T-maze alternation and prevents the amnestic effect of scopolamine administered systemically or intrahippocampally. Neurobiol Learn Mem. 1998;69:225–240. doi: 10.1006/nlme.1998.3820. [DOI] [PubMed] [Google Scholar]
- 228.Singh M, Meyer EM, Millard WJ, Simpkins JW. Ovarian steroid deprivation results in a reversible learning impairment and compromised cholinergic function in female Sprague Dawley rats. Brain Res. 1994;644:305–312. doi: 10.1016/0006-8993(94)91694-2. [DOI] [PubMed] [Google Scholar]
- 229.Daniel JM, Dohanich GP. Acetylcholine mediates the estrogen-induced increase in NMDA receptor binding in CA1 of the hippocampus and the associated improvement in working memory. J Neurosci. 2001;21:6949–6956. doi: 10.1523/JNEUROSCI.21-17-06949.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Lacreuse A, Wilson ME, Herndon JG. Estradiol, but not raloxifene, improves aspects of spatial working memory in aged ovariectomized rhesus monkeys. Neurobiol Aging. 2002;23:589–600. doi: 10.1016/s0197-4580(02)00002-7. [DOI] [PubMed] [Google Scholar]
- 231.Markowska AL, Savonenko AV. Effectiveness of estrogen replacement in restoration of cognitive function after long-term estrogen withdrawal in aging rats. J Neurosci. 2002;22:10985–10995. doi: 10.1523/JNEUROSCI.22-24-10985.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Daniel JM, Huist JL, Berbling JL. Estradiol replacement enhances working memory in middle-aged rats when initiated immediately after ovariectomy but not after a long-term period of ovarian deprivation. Endocrinology. 2006;147:607–614. doi: 10.1210/en.2005-0998. [DOI] [PubMed] [Google Scholar]
- 233.Terasawa E, Timiras PS. Electrical activity during the estrous cycle of the rat: cyclic changes in limbic structures. Endocrinology. 1968;83:201–216. doi: 10.1210/endo-83-2-207. [DOI] [PubMed] [Google Scholar]
- 234.Woolley CS, Weiland NG, McEwen BS, Schwartzkroin PA. Estradiol increases the sensitivity of hippocampal CA1 pyramidal cells to NMDA receptor-mediated synaptic input: correlation with dendritic spine density. J Neurosci. 1997;17:1848–1859. doi: 10.1523/JNEUROSCI.17-05-01848.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Woolley CS, Gould E, Frankfurt M, McEwen BS. Naturally occurring fluctuation in dendritic spine density on adult hippocampal pyramidal neurons. J Neurosci. 190;10:4035–4039. doi: 10.1523/JNEUROSCI.10-12-04035.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Gould E, Woolley CS, Frankfurt M, McEwen BS. Gonadal steroids regulate dendritic spine density in hippocampal pyramidal cells in adulthood. J Neurosci. 1990;10:1286–1291. doi: 10.1523/JNEUROSCI.10-04-01286.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Woolley CS, McEwen BS. Estrogen mediates fluctuation in hippocampal synapse density during the estrous cycle in the adult rat. J Neurosci. 1992;12:2549–2554. doi: 10.1523/JNEUROSCI.12-07-02549.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Adams MM, Shah Ra, Janssen WGM, Morrison JH. Different modes of hippocampal plasticity in response to estrogen in young and aged female rats. Proc Natl Acad Sci USA. 2001;98:8071–8076. doi: 10.1073/pnas.141215898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Murphy DD, Segal M. Regulation of dendritic spine density in cultured rat hippocampal neurons by steroid hormones. J Neurosci. 1996;16:4059–4068. doi: 10.1523/JNEUROSCI.16-13-04059.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Akama KT, McEwen BS. Estrogen stimulates postsynaptic density-95 rapid protein synthesis via the Akt/protein kinase B pathway. J Neurosci. 2003;23:2333–2339. doi: 10.1523/JNEUROSCI.23-06-02333.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Hao J, Janssen WGM, Tang W, Roberts JA, McKay H, Lasley B, Allen PB, Greengard P, Rapp PR, Kordower JH, Hof PR, Morrison JH. Estrogen increases the number of spinophilin immunoreactive spines in the hippocampus of young and aged female rhesus monkeys. J Comp Neurol. 2003;465:540–550. doi: 10.1002/cne.10837. [DOI] [PubMed] [Google Scholar]
- 242.Choi J, Romeo RD, Brake WG, Bethea CL, Rosenwaks Z, McEwen BS. Estradiol increases pre- and post-synaptic proteins in the CA1 region of the hippocampus in female rhesus macaques (Macaca mulatta) Endocrinology. 2003;144:4734–4738. doi: 10.1210/en.2003-0216. [DOI] [PubMed] [Google Scholar]
- 243.Leranth C, Petnehazy O, MacLusky NJ. Gonadal hormones affect spine synaptic density in the CA1 hippocampal subfield of male rats. J Neurosci. 2003;23:1588–1592. doi: 10.1523/JNEUROSCI.23-05-01588.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Lewis C, McEwen BS, Frankfurt M. Estrogen-induction of dendritic spines in ventromedial hypothalamus and hippocampus: effects of neonatal aromatase blockade and adult GDX. Dev Brain Res. 1995;87:91–95. doi: 10.1016/0165-3806(95)00052-f. [DOI] [PubMed] [Google Scholar]
- 245.Li CJ, Brake WG, Romeo RD, Dunlop JC, Gordon M, Buzescu R, Magarinos AM, Allen PB, Greengard P, Luine V, McEwen BS. Estrogen alters hippocampal dendritic spine shape and enhances synaptic protein immunoreactivity and spatial memory in female mice. Proc Natl Acad Sci USA. 2004;101:2185–2190. doi: 10.1073/pnas.0307313101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Zhao L, Chen S, Wang J, Brinton RD. 17Beta-estradiol induces Ca2+ influx, dendritic and nuclear Ca2+ rise and subsequent cyclic AMP response element-binding protein activation in hippocampal neurons: a potential initiation mechanism for estrogen neurotrophism . Neuroscience. 2005;132:299–311. doi: 10.1016/j.neuroscience.2004.11.054. [DOI] [PubMed] [Google Scholar]
- 247.Cordoba Montoya DA, Carrer HF. Estrogen facilitates induction of long term potentiation in the hippocampus of awake rats. Brain Res. 1997;778:430–438. doi: 10.1016/s0006-8993(97)01206-7. [DOI] [PubMed] [Google Scholar]
- 248.Foy MR, Xu J, Xie X, Brinton RD, Thompson RF, Berger TW. 17Beta-Estradiol enhances NMDA receptor-mediated EPSPs and long term potentiation. J Neurophysiol. 1999;81:925–929. doi: 10.1152/jn.1999.81.2.925. [DOI] [PubMed] [Google Scholar]
- 249.Smith CC, McMahon LL. Estrogen-induced increase in the magnitude of long-term potentiation occurs only when the ratio of NMDA transmission to AMPA transmission is increased. J Neurosci. 2005;25:7780–7791. doi: 10.1523/JNEUROSCI.0762-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Smith CC, McMahon LL. Estrogen-induced increase in the magnitude of long-term potentiation is prevented by blocking NR2B-containing receptors. J Neurosci. 2006;26:8517–8522. doi: 10.1523/JNEUROSCI.5279-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Good M, Day M, Muir JL. Cyclical changes in endogenous levels of oestrogen modulate the induction of LTD and LTP in the hippocampal CA1 region. Eur J Neurosci. 1999;11:4476–4480. doi: 10.1046/j.1460-9568.1999.00920.x. [DOI] [PubMed] [Google Scholar]
- 252.Warren SG, Humphreys AG, Juraska JM, Greenough WT. LTP varies across the estrous cycle: enhanced synaptic plasticity in proestrus rats. Brain Res. 1995;703:26–30. doi: 10.1016/0006-8993(95)01059-9. [DOI] [PubMed] [Google Scholar]
- 253.Leuner B, Shors TJ. New spines, new memories. Mol Neurobiol. 2004;29:117–130. doi: 10.1385/MN:29:2:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Segal M. Dendritic spines and long-term plasticity. Nature Rev Neurosci. 2005;6:277–284. doi: 10.1038/nrn1649. [DOI] [PubMed] [Google Scholar]
- 255.Barkai E. Dynamics of learning-indced cellular modifications in the cortex. Biol Cybern. 2005;92:360–366. doi: 10.1007/s00422-005-0564-0. [DOI] [PubMed] [Google Scholar]
- 256.Silva AJ. Molecular and cellular cognitive studies of the role of synaptic plasticity in memory. J Neurobiol. 2003;54:224–237. doi: 10.1002/neu.10169. [DOI] [PubMed] [Google Scholar]
- 257.Sweatt JD. Mitogen-activated protein kinases in synaptic plasticity and memory. Curr Opin Neurobiol. 2004;14:311–317. doi: 10.1016/j.conb.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 258.Leuner B, Falduto J, Shors TJ. Associative memory formation increases the observation of dendritic spines in the hippocampus. J Neurosci. 2003;23:659–665. doi: 10.1523/JNEUROSCI.23-02-00659.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Wallace M, Luine V, Arellanos A, Frankfurt M. Ovariectomized rats show decreased recognition memory and spine density in the hippocampus and frontal cortex. Brain Res. 2006;1126:176–182. doi: 10.1016/j.brainres.2006.07.064. [DOI] [PubMed] [Google Scholar]
- 260.Frick KM, Fernandez SM, Bulinski SC. Estrogen replacement improves spatial reference memory and increases hippocampal synaptophysin in aged female mice. Neuroscience. 2002;115:547–558. doi: 10.1016/s0306-4522(02)00377-9. [DOI] [PubMed] [Google Scholar]
- 261.Berman KF, Schmidt PJ, Rubinow DR, Danaceau MA, Van Horn JD, Esposito G, Ostrem JL, Weinberger DR. Modulation of cognition-specific cortical activity by gonadal steroids: a positronemission tomography study in women. Proc Natl Acad Sci USA. 1997;94:8836–8841. doi: 10.1073/pnas.94.16.8836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Shaywitz SE, Shaywitz BA, Pugh KR, Fulbright RK, Skudlarski P, Mencl WE, Constable RT, Naftolin F, Palter SF, Marchione KE, Katz L, Shankweiler DP, Fletcher JM, Lacadie C, Keltz M, Gore JC. Effect of estrogen on brain activation patterns in postmenopausal women during working memory tasks. JAMA. 199;281:1197–1202. doi: 10.1001/jama.281.13.1197. [DOI] [PubMed] [Google Scholar]
- 263.Joffe H, Hall JE, Gruber S, Sarmirnto IA, Cohen LS Yurgelun-Todd D, Martin KA. Estrogen therapy selectively enhances prefrontal cognitive processes: a randomized, double-blind, placebo-controlled study with functional magnetic resonance imaging in perimenopausal and recently postmenopausal women. Menopause. 2006;13:411–422. doi: 10.1097/01.gme.0000189618.48774.7b. [DOI] [PubMed] [Google Scholar]
- 264.Resnick SM, Maki PM, Golski S, Kraut MA, Zonderman AB. Effects of estrogen replacement therapy on PET Cerebral blood flow and neuropsychological performance. Hormones Behav. 1998;34:171–182. doi: 10.1006/hbeh.1998.1476. [DOI] [PubMed] [Google Scholar]
- 265.Dietrich T, Krings T, Neulen J, Willmes K, Erberich S, Thron A, Sturm W. Effects of blood estrogen level on cortical activation patterns during cognitive activation as measured by functional MRI. Neuroimage. 2001;13:425–432. doi: 10.1006/nimg.2001.0703. [DOI] [PubMed] [Google Scholar]
- 266.Keenan PA, Ezzat WH, Ginsburg K, Moore GJ. Prefrontal cortex as the site of estrogen’s effect on cognition. Psychoneuroendocrinology. 2001;26:577–590. doi: 10.1016/s0306-4530(01)00013-0. [DOI] [PubMed] [Google Scholar]
- 26.Moskovitch M. Memory and working with memory: Evaluation of a component model and comparisons with other models. In: Schacter D, Tulving E, editors. Memory Systems. MIT Press; Cambridge: 1994. [Google Scholar]
- 268.Baddely AD. Working Memory. Oxford University Press; New York: 1986. [Google Scholar]
- 269.Miller BL, Cummings JL. The Frontal Lobes: Functions and Disorders. Guilford Press; New York: 1999. [Google Scholar]
- 270.Duff SJ, Hampson E. A sex difference on a novel spatial working memory task in humans. Brain Cognition. 2001;47:470–493. doi: 10.1006/brcg.2001.1326. [DOI] [PubMed] [Google Scholar]
- 271.Duff SJ, Hampson E. A beneficial effect of estrogen on working memory in post-menopausal women taking hormone replacement. Horm Behav. 2000;38:262–276. doi: 10.1006/hbeh.2000.1625. [DOI] [PubMed] [Google Scholar]
- 272.Smith YR, Giordani B, Lajuness-O’Neill R, Zubieta JK. Long-term estrogen replacement is associated with improved nonverbal memory and attentional measures in postmenopausal women. Fertil Steril. 2001;76:1101–1107. doi: 10.1016/s0015-0282(01)02902-8. [DOI] [PubMed] [Google Scholar]
- 273.Dumas J, Hancur-Bucci C, Naylor M, Sites C, Newhouse P. Estrogen treatment effects on anticholinergic-induced cognitive dysfunction in normal postmenopausal women. Neuropsychopharmacology. 2006;31:2065–2078. doi: 10.1038/sj.npp.1301042. [DOI] [PubMed] [Google Scholar]
- 274.Rosenberg L, Park S. Verbal and spatial functions across the menstrual cycle in healthy young women. Psychoneuroendocrinology. 2002;27:835–841. doi: 10.1016/s0306-4530(01)00083-x. [DOI] [PubMed] [Google Scholar]
- 275.Grigorova M, Sherwin BB. No difference in performance on test of working memory and executive functioning between healthy elderly postmenopausal women using or not using hormone therapy. Climateric. 2006;9:181–194. doi: 10.1080/13697130600727107. [DOI] [PubMed] [Google Scholar]
- 276.File SE, Hartley DE, Elsabagh S, Duffy R, Wiseman H. Cognitive improvements after 6 weeks of soy supplements in postmenopausal women is limited to frontal lobe function. Menopause. 2005;12:193–201. doi: 10.1097/00042192-200512020-00014. [DOI] [PubMed] [Google Scholar]
- 277.Wiltgen BJ, Brown RA, Talton LE, Silva AJ. New circuits for old memories: the role of the neocortex in consolidation. Neuron. 2004;44:101–108. doi: 10.1016/j.neuron.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 278.Erickson KI, Colcombe SJ, Raz N, Korol DL, Scalf P, Webb A, Cohen NJ, McAuley E, Kramer AF. Selective sparing of brain tissue in postmenopausal women receiving hormone replacement therapy. Neurobiol Agng. 2005;26:1205–1213. doi: 10.1016/j.neurobiolaging.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 279.Tang Y, et al. Estrogen replacement increases spinophilin-immunoreactive spine number in the prefrontal cortex of female rhesus monkeys. Cereb Cortex. 2004;14:215–223. doi: 10.1093/cercor/bhg121. [DOI] [PubMed] [Google Scholar]
- 280.Hao, et al. Estrogen alters spine number and morphology in prefrontal cortex of aged female rhesus monkeys. J Neurosci. 2006;26:2571–2578. doi: 10.1523/JNEUROSCI.3440-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Khan MM, Dhandapani KM, Wakade C, De Sevilla LM, Mahesh VB, Brann DW. Regulation of synaptic proteins and spine density in the prefrontal and somatosensory cortex by 17β-estradiol: implications for synaptic remodeling in the brain. 2006; Neuroscience Meeting Planner; Washington, DC. 2005. Program No. 405.12. Online. [Google Scholar]
- 282.Shughrue PJ, Lane MV, Merchenthaler I. Comparative distribution of estrogen receptor-alpha and -beta mRNA in the rat central nervous system. J Comp Neurol. 1997;388:507–525. doi: 10.1002/(sici)1096-9861(19971201)388:4<507::aid-cne1>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 283.Mehra RD, Sharma K, Nyakas C, Vij U. Estrogen receptor alpha and beta immunoreactive neurons in normal and aged female rat hippocampus: a qualitative and quantitative study. Brain Res. 2005;1056:22–35. doi: 10.1016/j.brainres.2005.06.073. [DOI] [PubMed] [Google Scholar]
- 284.Azcoitia I, Sierra A, Garcia-Segura LM. Localization of estrogen receptor beta-immunoreactivity in astrocytes of the adult rat brain. Glia. 1999;26:260–267. [PubMed] [Google Scholar]
- 285.Wang J, Cheng C, Zhou J, Smith A, Weickert CS, Perlman WR, Becker KG, Powell D, Bondy CA. Estradiol alters transcription factor gene expression in primate prefrontal cortex. J Neurosci Res. 2004;76:306–314. doi: 10.1002/jnr.20076. [DOI] [PubMed] [Google Scholar]
- 286.Osterlund MK, Gustafsson J, Keller E, Hurd YL. Estrogen receptor β (ERβ) messenger ribonucleic acid (mRNA) expression within the human forebrain: distinct distribution pattern to ERα mRNA. J Clin Endo Metab. 2000;85:3840–3846. doi: 10.1210/jcem.85.10.6913. [DOI] [PubMed] [Google Scholar]
- 287.Weiland NG, Orikasa C, Hayashi S, McEwen BS. Distribution and hormone regulation of estrogen receptor immunoreactive cells in the hippocampus of male and female rats. J Comp Neurol. 1997;388:603–612. doi: 10.1002/(sici)1096-9861(19971201)388:4<603::aid-cne8>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 288.Adams MM, Fink SE, Shah RA, Janssen WG, Hayashi S, Milner TA, McEwen BS, Morrison JH. Estrogen and aging affect the subcellular distribution of estrogen receptor-alpha in the hippocampus of female rats. J Neurosci. 2002;22:3608–3614. doi: 10.1523/JNEUROSCI.22-09-03608.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.McEwen BS, Akama K, Alves S, Brake WG, Bulloch K, Lee S, Li C, Yuen G, Milner TA. Tracking the estrogen receptor in neurons: implications for estrogen-induced synapse formation. Proc Natl Acad Sci USA. 2001;98:7093–7100. doi: 10.1073/pnas.121146898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Gundiah C, Kohama SG, Mirkes SJ, Garyfallou VT, Urbanski HF, Bethea CL. Distribution of estrogen receptor beta (ERbeta) mRNA in hypothalamus, midbrain, and temporal lobes of sprayed macque: continued expression with hormone replacement. Brain Res Mol Brain Res. 2000;76:191–204. doi: 10.1016/s0006-8993(99)02475-0. [DOI] [PubMed] [Google Scholar]
- 291.Register TC, Shively CA, Lewis CE. Expression of estrogen receptor alpha and beta transcription in female monkey hippocampus and hypothalamus. Brain Res. 1998;788:320–322. doi: 10.1016/s0006-8993(98)00036-5. [DOI] [PubMed] [Google Scholar]
- 292.Milner TA, Ayoola K, Drake CT, Herrick SP, Tabori NE, McEwen BS, Warrior S, Alves SE. Ultrastructural localization of estrogen receptor beta immunoreactivity in the rat hippocampal formation. J Comp Neurol. 2005;491:81–95. doi: 10.1002/cne.20724. [DOI] [PubMed] [Google Scholar]
- 293.Kalita K, Szymczak S, Kaczmarek L. Non-nuclear estrogen receptor beta and alpha in the hippocampus of male and female rats. Hippocampus. 2005;15:404–412. doi: 10.1002/hipo.20066. [DOI] [PubMed] [Google Scholar]
- 294.Xu Y, Traystman RJ, Hurn PD, Wang MM. Neurite-localized estrogen receptor-alpha mediates rapid signaling by estrogen. J Neurosci Res. 2003;74:1–11. doi: 10.1002/jnr.10725. [DOI] [PubMed] [Google Scholar]
- 295.Murakami G, et al. Comparison between basal and apical dendritic spines in estrogen-induced rapid spinogenesis of CA1 principal neurons in the adult hippocampus. Biochem Biophys Res Commun. 2006;351:553–558. doi: 10.1016/j.bbrc.2006.10.066. [DOI] [PubMed] [Google Scholar]
- 296.McEwen BS, Tanapat P, Weiland NG. Inhibition of dendritic spine induction on hippocampal CA1 pyramidal neurons by a nonsteroidal estrogen antagonist in female rats. Endocrinology. 1999;140:1044–1047. doi: 10.1210/endo.140.3.6570. [DOI] [PubMed] [Google Scholar]
- 297.Fugger HN, Foster TC, Gustafson J, Rissman EF. Novel effects of estradiol and estrogen receptor α and β on cognitive function. Brain Res. 2000;883:258–264. doi: 10.1016/s0006-8993(00)02993-0. [DOI] [PubMed] [Google Scholar]
- 298.Day M, Sung A, Logue S, Bowlby M, Arias R. Beta estrogen receptor knockout (BERKO) mice present attenuated hippocampal CA1 long-term potentiation and related memory deficits in contextual fear conditioning. Behav Brain Res. 2005;164:128–131. doi: 10.1016/j.bbr.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 299.Weiland NG. Estradiol selectively regulates agonist binding sites on the N-methyl-D-aspartate receptor complex in the CA1 region of the hippocampus. Endocrinology. 1992;131:662–668. doi: 10.1210/endo.131.2.1353442. [DOI] [PubMed] [Google Scholar]
- 300.Cyr M, Thibault C, Morissette M, Landry M, Di Paolo T. Estrogen-like activity of tamoxifen and raloxifene on NMDA receptor binding and expression of its subunits in the brain. Neuropsychopharmacology. 2001;25:242–257. doi: 10.1016/S0893-133X(01)00233-0. [DOI] [PubMed] [Google Scholar]
- 301.Romeo RD, McCarthy JB, Wang A, Milner TA, McEwen BS. Sex differences in hippocampal estradiol-induced N-methyl-D-aspartic acid binding and ultrastructural localization of estrogen receptor-alpha. Neuroendocrinology. 2005;81:391–399. doi: 10.1159/000089557. [DOI] [PubMed] [Google Scholar]
- 302.Gazzaley AH, Weiland NG, McEwen BS, Morrison JH. Differential regulation of NMDAR1 mRNA and protein by estradiol in the rat hippocampus. J Neurosci. 1996;16:6830–3838. doi: 10.1523/JNEUROSCI.16-21-06830.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Adams MM, Fink SE, Janssen WG, Shah RA, Morrison JH. Estrogen modulates synaptic N-methyl-D-aspartate receptor subunit distribution in the aged hippocampus. J Comp Neurol. 2004;474:419–426. doi: 10.1002/cne.20148. [DOI] [PubMed] [Google Scholar]
- 304.Pozzo-Miller LD, Inoue T, Murphy DD. Estradiol increases spine density and NMDA-dependent Ca2+ transients in spines of CA1 pyramidal neurons from hippocampal slices. J Neurophysiol. 1999;81:1404–1411. doi: 10.1152/jn.1999.81.3.1404. [DOI] [PubMed] [Google Scholar]
- 305.Woolley CS, McEwen BS. Estradiol regulates hippocampal dendritic spine density via an N-methyl-D-aspartate receptor-dependent mechanism. J Neurosci. 1994;14:7680–7687. doi: 10.1523/JNEUROSCI.14-12-07680.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Rudick CN, Woolley CS. Estrogen regulates functional inhibition of hippocampal CA1 pyramidal cells in the adult female rat. J Neurosci. 2001;21:6532–6543. doi: 10.1523/JNEUROSCI.21-17-06532.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Murphy DD, Cole NB, Greenberger V, Segal M. Estradiol increases dendritic spine density by reducing GABA neurotransmission in hippocampal neurons. J Neurosci. 1998;18:2550–2559. doi: 10.1523/JNEUROSCI.18-07-02550.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Redmond L, Kashani AH, Ghosh A. Calcium regulation of dendritic growth via CaM kinase IV and CREB-Mediated transcription. Neuron. 2002;34:999–1010. doi: 10.1016/s0896-6273(02)00737-7. [DOI] [PubMed] [Google Scholar]
- 309.Miyamoto E. Molecular mechanism of neuronal plasticity: induction and maintenance of long-term potentiation in the hippocampus. J Pharmacol Sci. 2006;100:433–442. doi: 10.1254/jphs.cpj06007x. [DOI] [PubMed] [Google Scholar]
- 310.Lee SJ, Campomanes CR, Sikat PT, Greenfield AT, Allen PB, McEwen BS. Estrogen induces phosphorylation of cyclic AMP response element binding (pCREB) in primary hippocampal cells in a time-dependent manner. Neuroscience. 2004;124:549–560. doi: 10.1016/j.neuroscience.2003.11.035. [DOI] [PubMed] [Google Scholar]
- 311.Segal M, Murphy DD. CREB activation mediates plasticity in cultured hippocampal neurons. Neural Plast. 1998;6:1–7. doi: 10.1155/NP.1998.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Murphy DD, Segal M. morphological plasticity of dendritic spines in central neurons is mediated by activation of cAMP response element binding protein. Proc Natl Acad Sci USA. 1997;94:1482–1487. doi: 10.1073/pnas.94.4.1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Sawai T, Bernier F, Fukushima T, Hashimoto T, Ogura H, Nishizawa Y. Estrogen induces a rapid increase of calcium-calmodulin-dependent protein kinase II activity in the hippocampus. Brain Res. 2002;950:308–311. doi: 10.1016/s0006-8993(02)03186-4. [DOI] [PubMed] [Google Scholar]
- 314.Mannella P, Brinton RD. Estrogen receptor protein interaction with phosphatidylinositol 3-kinase leads to activation of phosphorylated Akt and extracellular signal-regulated kinase 1/2 in the same population of cortical neurons: a unified mechanism of estrogen action. J Neurosci. 2006;26:9439–9447. doi: 10.1523/JNEUROSCI.1443-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Kelleher RJ, Govindarajan A, Kang H, Tonegawa S. Translational control by MAPK signaling in long-term synaptic plasticity and memory. Cell. 2004;116:467–479. doi: 10.1016/s0092-8674(04)00115-1. [DOI] [PubMed] [Google Scholar]
- 316.Klann E, Dever TE. Biochemical mechanisms for translational regulation in synaptic plasticity. Nature Rev Neurosci. 2004;5:931–942. doi: 10.1038/nrn1557. [DOI] [PubMed] [Google Scholar]
- 317.Hojo Y, et al. Adult male rat hippocampus synthesizes estradiol from pregnenolone by cytochrome P45017alpha and P450 aromatase localized in neurons. Proc Natl Acad Sci USA. 2004;101:865–870. doi: 10.1073/pnas.2630225100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Kretz O, Fester L, Wehrenberg U, Zhou L, Brauckmann S, Zhao S, Prange-Kiel J, Naumann T, Jarry H, Frotscher M, Rune GM. Hippocampal synapses depend on hippocampal estrogen synthesis. J Neurosci. 2004;24:5913–5921. doi: 10.1523/JNEUROSCI.5186-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Murakami G, et al. Role of cytochrome p450 in synaptocrinology: endogenous estrogen synthesis in the brain hippocampus. Drug Metab Rev. 2006;38:353–369. doi: 10.1080/03602530600724068. [DOI] [PubMed] [Google Scholar]
- 320.Boon WC, Diepstraten J, van der Burg J, Jones ME, Simpson ER, van den Buuse M. Hippocampal NMDA receptor subunit expression and watermaze learning in estrogen deficient female mice. Brain Res Mol Brain Res. 2005;140:127–132. doi: 10.1016/j.molbrainres.2005.07.004. [DOI] [PubMed] [Google Scholar]